Back to Journals » Journal of Inflammation Research » Volume 9
The macrophages in rheumatic diseases
Authors Laria A, Lurati AM, marrazza M, mazzocchi D, re K, scarpellini M
Received 6 February 2015
Accepted for publication 12 May 2015
Published 9 February 2016 Volume 2016:9 Pages 1—11
DOI https://doi.org/10.2147/JIR.S82320
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Antonella Laria, Alfredomaria Lurati , Mariagrazia Marrazza , Daniela Mazzocchi, Katia Angela Re, Magda Scarpellini
Rheumatology Unit, Fornaroli Hospital, Magenta, Italy
Abstract: Macrophages belong to the innate immune system giving us protection against pathogens. However it is known that they are also involved in rheumatic diseases. Activated macrophages have two different phenotypes related to different stimuli: M1 (classically activated) and M2 (alternatively activated). M1 macrophages release high levels of pro-inflammatory cytokines, reactive nitrogen and oxygen intermediates killing microorganisms and tumor cells; while M2 macrophages are involved in resolution of inflammation through phagocytosis of apoptotic neutrophils, reduced production of pro-inflammatory cytokines, and increased synthesis of mediators important in tissue remodeling, angiogenesis, and wound repair. The role of macrophages in the different rheumatic diseases is different according to their M1/M2 macrophages phenotype.
Keywords: macrophage, rheumatic diseases
Introduction
Macrophages are cells of the innate immune system involved in immunological response against pathogens but also in autoimmune disorders such as rheumatic diseases. They play the role of antigen-presenting cells and they release many inflammatory cytokines and chemokines that contribute to cartilage, bone, and tissue destruction. We want to present several aspects of macrophage function in autoimmune diseases: the development of the two monocyte subsets and of the two macrophage phenotypes. Macrophages are the main cells in most tissues. Their numbers increase massively in inflammation, in autoimmunity diseases, and in cancers. Their progenitor cell is CD34+ in the bone marrow, that differentiates into monoblasts and then into pro-monocytes; finally into monocytes (M0), that are released into the blood. Monocytes circulate for 1–3 days in the blood and then they enter tissues to differentiate into mature resident macrophages;1 for example they become Kupffer cells in the liver, microglial cells in the brain, and Langerhans cells in the skin. All these resident macrophages, although different in some aspects, have capacity to influence normal cell turnover and tissue remodeling, to counteract microbial infections, and to facilitate repair in sites of injury.2 The presence or absence of the Fc receptor CD16 identifies two populations of human monocytes, as demonstrated by Passlick et al.3 It seems that CD16+ monocytes do not express the chemokine receptor CCR2 according to Weber et al4 and these cells have an enhanced capacity for trans-endothelial migration.5 It is known that there are two phenotypically and functionally distinct monocyte subsets: inflammatory and resident phenotype. The first one is characterized by CCR2+CD62L+ or CD14lowCD16+ phenotype, while the last one is characterized by CCR2-CD62L- or CD14+CD16-. The inflammatory phenotype is preferentially recruited to inflammatory lesions, while the resident one is hypothesized to be a source of tissue resident macrophage and dendritic cells (Figure 1).6
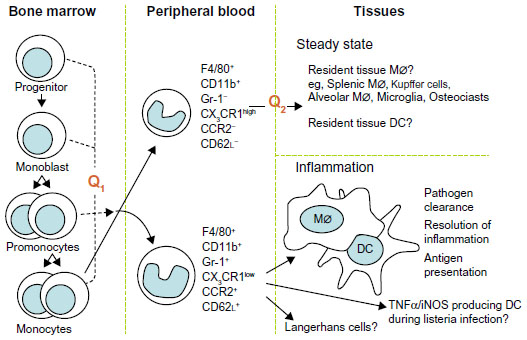 | Figure 1 Schematic representation of the development of the monocyte subsets. |
In the M1/M2 model described by Badylak and Gilbert, CD68 is a specific macrophage surface marker; CD80 and CCR7 identify pro-inflammatory and cytotoxic macrophage M1 phenotype, while CD163 is specific to M2 phenotype during the remodeling process.7
When macrophages are recruited into tissues, they become “activated macrophages” and they can have two different phenotypes related to different stimuli: M1 (classically activated) and M2 (alternatively activated) (Figure 2).8,9
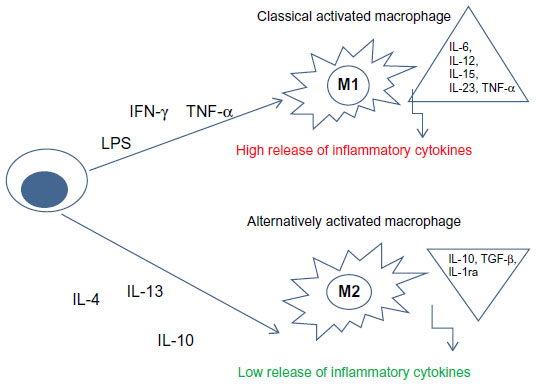 | Figure 2 Schematic representation of macrophage polarization. |
M1 macrophages are important in killing microorganisms and tumor cells; they release high levels of pro-inflammatory cytokines, reactive nitrogen and oxygen intermediates.
M2 macrophages are subdivided into three subpopulations in response to different cytokines and chemokines. IL-4 or IL-13 activates M2a; immune complexes (ICs) in combination with IL-1β or LPS activate M2b phenotype, while IL-10, TGF-β or glucocorticoids induce M2c macrophages. M2 macrophages are involved in resolution of inflammation through phagocytosis of apoptotic neutrophils, reduced production of pro-inflammatory cytokines, and increased synthesis of mediators important in tissue remodeling, angiogenesis, and wound repair (Figure 2).8,9
Macrophages and rheumatoid arthritis
Macrophages play a key role in the pathogenesis of rheumatoid arthritis (RA). They produce many pro-inflammatory cytokines and chemokines and then contribute to the cartilage and bone destruction.10 An increased number of macrophages are found in the synovial tissue; these cells can be activated to produce inflammatory cytokines. In Figure 3A we show an activated macrophage from synovial fluid of an RA patient, while Figure 3B and C illustrate macrophages and neutrophil granulocytes during phagocytosis of a small lymphocyte in the same patient.
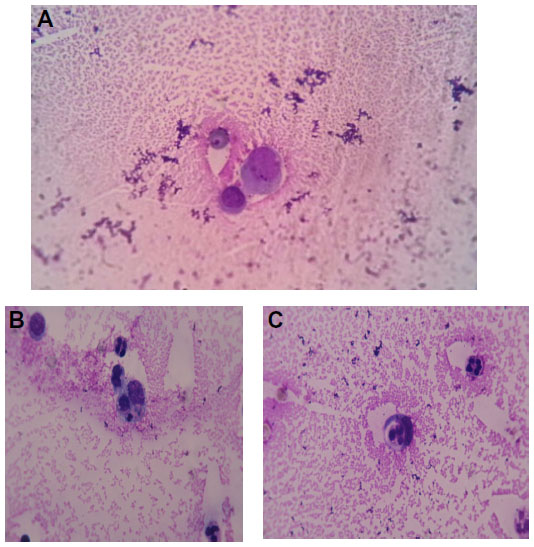 | Figure 3 Wright stain images of a patient with rheumatoid arthritis. |
Synovial sublining macrophage number can be used as a biomarker for disease severity as well as a predictor of responsiveness to disease-modifying antirheumatic drug (DMARD) therapy.11 It has demonstrated a strong correlation between number of macrophages, the mean change in the disease activity score,12 and the degree of joint erosion.13,14 M1 macrophages have a TNFhighIL-12highIL-10lowiNOS2high pro-inflammatory phenotype, while M2 macrophages have a TNFlowIL-12lowIL-10high- arginasehigh anti-inflammatory phenotype.15
From an in vitro polarization study using human monocytes, it was found that CD80 is the main phenotypic marker for human M1 macrophages polarized by IFN-γ, whereas CD200R is upregulated and CD14 is specifically downregulated on M2 macrophages polarized by IL-4, while CD163 and CD16 were found to be specific markers for macrophages polarized by IL-10 (Figure 4).16
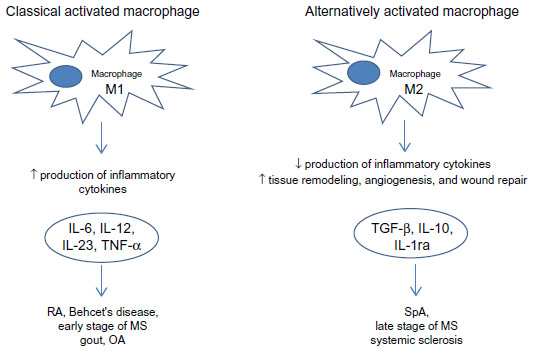 | Figure 4 Macrophages in rheumatic diseases. |
Polarization of RA synovial tissue or synovial fluid macrophages in M1 or M2 phenotype depends on “flare” of rheumatic disease. In fact, patients with highly active RA show a prevalence of M1 phenotype; on the other hand, RA patients with low disease activity score or clinical remission show a M2 phenotype. Furthermore, glucocorticoids induce the M2 state,8 and this “switching” is one of the pharmacological effects of this drugs class on RA. Other DMARDs (such as methotrexate and leflunomide) also have this mechanism of action on macrophage populations; moreover they inhibit cell replication and recruitment of immature and inflammatory monocytes to sites of inflammation.15 Elevated levels of GM-CSF and M-CSF are seen in synovial fluid from RA patients. GM-CSF has many actions on neutrophils, eosinophils, macrophages, and dendritic cells, whereas M-CSF acts more specifically on the macrophage lineage. It has been proposed that M-CSF in the absence of inflammation maintains steady-state levels of M2 macrophages, while GM-CSF induces pro-inflammatory M1 macrophages that release other inflammatory cytokines, such as TNF-α, IL-12, and IL-23. During development of arthritis, TNF-α and IL-1 stimulate fibroblasts and chondrocytes to release GM-CSF and M-CSF.17 CD68 and CD163 are two commonly used markers for identification of synovial macrophages in RA synovium. CD68 is a scavenger receptor that binds to oxidized low-density lipoprotein and may also be involved in the cell–cell interaction.17 It is localized on both cell surface and lysosomal membranes. Changes in the number of synovial sublining CD68+ macrophages can be used as a sensitive biomarker to predict the possible efficacy of anti-rheumatic therapy because this variation’s number correlates with disease activity score.17 CD163 is a type I trans-membrane protein that belongs to the group B scavenger receptor cysteine-rich superfamily. The major role of CD163 is its capacity to bind and internalize hemoglobin–haptoglobin complexes leading to the release of IL-10 and carbon oxide, which exert strong anti-inflammatory effects.17 CD163 has major advantages as a macrophage marker, as compared with CD68, in RA synovium. In fact CD163 discriminates between synovial macrophages and synovial intimal fibroblasts, which also stain positive for CD68 in diseased tissue.17 Moreover, soluble CD163 in sera is a promising diagnostic marker for untreated new-onset systemic juvenile idiopathic arthritis and macrophage activation syndrome, which is characterized by excessive expansion of T cells and hemophagocytic macrophages that induce a strong inflammatory reaction.18 Macrophage markers can help rheumatology specialists to choose the best biological DMARD for rheumatoid patients; in fact it is known in literature that in baseline synovial myeloid, but not lymphoid, gene signature expression is higher in patients with good clinical response to anti-TNF-α therapy.
Dennis et al observed that some serum molecules can predict clinical response to different biological DMARDs. In particular, high baseline serum sICAM1, associated with the myeloid phenotype, is associated with the highest American College of Rheumatologists (ACR) 50 response rate to anti-TNF-α treatment; while high serum CXCL13, associated with the lymphoid phenotype, is associated with greater ACR50 response to anti-IL6R treatment.19
Macrophages and angiogenesis
Angiogenesis has a key role in pathogenesis of several rheumatic diseases, such as RA, osteoarthritis (OA), ankylosing spondylitis, systemic sclerosis (SSc), systemic lupus erythematosus (SLE), and vasculitides. Among major cell types involved in angiogenesis, macrophages are known to produce numerous angiogenic factors, including VEGF, FGF, TGF-β, PDGF, TNF-α, MCP-1, IL-6, IL-8, and IL-18 (Figure 5).20,21
 | Figure 5 Chemokines, growth factors, and cytokines involved in the angiogenic activity of macrophages. |
Macrophages and SLE
SLE is an autoimmune disease with a broad spectrum of clinical symptoms. Autoreactive B and T lymphocytes,21 together with the innate immune system play a role in the pathogenesis of SLE. SLE macrophages are unable to clear apoptotic cells. In SLE, there is an altered pro-inflammatory/anti-inflammatory macrophage status that leads to an overproduction of inflammatory cytokines such as TNF-α, IL-6, IL-10, and antiviral type I IFNs (Figure 6).22,23 In an inflammatory context, SLE monocytes and macrophages present self-antigens to autoreactive T cells rather than the immunosilent presentation normally associated with material from apoptotic cells.22 Moreover, myeloid cells (including dendritic cells) induce overproduction of type I IFNs that lead to an overproduction of antibodies (classes IgG, IgA, IgM) and class switching from B cells.22 Recent studies focused attention on innate immune system as promoter of autoimmunity.
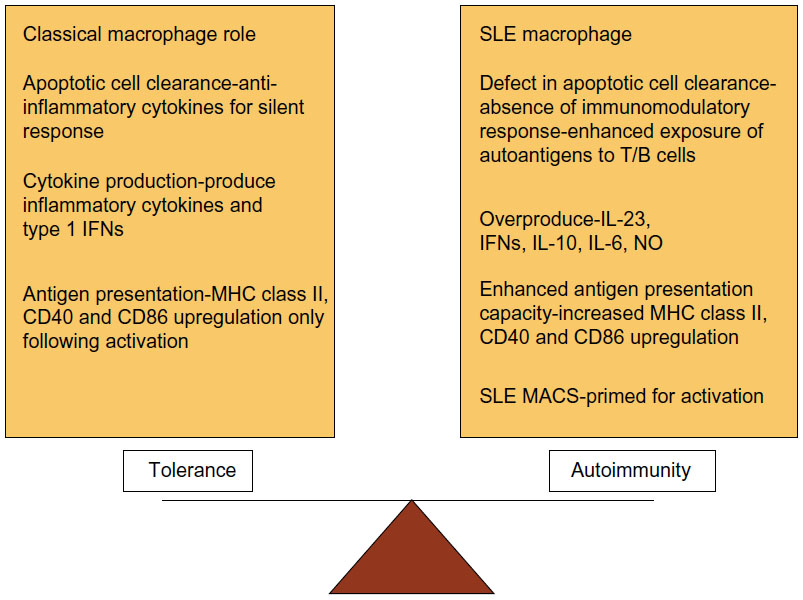 | Figure 6 Dysregulation of macrophage function in systemic lupus erythematosus (SLE). |
PRPs, present on immune cells, recognize microbes during the normal immune responses against infectious agents.24 TLRs, members of PRPs, control immune responses detecting common molecular motifs, including RNA ligands by TLR3, TLR7 and TLR8, DNA ligands by TLR9, and bacterial cell surface proteins such as LPS or endotoxin that is a ligand for TLR4.
After the link between molecular motifs and TLRs on immune cells (dendritic cells, monocytes/macrophages, and B cells), many pro-inflammatory cytokines are released.
TLRs are activated not only during response against host, but it is also known that they contribute to the pathogenesis of SLE.25 TLRs (in particular TLR7, TLR8, and TLR9) of SLE patients recognize the serum ICs formed by autoantibodies to nucleic acids or nucleic acid binding proteins.26 Dendritic and B cells can internalize these nucleic acid-containing ICs through Fc and surface immunoglobulin receptors, respectively.27,28 After the ICs’ internalization, nucleic acids activate TLRs present on endosomal compartment (RNA and DNA activate TLR7 and TLR9, respectively) and then dendritic cells are stimulated to produce IFN and B cells to undergo maturation. Plasmacytoid dendritic cells are the major source of IFN-α, under TLR7 or TLR9 activation. From these observations, it seems clear that innate immunity regulates key aspects of autoimmunity playing a key role in the pathogenesis of SLE but also in other autoimmunity diseases such as SSc. Moreover, it is known that specific B cell subtype in the marginal zone of the spleen, so-called marginal zone B cells (MZBs), are involved in SLE pathogenesis.29 MZBs are essential for early responses against pathogens and they are phenotypically characterized by high IgM and complement receptor expression.30 However, MZBs are also involved in self-reactivity.31 Autoreactive MZBs produce autoantibodies to self-antigens derived from apoptotic cells.32 In the marginal zone, the MZBs are in close contact with highly phagocytic macrophages called marginal zone macrophages (MZMs).33 The MZMs are characterized by high expression of the class A scavenger receptor MARCO and SR-A.33 MARCO and SR-A bind a variety of self- and foreign ligands, and through them MZMs play an important role in MZB activation.29
Macrophages and SSc
SSc is an autoimmune disease characterized by inflammation, endothelial damage, and fibrosis. Type I IFNs have been implicated in B cell maturation34,35 and they ameliorate fibrosis of SSc by blocking effect of TGF-β. Upon TLRs’ activation, dendritic cells and macrophages release cytokines such as IL-1, TNF, and IL-6 that stimulate inflammation and fibrosis in SSc. Maturation of autoreactive B lymphocytes is induced by TLR activation, also. It seems that TLR activation leads to fibrosis through TGF-β.36 Moreover data from literature suggest that TLR activation might directly stimulate fibroblast conversion to pro-fibrotic myofibroblasts through TLR337 or TLR9.38 Macrophages, in the dermis of scleroderma patients, become pro-fibrotic under activation of IL-1339 and they release many pro-fibrotic mediators, including PDGF and TGF-β. Th1 cytokine IFN-γ induces “classically activated” monocytes whereas Th2 cytokines IL-4 and IL-13 stimulate “alternatively activated” macrophages.40 Alternatively activated macrophages are proposed to be pro-fibrotic, possibly by activating TGF-β. We need future studies to explain this paradigm, to date not fully explored in SSc.
Macrophages and gout
Hyperuricemia can induce precipitation of monosodium urate monohydrate crystals in joints and soft tissues leading to release of many pro-inflammatory cytokines such as TNF, IL-1, and IL-6.41 It is well known that IL-1 plays a key role in the pathogenesis of gout. It is secreted in the molecular platform called inflammasome,41 that promotes the activation of CASP1, which then cleaves Pro-IL-1 (Figure 7). There are many different inflammasomes, but NALP3 inflammasome is the most important in gout. Monosodium urate monohydrate or calcium pyrophosphate dihydrate crystals or pathogen-associated molecular patterns or danger-associated molecular patterns bind NALP3 through an LRR domain (that is capable of recognizing microbial pathogen-associated molecular patterns).41 NALP3 possesses a NACHT domain to bind ATP and a pyrin domain, which is critical for homotypic protein–protein interactions (Figure 7). ASC, an adopter molecule, links NALP3 with CASP1 (Figure 6).42 These interactions lead to activation of CASP1 molecules, becoming capable of cleaving or processing Pro-IL-1 (Figure 7).41 So, it is clear that macrophage-derived IL-1β (M1-associated phenotype in gout) is a pivotal mediator of acute gout and could become a potential therapeutic target.9 In this way, colchicine is a drug used to treat gout, able to block the activation of inflammasomes and then the production of macrophagic activated IL-1β (Figure 7).
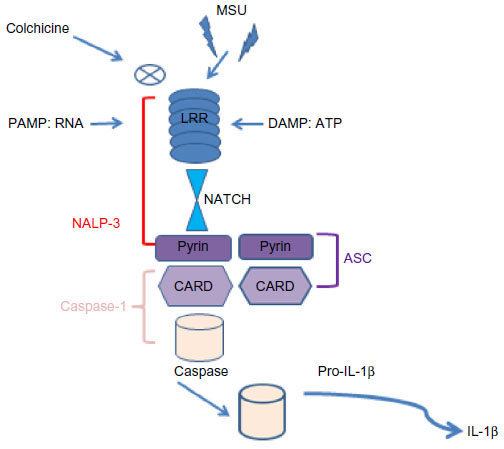 | Figure 7 Schematic representation of the role of the inflammasome in processing Pro-IL-1β. |
Macrophages and spondyloarthritis
Spondyloarthritis (SpA) includes different forms of arthritis, comprises ankylosing spondylitis, psoriatic arthritis (PsA), reactive arthritis, enteropathic-related arthritis, and undifferentiated SpA. When we refer to lymphocytic cells we know the different origins of RA and SpA; but the synovial infiltrate of patients with PsA and RA is comparable with regard to numbers of fibroblast-like synoviocytes and macrophages.43 Prominent swelling and inflammation of peripheral joints is a hallmark of both RA and SpA, with a preference for large joints of the lower extremities in SpA. Of interest, the total number of macrophages is similar in RA and SpA synovitis, but the subset expressing the M2 surface marker, CD163,44,45 is clearly increased in the latter.46,47 In accordance with this observation, it was recently described that synovial fluid from SpA patients promotes preferential expression of the M2 markers CD163 and CD200R in vitro. Interestingly, this was still observed even if synovial fluid levels of the prototypical M2-polarizing factors (IL-4, IL-13, and IL-10) were not increased as compared with those in RA synovial fluid.48 In this respect RA macrophages are associated with an M1 phenotype whereas SpA macrophages with an M2 phenotype.
From these literature data, we can assume that SpA synovitis is characterized from overexpression of alternatively activated macrophages (M2 phenotype) responsible for pro-fibrotic state and for bone overgrowth (syndesmophytes) through abnormal tissue remodeling.
Macrophages and OA
OA is characterized by loss of articular cartilage and modification of subchondral bone. Fluid accumulation (joint effusion), bone overgrowth (osteophytes), and weakness of tendons and muscles can also result from the degenerative process. OA commonly affects the hands, feet, spine, and large weight-bearing joints, such as the hips and knees. The pathogenesis of OA is largely unknown and under current investigation. Biomechanical stress such as cartilage integrity, maintained by a balance obtained from cytokine-driven anabolic and catabolic processes, are involved in the pathogenesis of OA.49 Cartilage erosion, induced by proteolytic enzymes that lead to a breakdown of the cartilage macromolecules, lead to synovial inflammation. Activated synoviocytes and mononuclear cells (eg, macrophages) release cytokines such as IL-1β and TNF-α and up-regulate MMP gene expression.50,51 Then, the synovial inflammation is implicated in many of the signs and symptoms of OA, including joint swelling and effusion.52,53
Histologically, the OA synovium shows hyperplasia with an increased number of lining cells and a mixed inflammatory infiltrate consisting mainly of macrophages.54
Synovitis in OA is likely to contribute to disease progression, by releasing inflammatory cytokines and enzymes that degrade cartilage and induce progression of structural changes in OA.55,56 However, the levels of pro-inflammatory cytokines in the OA synovitis are lower than in RA. In particular, TNF-α and IL-1 have been suggested to be key players in OA pathogenesis, both in synovial inflammation and in activation of chondrocytes and they stimulate synovial cells and chondrocytes to produce IL-6, IL-8, and LIF as well as stimulate protease and prostaglandin production.52,57 It is clear that RA macrophage is the main promoter of disease activity and that macrophage-produced TNF-α is a major therapeutic target. The role of macrophages in OA is not well studied to date. OA synovial macrophages produce both pro-inflammatory cytokines and VEGF52 and synovial macrophage differentiation differs between inflammatory and non-inflammatory OA.55 In experimental OA the involvement of macrophages in this pathology has been demonstrated.58,59 van der Kraan et al demonstrated that macrophages mediate osteophyte formation and fibrosis in the early stages of experimental OA.60 Moreover, these knee synovial macrophages were crucial in early MMP activity and appeared to mediate MMP production in synovium rather than cartilage.61,62 Blom et al63 showed that Wnt signaling pathway is involved in OA pathogenesis.
WISP-1 expression was strongly increased in the synovium and cartilage of human and experimental OA64 regulating chondrocyte and macrophage MMP and aggrecanase expression (molecules capable of inducing articular cartilage damage).64 In OA, macrophage-derived MMP and pro-inflammatory cytokines might contribute to cartilage destruction and are likely to play a crucial role in disease pathogenesis. To date it seems that M1 phenotype is involved in OA pathogenesis, but more future studies are necessary to define the role of macrophages, in particular, their phenotype.
Macrophages in Behcet’s disease and Sjogren’s syndrome
Behcet’s disease (BD) is a chronic, relapsing, inflammatory disorder which is clinically characterized by bipolar aphthosis and microvascular skin and ocular lesions.65 The synovitis of patients with BD in terms of CD68+ macrophage numbers is similar in the intimal lining layer as well as the synovial sublining to the synovitis of PsA patients.66 However, it has been reported that serum factor(s) are able to induce classical (pro-inflammatory) activation (M1 phenotype) of human peripheral blood macrophages in vitro in this disease67 suggesting that serum factor(s) might be responsible for inflammatory changes in BD.
Sjogren’s syndrome (SS) is a systemic autoimmune disease that affects woman targeting exocrine glands primarily. Chronic focal inflammatory infiltration is constituted by CD3+ T and CD19+ B lymphocytes as well as M1 and M2 macrophages and it increases inflammatory cytokines’ release. Baban et al demonstrated that B cells and M2 macrophages are more prominent than T cells and M1 macrophages. This result could be explained by temporal relation of immune cell infiltrates in salivary glands in SS.68,69
Macrophages and anti-neutrophil cytoplasmic antibodies-related vasculitis
The primary small vessel systemic vasculitides are disorders that target small blood vessels, inducing vessel wall inflammation and are associated with development of anti-neutrophil cytoplasmic antibodies (ANCAs). From data literature it is known that antibodies activate neutrophils inappropriately, leading to endothelial and vascular damage through cytokines such as TNF. The role of monocyte/macrophages in small vessel systemic vasculitides remain unknown to date, although some studies continue to suggest that down-regulating their activities can be beneficial. To support this, van der Veen et al showed that the use of a p38 MAPK inhibitor reduced ANCA-activation of neutrophils but also reduced glomerular macrophage accumulation and crescent formation.70 One feature of the accelerated apoptosis was perplexing. When a neutrophil becomes apoptotic, normally there is enhanced expression of phosphatidylserine molecules on its surface which allows it to be recognized and eaten by a phagocytic cell. ANCA-treated neutrophils did not show enhanced expression of phosphatidylserine on their surfaces. This suggested that the cell membrane changes were dissociated from the morphological and nuclear changes of apoptosis and, perhaps more importantly, that the apoptotic neutrophils might not be efficiently cleared by phagocytic cells such as macrophages. In vitro, human monocyte-derived macrophages were, as predicted, less efficient at removing ANCA-treated apoptotic neutrophils than untreated apoptotic neutrophils. The consequences of this failure of removal may be profound since late apoptotic neutrophils disintegrate with release of cell contents into the microenvironment. These observations may also explain the fragmentation of white blood cells seen in electron microscopy studies of early vasculitic lesions.70
Macrophages and multiple sclerosis
Multiple sclerosis (MS), or experimental autoimmune encephalomyelitis (EAE) in an animal model, is a debilitating neurological disorder of the central nervous system (CNS). This disorder is defined by inflammatory infiltration of the CNS, constituted by leukocytes and dendritic cells. In the early stage of MS, peripheral macrophages infiltrate the CNS, where, together with residential microglia, they participate in the induction and development of disease. During the early phase, microglia/macrophages are immediately activated to become classically activated macrophages (M1 cells), releasing pro-inflammatory cytokines and damaging CNS tissue. During the later phase, alternatively activated macrophage phenotype (M2 cells) is predominant in CNS inflammation, releasing anti-inflammatory cytokines, and they are responsible for inflammation resolution and tissue repair.71 The balance between activation and polarization of M1 cells and M2 cells in the CNS is important for disease progression. M1 cell polarization is driven by pro-inflammatory IFN-γ and IL-12, while IL-4 and IL-13 drive switch to M2 cell. Because macrophage polarization is reversible and dependent on cytokine environment, macrophage phenotypes in the CNS can be modulated by molecular intervention to obtain beneficial effects on disease progression.72
In EAE, an animal model of MS, the therapeutic effect of fasudil, a selective ROCK inhibitor was observed.72 Fasudil ameliorated the clinical severity of EAE with an improvement in demyelination and inhibition of inflammatory cells. One cause of its potential effect is due to induction of switch from inflammatory M1 to anti-inflammatory M2 macrophages. The polarization of M2 macrophages was associated with the decrease of inflammatory cytokine IL-1β, TNF-α, and MCP-1.73
Effect of treatments on macrophage lineage numbers
Different conventional and biological DMARDs may play a role through macrophage inhibition.
As expressed above, one other stimulus that favors the M2 state is glucocorticoids,14 and this “switching” may form part of the action of this class of drugs at the macrophage level. This is one of the effects of DMARDs (such as methotrexate and leflunomide) on macrophage populations, in addition to inhibition of cell replication and recruitment of immature and inflammatory monocytes to sites of inflammation.13
In RA, TNF-α acts as a positive feedback signal to further promote development and survival of macrophages. Under these circumstances, which are usually encountered in clinical practice, blockade of TNF-α signaling in macrophages can promote depletion of macrophages.74 Etanercept and infliximab have been shown to induce apoptosis in monocytes and macrophages in both synovial fluid and peripheral blood in vivo.75 Furthermore, treatment with TNF-α blockers reduces the number of infiltrating synovial granulocytes and macrophages, as well as reducing the expression of chemokines, IL-8, and MCP-1.76 Anti-TNF-α therapy has also been shown to potentially ameliorate arthritis by upregulating TGF-β and reversing the functional defect of regulatory T cells in RA, which can then inhibit the activity of inflammatory macrophages.77 Infliximab, a chimeric monoclonal antibody directed against TNF-α, in combination with methotrexate, leads to decreased synovial and skin VEGF expression in patients affected by PsA.78 The development of other biological therapies, such as the anti-IL-6 receptor antibody, tocilizumab, exhibits the therapeutic effect, in part, by inhibition of macrophage development and function.79 It also may reduce VEGF production in RA.80,81 Thalidomide, recently introduced in the treatment of RA and lupus, is responsible for angiogenesis and TNF-α inhibition.82,83 Blockade of GM-CSF or M-CSF inhibits the development of arthritis.84,85 The oral inhibitor of M-CSF receptor 27 reduced the arthritis progression by inactivating the tyrosine kinase, which can also be achieved by imatinib mesylate (Glivec®; Novartis International AG, Basel, Switzerland).86 Since depletion of both M-CSF and GM-CSF is effective, several therapies are in Phase I or II clinical trials or preclinical studies using GM-CSF-specific antibodies, GM-CSF receptor-specific antibodies, or antibodies directed at M-CSF or its receptor.87,88 JAK inhibitors, including tofacitinib, may also act to effect macrophage development.16,88 Macrophage polarization and related therapeutic targets and novel agents are summarized in Figure 8.
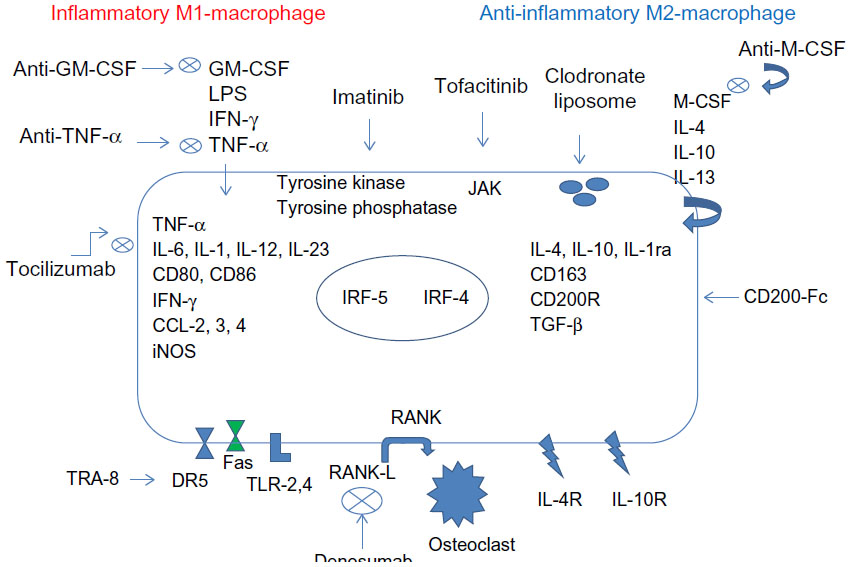 | Figure 8 Macrophage polarization in RA synovium and related therapeutic targets. |
Conclusion
Macrophages play a key role in rheumatic diseases. Their role in the different rheumatic diseases is different according to their M1/M2 macrophage phenotype (Figure 4).
Pro-inflammatory M1 phenotype expression is prevalently seen in RA, BD, early stage MS, gout, and OA in the presence of pro-inflammatory cytokines such as IFN-γ, TNF-α, IL-6, IL-1, IL-12, and IL-23. On the other hand, M2 macrophages are more predominant in SpA, late stage MS, and SS releasing anti-inflammatory cytokines such as IL-4, IL-10, IL-1ra, and TGF-β and contributing to tissue remodeling and angiogenesis. So, in the future, using specific targeting molecules we can modulate shift versus M1 or M2 phenotype and we can block progression of many rheumatic diseases.
Disclosure
The authors have no conflicts of interest to disclose.
References
Volkman A, Gowans JL. The Origin of Macrophages from Bone Marrow in the Rat. Br J Exp Pathol. 1965;46:62–70. | |
Burke B; Sumner S, Maitland N, Lewis CE. Macrophages in gene therapy: cellular delivery vehicles and in vivo targets. J Leukoc Biol. September 2002;72(3):417–428. | |
Passlick B, Flieger D, Ziegler-Heitbrock HW. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. 1989;74(7):2527–2534. | |
Weber C, Belge KU, von Hundelshausen P, et al. Differential chemokine receptor expression and function in human monocyte subpopulations. J Leukoc Biol. 2000;67:699–704. | |
Randolph GJ, Sanchez-Schmitz G, Liebman RM, Schakel K. The CD16(+) (FcgammaRIII(+)) subset of human monocytes preferentially becomes migratory dendritic cells in a model tissue setting. J Exp Med. 2002;196:517–527. | |
Serbina NV, Salazar Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. July 2003;19(1):59–70. | |
Badylak SF, Gilbert TW. Immune Response to Biologic Scaffold Materials. Semin Immunol. 2008;20(2):109–116. | |
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–555. | |
Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7(3):211–217. | |
Lebre MC, Tak PP. Macrophage Subsets in Immune-Mediated Inflammatory Disease: Lessons from Rheumatoid Arthritis, Spondyloarthritis, Osteoarthritis, Behçet’s Disease and Gout. The Open Arthritis Journal. 2010;3:18–23. | |
Gerlag DM, Tak PP. Novel approaches for the treatment of rheumatoid arthritis: lessons from the evaluation of synovial biomarkers in clinical trials. Best Pract Res Clin Rheumatol. 2008;22(2):311–323. | |
Van der Heijde DM, van’t Hof MA, van Riel PL, et al. Judging disease activity in clinical practice in rheumatoid arthritis: first step in the development of a disease activity score. Ann Rheum Dis. 1990; 49(11):916–920. | |
Mulherin D, FitzGerald O, Bresnihan B. Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheum. 1996;39(1):115–124. | |
Tak PP, Bresnihan B. The pathogenesis and prevention of joint damage in rheumatoid arthritis: advances from synovial biopsy and tissue analysis [review]. Arthritis Rheum. 2000;43(12):2619–2633. | |
Hamilton JA, Tak PP. The Dynamics of Macrophage Lineage Populations in Inflammatory and Autoimmune Diseases. Arthritis Rheum. 2009;60(5):1210–1221. | |
Ambarus CA, Krausz S, van Eijk M, et al. Systematic validation of specific phenotypic markers for in vitro polarized human macrophages. J Immunol Methods. 2011;375(1–2):196–206. | |
Li J, Hsu HC, Mountz JD. Managing Macrophages in Rheumatoid Arthritis by Reform or Removal. Curr Rheumatol Rep. 2012;14(5):445–454. | |
Bleesing J, Prada A, Siegel DM, et al. The diagnostic significance of soluble CD163 and soluble interleukin-2 receptor alpha-chain in macrophage activation syndrome and untreated new-onset systemic juvenile idiopathic arthritis. Arthritis Rheum. 2007;56(3):965–971. | |
Dennis G Jr, Holweg CT, Kummerfeld SK, et al. Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Res Ther. 2014;16(2):R90. | |
Maruotti N, Annese T, Cantatore FP, Ribatti D. Macrophages and angiogenesis in rheumatic diseases. Vasc Cell. 2013;5(1):11. | |
Byrne JC, Ní Gabhann J, Lazzari E, et al. Genetics of SLE: functional Relevance for Monocytes/Macrophages in disease. Clin Dev Immunol. 2012;2012:582352. | |
Kavai M, Szegedi G. Immune complex clearance by monocytes and macrophages in systemic Lupus erythematosus. Autoimmun Rev. 2007;6(7):497–502. | |
Sestak AL, Fürnrohr BG, Harley JB, Merrill JT, Namjou B. The genetics of systemic Lupus erythematosus and implications for targeted therapy. Ann Rheum Dis. 2011;70 Suppl1:i37–i43. | |
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. | |
Lafyatis R, Marshak-Rothstein A. Toll-like receptors and innate immune responses in systemic lupus erythematosus. Arthritis Res Ther. 2007; 9(6):222. | |
Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6(11):823–835. | |
Lau CM, Broughton C, Tabor AS, et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202(9):1171–1177. | |
Leadbetter EA, Rifkin IR, Hohlbaum AM, et al. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416(6881):603–607. | |
Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25(3):383–392. | |
Wermeling F, Chen Y, Pikkarainen T, et al. Class A scavenger receptors regulate tolerance against apoptotic cells, and autoantibodies against these receptors are predictive of systemic lupus. J Exp Med. 2007; 204(10):2259–2265. | |
Martin F, Kearney JF. Marginal-zone B cells. Nat Rev Immunol. 2002; 2(5):323–335. | |
Li Y, Li H, Ni D, Weigert M. Anti-DNA B cells in MRL/lpr mice show altered differentiation and editing pattern. J Exp Med. 2002;196(12):1543–1552. | |
Enzler T, Bonizzi G, Silverman GJ, et al. Alternative and classical NF-kappa B signaling retain autoreactive B cells in the splenic marginal zone and result in lupus-like disease. Immunity. 2006;25(3):403–415. | |
Kraal G, Mebius R. New insights into the cell biology of the marginal zone of the spleen. Int Rev Cytol. 2006;250:175–215. | |
Ronnblom L, Eloranta ML, Alm GV. The type I interferon system in systemic lupus erythematosus. Arthritis Rheum. 2006;54(2):408–420. | |
Seki E, De Minicis S, Osterreicher CH, et al. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13(11):1324–1332. | |
Sugiura H, Ichikawa T, Koarai A, et al. Activation of Toll-like receptor 3 augments myofibroblast differentiation. Am J Respir Cell Mol Biol. 2009;40(6):654–662. | |
Meneghin A, Choi ES, Evanoff HL, et al. TLR9 is expressed in idiopathic interstitial pneumonia and its activation promotes in vitro myofibroblast differentiation. Histochem Cell Biol. 2008;130(5):979–992. | |
Fisher ER, Rodnan GP. Pathologic observations concerning the cutaneous lesion of progressive systemic sclerosis: an electron microscopic histochemical and immunohistochemical study. Arthritis Rheum. 1960;3:536–545. | |
Munder M, Eichmann K, Modolell M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J Immunol. 1998;160(11):5347–5354. | |
Pope RM, Tschopp J. The Role of Interleukin-1 and the Inflammasome in Gout Implications for Therapy. Arthritis rheum. 2007;56(10):3183–3188. | |
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-. Mol Cell. 2002;10(2):417–426. | |
Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007;14(1):10–22. | |
van Kuijk AW, Reinders-Blankert P, Smeets TJ, Dijkmans BA, Tak PP. Detailed analysis of the cell infiltrate and the expression of mediators of synovial inflammation and joint destruction in the synovium of patients with psoriatic arthritis: implications for treatment. Ann Rheum Dis. 2006;65(12):1551–1557. | |
Buechler C, Ritter M, Orso E, Langmann T, Klucken J, Schmitz G. Regulation of scavenger receptor CD163 expression in human monocytes and macrophages by pro- and antiinflammatory stimuli. J Leukoc Biol. 2000;67(1):97–103. | |
Sulahian TH, Hogger P, Wahner AE, et al. Human monocytes express CD163, which is upregulated by IL-10 and identical to p155. Cytokine. 2000;12(9):1312–1321. | |
Baeten D, Demetter P, Cuvelier CA, et al. Macrophages expressing the scavenger receptor CD163: a link between immune alterations of the gut and synovial inflammation in spondyloarthropathy. J Pathol. 2002; 196(3):343–350. | |
Baeten D, Kruithof E, De RL, et al. Diagnostic classification of spondylarthropathy and rheumatoid arthritis by synovial histopathology: a prospective study in 154 consecutive patients. Arthritis Rheum. 2004;50(9):2931–2941. | |
Vandooren B, Noordenbos T, Ambarus C, et al. Absence of a classically activated macrophage cytokine signature in peripheral spondylarthritis, including psoriatic arthritis. Arthritis Rheum. 2009;60(4):966–975. | |
Warger T, Hilf N, Rechtsteiner G, et al. Interaction of TLR2 and TLR4 ligands with the N-terminal domain of Gp96 amplifies innate and adaptive immune responses. J Biol Chem. 2006;281(32):22545–22553. | |
Fernandes JC, Martel-Pelletier J, Pelletier JP. The role of cytokines in osteoarthritis pathophysiology. Biorheology. 2002;39(1–2):237–246. | |
Martel-Pelletier J. Pathophysiology of osteoarthritis. Osteoarthritis Cartilage. 2004;12 Suppl A:S31–S33. | |
Pelletier JP, Martel-Pelletier J, Abramson SB. Osteoarthritis, an inflammatory disease: potential implication for the selection of new therapeutic targets. Arthritis Rheum. 2001;44(6):1237–1247. | |
Benito MJ, Veale DJ, FitzGerald O, van den Berg WB, Bresnihan B. Synovial tissue inflammation in early and late osteoarthritis. Ann Rheum Dis. 2005;64(9):1263–1267. | |
Farahat MN, Yanni G, Poston R, Panayi GS. Cytokine expression in synovial membranes of patients with rheumatoid arthritis and osteoarthritis. Ann Rheum Dis. 1993;52(12):870–875. | |
Clark AG, Jordan JM, Vilim V, et al. Serum cartilage oligomeric protein reflects osteoarthritis presence and severity: the Johnston County Osteoarthritis Project. Arthritis Rheum. 1999;42(11):2356–2364. | |
Sowers M, Jannausch M, Stein E, Jamadar D, Hochberg M, Lachance L. C-reactive protein as a biomarker of emergent osteoarthritis. Osteoarthritis Cartilage. 2002;10(8):595–601. | |
Haywood L, McWilliams DF, Pearson CI, et al. Inflammation and angiogenesis in osteoarthritis. Arthritis Rheum. 2003;48(8):2173–2177. | |
Danks L, Sabokbar A, Gundle R, Athanasou NA. Synovial macrophage- osteoclast differentiation in inflammatory arthritis. Ann Rheum Dis. 2002;61(10):916–921. | |
van der Kraan PM, Vitters EL, van de Putte LB, van den Berg WB. Development of osteoarthritic lesions in mice by “metabolic” and “mechanical” alterations in the knee joints. Am J Pathol. 1989;135(6):1001–1014. | |
van Lent PL, Blom AB, van der KP, et al. Crucial role of synovial lining macrophages in the promotion of transforming growth factor beta-mediated osteophyte formation. Arthritis Rheum. 2004; 50(1):103–111. | |
Blom AB, van Lent PL, Holthuysen AE, et al. Synovial lining macrophages mediate osteophyte formation during experimental osteoarthritis. Osteoarthritis Cartilage. 2004;12(8):627–635. | |
Blom AB, Brockbank SM, van Lent PL, et al. Involvement of the Wnt signaling pathway in experimental and human osteoarthritis: prominent role of Wnt-induced signaling protein 1. Arthritis Rheum. 2009; 60(2):501–512. | |
Blom AB, van Lent PL, Libregts S, et al. Crucial role of macrophages in matrix metalloproteinase-mediated cartilage destruction during experimental osteoarthritis: involvement of matrix metalloproteinase 3. Arthritis Rheum. 2007;56(1):147–157. | |
Sakane T, Takeno M, Suzuki N, Inaba G. Behcet’s disease. N Engl J Med. 1999;341(17):1284–1291. | |
Canete JD, Celis R, Noordenbos T, et al. Distinct synovial immunopathology in Behcet disease and psoriatic arthritis. Arthritis Res Ther. 2009;11(1):R17. | |
Alpsoy E, Kodelja V, Goerdt S, Orfanos CE, Zouboulis C. Serum of patients with Behcet’s disease induces classical (proinflammatory) activation of human macrophages in vitro. Dermatology. 2003;206(3):225–232. | |
Baban B, Liu JY, Abdelsayed R, Mozaffari MS. Reciprocal relation between GADD153 and Del-1 in regulation of salivary gland inflammation in Sjogren syndrome. Exp Mol Pathol. 2013;95(3):288–297. | |
Roescher N, Lodde BM, Vosters JL, et al. Temporal changes in salivary glands of non-obese diabetic mice as a model for Sjogren’s syndrome. Oral Dis. 2012; 18(1):96–106. | |
van der Veen BS, Chen M, Muller R, et al. Effects of p38 mitogenactivated protein kinase inhibition on anti-neutrophil cytoplasmic autoantibody pathogenicity in vitro and in vivo. Ann Rheum Dis. 2011; 70(2):356–365. | |
Jiang Z, Jiang JX, Zhang GX. Macrophages: a double-edged sword in experimental autoimmune encephalomyelitis. Immunol Lett. 2014;160(1):17–22. | |
Miron VE, Boyd A, Zhao JW, et al. M2 microglia/macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16(9):1211–1218. | |
Liu C, Li Y, Yu J, et al. Targeting the shift from M1 to M2 macrophages in experimental autoimmune encephalomyelitis mice treated with fasudil. PLoS One. 2013;8(2):e54841. | |
Savage CO. The evolving pathogenesis of systemic vasculitis. Clin Med. 2002;2(5):458–464. | |
Parameswaran N, Patial S. Tumor necrosis factor-alpha signaling in macrophages. Crit Rev Eukaryot Gene Expr. 2010;20(2):87–103. | |
Catrina AI, Trollmo C, af Klint E, et al. Evidence that anti-tumor necrosis factor therapy with both etanercept and infliximab induces apoptosis in macrophages, but not lymphocytes, in rheumatoid arthritis joints: extended report. Arthritis Rheum. 2005;52(1):61–72. | |
Taylor PC, Peters AM, Paleolog E, et al. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor alpha blockade in patients with rheumatoid arthritis. Arthritis Rheum. 2000; 43(1):38–47. | |
Cornish AL, Campbell IK, McKenzie BS, Chatfield S, Wicks IP. G-CSF and GM-CSF as therapeutic targets in rheumatoid arthritis. Nat Rev Rheumatol. 2009;5(10):554–559. | |
Ehrenstein MR, Evans JG, Singh A, et al. Compromised function of regulatory t cells in rheumatoid arthritis and reversal by anti-tnfalpha therapy. J Exp Med. 2004;200(3):277–285. | |
Goedkoop AY, Kraan MC, Picavet DI, et al. Deactivation of endothelium and reduction in angiogenesis in psoriatic skin and synovium by low dose infliximab therapy in combination with stable methotrexate therapy. Arthritis Res Ther. 2004;6(4):R326–R334. | |
Nakahara H, Song J, Sugimoto M, et al. Anti-interleukin-6 receptor antibody therapy reduces vascular endothelial growth factor production in rheumatoid arthritis. Arthritis Rheum. 2003;48(6):1521–1529. | |
Szekanecz Z, Koch AE. Chemokines and angiogenesis. Curr Opin Rheumatol. 2001;13(3):202–208. | |
Szekanecz Z, Koch AE. Mechanism of disease: angiogenesis in inflammatory diseases. Nat Clin Pract Rheumatol. 2007;3(11):635–643. | |
Singh JA, Beg S, Lopez-Olivo MA. Tocilizumab for rheumatoid arthritis: a Cochrane systematic review. J Rheumatol. 2011;38(1):10–20. | |
Campbell IK, Rich MJ, Bischof RJ, Hamilton JA. The colony-stimulating factors and collagen-induced arthritis: Exacerbation of disease by m-csf and g-csf and requirement for endogenous m-csf. J Leukoc Biol. 2000;68(1):144–150. | |
Cook AD, Braine EL, Campbell IK, Rich MJ, Hamilton JA. Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (gm-csf): Requirement for gm-csf in the effector phase of disease. Arthritis Res. 2001;3(5):293–298. | |
Ando W, Hashimoto J, Nampei A, et al. Imatinib mesylate inhibits osteoclastogenesis and joint destruction in rats with collagen-induced arthritis (CIA). J Bone Miner Metab. 2006;24(4):274–282. | |
Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8(7):533–544. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.