Back to Journals » OncoTargets and Therapy » Volume 11
The investigation for potential modifier genes in patients with neurofibromatosis type 1 based on next-generation sequencing
Authors Yang F, Xu S ![]() , Liu RW, Shi T, Li XF, Li XB, Chen G, Liu HY, Zhou QH, Chen J
, Liu RW, Shi T, Li XF, Li XB, Chen G, Liu HY, Zhou QH, Chen J ![]()
Received 14 November 2017
Accepted for publication 20 December 2017
Published 21 February 2018 Volume 2018:11 Pages 919—932
DOI https://doi.org/10.2147/OTT.S156998
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Fan Yang,1,2,* Song Xu,1,2,* Renwang Liu,1,2 Tao Shi,3 Xiongfei Li,1 Xuebing Li,2 Gang Chen,1 Hongyu Liu,2 Qinghua Zhou,1,2 Jun Chen1,2
1Department of Lung Cancer Surgery, 2Tianjin Key Laboratory of Lung Cancer Metastasis and Tumor Microenvironment, Tianjin Lung Cancer Institute, 3Department of Pathology, Tianjin Medical University General Hospital, Tianjin, People’s Republic of China
*These authors contributed equally to this work
Introduction: Neurofibromatosis type 1 (NF1) is a common Mendelian multi-system disorder that is characterized by café-au-lait spots (CLS), axillary freckling, optic glioma and plexiform neurofibroma. Various mutations of the NF1 gene are widely accepted to be the main cause of this disease, while whether there are still certain other modifier genes that could influence the phenotypes of NF1 is our concern.
Patients and Methods: One proband and his father are involved, who are characterized by plexiform neurofibroma and cutaneous neurofibroma, respectively. Enhanced Computed tomography (CT) and Positron emission tomography-CT (PET-CT) were taken to collect the radiographic data, and the specimens of this neurofibroma as well as the blood samples from the father and son were sent for panel mutation screening of 295 tumor-related genes based on next-generation screening. Furthermore, the NF1 gene mutations were referred with Canis lupus familiaris, Rattus norvegicus, Gallus gallus, Danio rerio, and Drosophila melanogaster NF1 sequencing for evolutionary conservativeness and then analyzed in Condel databases for pathogenicity prediction.
Results: The radiography indicated that the benign plexiform neurofibroma only occurred in the son. Also, TP53, FANCA, BCL6, PIK3C2G, RNF43, FGFR4, FLT3, ERBB2, PAK7, NSD1, MEN1 and TSC1 were uniquely found mutated in the son, which could be candidates as new modifier genes; besides, RNF43 was also mutated in public neurofibroma seuquencing data. By KEGG pathway annotation, phosphoinositide-3-kinase-Akt pathway was altered in both the public plexiform neurofibroma sample and in our proband patient.
Conclusion: This study reexamined the background germline mutations and suggested their potential value as modifier genes that may influence the phenotype heterogenity.
Keywords: plexiform, neurofibroma type 1, mutation, modifier gene, next-generation sequencing
Introduction
Neurofibromatosis type 1 (NF1, MIM #162200) is one of the most prevalent heritable multisystem disorders; it is caused by the mutation of the NF1 gene, which produces a protein called neurofibromin. Nearly half of the NF1 cases are sporadic.1 Both the population prevalence and birth prevalence are ~1 in 3,500.2 In contrast with neurofibromatosis type 2, which is characterized by bilateral acoustic neuroma and meningioma in the central nervous system, the NF1 lesions generally appear in the peripheral nervous system or organs other than nerves. So far, consensus diagnosis criteria for NF1 have been established by the National Institutes of Health and accepted universally, and any individuals who meet at least two of the following criteria should be diagnosed with NF1:3
- Six or more café-au-lait spots (CLS) >5 mm in the greatest diameter in prepubertal individuals and >15 mm in the greatest diameter in postpubertal individuals.
- Two or more neurofibromas of any type or one plexiform neurofibroma (PNF).
- Freckling in the axillary or inguinal region.
- Two or more Lisch nodules (iris hamartomas).
- Optic glioma.
- A distinctive osseous lesion such as sphenoid dysplasia and thinning of long bone cortex, with or without pseudarthrosis.
- A first-degree relative (parent, sibling, or offspring) with NF1 by the above criteria.
PNF is referred to as a subtype of neurofibroma in which Schwann cells proliferate inside the nerve sheath, leading to an irregularly thickened, distorted, winding structure. It is also characterized by unencapsulated, poorly circumscribed neoplasms that infiltrate the nerves and adjacent fat and muscles.
In this study, we studied a young NF1 proband patient and his NF1 father, who presented with torso PNF and dermal neurofibroma, respectively. The somatic mutations of the plexiform lesions of the patients, as well as the germline mutations based on blood, were detected by whole-exon sequencing-based mutation panel detection. The aim of this study is to further understand the molecular characteristics of PNF and to unravel the reasons for the heterogeneities between familial NF1 patients.
Patients and methods
Patients
An 18-year-old Chinese male was admitted with complaints of multiple palpable radical nodules on both sides of the neck for 1 year. The nodules were detected by haphalgesia and were more obvious when the patient was agitated but he presented with no cough, nausea, vomiting, fever, giddiness, or fatigue. In addition, palpable nodules could be found in both inguinal regions without any haphalgesia. All palpable nodules had clear boundaries with nearby tissues. Freckling could be found in inguinal and auxiliary regions (Figure 1A), and café-au-lait macules were located on nearly all parts of the body, especially the back (Figure 1B), while no dermal neurofibroma was found in any area of skin.
 | Figure 1 The clinical manifestations of the proband NF1 son and his father. |
In addition to the proband young male, his 54-year-old father was a potential NF1 patient. The first noticeable abnormal lesion was a slowly growing brown lesion found on the front of the right thigh since his adolescence (Figure 1D), which had been surgically removed 23 years ago. There were also CLS all over the body, and afterward several slow growing nodules were detected, especially on the trunk, popliteal space, and face (Figure 1C and E). However, no lesions on the body showed haphalgesia, and no subcutaneous nodules like his son’s were found on either side of the neck or the inguinal regions.
Chest-enhanced computed tomography (CT)-revealed nodules were found in the subcutaneous region of the anterior chest wall, mediastinum, heart, right lung hilus, beneath pleura, spleen hilus, and abdominal aorta, with variable size and solid or cystic properties (Figure 2, enhanced CT of son). In addition, the nodules ran along the lymph nodes and all the way to the abdomen and were not enhanced by contrast medium. Afterward, the positron emission tomography (PET)–CT showed soft-tissue nodules in both sides of submental area, sternocleidomastoid regions, posterior cervical space and supraclavicular fossa, splenogastric ligament region, quadratus lumborum regions, psoas major regions, pelvic regions, sacral cavities, spleen hilus region, para-pancreatic region, para-abdominal aorta regions, mesenteric regions, presacral space, and right external abdominis muscle, none of which showed abnormal increased tracer uptake (Figure 3). All this evidence supported a diagnosis of benign PNF. To identify whether the father also suffered from similar plexiform lesions, a chest and abdomen CT scan was performed, which showed no plexiform lesions as with the proband patient’s (Figure 2, right column).
 | Figure 2 The CT scan and enhanced CT scan of the son and CT scan of the father. |
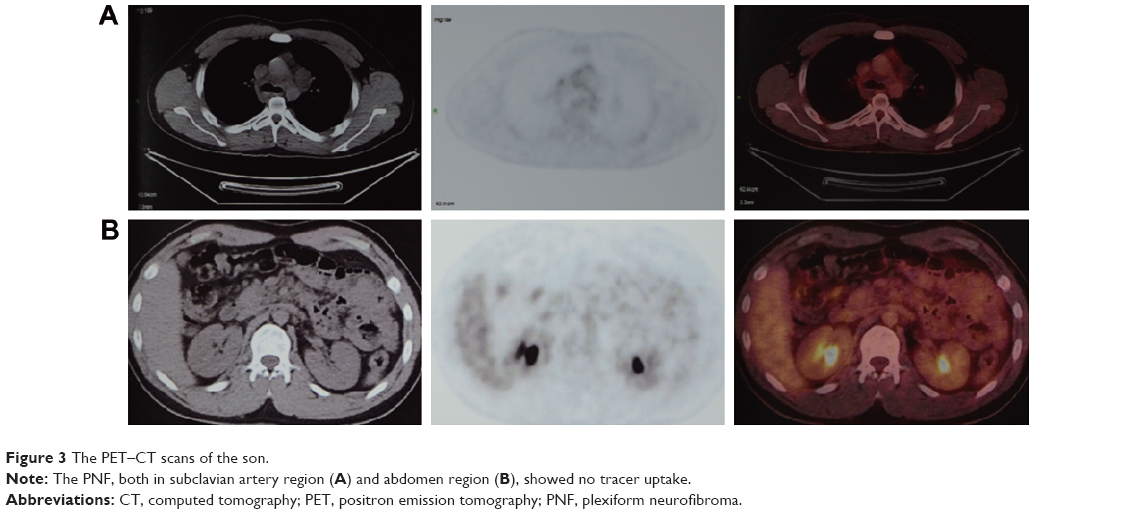 | Figure 3 The PET–CT scans of the son. |
Therefore, a right neck nodule biopsy was performed, and the specimen, with a volume of 0.8×0.3×0.4 mm3, was sent for pathological analysis. The pathology report showed that the nodule was in accord with neurofibromatosis. Immunohistochemistry showed that the specimens were positive for S100, B-cell lymphoma 2 (BCL2), and hematopoietic progenitor cell antigen (CD34) and negative for smooth muscle actin (SMA) and desmin.
Mutation analysis
The blood samples of both patients, as well as the neck nodule specimen, were sent to Burning Rock Biotech (Guangzhou, China) to detect the mutations of a panel of 295 tumor-related genes based on next-generation sequencing (OncoScreen™) and the germline mutations, while the tissue sample from the proband patient’s neck nodules was assessed for detection of the somatic mutations of PNF. Using the online multiple protein sequences’ alignment tool COBALT (https://www.ncbi.nlm.nih.gov/tools/cobalt/cobalt.cgi?CMD=Web), we analyzed the conserved domain among Canis lupus familiaris (XP_013971967.1 dog), Homo sapiens (NP_001035957.1 human), Rattus norvegicus (NP_036741.1 Norway rat), Gallus gallus (XP_015151338.1 chicken), Danio rerio, (XP_017206680.1 zebrafish), and Drosophila melanogaster (NP_733132.2 fruit fly) and then analyzed whether the NF1 mutations were located in the conserved region of human NF1 protein.
Next, the online Condel databases (http://bg.upf.edu/fannsdb/queries/) were used for batch prediction of the pathogenicity of the single-nucleotide substitution mutations (single-nucleotide variants [SNVs]) in a manner integrating the prediction powers of Sorting Intolerant from Tolerant (SIFT; http://sift.bii.a-star.edu.sg/), PolyPhen-2 (PPH2; http://genetics.bwh.harvard.edu/pph2/), FatHMM (http://fathmm.biocompute.org.uk/cancer.html), and MutationAssessor (MA; http://mutationassessor.org/r3/howitworks.php).4–8 As the Condel scores consist of a weighted average of the scores of MA and FatHMM, the Condel scores were used to represent an integrated score for MA and FatHMM. Next, all the SNVs, meeting either the criteria that SIFT scored no greater than 0.5, PPH2 scored no <0.5, and Condel scored no <0.3, or labeled with “deleterious”, were collected as the potential damaging/pathogenic mutations. Then, specific groups of mutated genes were functionally annotated on Database for Annotation, Visualization and Integrated Discovery (DAVID; https://david.ncifcrf.gov/) for KEGG pathway.
In order to reinforce the analysis for modifier genes, all the published data of accompanied mutations in PNF were screened from PubMed with the retrieval type as (((mutation) OR sequence) OR sequencing) AND ((((((von Recklinghausen [title/abstract]) OR neurofibroma [title/abstract]) OR neurofibromatosis type 1 [title/abstract]) OR neurofibromatosis [title/abstract]) OR Neurofibromatosis 1 [MeSH terms]) AND plexiform). Besides, only mutation data that met the following criteria were collected: 1) the patients should be diagnosed as NF1; 2) the patients should present with PNF, and all patients with malignant peripheral nerve sheath tumor (MPNST) are excluded; and 3) the mutations should be SNV instead of micro deletion or large chromatin truncation. Next, the published accompanied mutations were predicted for pathogenicity on Condel and functional annotated on DAVID as described earlier, and the ones which met the same requirements were intersected with the son-only mutation group to find the potential modifier genes. All the procedures in mutation analysis were concluded and are illustrated in the figure of work flow (Figure 4).
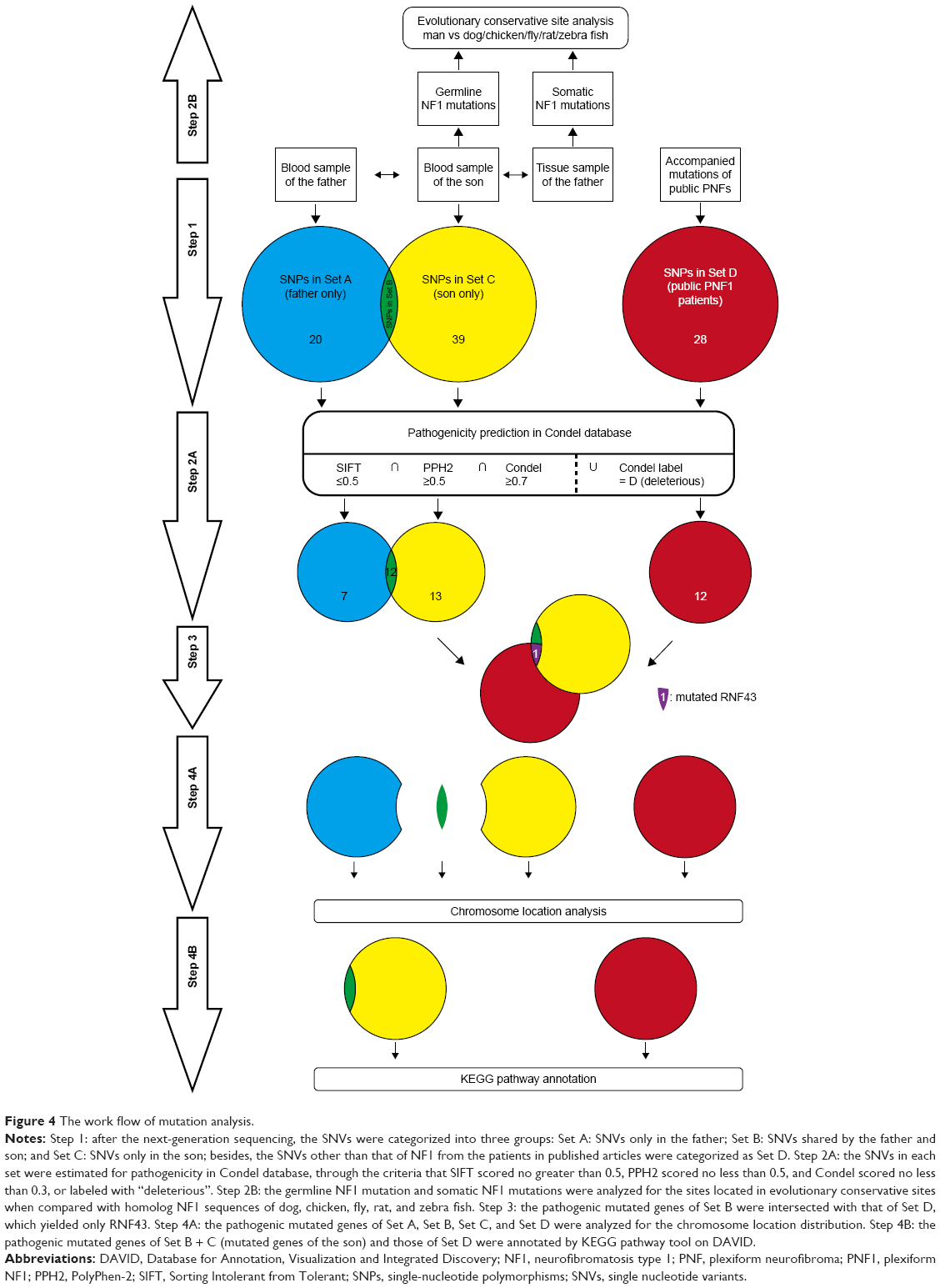 | Figure 4 The work flow of mutation analysis. |
Ethical approval and consent
All procedures performed in studies involving human participants were in accordance with the ethical standards of the clinical research ethical review board at the Tianjin Medical University General Hospital, and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. Written informed consents were obtained from the patients for publishing the detailed information of the two patients involved and the accompanying images.
Results
Here, two sets of germline mutations were acquired and intersected for the shared mutations (Set B), as well as father-only mutations (Set A) and son-only mutations (Set C). Meanwhile, the SNPs of the NF1 patients in published articles are categorized as Set D. In Set B, the NF1 mutations would be regarded as a potential dominant mutation for the two patients. Among the 104 germline mutations of the father and the 127 germline mutations of the son, 82 mutations are shared (Set B), of which 77 SNVs account for the majority, and the only NF1 mutation is a frameshift one. There are 20 and 29 SNVs in Set A and Set C, respectively. Using batch queries of the Condel database, seven, 12, and 13 SNVs were predicted as probably damaging in Set A, Set B, and Set C, respectively (Table 1). Similarly, from all 28 SNVs collected from published articles, which are regarded as Set D, 12 are predicted as pathogenic (Table 2).9–11 Interestingly, when intersecting the Set D with Set C, RNF43 was mutated in the proband of our study and in one PNF patient in the study by Pemov et al.9 Furthermore, the pathogenic mutated genes in Set D and those in Set C + Set B (pathogenic mutated genes of the son) were annotated in DAVID database for KEGG pathway analysis, as illustrated in Table S1, and the “phosphoinositide 3-kinase (PI3K)–Akt signaling pathway” is the only altered pathway shared by the abovementioned proband and published PNF sample PN9.
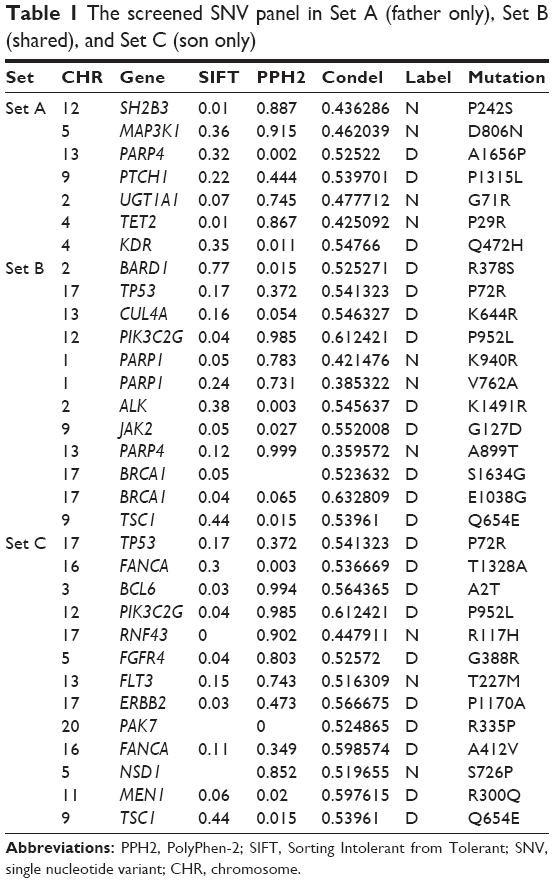 | Table 1 The screened SNV panel in Set A (father only), Set B (shared), and Set C (son only) |
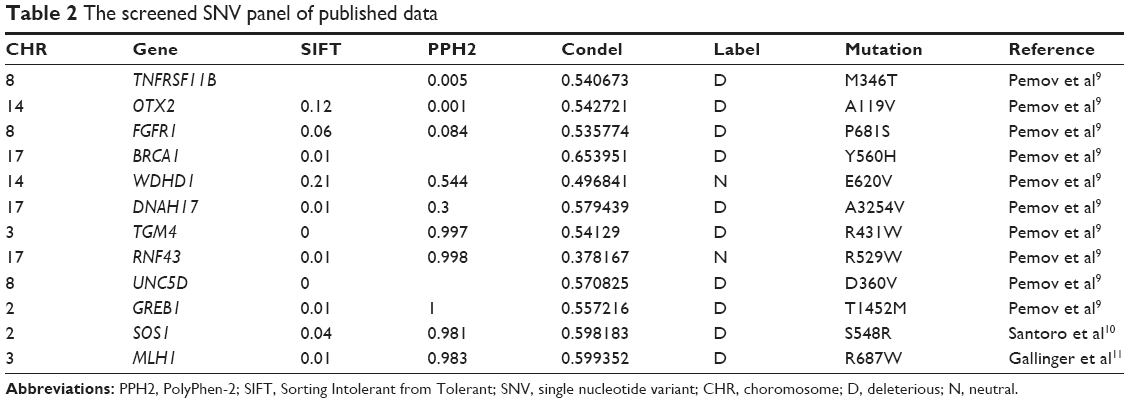 | Table 2 The screened SNV panel of published data |
Importantly, both cases contain an NF1 c.6852_6855delTTAC (p.Tyr2285fs) mutation, with the occurrences in both cases at ~50%. After aligning the six protein sequences for the conserved region, this shared NF1 frameshift mutation was discovered to be located only in a conserved region after a short variable sequence, which is “CLKGPDT” in humans (Figure S1C). This germline mutation is interpreted as “pathogenic” in the PubMed ClinVar database, with two submitted evidence for NF1 that are also reported in glioma and breast cancer in the COSMIC database (http://cancer.sanger.ac.uk/cosmic). Meanwhile, in the proband patient’s neurofibroma nodule specimen, only two somatic mutations, NF1 c.6435_6439delCAAGC (p.Lys2146fs) and NF1 c.6435_6439delCAAGC (p.Lys2146fs), were detected, with frequencies 9.3 and 3%, respectively, both of which also lie in the conserved regions in species-coding sequence comparison (Figure S1A and B). No tracking of these two NF1 mutations were found in COSMIC, and their low frequencies indicated that these two mutations may not be primary dominant initiating mutations in this PNF case.
Discussion
NF1 gene and neurofibromin protein
The NF1 gene was explored as the causative gene of NF1, and its location was identified in 1990 at 17q11.2.12 The NF1 gene spans ~282 kb of genomic sequence and contains 62 exons. It is expressed in nearly all kinds of health issues, but in some systems, including the central and peripheral nervous systems, endocrine glands, and female reproductive system, it is highly expressed (Figure S2). The protein that NF1 encodes is named neurofibromin, which so far has been found as three isoforms, isoform 1: NP_001035957.1 (2839 amino acids, aa), isoform 2: NP_000258.1 (2818 aa), and isoform 3: NP_001121619.1 (593 aa). NF1 has been widely accepted as a tumor suppressor, and neurofibromin plays an indispensable role in proliferation-related pathways such as the RAS/MAPK pathway and mTOR pathway.
So far, the public mutation database has collected 1,199 NF1-coding sequence mutations from all kinds of neoplasm samples. From the lollipop figure of mutations on neurofibromin generated by the MutationMapper, it can be concluded that the mutations are comparatively evenly distributed, without any apparent hot-spot regions (Figure 5B). As a matter of fact, the missense, synonymous, and nonsense mutations constitute the majority of all mutation types (Figure 5A). In our cases of NF1, the only NF1 germline mutation is c.6852_6855delTTAC on NM_001042492.2, which is not located in any known domains on neurofibromin isoform 1: cd05130 (RasGAP_Neurofibromin; Ras-GTPase activating domain of neurofibromin, location: 1198→1551), smart00323 (RasGAP; GTPase-activator protein for Ras-like GTPases, location: 1187→1557), cd13313 (PH_NF1; neurofibromin-1 pleckstrin homology-like domain, location: 1728→1837), or pfam13716 (CRAL_TRIO_2; divergent CRAL/TRIO domain, location: 1602→1728). This mutation is recorded in ClinVar with a variant ID 216866 and is interpreted as a pathogenic germline mutation. Therefore, it can be deduced that the alteration of this unknown evolutionary conservative site may also be detrimental to the neurofibromin function.
 | Figure 5 The NF1 mutation categories and distribution. |
Mechanism behind NF1
Up till now in the literature, the high allelic heterogeneity of the NF1 mutation could not explain the variability of phenotype, while it could still be concluded that the phenotype severity is correlated with the reading frame truncation degree.13–15 Interestingly, a group of patients with the same 3 bp inframe deletion of the NF1 gene (c.2970_2972delAAT) shared a common phenotype of the absence of cutaneous neurofibromas, indicating that an inframe deletion may lead to a comparatively mild form of NF1.16 Similarly, another study by Pinna et al17 demonstrated that unrelated proband patients with the heterozygous c.5425C>T missense variant (p.Arg1809Cys) universally showed multiple CLS and were frequently associated with skinfold freckling, but absence of discrete cutaneous or PNFs, Lisch nodules, typical NF1 osseous lesions, or symptomatic optic gliomas. Meanwhile, the gross deletions of NF1 are closely related to a more severe phenotype in NF1 patients, including learning disability, facial dysmorphia, and developmental delay.18–20
Although NF1 is a typical monogenic syndrome, the germline NF1 gene mutation could not explain all the disease heterogeneity and variable morbidity.21 Gender, other mutated modifier genes, and aging, together with the NF1 truncation itself, could give rise to the differences even among NF1 family members. From the NF1 genetically engineered mice (GEM) models, the optic glioma-associated vision disturbances are confirmed to be more severe in females, while spatial learning impairments occur only in NF1-CKO knockout male mice.22 Afterward, the cyclic AMP pathway was identified as the reason for NF1-related glioma tendency in females and polymorphisms in the human adenylate cyclase gene ADCY8 are in accord with glioma risk in a gender-specific manner.23 Therefore, the ADCY8 is regarded as a modifier gene of glioma risk in NF1.
The term modifier gene of NF1 was defined as “any gene unlinked to the NF1 locus whose genotype modulates one or several features of the NF1 phenotype”.24 The concept of modifier gene was first inspired by the phenomenon in which nearly three-quarters of NF1 families revealed remarkable phenotypic differences among individuals.25 Briefly, the following two strategies are established to explore the potential modifier genes in NF1: the phenotypic approach that started from morphological contrast, and hypothesis-driven approach that first focuses on a set of carefully chosen candidate genes. To investigate the contribution of the modifier genes to phenotypes, the following was hypothesized: if the variance is dominantly determined by NF1 mutation itself, the phenotype correlation for different individuals within the same family should be the same regardless of the blood relativeness; oppositely, the phenotypic correlation will decrease with the degree of relationship.24 The first modifier gene alteration was found to be a common deletion region in 9p21.3, including the CDKN2A-CDKN2B-ARF gene cluster and single-nucleotide polymorphism (SNP) rs2151280 located in ANRIL.26 The allele T of rs2151280 is statistically significantly associated with reduced levels of ANRIL transcript, a long noncoding RNA that interacts with polycomb repressive complex (PRC)-1 and -2, and silencing the genes in the gene cluster mentioned earlier.26
Aging is another factor influencing the NF1 clinical characteristics. Diagnosing small children is often challenging because nearly half of the sporadic NF1 cases fail to reach the National Institutes of Health diagnostic criteria when patients are younger than 1 year.27 Typical NF1 features such as axillary and inguinal freckles often begin to appear at the age of 3–5 years and are visible at the age of 7 years. Similarly, another NF1 feature, Lisch nodules, is not symptomatic until the patient reaches 5–6 years, while >90% of adult NF1 patients have Lisch nodules.24 As a Mendelian disorder, NF1 patients harboring a heterozygous germline mutation develop neurofibroma upon somatic mutation of the wild-type second allele, which leads to the loss of heterozygosity (LOH) of the whole NF1 gene and makes the NF1−/- Schwann cells the initiating cells for neurofibroma formation.28 Furthermore, it is reported that in plexiform NF1, LOH of the TP53 gene also plays a role in the oncogenesis, which is comparatively rare in simply neurofibroma.29 In conclusion, the development of NF1 can be summarized as follows: the NF1 is first initiated by the inherited germline mutation or early-year mosaic mutation of NF1, which somehow enhances the overall somatic mutation rate and then leads to the LOH of HF1 in Schwann cells, which initiates the neurofibroma formation in a favorable NF heterogeneous environment, and some accompanying alterations of modifier genes such as ANRIL, TP53, and ADCY8 also influence the clinical phenotypes or even boost the progress of tumorigenesis toward the malignant form of MPNST.
Potential modifier genes discovered in this study
In this study, a group of pathogenic mutated genes, including TP53, FANCA, BCL6, PIK3C2G, RNF43, FGFR4, FLT3, ERBB2, PAK7, NSD1, MEN1, and TSC1, were uniquely found in the son, which could be potential new modifier genes for the phenotype of PNF. Similarly, SH2B3, MAP3K1, PARP4, PTCH1, UGT1A1, TET2, and KDR were specifically only mutated in the father. In order to extend the range of potential modifier genes, published studies of plexiform neufibroma were reviewed and screened for the concomitant mutated genes, other than NF1. The inclusion criteria was defined as PNF only so as to decreasing misleading pathogenic mutations From the limited three studies, 28 genes were mutated, 12 of which were predicted as pathogenic in our defined method.9–11 Any recurrent mutations among public data and our sequencing data were believed to have more possibility to be dominant modifier genes. Here, during our attempt to explore potential modifier genes, we found three inspiring findings. First, RNF43, the key modulator of WNT–β-catenin pathway, was mutated in both the amino acid site 117 and 529 in our proband patient and in the PN9 sample in Pemov et al’s9 study. WNT–β-catenin pathway is a highly conserved pathway that dominants various key cellular procedures such as proliferation, differentiation, migration, apoptosis, and genetic stability; and ectopic WNT pathway is causative in multiple growth-related diseases and, more importantly, cancer.30,31 Next, by the KEGG pathway annotation, both the PN9 sample from published studies and the blood samples of the proband son shared only one common altered pathway, PI3K–Akt pathway. This is one of the main principle growth factor signaling transduction pathways, and once it is dysregulated, many biological procedures such as cell growth, proliferation, migration, metabolism, and survival are altered and lead to canceration.32 At last, the chromosome 17 seems to be the most frequent location of SNVs no matter in each set (Figure 5C). This phenomenon features in two PNF studies, both of which reported the loss of the long arm of chromosome 17.33,34 Based on these three findings, the following three possible characteristics of modifier genes in NF1 can be predicted:
- The modifier genes could be a set of genes that play vital roles in signaling pathways.
- The modifier genes may be involved in the pathways other than the NF1-Ras-mTOR pathway.
- The modifier gene locations may have some preference, especially in chromosome 17; bulk truncation on chromosome 17 could be another positive factor.
In this study, we started with the phenotypic heterogenity, categorized the germline SNV, predicted the pathogenicity in Condel for pathogenicity prediction, and annotated the mutated genes in DAVID. These are simple and intuitive methods attracting the attention on the non-NF1 germline-mutated genes, which could be underlying modifier genes, and are usually neglected in distinguishing the somatic mutations and regarded as “genetic background noise”. Our strategy of taking full advantage of both the germline mutation reports and public sequencing results is an economic way to explore potential factors of neoplasm phenotypes. However, we have to admit that the quantity of the sequencing data is limited and other methods such as SNPs chip and genome-wide association studies (GWASs) are also indispensible; moreover, in vitro and in vivo confirmative experiments are desperately needed. Although in its infancy, the “modifier gene” concept could help to decipher the mystery of tumor phenotype heterogeneity and greatly benefit personalized precision medicine.
Acknowledgments
The authors are grateful to the two patients for their participation in this study. This work was partially supported by grants from the National Natural Science Foundation of China (81773207, 81772464, and 81372306), the Science and Technology Support Key Program of Tianjin (17YFZCSY00840), and Tianjin Key Project of Natural Science Foundation (16JCZDJC34200, 16PTSYJC00160, and 17JCZDJC36200). The funders had no role in study design, data collection and analysis, decision to publish, or in the preparation of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Clementi M, Barbujani G, Turolla L, Tenconi R. Neurofibromatosis-1: a maximum likelihood estimation of mutation rate. Hum Genet. 1990;84(2):116–118. | ||
Evans DG, Howard E, Giblin C, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A. 2010;152A(2):327–332. | ||
Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol. 1988;45(5):575–578. | ||
González-Pérez A, López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88(4):440–449. | ||
Kumar D. Disorders of the genome architecture: a review. Genomic Med. 2008;2(3–4):69–76. | ||
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. | ||
Shihab HA, Gough J, Cooper DN, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34(1):57–65. | ||
Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39(17):e118. | ||
Pemov A, Li H, Patidar R, et al. The primacy of NF1 loss as the driver of tumorigenesis in neurofibromatosis type 1-associated plexiform neurofibromas. Oncogene. 2017;36(22):3168–3177. | ||
Santoro C, Giugliano T, Melone MAB, et al. Multiple spinal nerve enlargement and SOS1 mutation: further evidence of overlap between neurofibromatosis type 1 and Noonan phenotype. Clin Genet. 2018;93(1):138–143. | ||
Gallinger S, Aronson M, Shayan K, et al. Gastrointestinal cancers and neurofibromatosis type 1 features in children with a germline homozygous MLH1 mutation. Gastroenterology. 2004;126(2):576–585. | ||
Cawthon RM, O’Connell P, Buchberg AM, et al. Identification and characterization of transcripts from the neurofibromatosis 1 region: the sequence and genomic structure of EVI2 and mapping of other transcripts. Genomics. 1990;7(4):555–565. | ||
De Luca A, Schirinzi A, Buccino A, et al. Novel and recurrent mutations in the NF1 gene in Italian patients with neurofibromatosis type 1. Hum Mutat. 2004;23(6):629. | ||
Castle B, Baser ME, Huson SM, Cooper DN, Upadhyaya M. Evaluation of genotype-phenotype correlations in neurofibromatosis type 1. J Med Genet. 2003;40(10):e109. | ||
Sabbagh A, Pasmant E, Imbard A, et al. NF1 molecular characterization and neurofibromatosis type I genotype-phenotype correlation: the French experience. Hum Mutat. 2013;34(11):1510–1518. | ||
Upadhyaya M, Huson SM, Davies M, et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am J Hum Genet. 2007;80(1):140–151. | ||
Pinna V, Lanari V, Daniele P, et al. p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur J Hum Genet. 2015;23(8):1068–1071. | ||
Upadhyaya M, Ruggieri M, Maynard J, et al. Gross deletions of the neurofibromatosis type 1 (NF1) gene are predominantly of maternal origin and commonly associated with a learning disability, dysmorphic features and developmental delay. Hum Genet. 1998;102(5):591–597. | ||
Mautner VF, Kluwe L, Friedrich RE, et al. Clinical characterisation of 29 neurofibromatosis type-1 patients with molecularly ascertained 1.4 Mb type-1 NF1 deletions. J Med Genet. 2010;47(9):623–630. | ||
Pasmant E, Sabbagh A, Spurlock G, et al. NF1 microdeletions in neurofibromatosis type 1: from genotype to phenotype. Hum Mutat. 2010;31(6):E1506–E1518. | ||
Monroe CL, Dahiya S, Gutmann DH. Dissecting clinical heterogeneity in neurofibromatosis type 1. Annu Rev Pathol. 2017;12:53–74. | ||
Diggs-Andrews KA, Brown JA, Gianino SM, Rubin JB, Wozniak DF, Gutmann DH. Sex is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann Neurol. 2014;75(2):309–316. | ||
Warrington NM, Sun T, Luo J, et al. The cyclic AMP pathway is a sex-specific modifier of glioma risk in type I neurofibromatosis patients. Cancer Res. 2015;75(1):16–21. | ||
Upadhyaya M, Cooper DN. Neurofibromatosis Type 1: Molecular and Cellular Biology. New York: Springer; 2013. | ||
Carey JC, Baty BJ, Johnson JP, Morrison T, Skolnick M, Kivlin J. The genetic aspects of neurofibromatosis. Ann N Y Acad Sci. 1986;486:45–56. | ||
Pasmant E, Sabbagh A, Masliah-Planchon J, et al. Role of noncoding RNA ANRIL in genesis of plexiform neurofibromas in neurofibromatosis type 1. J Natl Cancer Inst. 2011;103(22):1713–1722. | ||
DeBella K, Szudek J, Friedman JM. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics. 2000;105(3 pt 1):608–614. | ||
Laycock-van Spyk S, Thomas N, Cooper DN, Upadhyaya M. Neurofibromatosis type 1-associated tumours: their somatic mutational spectrum and pathogenesis. Hum Genomics. 2011;5(6):623–690. | ||
Sant DW, Margraf RL, Stevenson DA, et al. Evaluation of somatic mutations in tibial pseudarthrosis samples in neurofibromatosis type 1. J Med Genet. 2015;52(4):256–261. | ||
Pai SG, Carneiro BA, Mota JM, et al. Wnt/beta-catenin pathway: modulating anticancer immune response. J Hematol Oncol. 2017;10(1):101. | ||
Nusse R, Clevers H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169(6):985–999. | ||
Rodgers SJ, Ferguson DT, Mitchell CA, Ooms LM. Regulation of PI3K effector signalling in cancer by the phosphoinositide phosphatases. Biosci Rep. 2017;37(1). pii: BSR20160432. | ||
Steinmann K, Kluwe L, Friedrich RE, Mautner VF, Cooper DN, Kehrer-Sawatzki H. Mechanisms of loss of heterozygosity in neurofibromatosis type 1-associated plexiform neurofibromas. J Invest Dermatol. 2009;129(3):615–621. | ||
De Luca A, Bernardini L, Ceccarini C, et al. Fluorescence in situ hybridization analysis of allelic losses involving the long arm of chromosome 17 in NF1-associated neurofibromas. Cancer Genet Cytogenet. 2004;150(2):168–172. |
Supplementary materials
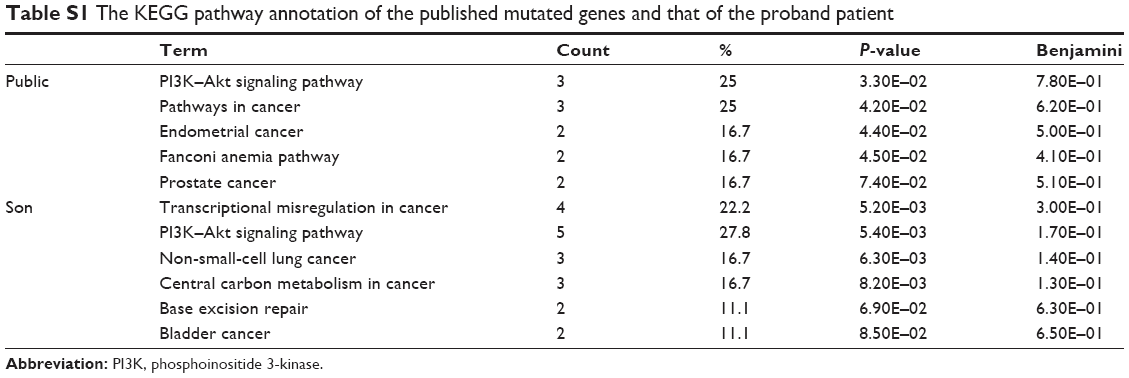 | Table S1 The KEGG pathway annotation of the published mutated genes and that of the proband patient |
 | Figure S1 The location of the two somatic frameshift mutations detected in the PNF (A and B) of the proband son and the germline NF1 mutation shared by the father and son (C). |
 | Figure S2 The NF1 transcription and expression levels in multiple organs as tracked by The Human Protein Atlas database. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.