Back to Journals » Neuropsychiatric Disease and Treatment » Volume 14
The interactive effects of ketamine and ethanol on dopamine expression in the ventral tegmental area of rats
Authors Zhang ZB, Xu TY, You DY, Yi S ![]() , Liu Q, Li HJ, Gu JY
, Liu Q, Li HJ, Gu JY ![]()
Received 23 January 2018
Accepted for publication 13 June 2018
Published 20 August 2018 Volume 2018:14 Pages 2105—2114
DOI https://doi.org/10.2147/NDT.S163449
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Wai Kwong Tang
Zhi-Bi Zhang,1 Tian-Yong Xu,2 Ding-Yun You,3 Shuai Yi,4 Qing Liu,4 Huifang-Jie Li,4 Jin-Yun Gu5
1Biomedical Engineering Center, Kunming Medical University, Kunming, People’s Republic of China; 2Experiment Center for Medical Science Research, Kunming, People’s Republic of China; 3Department of Science and Technology, Kunming Medical University, Kunming, People’s Republic of China; 4School of Forensic Medicine, Kunming Medical University, Kunming, People’s Republic of China; 5Zhanyi Branch of Qujing Public Security Bureau, Qujing, People’s Republic of China
Background: A number of studies have demonstrated the significant and rapid antidepressant effects of ketamine, which is also known as a neurotoxic and illicit drug. Ketamine and alcohol are increasingly used together in clubs by teenagers and young adults. Previous studies have proven that chronic ketamine consumption induces a delayed and persistent activation of the dopamine (DA) system. However, the rewarding properties of recreational ketamine abuse remain unclear, and the underlying mechanisms of the effects on the DA system after administration of ketamine with ethanol are yet to be explored.
Methods: Here, we evaluated the effects of two different doses of ketamine (30 mg/kg and 60 mg/kg) with and without ethanol (0.3156 g/kg) on DA concentration in the rat’s ventral tegmental area (VTA), a vital region in the reward and motivation system. We explored the effects of the combined drug treatment on the expression profiling of the DA metabolism genes, tyrosine hydroxylase, dopa decarboxylase, vesicular monoamine transporter 2, and synaptosomal-associated protein 25, as well as protein expression level of brain-derived neurotrophic factor in the rat’s VTA.
Results: We found that administration of ketamine with ethanol led to a significant increase of DA in the VTA associated with differential regulation of mRNA levels of the four DA metabolism genes and protein levels of brain-derived neurotrophic factor. Moreover, the rewarding properties of coadministration of ketamine and ethanol were related to dopaminergic neuron activation in the VTA.
Conclusion: These results indicated the possibility that combined drug treatment might positively affect the mesencephalic DA reward system.
Keywords: ketamine, ethanol, TH, DDC, dopamine, ventral tegmental area
Introduction
Recent research reported that chronic recreational ketamine consumption, which refers in particular to coadministration with alcohol, has a positive impact on the activity of the cerebellum in both mice and humans; furthermore, its toxic effects could cause liver fibrosis, deteriorated cardiac functions, and kidney dysfunction that leads to reduction in the size of the urinary bladder.1–4 DA system plays a critical role in motor function, motivated behavior, reward association, learning, and mood.5 Focusing on the DA system in rat brain, recently our group has shown that recreational ketamine abuse induces different regulation in the mRNA expression of four genes related to DA metabolism, especially for SNAP25, and protein function of BDNF in the cortex–striatum circuitry.6 We found that significant increase of DA concentration in rats’ striatum could result in drug-paired place preference exhibited by ketamine and ethanol coadministration. However, upregulation of DA-related genes, SNAP25, was observed in the PFC of rats. And behavioral effects of coadministration of ketamine and ethanol were also associated with upregulation of BDNF protein expression in both striatum and PFC. These interesting results helped us better understand the established conditioned place preference of rats and the elevated DA level in the cortex–striatum circuitry and also urged us to explore more.
Even though numerous aspects of neurological damage of drug abuse are needed to be explored, the common neurochemical mechanism of drugs causing abuse in human and other subjects is the increase of the DA released from the VTA to a region in the mesocorticolimbic part of the brain, for example, the NAc and the PEC.7,8 Therefore, we hypothesized that rapid activation of neural mechanism including neurobiological and molecular events in the VTA could be more susceptible to drug abuse. Administration of recreational drugs, such as amphetamine, cocaine, etc., increases DA release in the VTA, which is the core pathway of relapse that causes subsequent addiction after cessation of chronic drug use.9 Drug-seeking behavior, caused by decrease in dopaminergic neuronal activity after withdrawal of amphetamine, could be reversed by an injection of ketamine.10 The synaptic alteration in dopaminergic neurons in the VTA was reported 24 hours after a single injection of cocaine.11
In the CNS, DA levels are largely determined by DA-associated synthesis and transport genes, including TH, DDC, VMAT2, and SNAP25.12–14 A recent report showed that DDC, which encodes aromatic L-amino acid decarboxylase in the substantia nigra and VTA, was critically involved in motor function.15
Therefore, the current study aimed to assess whether administration of ketamine with or without ethanol at different dose induces molecular effects similar to or different from the results observed in the cortex–striatum circuitry. We explored the DA concentration, transcriptional output of the DA-related genes (TH, DDC, SNAP25, and VMAT2), BDNF protein in the VTA, and the activated dopaminergic neuron in the VTA to find the answer. These findings may open new doors to researching the mechanisms underlying ketamine and ethanol interaction in the DA system.
Methods
Animals
Mature male SD rats (200~250 g, Laboratory Animal Centre, KMU, People’s Republic of China) were housed at 22°C–24°C with 45%–55% humidity and a 12-hour alternating light–dark cycle, with access to food and water ad libitum. All animal experiments were performed according to the principles of laboratory animal care established under China’s law and were also approved by the Ethics Committee of KMU.
Drugs administration
Ketamine (Cat# H35020148, Ketamine Hydrochloride Injection, Fujian Gutian Pharmaceutical Co Ltd, Fujian, People’s Republic of China) was administered intraperitoneally once per day for two groups at the sub-anesthetic dose of 30 mg/kg (K30) and the higher dose of 60 mg/kg (K60), according to the suggested dosage.4,16 The other two experimental groups were administrated with ketamine intraperitoneally at the dose of 30 mg/kg (K30+E) and 60 mg/kg (K60+E), and 20% ethanol (Cat# GB/678-2002, Changshu Yangyuan Chemical Industry Co., Ltd, Changshu, People’s Republic of China, analytical reagent [AR]) immediately at a dose of 0.3156 g/kg once a day. The control groups received the same dose of ethanol and saline. The animal models were set as in Table 1. The above mentioned treatments were performed once a day for 30 days. Body weights of the rats were measured weekly and doses were adjusted appropriately. Sixteen rats in each group were decapitated under deep anesthesia on the 31st day. The VTA was dissected on ice and chosen randomly for evaluation of DA levels (n = 6), expression of DA-related genes (n = 3), expression of BDNF (n = 4), and dopaminergic neuron population (n = 3). These evaluations were made using ELISA, qRT-PCR, western blot, and IHC analysis.
 | Table 1 Animal models |
DA determination
DA concentrations were measured by the commercially available Rat DA Research ELISA kit (Cat# CSB-E08660r, Causation Biotech Company Ltd, Wuhan, People’s Republic of China) according to the user’s manual. Samples of fresh brain tissue (100 mg) were rinsed with 1×PBS, homogenized in 1 mL of 1×PBS and stored overnight at −20°C. Two freeze–thaw cycles were performed to break the cell membranes; and the homogenates were centrifuged for 5 minutes at 5,000 g at 2°C–8°C. The supernatant was assayed and stored at −80°C. The samples were centrifuged again after thawing before the assay, and repeated freeze–thaw cycles were avoided. The absorbance of standards and samples were measured using a micro plate reader (MK-3, Thermo Fisher Scientific, Waltham, MA, USA) at 450 nm and a correction wavelength of 570 nm. The concentration of each sample was determined by a four-parameter logistic (4-PL) regression model. The detection range for DA was 0.156–10 ng/mL and the sensitivity was 0.039 ng/mL. All data were obtained from a standard curve, with r > 0.999.
qRT-PCR
qRT-PCR was performed as previously described.17 Each group contained three samples, and each sample was analyzed in triplicate. The dissected brain VTA was flash frozen with liquid nitrogen and kept at −80°C. The frozen brain tissues (50–100 mg) were homogenized in 1 mL TRIzol (Cat# 15596-026, Thermo Fisher Scientific, Waltham, MA, USA), and total RNA was isolated using Small AxyPrep total RNA preparation kit (Cat# AP-MN-MS-RNA-50G, Axygen, Corning, NY, USA), according to the manufacturer’s manual, and the RNA concentration and purity (OD260/280) were measured by ND-1000 Spectrophotometer (Nanodrop Technologies, Inc., Wilmington, DE, USA). First strand cDNA was synthesized using a RevertAid™ First Strand cDNA synthesis Kit (Cat# K1622, Thermo Fisher Scientific) in a 20 μL reaction volume that included 1 μg of template RNA according to the user’s manual. RT-PCR reactions were performed in 96-well plates in a 7900HT Fast Real-Time PCR System (Cat# 4346906, Thermo Fisher Scientific) in a 25-μL mixture containing 12.5 μL Maxima® SYBR Green/ROX qPCR Master Mix (2×) (Cat# K0221, Thermo Fisher Scientific), 1 μL cDNA, 0.75 μL Forward Primer (10 μM), 0.75 μL Reverse Primer (10 μM) and 10 μL nuclease free water, in the following condition: an initial denaturation at 95°C for 10 minutes, followed by denaturation at 95°C for 15 seconds, and annealing/extension at 55°C for 60 seconds, for 35 cycles in total. The primer sequence is shown in Table 2. The primers of TH, DDC, SNAP25, VMAT2, and GAPDH were designed using Primer Premier 5.0 (Premier Biosoft International). Specificity of amplification was confirmed by a melting curve with one peak in PCR reactions. Gene expression changes between the drug and saline groups were calculated according to a 2−ΔΔCT method using GAPDH as an endogenous control, and relative quantifications of mRNA were performed.18
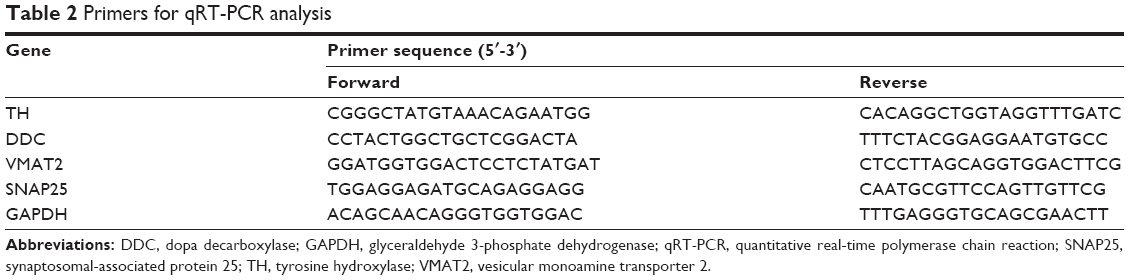 | Table 2 Primers for qRT-PCR analysis |
Western blot
The VTA of the brains of four rats from each group were separated on ice and homogenized in ice-cold radioimmunoprecipitation assay buffer containing 0.1% PMSF. The dissolved proteins were collected from the supernatant after centrifugation at 14,000 g at 4°C for 30 minutes. Protein concentrations were determined using Nanodrop ND1000 Spectrophotometer (Nanodrop Technologies, Inc.). A total of 20 μg of proteins were separated using 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis performed at 80–120 V for 60–80 minutes and then transferred onto a PVDF membrane. The membranes were blocked with 5% skim milk in Tris-buffered saline for 2 hours and then incubated at 4°C overnight with primary antibody: anti-BDNF antibody (1:1,000, Rabbit mAb-100 uL, Cat# 3160-1, Epitomics, Burlingame, CA, USA) and β-Actin (1:1,000, Cat# P30002, ABmart, Berkeley Heights, NJ, USA). Following three washes with TBST, the membranes were incubated with a horseradish peroxidase conjugated secondary antibody (1:3,000, Cat# L3012, Goat anti Rabbit IgG HRP-conjugated) for 2 hours at room temperature. Blots were developed using enhanced chemiluminescence detection reagents (Thermo Fisher Scientific); and the OD values of the protein bands were measured and quantified by a gel imaging system (Tanon Science & Technology Co., Ltd., Shanghai, People’s Republic of China), and the blots were normalized to the respective actin levels.
IHC
The rats were anesthetized and perfused transcardially with 0.9% saline followed by 4% paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.4). Brains were dissected and post-fixed with 4% paraformaldehyde for 24 hours and then embedded in paraffin wax. Sections of the VTA (5 μm each) were cut coronally from paraffin blocks. IHC was carried out as described in the user’s manual, and brain tissue sections were de-waxed and rehydrated before antigen retrieval with sodium citrate buffer (pH 6.0). Sections were treated with 0.3% hydrogen peroxide (H2O2) in methanol for 15 minutes to inactivate endogenous peroxidases. They were then blocked with 1% BSA and 10% normal rabbit serum at room temperature for 1 hour, and then incubated at 4°C overnight with rabbit anti-TH (Cat# AB152, EMD Millipore, Billerica, MA, USA) diluted 1:500 with 1% BSA. On the next day, sections were incubated with solvent 1 (Polymer Helper, Cat# PV900, ZSGB-BIO) at room temperature for 20 minutes, then incubated with solvent 2 (polyperoxidase-anti-mouse/rabbit IgG, Cat# PV900, ZSGB-BIO, Beijing, People’s Republic of China) diluted 1:1,000 in 1% BSA at room temperature for 30 minutes. All slides were washed thrice (2 minutes per wash) with PBS after incubation with H2O2, primary antibody, solvent 1, and solvent 2. Finally, the sections were developed with a DAB kit (Cat# Zli-9018, ZSGB-BIO) under a light microscope at room temperature for 3 minutes, and the reactions were quenched with running tap water. After staining, the sections were dehydrated through an alcohol series, cleared with xylene and mounted. TH immunoreactive cell counts were obtained using a confocal imaging system with 200× magnification (Olympus FV-1000; Olympus). The images were quantified using Image Pro Plus 6.0 software analysis system (Media Cybernetics, Rockville, MD, USA).
Statistical analysis
Simple randomization was used to ensure that each rat had an equal chance of receiving any of the treatments under study. Continuous variables were expressed as the mean ± SEM. Factorial design was used in our experiment, and a two-way ANOVA was used to analyze the effect of two different doses of ketamine administration on the DA system in the rat VTA. We also compared the effect of the different ketamine doses when administered with and without ethanol. Tukey’s multiple comparisons post hoc test was used to assess the differences in the DA concentration, gene and protein expression levels in the VTA of the different treatment groups compared with that of the controls. Analyses were performed using Prism 6.0 for Windows. p < 0.05 was considered statistically significant.
Results
Effects of coadministration of ketamine and ethanol on DA concentration in the VTA
As shown in Figure 1, the mean DA concentration of the saline-paired controls was 0.53 ± 0.018 ng/mL. Compared with the control groups, the DA concentration remarkably increased in the K30+E group by 255.9% (1.38 ± 0.307 ng/mL, 95% CI: −1.608 to −0.07666, p = 0.0266), which almost equaled to K60 group (1.56 ± 0.271 ng/mL, 95% CI: −1.794 to −0.2621, p = 0.0051). However, the combined administration using a higher dose of ketamine (60 mg/kg) and 20% ethanol (K60+E) did not affect the DA concentration. These results correspond with our previous publication, in which a lower dose of ketamine (30 mg/kg) can synergistically interact with 20% ethanol to promote the DA release in the striatum,6 and in present work, K30+E treatment significantly increased the DA concentration in the VTA.
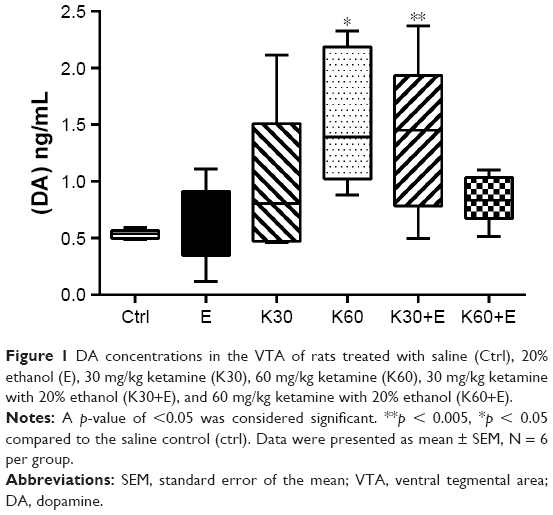 | Figure 1 DA concentrations in the VTA of rats treated with saline (Ctrl), 20% ethanol (E), 30 mg/kg ketamine (K30), 60 mg/kg ketamine (K60), 30 mg/kg ketamine with 20% ethanol (K30+E), and 60 mg/kg ketamine with 20% ethanol (K60+E). |
Effects of coadministration of ketamine and ethanol on mRNA levels of DA-related genes in the VTA
In the quantitative analysis of the mRNA expression levels of DA-associated synthesis genes (TH and DDC) and transportation genes (VMAT2 and SNAP25) in the VTA using qRT-PCR as shown in Figure 2. The K30+E treatment significantly upregulated TH expression in the VTA by 466.67% (3.279 ± 0.78, 95% CI: −4.161 to −0.9918, p = 0.0014) compared with the control treatment. The average TH expression induced by K30+E treatment was higher than any other treatments. This suggested that a low dose of ketamine (30 mg/kg) synergistically worked with 20% ethanol to increase TH expression in the VTA.
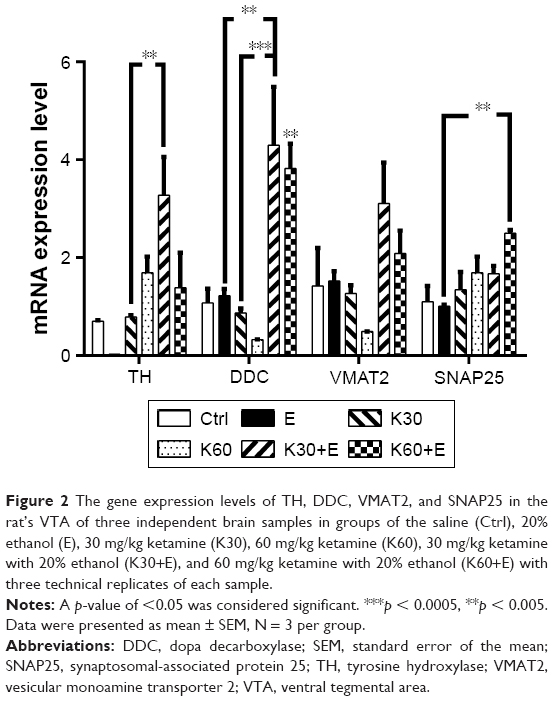 | Figure 2 The gene expression levels of TH, DDC, VMAT2, and SNAP25 in the rat’s VTA of three independent brain samples in groups of the saline (Ctrl), 20% ethanol (E), 30 mg/kg ketamine (K30), 60 mg/kg ketamine (K60), 30 mg/kg ketamine with 20% ethanol (K30+E), and 60 mg/kg ketamine with 20% ethanol (K60+E) with three technical replicates of each sample. |
DDC expression was not altered by ethanol administration and was even downregulated by individual treatment of ketamine both at low (30 mg/kg) and high dose (60 mg/kg). However, it was dramatically upregulated in the K30+E by 398.96% (4.30 ± 1.19, 95% CI: −5.180 to −1.263, p = 0.0012) and in the K60+E by 354.82% (3.824 ± 0.502, 95% CI: −4.705 to −0.7878, p = 0.005) compared with that in the control treatment. This suggested that combined treatment (both K30+E and K60+E) clearly altered DDC expression in the VTA, which, however, was inhibited when a high dose of ketamine was used. These findings indicates that coadministration of ketamine and ethanol clearly alters DDC expression in VTA, which differentiate the present study from our previous publication, in which the same treatment did not interfere DDC expression in PFC and striatum.6
Only did K60+E treatment significantly upregulate SNAP25 expression by 226.20% (2.499 ± 0.066, 95% CI: −2.373 to −0.4151, p = 0.0044) compared with that in the control group. These results were similar to our previous findings that combined treatment of ketamine at a high dose (60 mg/kg) with 20% ethanol promoted SNAP25 expression in the PFC, and other ketamine treatment with or without ethanol did not alter SNAP25 transcript levels.6,8
We also noted that VMAT2 expression in the VTA was not altered by any treatments. TH and DDC transcript levels were exclusively upregulated by K30+E treatment. In conclusion, mRNA expression of the four DA-related genes responded to coadministration of ketamine at a low dose (30 mg/kg) with 20% ethanol, especially the TH and DDC expression.
Effects of coadministration of ketamine and ethanol on BDNF protein levels in the VTA
In order to better understand the effects of the combined ketamine and ethanol on the DA system in the VTA, we explored the BDNF protein expression (Figure 3). BDNF levels were downregulated by K30 treatment, however, K30+E treatment significantly elevated the protein expression by 255.53% (2.272 ± 0.203, 95% CI: 0.03040 to 2.486, p = 0.0432) compared with the control. And BDNF expression was significantly upregulated by combined treatment of ketamine in a dose-dependent manner in the VTA, K60+E treatment enhanced protein expression by 261.44% (2.634 ± 0.008, 95% CI: 0.3921 to 2.848, p = 0.0067) compared with the control. This demonstrated a dose-dependent upregulation of BDNF abundance by combined treatment of ketamine and ethanol. Besides, we also noted that ethanol treatment upregulated the BDNF protein expression by 206.78% (2.067 ± 0.600, 95% CI: −0.1744 to 2.281, p = 0.1110) compared with the control, even though it did not reach statistical significance, which was inferior to the coadministration treatment, as shown by the representative blot images in the Figure 3.
 | Figure 3 BDNF protein expression in the rat’s VTA treated with the saline (Ctrl), 20% ethanol (E), 30 mg/kg ketamine (K30), 60 mg/kg ketamine (K60), 30 mg/kg ketamine with 20% ethanol (K30+E), and 60 mg/kg ketamine with 20% ethanol (K60+E). |
Effects of coadministration of ketamine and ethanol on dopaminergic neurons in the VTA
We evaluated the alteration of dopaminergic neurons in the VTA after different exposure to ketamine using TH IHC. As shown in Figure 4A, in the immunostaining of sections from the VTA in rat brains, TH positive cells were observed, especially in K30, K60, K30+E, and K60+E groups. Average OD value in K30+E and K60+E were higher than administration of ketamine or ethanol only and control (Figure 4B). These results were consistent with BDNF protein expression in the VTA, suggesting that cellular protein abundance of BDNF probably has corresponding effects with activation of dopaminergic neuron. Coadministration of ketamine with ethanol (K30+E and K60+E) significantly increased dopaminergic neurons compared with the other groups.
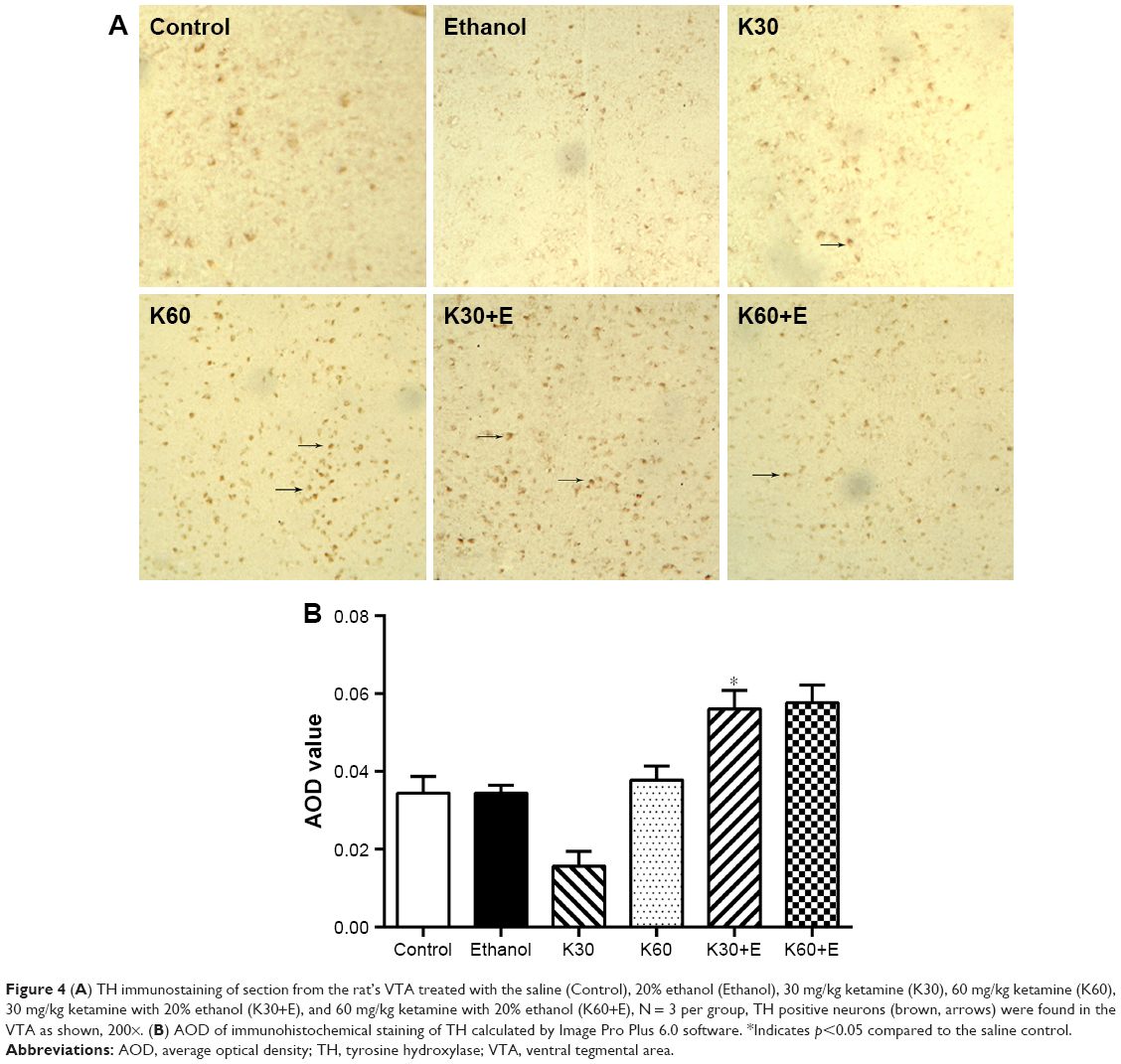 | Figure 4 (A) TH immunostaining of section from the rat’s VTA treated with the saline (Control), 20% ethanol (Ethanol), 30 mg/kg ketamine (K30), 60 mg/kg ketamine (K60), 30 mg/kg ketamine with 20% ethanol (K30+E), and 60 mg/kg ketamine with 20% ethanol (K60+E), N = 3 per group, TH positive neurons (brown, arrows) were found in the VTA as shown, 200×. (B) AOD of immunohistochemical staining of TH calculated by Image Pro Plus 6.0 software. *Indicates p<0.05 compared to the saline control. |
Discussion
Besides an antidepressant, ketamine is also a recreational drug. Drug abusers take ketamine by inhaling (60–250 mg), consuming orally (200–300 mg), injecting intramuscularly (75–125 mg), or intravenously (50–100 mg).19 Available data indicate that recreational dose of ketamine orally consumed can start at approximately 100 mg20 and long-term recreational dose increases based on the abuser’s psychological dependence and tolerance.21
The dopaminergic neurons in the VTA play a critical role in the reward system, and the glutamatergic–dopaminergic pathway interactions are believed to contribute to addiction-related behavior in ketamine abusers.22 Self-administration of ketamine significantly decreases the ratio of p-GSK-3β and t-GSK-3β in the VTA, which is thought to be implicated in the neurochemical mechanism underlying ketamine-induced neuronal toxicity and behavioral disturbance.23 Considering that ketamine and ethanol are often consumed together, sufficient exploration of their direct and indirect effects on the neural pathways in the VTA is needed.
It was reported that cocaine (10 mg/Kg i.p.) induced an increase of DA extracellular levels in VTA of control rats;24 and withdrawal of cocaine for 7 consecutive days induced a reduction in DA extracellular levels in VTA and substantia nigra pars compacta in response to a cocaine challenge.25 Similarly, in vivo microdialysis experiments demonstrated that appetitive rewards promoted DA release in the NAc via the pathway receiving projections from the VTA.26 From the present study, we noted a significant increase of DA concentration in the VTA after being treated with K30+E (Figure 1), which correlated with our published findings that coadministration of 30 mg/kg ketamine and 20% ethanol elevated the DA concentration in the striatum of rats,6 suggesting combined treatment of ketamine at a low dose and ethanol probably induced dopaminergic hyperactivity. However, few effects of ethanol on the alteration of the DA concentration were observed. Application of ethanol may increase the phosphorylation levels of NMDA receptors mediated by activating signaling pathways, and acute ethanol exposure might affect the inhibitory effects of ketamine.27 Although ethanol did not induce DA efflux at this dose (0.3156 g/kg),28 ketamine and ethanol did have overlapping mechanisms of activation of midbrain dopaminergic transmission. And effects of an anesthetic dose of ketamine on glutamate were enhanced when ketamine was administered together with ethanol.
TH is a rate-limiting enzyme in DA biosynthesis and its activity plays an important role in determining DA concentrations.29 Upregulation of TH expression was found in animals either acutely or chronically treated with other abusive drugs such as morphine and phencyclidine.30,31 Repeated exposure to ethanol increased levels of TH and enhanced the firing rate of the VTA DA neurons via an unknown mechanism.12 In this study, we observed that there was a significant upregulation of TH transcriptional expression (4.6 folds) in the VTA following the K30+E treatment (Figure 2), which corresponded with our previous findings that the K30+E treatment elevated the TH expression by 8.16 folds in the PFC of rats.6 The findings suggested that the increase of DA concentration in the VTA after K30+E treatment probably correlated with altered expression of DA synthesis genes.
This hypothesis was further supported by our results that the mRNA levels in DDC gene expression increased more (4.0 fold for K30+E group; 3.5 fold for K60+E group) in the rat’s VTA. Very early reports showed that the L-aromatic amino acid decarboxylase in limbic system structures was involved in the intake of alcohol and could comprise a part of the mechanism underlying the introduction of drinking.32 It was also observed that the brain DA concentration increased in an alcohol-preferring rat model via activation of TH and DDC.33 As research showed, the DDC gene was more susceptible to ethanol than ketamine, also the TPH2 and DDC were suggested as the potential mediators of the serotonin synthesis pathway in alcohol consumption.34 The inhibition of DDC gene expression after coadministration of ketamine at a higher dosage with ethanol mainly explained attenuation of DA release under this dose.
The significant increase of DA levels in the VTA of the K30+E treatment (Figure 1) might also be explained by high BDNF protein expression in the VTA of the same group (Figure 3). BDNF is present in the VTA,35 and its trkB receptors are present in both GABA neurons and DA neurons in the VTA.36,37 Chronic drug abuse upregulates BDNF protein levels in the VTA neurons,35 and a single infusion of BDNF into the VTA shifts the opiate reward system from DA-independent to DA-dependent in opiate-naïve rat model.38 Therefore, we observed a significant increase of BDNF protein levels in the VTA for the K30+E group, suggesting that the BDNF pathway may contribute to drug treatment-induced DA efflux in the VTA. We also noticed that decreased BDNF signaling may participate in NMDAR antagonist (ketamine 75 mg/kg, i.p. or MK801 0.5 mg/kg, i.p.) induced neurotoxicity and neuroapoptosis,39,40 in line with these findings, K30 treatment downregulated the BDNF protein level in the VTA, which indicated neuroapoptosis might occurred after 30 days’ ketamine treatment. The upregulation of BDNF expression induced by combined treatment (2.2 folds for K30+E, 2.6 folds for K60+E) was higher than only ethanol or ketamine given (2.0 folds for E and K60, 0.5 folds for K30), which further explained the interactive effects of ketamine and ethanol in the VTA’s DA system.
We visually evaluated the alteration of dopaminergic neurons in the VTA using TH IHC (Figure 4). Dopaminergic neurons in the VTA are believed to play a critical role in the reward and motivation systems, as the addictive properties of psychostimulant drugs depend on the activation of dopaminergic neurons. As reported, acute withdrawal of amphetamine causes the DA neuronal activity in the VTA to decrease. Whereas, the DA activity in the VTA is enhanced after acute administration of drugs, which is proposed to predispose an individual to seeking and taking drugs.10 It was also reported that DA dysfunctions contributed to anhedonia, a decreased sensitivity to rewarding events,41 and were observed in addicted individuals or animal models of long-term withdrawal.42,43 In the present study, the activation of dopaminergic neurons was also observed after the K30+E treatment, which indicated that the enhanced rewarding properties of ketamine recreational abuse might be related to activation of dopaminergic neurons in the VTA.
However, additional investigation into the NAc core after drug administration is necessary to examine the projection from the VTA dopaminergic neurons. Furthermore, to thoroughly understand the reward properties of ketamine, it is crucial to also investigate how different dosages of ketamine with or without ethanol and different durations of treatment might affect the reward system.
Conclusion
In summary, in the present study, we showed that there was a possible positive activation of the DA neurons in the VTA mediated by upregulation of TH and DDC genes and BDNF protein expression in the K30+E group. Different coadministration effects on the DA system in the cortex–striatum circuitry6 and the VTA indicated that there were different signaling pathways associated with abusive use of ketamine with or without ethanol. Due to the increasing damage induced by ketamine recreational abuse, more detailed mechanisms underlying the interactive effects of ketamine and ethanol on the CNS need to be explored.
Abbreviations
BDNF, brain-derived neurotrophic factor; BSA, bovine serum albumin; CNS, central nervous system; DA, dopamine; DDC, dopa decarboxylase; GABA, γ-aminobutyric acid; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; IHC, immunohistochemistry; KMU, Kunming Medical University; NAc, nucleus accumbens; NMDA, N-methyl-D-aspartic acid; OD, optical density; PFC, prefrontal cortex; PMSF, phenylmethyl sulfonyl fluoride; qRT-PCR, quantitative real-time polymerase chain reaction; SD, Sprague–Dawley; SEM, standard error of the mean; SNAP25, synaptosomal-associated protein 25; TBST, tris-buffered saline and Tween 20; TH, tyrosine hydroxylase; TPH2, tryptophan hydroxylase 2; trkB, tropomyosin receptor kinase B; VMAT2, vesicular monoamine transporter 2; VTA, ventral tegmental area.
Acknowledgments
This work was supported by the National Nature Science Foundation of China (No 81460213) and the Kunming Medical University Creative Group Program (CXTD201604).
Author contributions
Zhi-Bi Zhang, Tian-Yong Xu, Ding-Yun You, Shuai Yi, and Qing Liu participated in literature search, experimental validation, datamining, bioinformatics analysis and interpretation, creating figures and tables. Huifang-Jie Li and Jin-Yun Gu supervised the study design, critically wrote and edited the manuscript. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work. All authors approved the final version for publication.
Disclosure
The authors report no conflicts of interest in this work.
References
Wong YW, Lam LH, Tang HC, Liang Y, Tan SJ, Yew DT. Intestinal and liver changes after chronic ketamine and ketamine plus alcohol treatment. Microsc Res Tech. 2012;75(9):1170–1175. | ||
Chan WM, Liang Y, Wai MS, Hung AS, Yew DT. Cardiotoxicity induced in mice by long term ketamine and ketamine plus alcohol treatment. Toxicol Lett. 2011;207(2):191–196. | ||
Chan WM, Xu J, Fan M, et al. Downregulation in the human and mice cerebella after ketamine versus ketamine plus ethanol treatment. Microsc Res Tech. 2012;75(3):258–264. | ||
Wai MS, Chan WM, Zhang AQ, Wu Y, Yew DT. Long-term ketamine and ketamine plus alcohol treatment produced damages in liver and kidney. Hum Exp Toxicol. 2012;31(9):877–886. | ||
Barrot M. The ventral tegmentum and dopamine: a new wave of diversity. Neuroscience. 2014;282:243–247. | ||
Liu Q, Xu TY, Zhang ZB, et al. Effects of co-administration of ketamine and ethanol on the dopamine system via the cortex-striatum circuitry. Life Sci. 2017;179:1–8. | ||
Filip M, Zaniewska M, Frankowska M, Wydra K, Fuxe K. The importance of the adenosine A(2A) receptor-dopamine D(2) receptor interaction in drug addiction. Curr Med Chem. 2012;19(3):317–355. | ||
Morales M, Margolis EB. Ventral tegmental area: cellular heterogeneity, connectivity and behaviour. Nat Rev Neurosci. 2017;18(2):73–85. | ||
Bardo MT, Valone JM, Bevins RA. Locomotion and conditioned place preference produced by acute intravenous amphetamine: role of dopamine receptors and individual differences in amphetamine self-administration. Psychopharmacology (Berl). 1999;143(1):39–46. | ||
Belujon P, Jakobowski NL, Dollish HK, Grace AA. Withdrawal from acute amphetamine induces an amygdala-driven attenuation of dopamine neuron activity: reversal by ketamine. Neuropsychopharmacology. 2016;41(2):619–627. | ||
Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411(6837):583–587. | ||
Stuber GD, Hopf FW, Hahn J, Cho SL, Guillory A, Bonci A. Voluntary ethanol intake enhances excitatory synaptic strength in the ventral tegmental area. Alcohol Clin Exp Res. 2008;32(10):1714–1720. | ||
Kaplan GB, Leite-Morris KA, Joshi M, Shoeb MH, Carey RJ. Baclofen inhibits opiate-induced conditioned place preference and associated induction of Fos in cortical and limbic region. Brain Res. 2003;987(1):122–125. | ||
Moaddab M, Haghparast A, Hassanpour-Ezatti M. Effects of reversible inactivation of the ventral tegmental area on the acquisition and expression of morphine-induced conditioned place preference in the rat. Behav Brain Res. 2009;198(2):466–471. | ||
San Sebastian W, Kells AP, Bringas J, et al. Safety and tolerability of MRI-guided infusion of AAV2-hAADC into the mid-brain of nonhuman primate. Mol Ther Methods Clin Dev. 2014;3:14049. | ||
Tan SJ, Chan WM, Wai MS, et al. Ketamine effects on the urogenital system – changes in the urinary bladder and sperm motility. Microsc Res Tech. 2011;74(12):1192–1198. | ||
Tan S, Lam WP, Wai MS, Yu WH, Yew DT. Chronic ketamine administration modulates midbrain dopamine system in mice. PLoS One. 2012;7(8):e43947. | ||
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. | ||
Kalsi SS, Wood DM, Dargan PI. The epidemiology and patterns of acute and chronic toxicity associated with recreational ketamine use. Emerg Health Threats J. 2011;4:7107. | ||
Shram MJ, Sellers EM, Romach MK. Oral ketamine as a positive control in human abuse potential studies. Drug Alcohol Depend. 2011;114(2–3):185–193. | ||
Muetzelfeldt L, Kamboj SK, Rees H, Taylor J, Morgan CJ, Curran HV. Journey through the K-hole: phenomenological aspects of ketamine use. Drug Alcohol Depend. 2008;95(3):219–229. | ||
Aalto S, Hirvonen J, Kajander J, et al. Ketamine does not decrease striatal dopamine D2 receptor binding in man. Psychopharmacology (Berl). 2002;164(4):401–406. | ||
Huang X, Huang K, Zheng W, et al. The effects of GSK-3β blockade on ketamine self-administration and relapse to drug-seeking behavior in rats. Drug Alcohol Depend. 2015;147:257–265. | ||
Sotomayor-Zárate R, Abarca J, Araya KA, Renard GM, Andres ME, Gysling K. Exposure to repeated immobilization stress inhibits cocaine-induced increase in dopamine extracellular levels in the rat ventral tegmental area. Pharmacol Res. 2015;101:116–123. | ||
Zhang Y, Loonam TM, Noailles PA, Angulo JA. Comparison of cocaine- and methamphetamine-evoked dopamine and glutamate overflow in somatodendritic and terminal field regions of the rat brain during acute, chronic, and early withdrawal conditions. Ann N Y Acad Sci. 2001;937:93–120. | ||
Roitman MF, Wheeler RA, Wightman RM, Carelli RM. Real-time chemical responses in the nucleus accumbens differentiate rewarding and aversive stimuli. Nat Neurosci. 2008;11(12):1376–1377. | ||
Keng NT, Lin HH, Lin HR, Hsieh WK, Lai CC. Dual regulation by ethanol of the inhibitory effects of ketamine on spinal NMDA-induced pressor responses in rats. J Biomed Sci. 2012;19:11. | ||
Liu W, Thielen RJ, Rodd ZA, McBride WJ. Activation of serotonin-3 receptors increases dopamine release within the ventral tegmental area of Wistar and alcohol-preferring (P) rats. Alcohol. 2006;40(3):167–176. | ||
Flatmark T, Stevens RC. Structural insight into the aromatic amino acid hydroxylases and their disease-related mutant forms. Chem Rev. 1999;99(8):2137–2160. | ||
Boundy VA, Gold SJ, Messer CJ, et al. Regulation of tyrosine hydroxylase promoter activity by chronic morphine in TH9.0-LacZ transgenic mice. J Neuro sci. 1998;18(23):9989–9995. | ||
du Bois TM, Hsu CW, Li Y, Tan YY, Deng C, Huang XF. Altered dopamine receptor and dopamine transporter binding and tyrosine hydroxylase mRNA expression following perinatal NMDA receptor blockade. Neurochem Res. 2008;33(7):1224–1231. | ||
Miñano FJ, McMillen BA, Myers RD. Interaction of tetrahydropapaveroline with inhibition of dopa-decarboxylase by Ro 4-4602 in brain: effects on alcohol drinking in the rat. Alcohol. 1989;6(2):133–137. | ||
Kiianmaa K, Stenius K, Sinclair JD. Determinants of alcohol preference in the AA and ANA rat lines selected for differential ethanol intake. Alcohol Suppl. 1991;1:115–120. | ||
Agrawal A, Lynskey MT, Todorov AA, et al. A candidate gene association study of alcohol consumption in young women. Alcohol Clin Exp Res. 2011;35(3):550–558. | ||
Bolaños CA, Nestler EJ. Neurotrophic mechanisms in drug addiction. Neuromolecular Med. 2004;5(1):69–83. | ||
Numan S, Seroogy KB. Expression of trkB and trkC mRNAs by adult midbrain dopamine neurons: a double-label in situ hybridization study. J Comp Neurol. 1999;403(3):295–308. | ||
Hyman C, Juhasz M, Jackson C, Wright P, Ip NY, Lindsay RM. Overlapping and distinct actions of the neurotrophins BDNF, NT-3, and NT-4/5 on cultured dopaminergic and GABAergic neurons of the ventral mesencephalon. J Neurosci. 1994;14(1):335–347. | ||
Vargas-Perez H, Ting-A Kee R, Walton CH, et al. Ventral tegmental area BDNF induces an opiate-dependent-like reward state in naive rats. Science. 2009;324(5935):1732–1734. | ||
Li J, Wang B, Wu H, Yu Y, Xue G, Hou Y. 17β-estradiol attenuates ketamine-induced neuroapoptosis and persistent cognitive deficits in the developing brain. Brain Res. 2014;1593:30–39. | ||
Hansen HH, Briem T, Dzietko M, et al. Mechanisms leading to disseminated apoptosis following NMDA receptor blockade in the developing rat brain. Neurobiol Dis. 2004;16(2):440–453. | ||
Der-Avakian A, Markou A. The neurobiology of anhedonia and other reward-related deficits. Trends Neurosci. 2012;35(1):68–77. | ||
Leventhal AM, Kahler CW, Ray LA, et al. Anhedonia and amotivation in psychiatric outpatients with fully remitted stimulant use disorder. Am J Addict. 2008;17(3):218–223. | ||
Der-Avakian A, Markou A. Withdrawal from chronic exposure to amphetamine, but not nicotine, leads to an immediate and enduring deficit in motivated behavior without affecting social interaction in rats. Behav Pharmacol. 2010;21(4):359–368. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.