Back to Journals » Journal of Pain Research » Volume 13
The Interaction Between Spinal PDGFRβ and μ Opioid Receptor in the Activation of Microglia in Morphine-Tolerant Rats
Authors Li Z, Jia X, Peng X, Gao F
Received 24 March 2020
Accepted for publication 7 July 2020
Published 17 July 2020 Volume 2020:13 Pages 1803—1810
DOI https://doi.org/10.2147/JPR.S255221
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Überall
Zheng Li, Xiaoqian Jia, Xiaoling Peng, Feng Gao
Department of Anesthesiology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China
Correspondence: Feng Gao
Department of Anesthesiology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, 1095 Jiefang Ave, Wuhan 430030, People’s Republic of China
Email [email protected]
Purpose: Opioid tolerance remains a challenging problem, which limits prolonged drug usage in clinics. Previous studies have shown a fundamental role of platelet-derived growth factor receptor β submit (PDGFRβ) in morphine tolerance. The aim of this study was to investigate the mechanisms of spinal PDGFRβ activation in morphine tolerance.
Methods: Rats were treated with morphine for 7 days and the effect of drug was evaluated by tail-flick latency test. By using Western blot and real-time PCR, the interaction between μ opioid receptor (MOR) and PDGFRβ in microglia activation, as well as related signaling pathways during morphine tolerance were investigated.
Results: Chronic PDGFRβ agonist could induce microglia activation in spinal cord and decrease the analgesic effect of morphine. PDGFRβ inhibitor suppressed microglia activation during the development of morphine tolerance. Furthermore, antagonizing MOR could effectively inhibit the phosphorylations of PDGFRβ and JNK. Blocking PDGFRβ had no influence on JNK signaling, while JNK inhibitor could decrease the phosphorylation of PDGFRβ.
Conclusion: These results provide direct evidence that repeatedly activating MOR by morphine could induce the transactivation of PDGFRβ via JNK MAPK in spinal cord, which leads to microglia activation during the development of morphine tolerance.
Keywords: mu opioid receptor, platelet-derived growth factor receptor β, microglia, JNK signaling, morphine tolerance
Introduction
Morphine is the most effective and frequently used analgesic for acute and chronic pain. However, prolonged administration of morphine always leads to drug tolerance, in which a higher dose of morphine would be needed to achieve the same analgesic effect. For decades, increasing studies have intended to explore the mechanisms of morphine tolerance, including different signaling pathways,1 opioid receptor desensitization,2 and specific endogenous neuropeptides.3 However, more efficient therapeutic strategies to prevent, attenuate, or reverse morphine tolerance are still needed to be explored.
Microglia plays a significant role in neurodegenerative disorder, neuropathic pain,4 and spinal cord injury.5 During the development of morphine tolerance, microglia could also be activated by various stimuli, such as opioid receptor agonist,6 cytokines,7 and mitogen-activated protein kinases (MAPKs) signaling.8 Previous studies showed that platelet-derived growth factor receptor β submit (PDGFRβ) could be phosphorylated during chronic morphine treatment.9 Although PDGF-BB could activate microglia through PDGFRβ and produce tactile allodynia,10 whether there is a link between activations of microglia and PDGFRβ in the mechanism of morphine tolerance is still unknown.
PDGFR is one of the receptor tyrosine kinases (RTKs) which belong to the enzyme-linked receptors family. RTKs have been reported to be involved in several cellular processes including proliferation, differentiation, migration, and survival.11 Epidermal growth factor (EGF) is another RTK.12 The transactivation of EGF receptor participates in μ opioid receptor (MOR)-mediated activation of extracellular signal-regulated protein kinase.13 Adaptive changes in MOR and EGF receptor signal systems could be both detected after chronic morphine treatment.14 The evidence of PDGFRβ phosphorylation by morphine15 raises the possibility that PDGFRβ might also interact with MOR during the development of morphine tolerance.
In this study, we sought to investigate the interaction between MOR and PDGFRβ and their contributions to microglia activation during the development of morphine tolerance.
Materials and Methods
Animals
Adult male Sprague-Dawley rats (220–240 g) were purchased from Laboratory Animal Center, Tongji Medical College, Huazhong University of Science and Technology. A total of 174 rats were used in this study. Rats were housed in a 22°C±0.5°C, relative humidity 40–60%, standard 12-hour light/12-hour dark cycle, temperature- and humidity-controlled environment with food and water ad libitum. All experimental protocols and procedures were reviewed and approved by the Institutional Animal Care and Use Committee, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, and experiments were carried out in accordance with the National Institutes and Health Guidelines for the Care and Use of Laboratory Animals.
Intrathecal Catheter Implantation
For drug administration, intrathecal catheter was implanted using a lumbar approach, as described previously.16 The sterile polyethylene tube (PE-10; outer diameter 0.5 mm, inner diameter 0.3 mm; Anilab Software & Instruments, China) filled with saline was inserted through L4/L5 intervertebral space, and the tip of the tube was placed at the spinal lumbar enlargement level. After surgery, rats were individually housed, monitored postoperatively, and subjected to infection management. A 7-day recovery period was allowed before the following experiments. Correct intrathecal catheter placement was verified by a temporary motor block of both hind limbs after intrathecal injection of 10 μL of 2% lidocaine. Rats presenting with hind limb paralysis or paresis after surgery were excluded and euthanized by overdosed pentobarbital sodium.
Drug Administration
The drugs used in this study were prepared as follows. Morphine hydrochloride (10 μg/5 μL, Shenyang First Pharmaceutical Factory, China) and PDGFRβ inhibitor imatinib (10 μg/10 μL, LC Laboratories, USA)15 were diluted in saline (Northeast Pharmaceutical Group, China), respectively. Specific JNK MAPK inhibitor SP600125 (50 μg/10 μL, MedChem Express, China)8 and selective MOR antagonist naloxone (10 μg/10 μL, Selleckchem, USA)17 were dissolved in 20% dimethyl sulfoxide (DMSO, Sigma, USA), respectively. PDGF-BB (10 pmol/10 μL, R&D Systems, USA) was dissolved in PBS with 0.1% BSA.15 Imatinib (10 μg), SP600125 (50 μg), or naloxone (10 μg) was intrathecally injected 30 minutes before morphine administration, respectively. PDGF-BB (10 pmol) was intrathecally injected alone, then 10 μL of saline was used to flush the catheter after intrathecal administration of drugs.
Induction of Morphine Tolerance and Behavioral Assessment
Rats were intrathecally administered with morphine (10 μg/5 μL, twice daily) for 7 days to induce chronic morphine tolerance. An equivalent volume of saline was administered to rats in the control group at the same time points. To determine the development of morphine tolerance, thermal pain thresholds in rats were measured by a tail-flick latency test before drug administration and 30 minutes after morphine administration on days 1, 3, 5, and 7.16 The test was repeated three times with an interval of 5 minutes and the mean of three trials was considered as the final latency. The percentage of maximal possible antinociceptive effect (%MPE) was calculated by comparing the test latency before (baseline, BL) and after drug administration (TL) using the following equation: %MPE=[(TL−BL)/(cutoff time−BL)]×100. Rats were handled under similar conditions and tested by a pain testing device three times over a week before commencing experiments.
Western Blots
Under deep anesthesia with 60 mg/kg of intraperitoneal pentobarbital sodium, L3–L5 spinal cord segments of rats were rapidly removed. Proteins were extracted from tissue by using RIPA lysis buffer combined with a mixture of proteinase inhibitors.16 Protein concentration was measured by using the bicinchoninic acid kit. Then 30 μg proteins from each sample were separated on 10% SDS polyacrylamide gel. Electrophoresis was conducted at 60 V constant voltage for each gel. Then proteins were electro-transferred (250 mA, 30 or 120 minutes) to PVDF membranes (IPVH00010, Millipore, Billerica, MA, USA). The membranes were blocked with 5% bovine serum albumin for 2 hours at room temperature and incubated overnight at 4°C with mouse anti-ionized calcium-binding adapter molecule 1 (Iba1) antibody (1:200; Santa Cruz, USA), rabbit anti-p-JNK (Thr180/Tyr183) (1:1000; CST, USA), rabbit anti-phospho-PDGFRβ (1:1000; CST), rabbit anti-p-c-JUN (1:1000; CST), or rabbit anti-GAPDH (1:2000; Aspen, China). After being washed, the membranes were incubated with HRP-conjugated goat anti-rabbit IgG (1:5000, Aspen) or HRP-conjugated goat anti-mouse IgG (1:5000, Aspen) for 2 hours at room temperature. Proteins were finally detected by SuperLumia ECL reagents (Abbkine, China) and a computerized image analysis system (Bio-Rad, ChemiDoc XRSC, USA). Image Lab software (Bio-Rad Laboratories) was used to quantify the intensity of protein blots.
Quantitative Real‑Time Polymerase Chain Reaction (qRT‑PCR)
Under deep anesthesia with 60 mg/kg of intraperitoneal pentobarbital sodium, L3–L5 spinal cord segments of rats were rapidly removed and qRT-PCR was performed as described previously.16 In brief, total RNA was extracted from spinal cord using RNAiso Plus (Takara, Shiga, Japan) according to the manufacturer’s instructions and the reverse transcription procedure was performed to synthesize cDNA. The mRNA expression was examined following the protocols of SYBR Premix Ex TaqTM kit (Takara) on StepOne Real-Time PCR System (Applied Biosystems, USA). Each reaction was performed in triplicate. The mRNA expression was normalized to GAPDH expression. Specific primers for rat Iba1 (Forward: 5ʹ-CAGAAGCCAACTGGTCCCC-3ʹ; Reverse: 5ʹ-TGTCATTAGAAGGTCCTCGGTC-3ʹ) and housekeeping gene GAPDH (Forward: 5ʹ-CGCTAACATCAAATGGGGTG-3ʹ, Reverse: 5ʹ-TGCTGACAATCTTGAGGGAG-3ʹ) were obtained from GeneCopoeia Company (USA). Relative quantification of mRNA was performed by 2−ΔΔCt method.
Statistical Analysis
All data were expressed as mean±SEM. The sample number was selected based on the previous study that using PDGFRβ inhibitor imatinib could reverse morphine tolerance.15 No data point was excluded from statistical analysis in any experiments. All the groups for each experiment signified independent values, and statistical analysis was performed using these independent values. Animal behavior data were analyzed by two-way repeated measure ANOVA (treatment group×time) followed by Bonferroni’s test. These post hoc tests were run only if F-value achieved the level of statistical significance and there was no significant variance inhomogeneity. The results of qRT-PCR and Western blots were analyzed by one-way ANOVA. P<0.05 was considered to be statistically significant. Statistical analyses were performed with IBM SPSS 19.0 (IBM Corporation, USA) and figures were drawn with GraphPad Prism 6 (GraphPad Software Inc. USA).
Results
Chronic Morphine Administration Activates Microglia in Spinal Cord
Rats were intrathecally administered with morphine (10 μg/5 μL) or saline (5 μL) twice daily for 7 days consecutively. Behavioral tests were conducted before drug administration and 30 minutes after the last drug administration on days 1, 3, 5, and 7. The results showed that rats receiving morphine exhibited higher %MPE on days 1 and 3 when compared to the saline-treated rats, while there was no significant difference in %MPE level between morphine-treated and saline-treated rats on days 5 and 7 (Figure 1A).
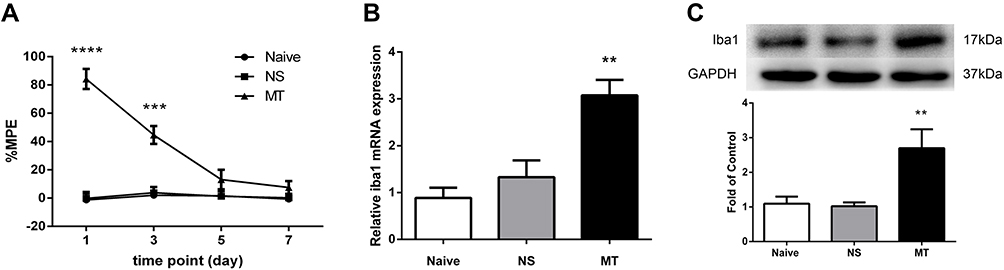 |
Figure 1 Expression of Iba1 in spinal cord of rats. (A) Thermal pain threshold of rats was assessed using the percentage of maximal possible antinociceptive effect (%MPE) according to the tail-flick latency. The %MPE in rats receiving morphine (10 μg, twice daily, intrathecally) on days 5 and 7 were decreased compared with the baseline on day 1. ***P<0.001, ****P<0.0001, vs NS rats. (B, C) The expressions of Iba1 mRNA (B) and protein (C) were significantly increased in morphine-tolerant rats measured by real-time PCR and Western blots, respectively. **P<0.01, vs NS rats. Values represent mean±SEM. n=6 in each group. Abbreviations: NS, normal saline; MT, morphine tolerance; %MPE, the percentage of maximal possible antinociceptive effect. |
To investigate the effect of morphine on microglia activation, the expression of Iba1 in spinal cord was examined. Figure 1B and C show that the levels of Iba1 mRNA and protein in morphine-treated rats were increased when compared to those in saline-treated rats.
PDGFRβ Mediates Microglia Activation During the Development of Morphine Tolerance
To investigate the effect of PDGFRβ activation on microglia, rats were intrathecally administered with PDGF-BB (10 pmol/10 μL) or PBS (10 μL) once daily for 5 days consecutively. Behavioral tests were conducted before drug administration and 30 minutes after the last drug administration. Figure 2A shows that PDGF-BB had no influence on the pain threshold of rats. And the phosphorylation of PDGFRβ and expressions of Iba1 in spinal cord were both increased in rats receiving PDGF-BB (Figure 2B–D), compared to those in PBS-treated rats.
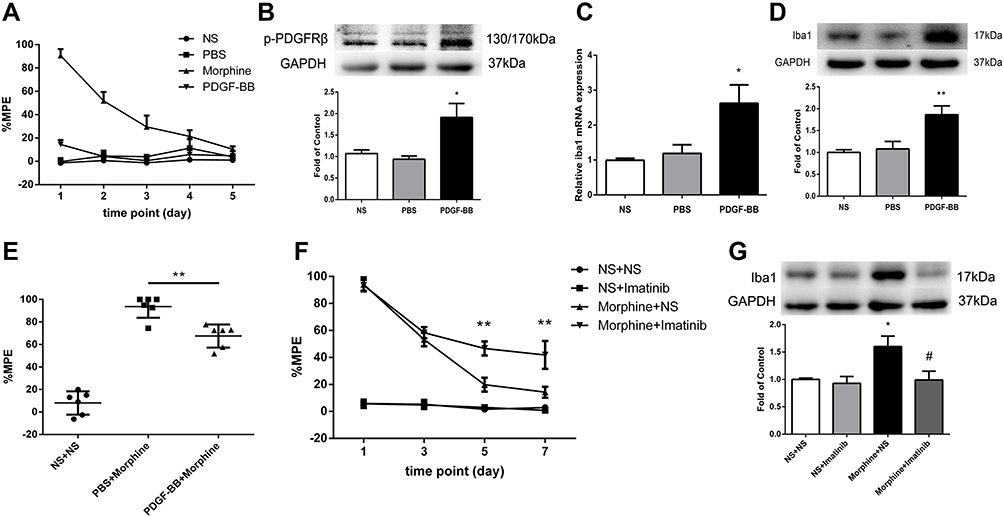 |
Figure 2 The effect of PDGFRβ agonist PDGF-BB on Iba1 expression. (A) PDGF-BB had no influence on %MPE of rats. (B–D) The phosphorylation of PDGFRβ (B) and the expressions of Iba1 mRNA (C) and protein (D) were significantly increased in rats received PDGF-BB measured by real-time PCR and Western blots, respectively. *P<0.05, **P<0.01 vs PBS rats. (E) Rats were intrathecally injected with PDGF-BB or PBS once daily for 4 days, followed by a single dose of morphine on day 5. The %MPE in rats receiving PDGF-BB and morphine was decreased. **P<0.01, vs PBS+Morphine rats. (F) The %MPE in rats receiving imatinib 30 minutes before morphine administration were higher than those in morphine-tolerant rats from day 5 to 7. **P<0.01, vs Morphine+NS rats. (G) Pretreatment with imatinib inhibited the increased expression of Iba1 induced by morphine measured by Western blots. *P<0.05, vs NS+NS rats; #P<0.05, vs Morphine+NS rats. Values represent mean±SEM. n=6 in each group. Abbreviations: NS, normal saline; PBS, phosphate buffered saline; %MPE, the percentage of maximal possible antinociceptive effect. |
We next investigated whether microglia activation induced by PDGF-BB has any influence on the antinociceptive effect of morphine. After intrathecal administration of PDGF-BB (10 pmol/10 μL) or PBS (10 μL) once daily for 4 days consecutively, rats were intrathecal injected with a single dose of morphine on day 5. The %MPE in rats receiving PDGF-BB and morphine was lower than that in rats receiving PBS and morphine (Figure 2E).
To verify the effect of PDGFRβ activation on microglia in morphine tolerance, rats were intrathecally injected with PDGFRβ inhibitor imatinib (10 μg/10 μL) 30 minutes before morphine administration for 7 days. A single dose of imatinib did not affect the antinociceptive effect of morphine on day 1, while consecutive administration of imatinib could significantly attenuate the development of morphine tolerance (Figure 2F). Besides, the increased expression of Iba1 in spinal cord induced by morphine was inhibited by imatinib pretreatment (Figure 2G).
Repeatedly Activating MOR Promotes PDGFRβ Activation via JNK MAPK in Morphine Tolerance
To investigate the interaction between MOR and PDGFRβ, rats were intrathecally injected with MOR antagonist naloxone (50 μg/10 μL) 30 minutes before morphine administration for 7 days. The %MPE in rats treated with naloxone and morphine was higher than those in morphine-tolerance rats from day 5 to 7 (Figure 3A). The phosphorylation of PDGFRβ and the increased expression of Iba1 induced by morphine were both inhibited by naloxone (Figure 3B and C).
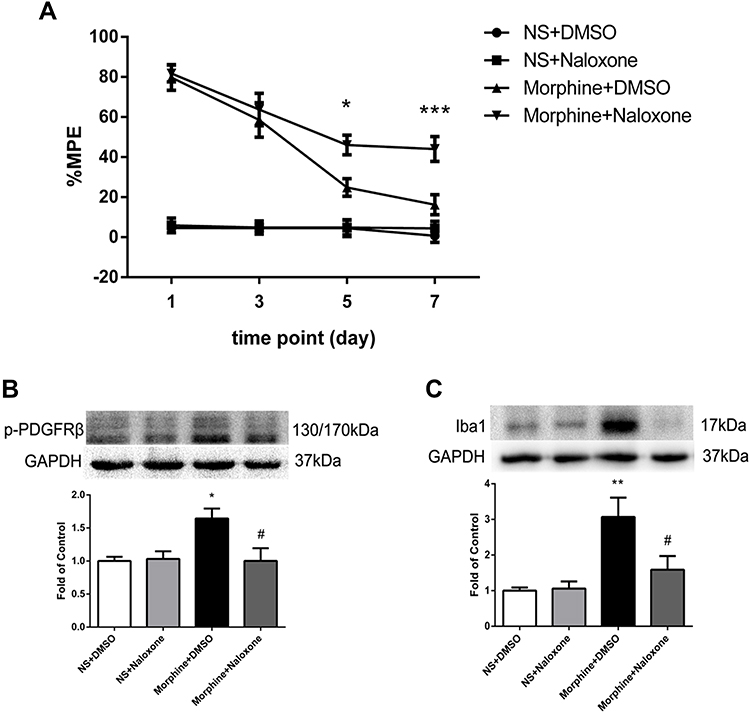 |
Figure 3 The effect of MOR antagonist naloxone on activations of PDGFRβ and microglia. (A) The %MPE in rats receiving naloxone 30 minutes before morphine administration were higher than those in morphine-tolerant rats from day 5 to 7. *P<0.05, ***P<0.001, vs Morphine+DMSO rats. (B, C) Pretreatment with naloxone inhibited the phosphorylation of PDGFRβ (B) and the increased expression of Iba1 (C) induced by morphine measured by Western blots. *P<0.05, **P<0.01, vs NS+DMSO rats; #P<0.05, vs Morphine+DMSO rats. Values represent mean±SEM. n=6 in each group. Abbreviations: NS, normal saline; DMSO, dimethyl sulfoxide; %MPE, the percentage of maximal possible antinociceptive effect. |
To explore the mechanism of MOR-mediated PDGFRβ activation, we then examined the expression of JNK in spinal cord. Figure 4A shows that the phosphorylation of JNK induced by morphine was inhibited by naloxone when compared to that in morphine-tolerant rats.
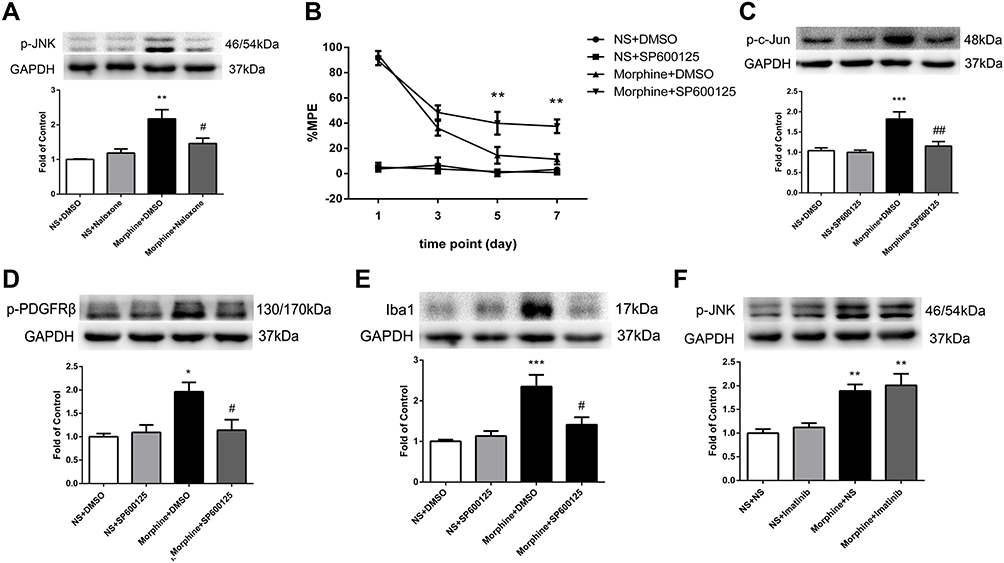 |
Figure 4 Involvement of JNK signaling in MOR-induced PDGFRβ activation in morphine tolerance. (A) Pretreatment with naloxone 30 minutes before morphine administration inhibited the phosphorylation of JNK induced by morphine measured by Western blots. **P<0.01, vs NS+DMSO rats; #P<0.05, vs Morphine+DMSO rats. (B) The %MPE in rats receiving JNK inhibitor SP600125 30 minutes before morphine administration were higher than those in morphine-tolerant rats from day 5 to 7. **P<0.01, vs Morphine+DMSO rats. (C–E) Pretreatment with SP600125 reduced increased expressions of p-c-Jun (C), p-PDGFRβ (D), and Iba1 (E) induced by morphine measured by Western blots. *P<0.05, ***P<0.001, vs NS+DMSO rats; #P<0.05, ##P<0.01, vs Morphine+DMSO rats. (F) Pretreatment with imatinib had no influence on the increased expression of p-JNK induced by morphine measured by Western blots. **P<0.01, vs NS+NS rats. Values represent mean±SEM. n=6 in each group. Abbreviations: NS, normal saline; DMSO, dimethyl sulfoxide; %MPE, the percentage of maximal possible antinociceptive effect. |
Next, rats were intrathecally injected with SP600125 (50 μg/10 μL, twice daily), a specific inhibitor of JNK, 30 minutes before morphine administration for 7 days. SP600125 had no effect on baseline tail-flick responses of rats and did not affect the antinociceptive effect of morphine on day 1. While consecutive administration of SP600125 could significantly attenuate the development of morphine tolerance (Figure 4B). SP600125 inhibited the increased expressions of p-c-Jun and Iba1 induced by morphine (Figure 4C and E), as well as the phosphorylation of PDGFRβ (Figure 4D).
A previous study reported that activation of PDGFRβ could induce vascular proliferation via JNK MAPK.18 To investigate whether PDGFRβ has any influence on JNK in morphine tolerance, rats were treated with imatinib (10 μg/10 μL) 30 minutes before morphine administration for 7 days. Figure 4F shows that the increased p-JNK induced by chronic morphine treatment was not affected by imatinib pretreatment.
Discussion
In this study, we found that spinal microglia was activated by chronic morphine treatment or PDGF-BB injection. Inhibiting PDGFRβ could suppress the activation of microglia. Moreover, the activations of PDGFRβ and microglia induced by morphine could be inhibited by MOR antagonist or JNK inhibitor. Taken together, our results provide direct evidence that repeatedly activating MOR by morphine could induce the transactivation of PDGFRβ via JNK MAPK, which leads to microglia activation in morphine tolerance.
Morphine exerts its antinociceptive effect mainly through binding to MOR. The roles of MOR desensitization and endocytosis in morphine tolerance have been well reported.2,19 A broad range of central nuclei are involved in the mechanisms of morphine tolerance, including locus coeruleus, paragigantocellularis, ventral tegmental areas, etc. The activity of specific endogenous neuropeptides has recently been found to be critical in regulation of opioid tolerance within both spinal and supraspinal areas.3 Moreover, the mechanism in spinal level play an essential role in morphine tolerance. Microglia in spinal cord could be activated by long-term morphine exposure,20 and participates in the development of morphine tolerance via releasing nitric oxide,21 proinflammatory cytokines,22 and tumor necrosis factor-α.23 Furthermore, MOR can mediate the modulation of microglia activity through the AKT/KATP/ERK/HSP70 pathway in morphine tolerance.6 A similar phenomenon was detected in our study that chronic morphine treatment increased spinal microglia activation, which could be suppressed by MOR antagonist. These provided fundamental information for the following experiments in this study.
PDGF is involved in the development of bone cancer pain through AKT-ERK signaling pathway.24 Recently, PDGFRβ in spinal cord is activated after chronic morphine administration, and antagonizing PDGFRβ could reverse morphine tolerance.15 Here, we explored the intrinsic mechanism of this phenomenon and provided the first evidence of the contribution of PDGFRβ to microglia activation in morphine tolerance. PDGF-BB could induce tactile allodynia by mediating spinal microglia.10 A previous study has reported that inhibiting PDGFR had an anti-inflammatory effect.25 It has been widely accepted that neuroinflammation plays a key role in pathophysiological processes in the central nervous system. Moreover, neuroinflammation caused by microglia activation is an important mechanism of morphine tolerance.22 Our results indicate that PDGFβ-mediated microglia activation, which leads to the occurrence of neuroinflammation in the spinal cord, might be a novel mechanism of morphine tolerance.
PDGFR is one of receptor tyrosine kinases (RTKs), and many studies have investigated the interaction between receptor tyrosine kinases (RTKs) (ie, EGFR and PDGFR) and G protein-coupled receptors (GPCRs) (ie, MOR, NMDAR, and dopamine receptor).26,28 The transactivation of MOR to PDGFRβ had a compounding effect on angiogenic signaling in mouse retinal endothelial cells.26 The co-activation between MOR and PDGFRβ in the kidney was also found in sickle mice.27 Given the influences of MOR and PDGFRβ on microglia, we speculated that the activations of MOR and PDGFRβ during the development of morphine tolerance might share a similar molecular mechanism. Moreover, in addition to the activation of PDGFRβ induced by morphine, MOR antagonist inhibited PDGFRβ phosphorylation induced by chronic morphine administration. These results confirm that PDGFRβ could be transactivated by MOR, which mediates microglia activation in morphine tolerance. Since our preliminary study found that phosphorylated PDGFRβ was co-localized with microglia marker Iba1 and neuronal marker NeuN in the spinal cord after chronic morphine treatment (Feng Gao et al, unpublished data), the interaction between MOR and PDGFRβ might be an intracellular and/or intercellular effect.
JNK is the intracellular mediator of morphine analgesia after MOR activation. The roles of the MAPKs family were confirmed in the mechanism of morphine tolerance.1,8 In our study, the JNK inhibitor attenuated the development of morphine tolerance, and JNK phosphorylation could be inhibited by naloxone, indicating the participation of JNK in MOR-mediated morphine tolerance. In addition, the JNK inhibitor reduced the activation of PDGFRβ induced by chronic morphine treatment, while JNK phosphorylation was not affected by PDGFRβ inhibitor, indicating the regulatory effect of JNK on PDGFRβ. JNK participates in the activation of RTKs signaling cascade12 and also PDGFR-mediated cellular proliferation, migration, and gene expression.18 JNK activation could up-regulate the expression of PDGFR in drosophila.29 In rat brain astrocytes, transactivation of PDGFR dependents on activated c-Src,30 which could be regulated by JNK.31 Taken together, JNK might be the mediator of the transactivation of PDGFRβ in morphine tolerance. However, the source of endogenous PDGFRβ agonist15 and the mechanism of PDGFRβ-mediated microglia activation in morphine tolerance still need to be further explored. Besides the effect of JNK, we cannot exclude the existence of heterodimers between MOR and PDGFRβ.
Conclusion
Our study provides the first evidence that activated MOR could increase the activity of microglia through PDGFRβ transactivation during the development of morphine tolerance, and JNK MAPK might participate in the initiation of PDFGRβ activation. These findings enrich the mechanisms of interaction between MOR and PDGFRβ, and implicate a significant therapeutic strategy in prophylactically inhibiting morphine tolerance. However, further studies are needed to elucidate detailed interaction between these receptors.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Guo RX, Zhang M, Liu W, et al. NMDA receptors are involved in upstream of the spinal JNK activation in morphine antinociceptive tolerance. Neurosci Lett. 2009;467(2):95–99. doi:10.1016/j.neulet.2009.10.013
2. Mohammad Ahmadi Soleimani S, Azizi H, Pachenari N, Mirnajafi-Zadeh J, Semnanian S. Enhancement of μ-opioid receptor desensitization by orexin-A in rat locus coeruleus neurons. Neuropeptides. Jun. 2017;63:28–36.
3. Ahmadi-Soleimani SM, Azizi H, Gompf HS, Semnanian S. Role of orexin type-1 receptors in paragiganto-coerulear modulation of opioid withdrawal and tolerance: A site specific focus. Neuropharmacology. 2017;126:25–37. doi:10.1016/j.neuropharm.2017.08.024
4. Luo X, Tai WL, Sun L, et al. Crosstalk between astrocytic CXCL12 and microglial CXCR4 contributes to the development of neuropathic pain. Mol Pain. 2016;1:12.
5. David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci. 2011;12(7):388–399.
6. Qu J, Tao XY, Teng P, et al. Blocking ATP-sensitive potassium channel alleviates morphine tolerance by inhibiting HSP70-TLR4-NLRP3-mediated neuroinflammation. J Neuroinflammation. 2017;14(1):228. doi:10.1186/s12974-017-0997-0
7. Eidson LN, Inoue K, Young LJ, Tansey MG, Murphy AZ. Toll-like Receptor 4 Mediates Morphine-Induced Neuroinflammation and Tolerance via Soluble Tumor Necrosis Factor Signaling. Neuropsychopharmacology. 2017;42(3):661–670.
8. Chen Y, Geis C, Sommer C. Activation of TRPV1 contributes to morphine tolerance: involvement of the mitogen-activated protein kinase signaling pathway. J Neurosci. 2008;28(22):5836–5845. doi:10.1523/JNEUROSCI.4170-07.2008
9. Yuill MB, Zee ML, Marcus D, Morgan DJ. Tolerance to the antinociceptive and hypothermic effects of morphine is mediated by multiple isoforms of c-Jun N-terminal kinase. Neuroreport. 2016;27(6):392–396. doi:10.1097/WNR.0000000000000551
10. Masuda J, Tsuda M, Tozaki-Saitoh H, Inoue K. Intrathecal delivery of PDGF produces tactile allodynia through its receptors in spinal microglia. Mol Pain. 2009;5:23. doi:10.1186/1744-8069-5-23
11. Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103(2):211–225. doi:10.1016/S0092-8674(00)00114-8
12. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141(7):1117–1134. doi:10.1016/j.cell.2010.06.011
13. Belcheva MM, Szucs M, Wang D, Sadee W, Coscia CJ. mu-Opioid receptor-mediated ERK activation involves calmodulin-dependent epidermal growth factor receptor transactivation. J Biol Chem. 2001;276(36):33847–33853. doi:10.1074/jbc.M101535200
14. Zhao H, Wu G, Cao X. EGFR dependent subcellular communication was responsible for morphine mediated AC superactivation. Cell Signal. 2013;25(2):417–428. doi:10.1016/j.cellsig.2012.10.016
15. Wang Y, Barker K, Shi S, Diaz M, Mo B, Gutstein HB. Blockade of PDGFR-beta activation eliminates morphine analgesic tolerance. Nat Med. 2012;18(3):385–387. doi:10.1038/nm.2633
16. Liu D, Zhou Y, Peng Y, et al. Endoplasmic Reticulum Stress in Spinal Cord Contributes to the Development of Morphine Tolerance. Front Mol Neurosci. 2018;11:72. doi:10.3389/fnmol.2018.00072
17. Mao J, Sung B, Ji RR, Lim G. Chronic morphine induces downregulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J Neurosci. 2002;22(18):8312–8323. doi:10.1523/JNEUROSCI.22-18-08312.2002
18. Zhan Y, Kim S, Izumi Y, et al. Role of JNK, p38, and ERK in platelet-derived growth factor-induced vascular proliferation, migration, and gene expression. Arterioscler Thromb Vasc Biol. 2003;23(5):795–801. doi:10.1161/01.ATV.0000066132.32063.F2
19. Arttamangkul S, Heinz DA Cellular tolerance at the µ-opioid receptor is phosphorylation dependent. March 28 2018;7.
20. Jokinen V, Sidorova Y, Viisanen H, et al. Differential Spinal and Supraspinal Activation of Glia in a Rat Model of Morphine Tolerance. Neuroscience. 2018;375:10–24. doi:10.1016/j.neuroscience.2018.01.048
21. Stefano GB. Autoimmunovascular regulation: morphine and anandamide and ancondamide stimulated nitric oxide release. J Neuroimmunol. 1998;83(1–2):70–76. doi:10.1016/S0165-5728(97)00223-3
22. Peterson PK, Molitor TW, Chao CC. The opioid-cytokine connection. J Neuroimmunol. 1998;83(1–2):63–69. doi:10.1016/S0165-5728(97)00222-1
23. Chao CC, Gekker G, Sheng WS, Hu S, Tsang M, Peterson PK. Priming effect of morphine on the production of tumor necrosis factor-alpha by microglia: implications in respiratory burst activity and human immunodeficiency virus-1 expression. J Pharmacol Exp Ther. 1994;269(1):198–203.
24. Xu Y, Liu J, He M, et al. Mechanisms of PDGF siRNA-mediated inhibition of bone cancer pain in the spinal cord. Sci Rep. 2016;6(1):27512. doi:10.1038/srep27512
25. Yang P, Manaenko A, Xu F, et al. Role of PDGF-D and PDGFR-beta in neuroinflammation in experimental ICH mice model. Exp Neurol. 2016;283(Pt A):157–164. doi:10.1016/j.expneurol.2016.06.010
26. Chen C, Farooqui M, Gupta K. Morphine stimulates vascular endothelial growth factor-like signaling in mouse retinal endothelial cells. Curr Neurovasc Res. 2006;3(3):171–180. doi:10.2174/156720206778018767
27. Weber ML, Chen C, Li Y, et al. Morphine stimulates platelet-derived growth factor receptor-beta signalling in mesangial cells in vitro and transgenic sickle mouse kidney in vivo. Br J Anaesth. 2013;111(6):1004–1012. doi:10.1093/bja/aet221
28. Kotecha SA, Oak JN, Jackson MF, et al. A D2 class dopamine receptor transactivates a receptor tyrosine kinase to inhibit NMDA receptor transmission. Neuron. 2002;35(6):1111–1122. doi:10.1016/S0896-6273(02)00859-0
29. Ohsawa S, Sugimura K, Takino K, Xu T, Miyawaki A, Igaki T. Elimination of oncogenic neighbors by JNK-mediated engulfment in Drosophila. Dev Cell. 2011;20(3):315–328. doi:10.1016/j.devcel.2011.02.007
30. Yang CM, Lin CC, Lee IT, et al. Japanese encephalitis virus induces matrix metalloproteinase-9 expression via a ROS/c-Src/PDGFR/PI3K/Akt/MAPKs-dependent AP-1 pathway in rat brain astrocytes. J Neuroinflammation. 2012;9:12. doi:10.1186/1742-2094-9-12
31. Samak G, Chaudhry KK, Gangwar R, Narayanan D, Jaggar JH, Rao R. Calcium/Ask1/MKK7/JNK2/c-Src signalling cascade mediates disruption of intestinal epithelial tight junctions by dextran sulfate sodium. Biochem J. 2015;465(3):503–515. doi:10.1042/BJ20140450
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.