")
Back to Journals » Journal of Pain Research » Volume 11
The increased release of amino acid neurotransmitters of the primary somatosensory cortical area in rats contributes to remifentanil-induced hyperalgesia and its inhibition by lidocaine
Authors Wang S, Cui W , Zeng M, Ren Y, Han S , Li J
Received 13 March 2018
Accepted for publication 15 May 2018
Published 14 August 2018 Volume 2018:11 Pages 1521—1529
DOI https://doi.org/10.2147/JPR.S168008
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor E Alfonso Romero-Sandoval

Shanshan Wang,1,* Weihua Cui,1,* Min Zeng,1 Yi Ren,1 Song Han,2 Junfa Li2
1Department of Anesthesiology, Beijing Tian Tan Hospital, Capital Medical University, Beijing, People’s Republic of China; 2Department of Neurobiology, Beijing Institute for Neuroscience, Capital Medical University, Beijing, People’s Republic of China
*These authors contributed equally to this work
Background: Studies have confirmed that activation of the neurons of primary somatosensory cortex (S1) is involved in the process of remifentanil (Remi)-induced hyperalgesia (RIH), which can be suppressed by lidocaine (Lido). A total intravenous anesthesia model of rats mimicking clinical Remi-based anesthesia was set up to explore the release of amino acid neurotransmitters of S1 cortex in RIH and its inhibition by Lido in this study.
Materials and methods: Sprague Dawley rats were randomly divided into the following four groups: propofol (Pro), Remi, Remi combined Lido, and Lido groups. Mechanical hyperalgesia was evaluated by von Frey test; the amino acid neurotransmitters in the microdialysates of S1 area were detected by high-performance liquid chromatography (HPLC)-fluorescence, and conventional protein kinase C (cPKC)γ levels in the whole-cell lysates and membrane lipid rafts (MLRs) were determined by Western blotting.
Results: The von Frey test showed that co-administration of Lido significantly inhibited a Remi-induced decrease in the threshold of the paw withdrawal response in Remi group at 2 h postinfusion. Meanwhile, the Remi-induced increases in both the excitatory and inhibitory amino acid releases in S1 were suppressed by co-administrating Lido within 5 h postinfusion. Western blotting showed that the increased cPKCγ level in the membrane lipid rafts (MLR) induced by Remi was also inhibited by Lido.
Conclusion: The increased release of amino acid neurotransmitters and the translocation of cPKCγ in MLR suggest the activation of S1 neurons, which may be one of the mechanisms underlying RIH. Lido reduces the release of amino acid neurotransmitters in S1 neurons and the translocation of cPKCγ in MLRs after stopping Remi, which may be one of its antihyperalgesic mechanisms.
Keywords: hyperalgesia, remifentanil, lidocaine, amino acid neurotransmitter, protein kinase C
Introduction
Clinical and experimental research indicates that severe postoperative pain after the intraoperative use of remifentanil (Remi) has been linked to the development of Remi-induced hyperalgesia (RIH).1–4 Co-administration of lidocaine (Lido) and ketamine has been proved to effectively overcome RIH through several different mechanisms.5,6
The primary somatosensory cortex (S1), an important structure for pain perception, has also been found to be involved in the modulation of pain.7–10 Our previous study has shown that CaMKII phosphorylation in the S1 area is involved in RIH, which can be suppressed by the systemic application of Lido.4 Similarly, a recent study also demonstrated that S1 cortex participates in gabapentin’s antiallodynic effect on neuropathic pain.11
One of the important mechanisms of RIH is the activation of glutamatergic system.12 Studies have also demonstrated that Lido alleviates neuropathic and acute pain by suppressing the activation of glutamatergic system.13 However, most of them focus on the spinal cord. Studies on humans and rats with chronic pain have shown that neurotransmitter abnormalities, including increases in glutamate and decreases in γ-aminobutyric acid (GABA), could be responsible for the cortical hyperactivity and hyperalgesia.14–16 It suggests that excitatory and inhibitory amino acid neurotransmitter imbalance may be a mechanism of hyperalgesia. However, excitatory and inhibitory amino acid release characteristics are not yet clear in opioid-induced hyperalgesia. Therefore, using a Remi administration model, this study explored the changes in hyperalgesia-related amino acid neurotransmitters in S1 region during and after the discontinuation of Remi infusion, as well as the impact of Lido.
Membrane lipid rafts (MLRs), also known as detergent-resistant membrane fractions, are rich in cholesterol and serve as the discrete domains of sphingolipids with a flat boat shape and diameters <100 nm. Lipid raft is not only the critical subcellular structure for the execution of neurotransmitter release and exocytosis in the presynaptic membrane but also the region implicated in the congregation of postsynaptic membrane receptors and signaling proteins, thereby playing an important role in the process of synaptic signal transduction.17,18 Conventional protein kinase Cγ (cPKCγ) is a protein kinase C isoform specifically distributed in neurons, which is involved in the modulation of pain signals. Studies have shown that PKC promotes neurotransmitter release by adjusting the function of signaling proteins in MLRs.19,20 Moreover, it enhances the function of proteins involved in postsynaptic potentiation, which are located in MLRs, such as N-methyl-D-aspartic acid (NMDA) receptors.21 Translocation level assays have classically been used to screen the individual isoforms of PKC for the production of active PKC-membrane complexes. Thus, we further determined the changes in cPKCγ translocation level in MLRs in S1 region to provide more evidence for increasing the release of amino acid neurotransmitter and enhanced synaptic transmission.
Materials and methods
One hundred and three male Sprague Dawley rats weighing 240–260 g (provided by the Department of Laboratory Animals, Capital Medical University, Beijing, People’s Republic of China) were used for the experiment. Prior to the experiment, the rats were reared in individual cages (five per cage) with free access to food and water. The ambient temperature (20–25°C) and the circadian rhythm (12 h day/12 h night) remained stable. The Animal Care and Use Committee of Capital Medical University approved the design of this study, in line with the related ethical guidelines for investigations of experimental pain in conscious animals by International Association for the Study of Pain.22
Animal grouping and anesthesia regimen
The rats were randomly divided into four groups. Individual rats were fixed in separate plastic holders. Tail-vein intravenous access was established by using a 24 G trocar. The total duration of anesthesia is 3 h. The dosing regimens (Table 1) used in this study were determined based on the US Food and Drug Administration empirical formula. The human dosage per body weight unit was converted into the equivalent dose per body weight unit (mg [μg]/kg) for a rat, with the calculation of the human equivalent dose (HED) = rat dose ×0.16. The HED was equivalent to the common clinical dose of Remi and an antiarrhythmic dose of Lido. Infusion of Remi and/or Lido without hypnotic drugs cannot achieve the anesthetized status in which mice are unconsciousness and no body movement, so that propofol (Pro), a commonly used hypnotic drug, was used to achieve adequate anesthesia in this research. Pro only group served as sham group. To keep all mice in the same depth of anesthesia, the dose of Pro was lowered when co-administering with Remi and/or Lido. All anesthetized animals breathed spontaneously at ~60 breaths/min and maintained rectal temperature at 37.5±0.5°C during anesthesia. After the discontinuation of the infusion, all the animals awoke and resumed regular activity within 15 min. To show the effect of Pro on PKCγ translocation to lipid rafts, control (Ctrl) group was added in Western blot analysis.
 | Table 1 Animal grouping and dosing regimen Notes: Pro: the propofol group, in which the rats were anesthetized with propofol only; Remi: the remifentanil group, in which the rats were anesthetized with propofol and remifentanil; Remi–Lido: the remifentanil and lidocaine group, in which the rats were anesthetized with propofol, remifentanil, and lidocaine; Lido: the lidocaine group, in which the rats were anesthetized with propofol and lidocaine. All rats were spontaneously breathing during anesthesia. The experimental drugs were as follows: propofol (AstraZeneca SpA, Caponago, Italy), remifentanil (Yichang Humanwell Pharmaceutical, Co., Ltd., Yichang, People’s Republic of China), and lidocaine (Beijing Yimin Pharmaceutical Co., Ltd., Beijing, People’s Republic of China). |
Test of the mechanical pain threshold
A total of 40 rats were included in the test of the mechanical pain threshold. Grouping information was described as earlier (Table 1, n=10 per group). Researchers who carried out the test were blinded to grouping. An electronical von Frey anesthesiometer (IITC Life Science, Woodland Hills, CA, USA) mounted with rigid tip 1 mm in diameter was used for measuring PWT instead of traditional von Frey hairs.4 The test area is between the second and third metatarsus. An active retraction of hind paw was recognized as a withdrawal behavior but was not a passive push up by the rigid tip. A transparent test box with a mesh bottom was used in this behavior test. All animals were allowed to adapt for 15 min before test. The test time points were prior to administration (baseline Ctrl), 0.5 h, 2 h, 5 h, and 24 h after discontinuing the administration. The paw withdrawal threshold (PWT) was recognized as the pressure applied on plantar causing animal paw withdrawal. Each animal was measured three times at 60 s intervals, and the mean value was obtained. To avoid injury, the test was stopped when the reading of the von Frey device reached 120 g. This termination value (120 g) was recorded as the measured PWT regardless the response of rats.
Microdialysis
A total of 28 rats were randomly divided into four groups according to the above scheme (Table 1, n=7 per group). After rat was anesthetized, a dialysis probe (CMA 12, with a membrane length of 2 mm, a diameter of 0.5 mm, and a molecular weight cutoff of 20 kDa; CMA, Stockholm, Sweden) was placed on the right side of the S1 cortex (1.7 mm anterior to the fontanelle, 4.5 mm lateral, and 3.5 mm deep) and properly fixed. Thirty minutes after anesthesia induction, the dialysis perfusion system was initiated and the flow rate of the pump was adjusted to constant 2 μL/min. Sampling was performed when the perfusion with Ringer’s solution (NaCl, 145 mM; KCl, 4 mM; CaCl2, 1.3 mM, pH, 7.2–7.4) reached equilibrium until 150 min after starting the anesthesia. Each sample was collected for 30 min, obtaining a total of 60 μL. The administration was stopped after collecting the first sample under anesthesia. Subsequently, samples were collected at the time points of 0.5, 1.5, 4.5, and 23.5 h after discontinuing the administration. The concentration of the amino acid neurotransmitters in the dialysates, including glutamate, aspartic acid, GABA, and glycine, was detected using the method of high-performance liquid chromatography (HPLC) fluorescence. Before using the probe, its recovery was determined with the standard mixture (glutamate, aspartic acid, GABA, and glycine, 0.25 μg/mL) under the identical perfusion conditions as described earlier. The concentration of extracellular amino acid was equal to the concentration of amino acid in the sample divided by in vitro recovery. After collecting the microdialysis samples, the brains were taken for verification of the probe position by Nissl staining.
Preparation of the whole-cell lysate
A total of 35 rats were divided into the following five groups: Ctrl group and another four groups described earlier (Table 1, n=7 per group). Two hours after discontinuing the administration, the rats were scarified by decapitation and S1 cortex was dissected carefully on ice. The tissue samples were homogenized at 4°C with carbonate lysis buffer (500 mM sodium carbonate, pH 11.0, containing protease inhibitor and phosphatase inhibitor). After homogenization, the samples were sonicated for 3×15 s. The nuclear components were removed by centrifugation at 1000× g for 10 min, and the protein concentration was adjusted for later use.
Extraction of the lipid rafts
Sucrose was dissolved in 2-morpholino-ethanesulfonic acid (MES) saline buffer (25 mM MES and 150 mM NaCl, pH 6.5) to the concentrations of 80, 35, and 5%. To find the sucrose density gradient, 1 mL of 80% sucrose and 1 mL of whole-cell lysate (after adjusting the protein concentration) were mixed and placed at the bottom of a centrifuge tube, followed by subsequent slow addition of 6 mL of 35% sucrose and 4 mL of 5% sucrose. Ultracentrifugation at 39,000 rpm (190,000× g) at 4°C for 3 h was performed on the gradient medium (SW-41 rotor; Beckman, Indianapolis, IN, USA). After centrifugation, the factions of the gradient medium were collected from top to bottom in 1 mL/stratified layer. The extracts of the fourth and fifth layers, which contained the floating debris of the lipid rafts, were mixed 1:1 for later use.
Western blot
The whole-cell and the suspended lipid-raft samples were electrophoresed in 10% sodium dodecyl sulfate polyacrylamide gel electropheresis (SDS-PAGE) and then transferred onto polyvinylidene diflouride (PVDF) membrane. Membranes were blocked in 20 mM Tris-buffered Saline (TBS)-Tween with 5% albumin from bovine serum (BSA) and incubated overnight with primary antibodies for cPKCγ (1:1000, # 98952; Santa Cruz Biotechnology Inc., Dallas, TX, USA) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:1000, # 37168; Abcam, Cambridge, MA, USA). Primary antibodies were visualized with species-specific secondary antibodies conjugated to Horseradish Peroxidase (HRP) and Enhanced Chemiluminescence (ECL) lumigen solution. Gel Doc gel-imaging system (Version 4.2.3; Bio-Rad Laboratories Inc., Hercules, CA, USA) was used to image. GAPDH served as the internal reference by which to standardize the cPKCγ band for the whole-cell samples. The ratio of the cPKCγ band in the suspended debris sample compared to the cPKCγ band in the corresponding whole-cell sample was used to present the translocation level of cPKCγ. The calculated ratio of the Ctrl group was normalized to 100%, and the comparisons of other groups with the Ctrl group were represented as percentage.
Statistical analyses
The PWTs to mechanical stimulation at different time points and the concentrations of the amino acid neurotransmitters for the rats in different groups were compared using the two-way analysis of variance, with Tukey’s test for further multiple comparisons. Power analysis for the behavioral test was performed using the G power (Version 3.1.9.2; Heinrich-Heine University, Düsseldorf, Germany). Univariate analysis of variance was used, with the Bonferroni test being carried out for further multiple comparisons. Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA) was used for the statistics and plotting. A difference with p<0.05 was considered statistically significant.
Results
Lido inhibited Remi-induced mechanical hyperalgesia
All the animals had baseline PWT measurements prior to treatment. As shown in Figure 1, there was no difference in the baseline PWT value among groups. The PWT of the rats in group Pro also showed no difference in each time point after anesthesia compared to its baseline. The PWT of group Remi at 2 h after discontinuation of the infusion was significantly lower than its corresponding baseline value (P<0.05, n=10 per group), whereas the PWT of group Remi–Lido at each time point showed no significant differences compared to its baseline. The PWT of the rats in group Remi at 2 h after discontinuing the infusion was also significantly lower than the other three groups (P<0.05, n=10 per group). Given a=0.05, calculated effect size f=0.71, total sample size =40, and number of groups =4, the post hoc power (1-b) of PWT at 2 h after discontinuation calculated by G power is 0.96.
 | Figure 1 The von Frey test of the mechanical paw withdrawal response threshold showed that lidocaine could inhibit remifentanil-induced hyperalgesia. Notes: Pro: the propofol group, in which the rats were anesthetized with propofol only; Remi: the remifentanil group, in which the rats were anesthetized with propofol and remifentanil; Remi–Lido: the remifentanil and lidocaine group, in which the rats were anesthetized with propofol, remifentanil, and lidocaine; Lido: the lidocaine group, in which the rats were anesthetized with propofol and lidocaine. The data are represented as mean ± standard errors. *The PWT of group Remi was significantly lower than that of group Pro, P<0.05. #The PWT of group Remi was significantly lower than baseline, P<0.05; n=10 per group. Abbreviation: PWT, paw withdrawal threshold. |
Lido infusion suppressed Remi-induced amino acid neurotransmitters’ release in S1 cortex
The dynamic changes in glutamate, aspartic acid, GABA, and glycine release in the dialysates of S1 cortex under anesthesia and after discontinuing the infusion are shown in Figure 2. Group Pro served as Ctrl. Under anesthesia, the inhibitory amino acid neurotransmitters (glycine and GABA) of group Remi were significantly higher than those of group Pro, while other groups did not show much difference compared to group Pro (P<0.05, n=7 per group). There was no difference in excitatory amino acid neurotransmitters (glutamate and aspartic acid) among groups under anesthesia (P<0.05, n=7 per group). Within 5 h after stopping the infusion, both the excitatory and inhibitory amino acid levels of group Remi were higher than those of group Pro at multiple time points (P<0.05, n=7 per group). However, groups Remi–Lido and Lido did not show any difference compared to group Pro. At 24 h after stopping the infusion, the amino acid neurotransmitter levels of group Remi returned to levels similar to those of group Pro (P>0.05, n=7 per group). No significant difference was found in both the excitatory and inhibitory amino acid levels among group Pro, Remi–Lido and Lido at each time point. As shown in Figure 3, the representative Nissl staining image demonstrated that the microdialysis probe was located at the S1 cortex.
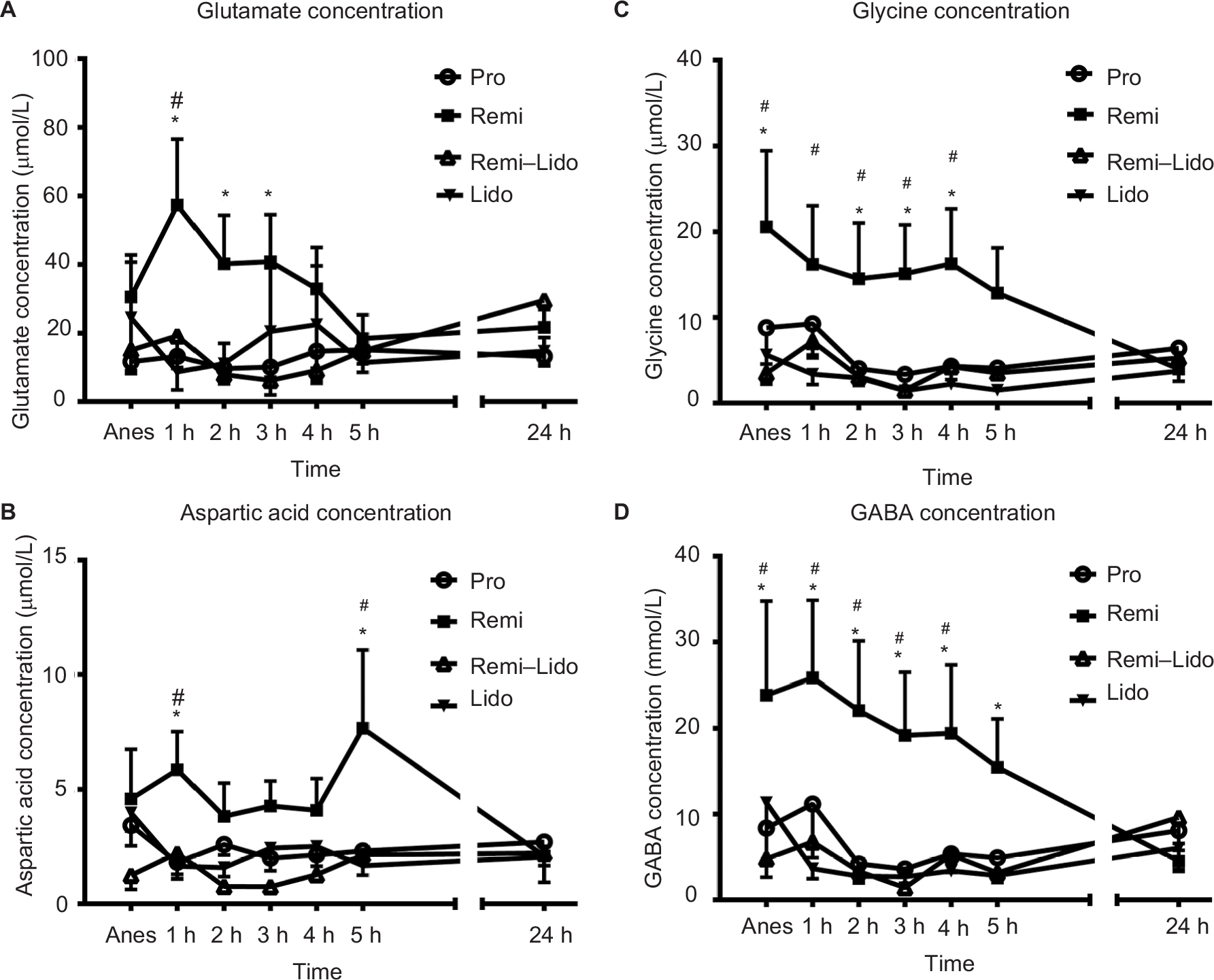 | Figure 2 Remifentanil increased the levels of amino acid neurotransmitters in S1 inhibited by systemic injection of lidocaine. Notes: (A) Glutamate concentration; (B) Aspartic acid concentration; (C) Glycine concentration; (D) GABA concentration. Pro: the propofol group, in which the rats were anesthetized with propofol only; Remi: the remifentanil group, in which the rats were anesthetized with propofol and remifentanil; Remi–Lido: the remifentanil and lidocaine group, in which the rats were anesthetized with propofol, remifentanil, and lidocaine; Lido: the lidocaine group, in which the rats were anesthetized with propofol and lidocaine. The data are represented as mean ± standard error. *The results of group Remi were significantly higher than those of group Pro, P<0.05. #The results of group Remi were significantly lower than those of 24 h after discontinuation of anesthetics infusion, P<0.05; n=7 per group. Abbreviations: Anes, during anestheisa; GABA, γ-aminobutyric acid; S1, primary somatosensory cortex. |
 | Figure 3 The position of the microdialysis probe. Notes: The dialysis probe was placed on the right side of the primary somatosensory cortex (S1) cortex (1.7 mm anterior to the fontanelle, 4.5 mm lateral, and 3.5 mm deep). (A) Schematic diagram of probe position. Light green area indicates S1 cortex, and black rectangle represents microdialysis probe. Adapted from The Rat Brain in Stereotaxic Coordinates, 5th edition. (B) The representative Nissl staining image verified the microdialysis probe position. |
Lido infusion suppressed Remi-induced cPKCγ translocation to the lipid rafts of S1 cortex
The typical Western blot and quantitative analyses’ results are shown in Figure 4, and no significant changes in cPKCγ protein levels were observed in the whole-cell lysate of S1 cortex from normal Ctrl, Pro, Remi, Remi–Lido, and Lido groups (Figure 4A, P>0.05, n=7 per group). However, the cPKCγ levels in the lipid rafts of S1 cortex increased significantly in group Remi when compared with that of Ctrl (Figure 4B, P<0.05, n=7 per group). However, this Remi-induced increase in cPKCγ translocation was significantly inhibited by co-administering Lido in group Remi–Lido when compared to group Ctrl (Figure 4B, P>0.05, n=7 per group).
 | Figure 4 Changes in cPKCγ levels in the lipid rafts of S1 cortex after discontinuation of remifentanil and lidocaine. Notes: (A) cPKCr levels in whole cell lysate. (B) cPKCr levels on lipid rafts. Ctrl: control group, which received the same amount of saline; Pro: the propofol group, in which the rats were anesthetized with propofol only; Remi: the remifentanil group, in which the rats were anesthetized with propofol and remifentanil; Remi–Lido: the remifentanil and lidocaine group, in which the rats were anesthetized with propofol, remifentanil, and lidocaine; Lido: the lidocaine group, in which the rats were anesthetized with propofol and lidocaine. The data are represented as mean ± standard error. *The result of group Remi was significantly higher than that of Ctrl, P<0.05; n=7 per group. Abbreviations: cPKCγ, conventional protein kinase Cγ; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; MLR, membrane lipid rafts; S1, primary somatosensory cortex. |
Discussion
A total intravenous anesthesia model of rats mimicking clinical Remi-based anesthesia was set up to explore underlying the mechanism of RIH in this study. As shown in behavior test, no significant change was observed in the PWT values of group Pro at each time point compared to the baseline, which means that the Pro itself did not change rats’ PWT value in this experiment setting. The PWT of group Remi was significantly lower than those of the other groups at 2 h after discontinuing the infusion, while group Remi–Lido showed no significant differences compared to group Ctrl, indicating that Remi can induce hyperalgesia, which was inhibited by systemic infusion of Lido. These results are consistent with our previous findings using Remi and Lido in isolation without Pro in the waking state.23
As the highest component of the somatosensory central nervous system, which receives upstream non-nociceptive and nociceptive afferent impulses, S1 cortex plays a key role in pain perception. Considering the results observed on humans by using functional magnetic resonance (fMRI) that showed significant activation in primary somatosensory cortex (S1) during temporal summation of pain sensation,24 we hypothesize that activation of the S1 area is also involved in RIH. In addition to receiving upstream non-nociceptive and nociceptive afferent impulses, the S1 cortex is involved in complex excitatory and inhibitory neuronal connections, both inside and outside the region. Different from the studies observing activated aspartatergic/glutamatergic system along with suppressed GABAergic/glycinergic system in spinal dorsal horn during pain state,25,26 a study using the chronic pain model showed that both excitatory and inhibitory neuronal activities in the S1 region are enhanced. However, the increased inhibition is insufficient to completely counterbalance the increased excitation and thus alleviate the symptoms of chronic pain.27 This region is also being found heavily implicated in the formation of hyperalgesia.7,8 Our previous studies have shown that, at 2 h after discontinuing a Remi infusion, the phosphorylation level of the CaMKII in the S1 cortex is increased but can be inhibited by the conjoint infusion of Lido.4 Moreover, this observation coincides with the occurrence of mechanical hyperalgesia after discontinuing the drug when testing the PWT of rats.4 According to these findings, we hypothesized that the increased release of neurotransmitters in S1 area, especially amino acids, which can activate key receptors, such as NMDA receptors, may also be involved in the process of RIH. Thus, we further explored amino acid neurotransmitter levels in S1 region after the infusion of Remi and Lido in this study. By using microdialysis, we found that the neurotransmitter levels of both excitatory amino acids (glutamate and aspartic acid) and inhibitory amino acids (glycine and GABA) of group Remi were significantly higher than other groups at multiple time points within 5 h following discontinuation of the drug; this also coincided with the timing of the decrease of the mechanical PWT after stopping the infusion of Remi. These findings suggest that the synapses releasing excitatory and inhibitory amino acid neurotransmitters were activated to varying degrees after discontinuing the administration of Remi. The unbalanced release of excitatory and inhibitory amino acids may be one of the mechanisms underlying RIH. The systemic application of Lido suppressed the increased release of amino acid neurotransmitters after discontinuing Remi, which may be one of the neurobiological bases for the relief of RIH by Lido. As the density of opioid receptors in S1 is relatively low compared to several other brain regions,28 the increased release of amino acid neurotransmitters in this region after the withdrawal of Remi may be not a consequence of the direct effect of Remi on the opioid receptors in S1 region but secondary changes caused by the increased afferent impulses from other brain areas with dense distributions of opioid receptors, which can be activated by Remi. The present study is a qualitative research exploring the changes in amino acid neurotransmitters in S1 region in the process of RIH and also the effect of Lido on these changes. However, the limitation of this study is that we have not investigated the dose–effect relationship between the concentration of Lido and RIH. Further studies on determining the minimum concentration of Lido needed to reverse RIH should be done with a more precise study design.
Lipid rafts are the important cellular substructures related to synaptic signal transduction.29 Previous studies have shown that the translocation of PKC in lipid rafts may enhance the functions of proteins associated with exocytosis, including GAP4330 and the L-type calcium channel.31 Moreover, translocation of PKC in lipid rafts has been found to activate the protein that is involved in synaptic signaling mediated by amino acid neurotransmitters, including the glycine transporter32 and the NMDA receptor.21 Since the present study showed that Lido can partly inhibit Remi-induced increase in the release of excitatory neurotransmitters in S1 area, combined with our previous study that showed membrane translocation of cPKCγ in the spinal cord dorsal horn is involved in RIH,3 we hypothesized that the translocation of cPKCγ in lipid rafts of S1 may be also involved in RIH and its inhibition by Lido. Our results showed that, at 2 h after the withdrawal of anesthesia, the translocation level of cPKCγ in lipid rafts of S1 in group Remi was higher than the other three groups. In group Remi, the increased translocation of cPKCγ in lipid rafts and the increased release of amino acid neurotransmitters in S1 were time-correlated with the reduced mechanical pain threshold level. This correlation may partly explain the role of S1 area in the formation of RIH. There are two possible explanations for the time correlation between the increased translocation of cPKCγ in lipid rafts and the release of amino acids in group Remi. First, after Remi interacting with the opioid receptors in S1, cPKCγ translocation in lipid rafts was promoted by certain intracellular mechanisms, which enhanced the function of neurotransmitter release-associated proteins and led to increasing release of the presynaptic neurotransmitters in S1. Another possibility is that the binding of Remi with opioid receptors in other brain regions of the nociceptive pathway caused changes in the afferent impulse to the S1 region, which further led to the activation of S1. The increased translocation of cPKCγ in lipid rafts and the release of amino acids in this region were the results promoted by certain mechanisms worth exploring. The intravenous injection of Lido may nonspecifically inhibit intracellular signal transduction, leading to an overall reduction in the release of amino acid neurotransmitters, thereby playing a role in reducing the hyperalgesia. The detailed mechanism upstream of cPKCγ will be the focus of further study.
Conclusion
The increased release of both excitatory and inhibitory amino acid neurotransmitters in S1 is correlated with behavioral changes and may serve as one of the mechanisms underlying RIH. The increased translocation level of neuron-specific cPKCγ in the lipid rafts suggested the activation of neurons in S1 and the increased synaptic signal transduction after Remi infusion. The reductions in amino acid neurotransmitter release in S1 and cPKCγ translocation in lipid rafts by Lido may be one of the mechanisms behind the antihyperalgesia effect of Lido.
Acknowledgments
This study was supported by the following grants: The Clinical-Basic Medicine Cooperation Fund of Capital Medical University (16JL51), the National Natural Science Foundation of China (81100823), and Beijing Natural Science Foundation (7173255).
Author contributions
SW was involved in the microdialysis. WC is a principal investigator and was involved in the manuscript composing. MZ carried out Western blotting. YR was involved in the animal anesthesia. SH carried out behavioral test. JL contributed to the data processing and statistical analysis. All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Comelon M, Raeder J, Stubhaug A, Nielsen CS, Draegni T, Lenz H. Gradual withdrawal of remifentanil infusion may prevent opioid-induced hyperalgesia. Br J Anaesth. 2016;116(4):524–530. | ||
Angst MS. Intraoperative use of remifentanil for TIVA: postoperative pain, acute tolerance, and opioid-induced hyperalgesia. J Cardiothorac Vasc Anesth. 2015;29(suppl 1):S16–S22. | ||
Cui W, Li Y, Li S, et al. Systemic lidocaine inhibits remifentanil-induced hyperalgesia via the inhibition of cPKCgamma membrane translocation in spinal dorsal horn of rats. J Neurosurg Anesthesiol. 2009;21(4):318–325. | ||
Cui W, Wang S, Han R, Wang Q, Li J. CaMKII phosphorylation in primary somatosensory cortical neurons is involved in the inhibition of remifentanil-induced hyperalgesia by lidocaine in male Sprague-Dawley rats. J Neurosurg Anesthesiol. 2016;28(1):44–50. | ||
Cui W, Li Y, Li S, Wang R, Li J. Systemic administration of lidocaine reduces morphine requirements and postoperative pain of patients undergoing thoracic surgery after propofol-remifentanil-based anaesthesia. Eur J Anaesthesiol. 2010;27(1):41–46. | ||
Wu L, Huang X, Sun L. The efficacy of N-methyl-D-aspartate receptor antagonists on improving the postoperative pain intensity and satisfaction after remifentanil-based anesthesia in adults: a meta-analysis. J Clin Anesth. 2015;27(4):311–324. | ||
Xie YF, Huo FQ, Tang JS. Cerebral cortex modulation of pain. Acta Pharmacol Sin. 2009;30(1):31–41. | ||
Canavero S, Bonicalzi V. Role of primary somatosensory cortex in the coding of pain. Pain. 2013;154(7):1156–1158. | ||
Frot M, Magnin M, Mauguiere F, Garcia-Larrea L. Cortical representation of pain in primary sensory-motor areas (S1/M1) – a study using intracortical recordings in humans. Hum Brain Mapp. 2013;34(10):2655–2668. | ||
Peyron R, Schneider F, Faillenot I, et al. An fMRI study of cortical representation of mechanical allodynia in patients with neuropathic pain. Neurology. 2004;63(10):1838–1846. | ||
Alles SRA, Bandet MV, Eppler K, et al. Acute anti-allodynic action of gabapentin in dorsal horn and primary somatosensory cortex: correlation of behavioural and physiological data. Neuropharmacology. 2017;113(pt A):576–590. | ||
Roeckel LA, Le Coz GM, Gaveriaux-Ruff C, Simonin F. Opioid-induced hyperalgesia: cellular and molecular mechanisms. Neuroscience. 2016;338:160–182. | ||
Kurabe M, Furue H, Kohno T. Intravenous administration of lidocaine directly acts on spinal dorsal horn and produces analgesic effect: an in vivo patch-clamp analysis. Sci Rep. 2016;6:26253. | ||
Watson CJ. Insular balance of glutamatergic and GABAergic signaling modulates pain processing. Pain. 2016;157(10):2194–2207. | ||
Foerster BR, Nascimento TD, DeBoer M, et al. Excitatory and inhibitory brain metabolites as targets of motor cortex transcranial direct current stimulation therapy and predictors of its efficacy in fibromyalgia. Arthritis Rheumatol. 2015;67(2):576–581. | ||
Harris RE, Clauw DJ. Imaging central neurochemical alterations in chronic pain with proton magnetic resonance spectroscopy. Neurosci Lett. 2012;520(2):192–196. | ||
Rosa P, Fratangeli A. Cholesterol and synaptic vesicle exocytosis. Commun Integr Biol. 2010;3(4):352–353. | ||
Salaun C, James DJ, Chamberlain LH. Lipid rafts and the regulation of exocytosis. Traffic. 2004;5(4):255–264. | ||
Deng PY, Xiao Z, Jha A, et al. Cholecystokinin facilitates glutamate release by increasing the number of readily releasable vesicles and releasing probability. J Neurosci. 2010;30(15):5136–5148. | ||
De Chiara V, Motta C, Rossi S, et al. Interleukin-1beta alters the sensitivity of cannabinoid CB1 receptors controlling glutamate transmission in the striatum. Neuroscience. 2013;250:232–239. | ||
Besshoh S, Bawa D, Teves L, Wallace MC, Gurd JW. Increased phosphorylation and redistribution of NMDA receptors between synaptic lipid rafts and post-synaptic densities following transient global ischemia in the rat brain. J Neurochem. 2005;93(1):186–194. | ||
Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16(2):109–110. | ||
Wang Q, Zhao X, Li S, Han S, Peng Z, Li J. Phosphorylated CaMKII levels increase in rat central nervous system after large-dose intravenous remifentanil. Med Sci Monit Basic Res. 2013;19:118–125. | ||
Perrotta A, Chiacchiaretta P, Anastasio MG, et al. Temporal summation of the nociceptive withdrawal reflex involves deactivation of posterior cingulate cortex. Eur J Pain. 2017;21(2):289–301. | ||
Todd AJ. Plasticity of inhibition in the spinal cord. Handb Exp Pharmacol. 2015;227:171–190. | ||
Takazawa T, Choudhury P, Tong CK, et al. Inhibition mediated by glycinergic and GABAergic receptors on excitatory neurons in mouse superficial dorsal horn is location-specific but modified by inflammation. J Neurosci. 2017;37(9):2336–2348. | ||
Eto K, Ishibashi H, Yoshimura T, et al. Enhanced GABAergic activity in the mouse primary somatosensory cortex is insufficient to alleviate chronic pain behavior with reduced expression of neuronal potassium-chloride cotransporter. J Neurosci. 2012;32(47):16552–16559. | ||
Goldberg JS. Chronic opioid therapy and opioid tolerance: a new hypothesis. Pain Res Treat. 2013;2013:407504. | ||
Head BP, Patel HH, Insel PA. Interaction of membrane/lipid rafts with the cytoskeleton: impact on signaling and function: membrane/lipid rafts, mediators of cytoskeletal arrangement and cell signaling. Biochim Biophys Acta. 2014;1838(2):532–545. | ||
Denny JB. Molecular mechanisms, biological actions, and neuropharmacology of the growth-associated protein GAP-43. Curr Neuropharmacol. 2006;4(4):293–304. | ||
Park Y, Kim KT. Dominant role of lipid rafts L-type calcium channel in activity-dependent potentiation of large dense-core vesicle exocytosis. J Neurochem. 2009;110(2):520–529. | ||
de Juan-Sanz J, Zafra F, Lopez-Corcuera B, Aragon C. Endocytosis of the neuronal glycine transporter GLYT2: role of membrane rafts and protein kinase C-dependent ubiquitination. Traffic. 2011;12(12):1850–1867. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.