Back to Journals » Journal of Pain Research » Volume 14
The Inclusion of Tolfenamic Acid into Cyclodextrins Stimulated by Microenvironmental pH Modification as a Way to Increase the Anti-Migraine Effect
Authors Stasiłowicz A ![]() , Tykarska E
, Tykarska E ![]() , Rosiak N
, Rosiak N ![]() , Sałat K, Furgała-Wojas A
, Sałat K, Furgała-Wojas A ![]() , Plech T
, Plech T ![]() , Lewandowska K, Pikosz K, Pawłowicz K, Cielecka-Piontek J
, Lewandowska K, Pikosz K, Pawłowicz K, Cielecka-Piontek J
Received 15 December 2020
Accepted for publication 25 February 2021
Published 14 April 2021 Volume 2021:14 Pages 981—992
DOI https://doi.org/10.2147/JPR.S295795
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor E Alfonso Romero-Sandoval
Anna Stasiłowicz,1 Ewa Tykarska,2 Natalia Rosiak,1 Kinga Sałat,3 Anna Furgała-Wojas,3 Tomasz Plech,4 Kornelia Lewandowska,5 Katarzyna Pikosz,1 Kamil Pawłowicz,1 Judyta Cielecka-Piontek1
1Department of Pharmacognosy, Poznan University of Medical Sciences, Poznan, Poland; 2Department of Chemical Technology of Drugs, Poznan University of Medical Sciences, Poznan, Poland; 3Department of Pharmacodynamics, Jagiellonian University Medical College, Krakow, Poland; 4Department of Pharmacology, Medical University of Lublin, Lublin, Poland; 5Institute of Molecular Physics, Polish Academy of Sciences, Poznan, Poland
Correspondence: Judyta Cielecka-Piontek
Department of Pharmacognosy, Poznan University of Medical Sciences, Swiecickiego 4, 61– 781, Poznan, Poland
Tel +48 61 854 67 09
Fax +48 61 854 67 01
Email [email protected]
Purpose: The poorly soluble nonsteroidal anti-inflammatory drug (NSAID), tolfenamic acid (TA), was studied to maximize its solubility, permeability through biological membranes, and pharmacological activity.
Methods: A mixture with magnesium stearate (MS) – microenvironment pH-modifier was prepared, as well as systems additionally containing incorporating substances methyl-β-cyclodextrin (M-β-CD) and 2-hydroxypropyl-β-cyclodextrin (HP-β-CD). The identification of TA-MS-CD systems was confirmed using experimental methods: X-ray powder diffraction (XRPD) and Fourier transform infrared spectroscopy (FT-IR) with the theoretical support. Apparent solubility study was performed using the paddle apparatus, while in vitro gastrointestinal tract (GIT) and blood-brain barrier (BBB) permeability were conducted by using PAMPA (Parallel Artificial Membrane Permeability Assay). The in vivo part of the study used the mouse nitroglycerin (NTG)-induced migraine pain model.
Results: From practically insoluble substance, TA in TA-MS-M-β-CD system dissolved up to 80.13% ± 2.77%, and in TA-MS-HP-β-CD up to 92.39% ± 3.25% in 180 minutes. An increase in TA permeability was also obtained in the TA-MS-M-β-CD and TA-MS-HP-β-CD systems through GIT membranes (Papp values 2.057 x 10− 5 cm s− 1 and 2.091 x 10− 5 cm s− 1, respectively) and through BBB (Papp values 3.658 x 10− 5 cm s− 1 and 3.629 x 10− 5 cm s− 1, respectively). The enlargement of the solubility and permeability impacted analgesia. The dose 25 mg/kg of both TA-MS-HP-β-CD and TA-MS-M-β-CD was almost equally effective and only slightly less effective than the dose 50 mg/kg of pure TA. Both TA-MS-HP-β-CD and TA-MS-M-β-CD used at 50 mg/kg more effectively attenuated tactile allodynia in NTG-treated mice than the same dose of pure TA. None of TA forms influenced heat hyperalgesia.
Conclusion: Increasing solubility of TA caused an increase of its analgesic effect in an animal model of migraine pain.
Keywords: tolfenamic acid, migraine pain, cyclodextrins, permeability, gastrointestinal tract, blood-brain barrier
Introduction
Ineffective pain management is still an unsolved clinical problem. For pain localized in the central nervous system (CNS), the difficulty is to achieve an effective drug concentration. Migraine is one of the medical cases requiring central analgesia. Migraine belongs to the most prevalent neurological disorders worldwide and is the main cause of disability within a patient population. Inflammation, in particular neurogenic inflammation, is still regarded as an essential contributor to this disorder.1 Therefore, in addition to triptans – agonists of serotonergic 5-HT1B/D receptors, the pharmacological approach to treat migraine attacks includes the use of non-steroidal anti-inflammatory drugs (NSAIDs). These drugs act via the inhibition of cyclooxygenases (COXs) – enzymes responsible for the synthesis of prostaglandins and they are still the most commonly prescribed drugs for analgesic, antipyretic and anti-inflammatory purposes.2 Prostaglandins (PGs) are widely distributed in the trigeminal-vascular system, and their receptors are localized in the trigeminal ganglion and the trigeminal nucleus caudalis.3 Sterile inflammatory state within the intracranial meninges, also referred to as neurogenic inflammation is a key phenomenon underlying migraine. It results from the sensitization and activation of trigeminal meningeal nociceptors and is characterized by the release of neuropeptides (eg, substance P, CGRP) from the trigeminal innervation, which leads to vasodilation, plasma extravasation, edema, and mast cell degranulation.4 Moreover, the synthesis of vasodilatory prostaglandins can be increased by catecholamines and indolamine, and this results in nociceptor sensitization and the development of hyperalgesia in migraine.5 Prostaglandin-induced vasodilatation, edema, and hyperalgesia in migraine are strongly associated with inflammation. Therefore, NSAIDs are administered alone or in combination with triptans in migraine to improve patient outcomes.6 Although not many substances can cross the blood-brain barrier (BBB), the research indicates the NSAIDs’ capacity to penetrate this barrier.7,8 Tolfenamic acid (TA) (N-(2-methyl-3-chlorophenyl)-anthranilic acid) is one of the most commonly used NSAID in this neurological disorder, both as a preventive and interventional drug with high analgesic efficacy and a good safety profile.9 It has been proven that, compared to other NSAIDs, TA exhibits low side effects and minor toxicity; in particular, it causes fewer gastrointestinal complications.10–12 It also has a beneficial effect on the maintenance of pregnancy in mice.13 Animal studies suggest that TA may penetrate the CNS, as demonstrated in the context of the chemopreventive effect, it exhibits neuroprotective effect, it might slow down the progression of Alzheimer’s disease.14–16 The most significant limitation of the wide use of TA is its poor water solubility. However, solubility of TA might vary among different polymorphic forms.17 Bioavailability of TA after oral administration is about 75%. As a highly permeable substance with low solubility, TA belongs to class II in Biopharmaceutics Classification System (BCS).18
It has been reported in the literature that a combination of TA with cyclodextrins (CDs) and suitable excipients improves its solubility. CDs are polymers that might enlarge the solubility of substances by incorporating them into the caves. Preparation of inclusion complexes of TA with hydroxypropyl-β-cyclodextrin (HP-β-CD) and methyl-β-cyclodextrin (M-β-CD) in a (1:2) ratio resulted in a two-fold increase in TA solubility for the system with M-β-CD and 30 times for the system with HP-β-CD.19 The preparation of inclusion complexes of TA with β-CD and HP-β-CD in the ratio 1:1 in the solid-state by the freeze-drying technique also resulted in better solubility of TA.20 Empirical research has been extended to in silico simulations. Hitherto, the inclusion complex of TA and β-cyclodextrin (1:1) was subjected to quantum chemical calculations to define the geometry of the system. The results showed that TA is introduced into the cavity in two orientations: (1) COOH group of TA points toward a wider rim of the β-CD, and (2) the COOH group of TA points toward the narrow rim of the β-CD.21
The present study aimed to confirm that prepared CD systems, that cause the increase in TA solubility by microenvironmental pH-modification, have a stronger analgesic effect in migraine than pure TA. Our studies involved three areas: (i) preparation and identification of TA-MS-CD systems, (ii) study of the effects of increasing TA solubility in regards to permeability through biological membranes simulating the GIT and BBB, (iii) evaluation of the analgesic effect of TA-MS-CD systems in a mouse model of migraine-like pain.
Experimental Part
Preparation, Identification, Solubility and Permeability of TA-MS-CD Systems
Materials
For physicochemical studies, TA (purity > 98%) in bulk substance, M-β-CD and HP-β-CD were obtained from Sigma-Aldrich (Poland). Sodium hydroxide and potassium dihydrogen phosphate were supplied by Avantor Performance Materials (Poland). Prisma HT, GIT Lipid solution, Acceptor Sink Buffer, BBB liquid solution, and Brain Sink Buffer were obtained from Pion Inc (United Kingdom). High-quality pure water was prepared using an Eliux S A 67120 Millipore purification system (Poland).
For in vivo assays, the test compounds were prepared in 1% Tween 80 solution (Avantor Performance Materials, Poland), and they were administered by the oral route. To perform a quantitative comparison of antiallodynic and antihyperalgesic properties of TA-MS-M-β-CD and TA-MS-HP-β-CD systems, we selected two doses: 25 and 50 mg/kg. The latter was the starting dose for our study as it is the most widely used dose of TA in various in vivo assays (oral route).22 Control mice received 1% Tween 80. To induce migraine-like pain, nitroglycerin (NTG) (Perlinganit, 1mg/1mL, UCB Pharma GmbH, Germany) was administered by the intraperitoneal route 60 min before further behavioral tests.23
Behavioral tests were performed at the Department of Pharmacodynamics, Faculty of Pharmacy, Jagiellonian University Medical College in Cracow, Poland. In in vivo tests, 63 adult male Albino Swiss (CD-1) mice weighing between 18 and 22 g were used. The animals were housed in groups of 10 mice per cage at room temperature of 295.15 ± 2 K, under light/dark (12:12) cycle. The animals had free access to food and tap water before experiments. The ambient temperature of the experimental room and humidity (50 ± 10%) were kept consistent throughout all the tests. For behavioral experiments, the animals were selected randomly. Each experimental group consisted of 6–10 animals/dose. The experiments were performed between 9 AM and 2 PM. To avoid potential bias in data recording, the investigators who were involved in behavioral assays were blinded to the experimental groups. Immediately after the in vivo assay, the animals were euthanized by cervical dislocation. The 1st Local Ethics Committee of the Jagiellonian University in Krakow (ethical approval No. 290/2019, 26.06.2019) approved all procedures, and the treatment of animals was in full accordance with ethical standards laid down in respective Polish and EU regulations (Directive No. 86/609/EEC). Animal studies are reported in compliance with the ARRIVE guidelines.24
Preparation of TA-MS-CD Systems by Co-Precipitation Method
150.0 mg of TA and 150.0 mg of magnesium stearate (MS) (mass ratio 1:1) were ground in a mortar for 15 minutes. Next, they were dissolved in a small amount of methanol. Independently there were prepared solutions of CDs (M-β-CD and HP-β-CD) in a small amount of water. Both solutions were mixed in a 1:1 molar ratio (TA:CD) and of the solvents were evaporated to dryness for 24 hours at 310 ± 0.5 K and 50 rotations per minute (MaxQ 4450 – Thermo Scientific).
The Identification of TA-MS-CD Systems
Identification of the CD systems of TA with MS and M-β-CD or HP-β-CD prepared by co-precipitation method was conducted basing on experimental methods such as X-ray powder diffraction (XRPD) and Fourier transform infrared spectroscopy (FT-IR). TA in free form was used as the reference substance in both analyzes.
XRPD
The powder X-ray diffraction patterns of TA, MS, M-β-CD, HP-β-CD, their physical mixtures (TA+MS, TA+MS+M-β-CD and TA+MS+HP-β-CD) and the binary TA-MS and ternary TA-MS-CD’s systems were collected on a Bruker AXS D2 Phaser diffractometer with LynxEye XE-T 1-dim detector using Cu Kα radiation (λ = 1.54060 Å). The machine operated with a generator voltage and current of 30 kV and 10mA, respectively. The measurements were performed at the 2θ scanning range between 5° and 40° with a step size of 0.02° and a counting rate of 2s/step. The program DIFFRAC.EVA was used to analyze the acquired data.25
FT-IR Spectroscopy
FT-IR spectroscopy was performed on a FT-IR Bruker Equinox 55 spectrometer. The samples were first mixed with KBr (IR grade) and then turned into pellets by using in the hydraulic press. The spectra were recorded in a frequency range of 400–4000 cm−1. Identification of bands, their intensity, and location on FT-IR spectrum of TA was conducted in comparison with FT-IR theoretical spectrum, which was obtained by using Density Functional Theory (DFT). The geometry was optimized using DFT with Becke, three-parameter, Lee-Yang-Parr hybrid functional, and 6–31G (d, p) basis set.
Studies of Solubility and Permeability of TA-MS-CD Systems Through Biological Membranes Simulating Gastrointestinal Walls and BBB
The changes of the TA solubility after its introduction into CD cavities during studies of apparent solubility and permeability through membranes simulating gastrointestinal walls and BBB were measured spectrophotometrically at 288 nm.
Apparent solubility study was conducted using the paddle Agilent 708-DS Dissolution Apparatus with a 500 mL dissolution medium at 310 K and 50 rpm. The TA+MS mixture and TA-MS-CD systems (containing 5 mg of TA) were weighed into gelatin capsules, which were later placed in metal springs to prevent flotation on the medium surface. The phosphate buffer with pH of 6.8 was used as the medium. The test was performed under sink conditions. At specific time intervals, 6.0 mL samples were removed and filtered. At the same time, equal volumes of the medium at the temperature 310 K was supplemented to the vessels. The changes in concentration of TA were measured using UV spectroscopy at 288 nm.
In vitro gastrointestinal tract (GIT) and BBB permeability were studied using PAMPA (Parallel Artificial Membrane Permeability Assay).26–28 The permeability of the TA+MS mixture was taken as reference. The system is based on an artificial membrane – a 120-μm-thick microfilter disc coated with a 20% (w/v) dodecane solution of a lecithin mixture (Pion, Inc.) – which separates two compartments – donor and acceptor in two 96-well plates. The tests were conducted in two environments: for GIT, the donor solution was adjusted to pH ≈ 6.8, and for BBB, the pH was ≈ 7.4 according to manufacturer’s recommendations. After adding samples to donor compartments, the plates were combined and then incubated for 3 hours for GIT model and 4 hours for BBB model in a humidity-saturated atmosphere at the temperature set at 310 K. The apparent permeability coefficient (Papp) was calculated according to the formula:
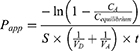
where VD – donor volume, VA – acceptor volume, Cequilibrium – equilibrium concentration 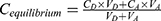 , S – membrane area, t – incubation time (in seconds).
, S – membrane area, t – incubation time (in seconds).
In vivo Studies of Analgesic Activity of TA-MS-CD Systems
Behavioral Testing Protocol and Studies of Analgesic Effect of TA-MS-CD Systems
In order to develop migraine-like pain in mice, the animals were injected intraperitoneally with a single dose of NTG (10 mg/kg).29 To assess the effect of the test compounds on tactile allodynia and heat hyperalgesia, mechanical and thermal pain sensitivity threshold of experimental animals was assessed using von Frey and hot plate tests, respectively. Based on our previous research measurements were collected before NTG administration (referred to as “before nitroglycerin”), 60 min after its injection (baseline, pre-drug measurements), and then, 60 min after TA systems administration (post-drug measurements).23 This time point for measuring post-drug effects of TA forms was selected based on the available literature data which showed that for TA used orally the mean time to reach the peak plasma concentration (Tmax) was approx.1 h.30
Mechanical hypersensitivity (tactile allodynia) in mice was assessed using the electronic von Frey unit (Bioseb, France).31 This device is supplied with a single flexible filament applying increasing force (from 0 to 10 g) against the plantar surface of the hind paw of a mouse. The paw withdrawal response automatically turns off the stimulus, and the mechanical pressure that evokes the response is recorded.
In this test, the mice were placed individually in test compartments with a wire mesh bottom for 30-min habituation. Then, each mouse was tested 3 times alternately in each hind paw, allowing at least 30 s between each measurement. Subsequently, the mice were pretreated with NTG solution, and 60 min later, the animals were tested again for their pain sensitivity threshold (pre-drug measurements). This procedure was also repeated 60 min after TA administration (post-drug measurements).
Antihyperalgesic properties of TA forms were assessed in the hot plate test using the hot plate apparatus (Hot/cold plate, Bioseb, France) according to a slightly modified method previously described.32 In the hot plate apparatus, a temperature-controller maintains the surface temperature to a set point (here 328–329 K). In this assay, the animals were tested first to obtain baseline latencies to pain reaction (licking hind paws or jumping) before NTG injection (referred to as latencies “before nitroglycerin”). Then, NTG was injected, and latencies to pain reaction were measured again (referred to as ‘pre-drug latencies’). After that, TA systems were administered, and post-drug latencies were measured again. In this assay, a cut-off time of 60 s was established to avoid potential paw tissue damage, and animals not responding within 60 s were removed from the apparatus and assigned a score of 60 s.
Data analysis of the results obtained in behavioral tests was provided by GraphPad Prism Software (v.8, CA, USA). Numerical results obtained in behavioral tests are expressed as the mean ± SEM. Statistical analysis was performed using the Shapiro–Wilk normality test, followed by the analysis of variance (ANOVA) of repeated measures of two factors (treatment x time) and Tukey’s or Dunnett’s post hoc comparison. P < 0.05 was considered significant.
Results
In the first of our studies, we prepared the TA-MS-CD systems by using the co-precipitation method. The mixture of TA and MS (1:1 mass ratio) was dissolved as starting reagents for the preparation of the TA-MS-CD systems. Simultaneously, aqueous solutions of CDs: M-β-CD and HP-β-CD were prepared. Next, the solutions were mixed, the solvents were evaporated, and the TA-MS-CD systems were obtained. Their formation was spontaneous and repetitive. The TA-MS-CDs systems were characterized by XRPD and FT-IR spectroscopy.
Pure TA, MS, M-β-CD, HP-β-CD, their mixtures obtained by grinding (TA+MS, TA+MS+β-CD’s), or systems prepared by recrystallization (TA-MS) and co-precipitation (TA-MS-β-CD’s) were studied by powder XRPD. Diffraction patterns of the pure TA, MS, M-β-CD, HP-β-CD are shown in Figure 1A–D. The experimental TA diffraction patterns were compared with the diffraction patterns in the Cambridge Structural Database (CSD) (Supplementary Materials, Figure S2, S1). Diffraction patterns of the TA+MS, as well as the TA+MS+β-CD’s are generally composed of the sum of the contributions of all constituent phases (Figure 1E–G) showing that mixtures obtained by grinding are physical mixtures. A different situation is observed for the TA-MS system obtained by recrystallization, where the change in the number and position of the peaks on the diffractogram indicates the formation of a new solid phase (Figure 1H). The most intense peaks of this phase occur at 2θ angles 10.4°, 13.3°, 13.8°, 17.6°, 20.0°, 23.6°, 25.5° and 26.5°. The peaks of a new TA-MS phase superimposed with the humps of amorphous CD’s are observed in XRPD patterns of ternary TA-MS-M-β-CD and TA-MS-HP-β-CD systems obtained by co-precipitation (Figure 1I and J).
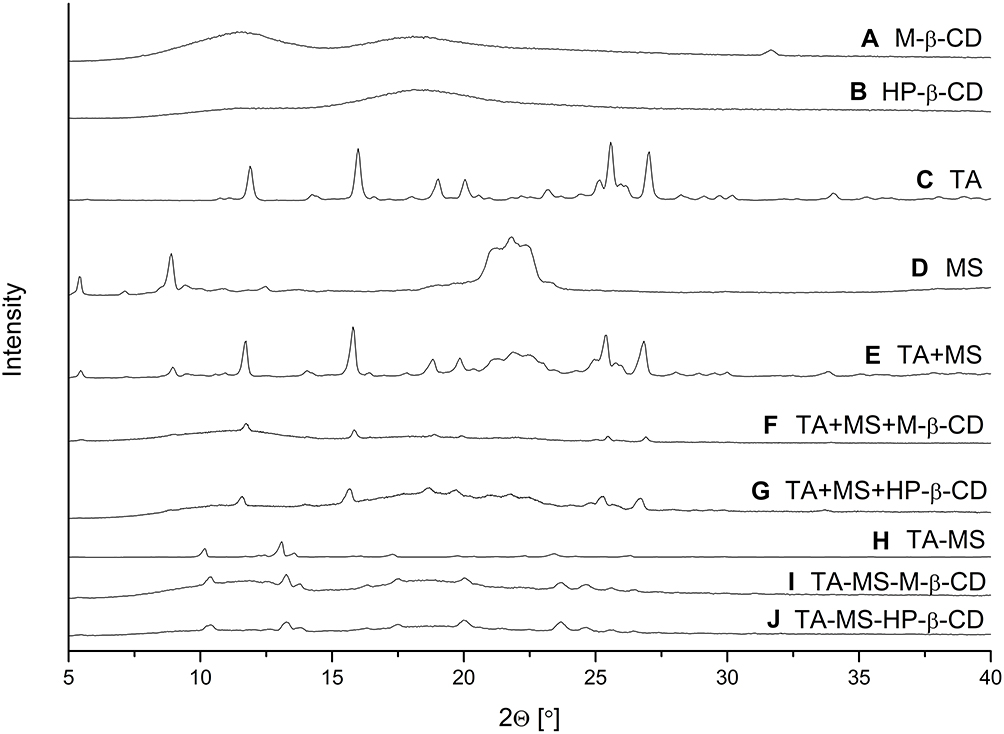 |
Figure 1 XRPD diffraction patterns of M-β-CD (A), HP-β-CD (B), TA (C), MS (D), TA+MS mixture (E), TA+MS+M-β-CD mixture (F), TA+MS+HP-β-CD mixture (G), TA-MS system (H), TA-MS-M-β-CD system (I), and TA-MS-HP-β-CD system (J). |
The characteristic FT-IR band assignments for the TA spectrum are presented in supplementary materials (Table S1, Figure S3). Identification of bands, their location and intensity on FT-IR spectrum of the TA was conducted in comparison with FT-IR theoretical spectrum calculated using DFT-B3LYP functional and 6–31G (d, p) as a basis set.
Comparison of FT-IR spectra of the TA and the TA-MS showed no changes in position and intensity of the band. Additional bands related to the presence of the carboxylate ion did not appear in the TA+MS spectra. In the result of FT-IR of TA and TA+MS did not shown differences. The FT-IR spectra of TA-MS-CDs systems were characterized by differences with respect to the spectrum of the TA+MS and TA+MS+CDs mixture (Figure 2). The strongest differences involved changes in the intensity of the bands in the range of 1110–1270 cm−1, a decrease in the intensity of the characteristic bands at about 1640 cm−1 and a change in the intensity of the bands at about 2900 cm−1 and at 3450 cm−1. Changes in band intensity in the range of 1100–1270 cm−1 are associated with changes of vibrations for the group C=O, changes of stretching vibrations (1079 cm−1) for the bond of C-O in the COOH group and rocking vibrations in the phenyl ring (1164 cm−1). Changes in band intensity at about 1265 cm−1 are combined with C-O-H bending vibrations and C-C stretching vibrations of the aromatic ring. The strong change in vibration intensity for the C = O carbonyl domain (1664 cm−1) is caused by its interaction with CDs. Changes in band intensity in the area of about 3420 cm−1 are associated with differences in the stretching vibrations of the O-H in the COOH group. It is worth noting that the results of system identity studies using FT-IR technique exclude the formation of stable salts while confirming the creation of hydrogen-bonded TA-MS-CD’ systems. XRPD studies confirmed the presence of a new crystalline phase of the systems.
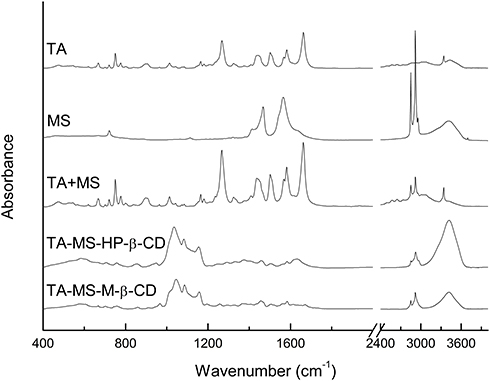 |
Figure 2 Experimental FT-IR absorption spectra of TA, TA+MS mixture, TA-MS-HP-β-CD and TA-MS-M-β-CD systems. |
Studies of apparent solubility and permeability were performed in the water environment. The differences in concentrations of pure TA during those experiments were undetectable according to its low solubility. Thus, the results for TA were excluded.
Studies on the changes of solubility of the incorporation of the TA into CDs began with the preparation of a mixture of TA and MS (1:1 weight ratio). As a result of the pH-modification, practically insoluble TA, was dissolved in 180 minute up to 63.21% ± 0.70% (C= 5.98 x 10−3 mg/mL). Systems with CDs: M-β-CD, HP-β-CD allowed for additional improvement of solubility up to 80.13% ± 2.77% (C= 7.69 x 10−3 mg/mL), and 92.39% ± 3.25% (C= 8.66 x 10−3 mg/mL), respectively (Figure 3).
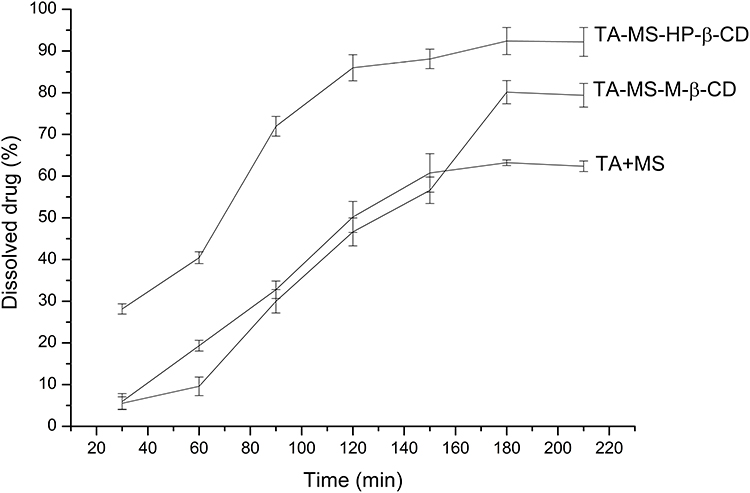 |
Figure 3 Apparent solubility study of TA+MS mixture and the systems with M-β-CD and HP-β-CD. |
The studies of the TA permeability through membranes simulating the gastrointestinal walls and the BBB were conducted using the PAMPA model with appropriate modifications (Figure 4). Changes in the TA concentration in acceptor and donor fluids were evaluated spectrophotometrically. As a result of its combination with MS, pH-modification of the TA solubility allowed its dissolution, which ensured proper permeability through gastrointestinal membranes. The effect of the increased solubility of the TA was better permeability through the system of artificial biological membranes simulating the gastrointestinal system. The permeability of the TA+MS mixture was described as 1.825 x 10−5 ± 2.496 x 10−6 cm s−1. The preparation of systems with CDs resulted in increased permeability: for the system with M-β-CD Papp value was 2.057 x 10−5 ± 3.949 x 10−6 cm s−1 and for the system with HP-β-CD Papp value was 2.091 x 10−5 ± 3.183 x 10−6 cm s−1. According to the guidelines, all tested systems can be treated as well-permeable API, because the Papp value in the gastrointestinal model greater than ≥1 x 10−6 cm s−1 appears to be highly permeable.33 The results of the BBB barrier permeation study allowed the classification of all tested systems also as well-permeable according to the guidelines that compounds having the Papp value bigger than 4.0 x 10−6 cm s−1 are highly permeable.34 The permeability in the BBB model showed that CD impacts the permeability of TA. The Papp values for the TA+MS mixture and the systems with M-β-CD and HP-β-CD were 3.014 x 10−5 ± 3.693 x 10−6 cm s−1, 3.658 x 10−5 ± 2.400 x 10−6 cm s−1 and 3.629 x 10−5 ± 1.614 x 10−6 cm s−1, respectively.
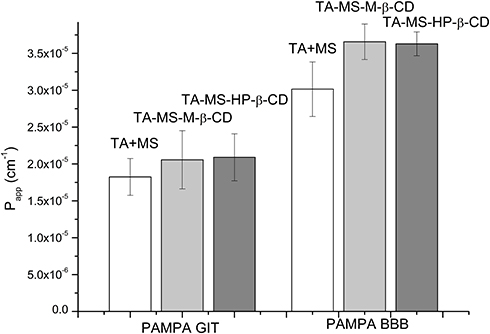 |
Figure 4 Permeability study diagram for TA+MS mixture, TA-MS-M-β-CD and TA-MS-HP-β-CD systems in the GIT model and the blood-brain barrier model. |
The in vivo model study aimed to assess potential changes in the analgesic activity of TA-MS-HP-β-CD and TA-MS-M-β-CD systems in relation to pure TA action by assessing their effects on tactile allodynia and heat hyperalgesia. In the von Frey test, repeated measures ANOVA revealed an overall effect of treatment (F[6.165]=30.62), p < 0.0001). Time also affected the results significantly (F[2.165]=499.8), p < 0.0001) and the drug x time interaction was significant (F[12.165]=18.55), p < 0.0001). As shown in Figure 5A, the paw withdrawal threshold before NTG administration was similar in all experimental groups. After NTG administration, a statistically significant difference in the paw withdrawal threshold was observed between vehicle-treated mice not receiving NTG and mice that received NTG (p < 0.0001). The comparison of post-drug paw withdrawal threshold of the control group that was injected with NTG and all other groups tested revealed significantly (p < 0.0001) lower post-drug paw withdrawal threshold in the former group compared to all other experimental groups, which indicates that the test TA systems effectively attenuated tactile allodynia in the mouse model of migraine pain.
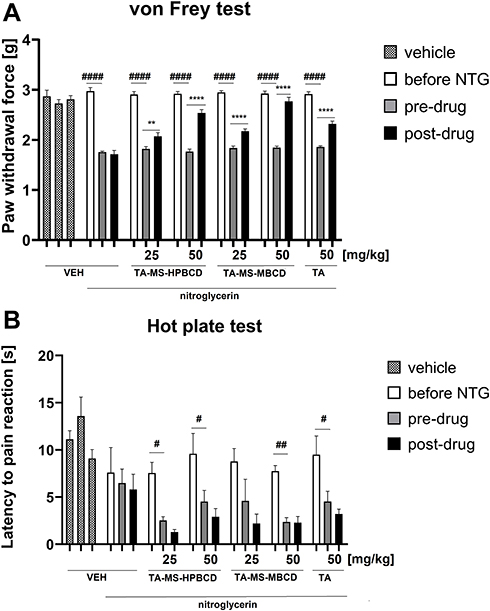 |
Figure 5 Influence of TA forms: TA-MS-HP-β-CD, TA-MS-M-β-CD and pure TA, administered orally 60 min before pain tests, on tactile allodynia measured in the von Frey test (A) and thermal hyperalgesia assessed in the hot plate test (B) in a mouse model of migraine pain induced by NTG administration. NTG was injected 60 min before pre-drug measurements of pain sensitivity were collected. Results are shown as mean paw withdrawal force (±SEM) measured in the von Frey test (A), or latency time to pain reaction (±SEM) measured in the hot plate test (B) for n = 6–10. Statistical analysis: repeated measures ANOVA followed by Dunnett’s or Tukey’s post hoc test. Significance vs paw withdrawal threshold or latency to pain reaction before NTG administration: #p < 0.05, ##p < 0.01, ####p < 0.0001; and significance vs pre-drug paw withdrawal threshold or pre-drug latency to pain reaction in the individual group: **p < 0.01, ****p < 0.0001. |
The comparison of pre-drug and post-drug paw withdrawal threshold in each particular group demonstrated no significant differences in the paw withdrawal threshold in control mice not treated with NTG, or in control mice that were treated with NTG. In contrast to this, in all other NTG-treated groups, a statistically significant elevation of post-drug paw withdrawal threshold compared to the pre-drug measurement was noted (p < 0.01 for NTG+TA-MS-HP-β-CD 25 mg/kg-treated mice, and p < 0.0001 for all other NTG+TA forms-treated mice; Figure 5A).
The comparison of the antiallodynic efficacy of all three TA forms revealed that the dose 25 mg/kg of TA-MS-HP-β-CD was almost equally effective as the same dose of TA-MS-M-β-CD. The dose of 25 mg/kg of these forms of TA, in particular, that of TA-MS-M-β-CD, was only slightly less effective than the dose 50 mg/kg of pure TA and therefore we decided not to test a lower dose (ie, 25 mg/kg) of pure TA. Importantly, both TA-MS-HP-β-CD and TA-MS-M-β-CD used at 50 mg/kg more effectively attenuated tactile allodynia in NTG-treated mice than the same dose of pure TA. TA-MS-M-β-CD at 50 mg/kg was more effective than the corresponding dose of TA-MS-HP-β-CD.
In the hot plate test, an overall effect of treatment was observed (F[6.168]=7.952, p < 0.0001). Time also affected the results significantly (F[2.168]=24.54, p < 0.0001), but the drug x time interaction was not significant (F[12.168]=1.119, p > 0.05). Post hoc analysis showed no significant differences in the latency to pain reaction among tested groups before NTG administration but there was a significantly (p < 0.01) prolonged latency to pain reaction in control mice not treated with NTG compared to control mice after they were injected with this drug. Importantly, significantly reduced latency to pain reaction was noted in mice after NTG administration (p < 0.05 vs latency before NTG injection; Figure 5B).
The comparison of pre-drug and post-drug latencies to pain reaction measured with in each particular group demonstrated no significant differences in this parameter in control mice not treated with NTG, or in mice that were treated with NTG and vehicle, or NTG and test compounds. None of the tested TA forms was effective as an antihyperalgesic agent in the hot plate test performed in NTG-treated mice (Figure 5B).
Discussion
Poor solubility of active pharmaceutical ingredients (API) is a crucial limitation of their clinical use. Solubility might be modified by alterations in the chemical structure (eg, salt formation), changes in the physical structure (eg, obtaining amorphous dispersions), or through pre-formulation and formulation works. For APIs with very poor solubility but significant clinical potential, it is worth combining several techniques to obtain better soluble APIs. For lipophilic APIs, having to achieve the required concentration within the CNS, it is essential to increase the dynamics of crossing the BBB. One strategy may include increasing the intra-body concentration to provide a greater diffusion gradient for the BBB. This approach was assumed for TA, which is practically insoluble in water, but it has significant analgesic potential, especially in migraine.14
In the first step, TA was combined with MS. As the result, there was obtained about 63.21% higher solubility than in the case of the TA in a free form. By carrying out FT-IR analysis, the presence of carboxylate ion bands was excluded, and the carboxyl group should be indicated as the domain involved in the interaction with the hydrophobic interior of CDs. Those observations are in agreement with changes in the intensity of stretching band of C-O and bending band of O-H (at 1079 cm−1, 1664 cm−1 and 1265 cm−1, respectively). A similar approach where changes of solubility were induced by pH-modification was applied for other NSAIDs. In the study of Abhijeet K. and Namita D., the solubility of aceclofenac was increased using melt granulation and micro-environmental pH modulation by sodium hydrogen carbonate.35 Piyush Gupta and Arvind K. Bansal co-processed celecoxib with polyvinylpyrrolidone, and pH-adjusting meglumine with the use of spray drying method which allowed to obtain an amorphous system with increased solubility about 10 times and increased stability.36
In the further part of the TA solubility optimization, TA was incorporated into the CDs: M-β-CD and HP-β-CD. As a result, systems ensuring TA solubility allow to obtain a solubility rate of TA 80.13% (C=7.69 x 10−3 mg/mL) and 92.39% (C= 8.66 x 10−3 mg/mL), respectively.
Regarding the initial research hypothesis that better API solubility after introduction into CD cavity was to induce an increase in permeability, the solubility studies of TA-MS-CD systems were conducted in the acceptor fluid at pH 6.8, corresponding to the absorption site of analogs from the NSAID group.37 For all modifications used, an increase in TA solubility was noted before 180 min (Figure 3). It is noteworthy that regardless of the solubility time, the HP-β-CD modified the solubility of TA stronger than M-β-CD. The observed increase in the TA solubility after introduction into the CD is the result of blocking the carboxylic groups. Blocking of the hydrophilic carboxyl group is, however, compensated by the presence of hydroxyl groups derived from CD. The number of hydroxyl groups in the case of TA-MS-HP-β-CD is greater than TA-MS-M-β-CD, which may explain the differences in the TA solubility when introduced into CD systems. The blocking of the hydrophilic group of the TA but at the same time an increase in solubility has also been reported for CD combinations with other APIs. Huang et al research show that complexation ursolic acid and oleanolic acid with β-CD increased their solubility, the complexes were formed in the ratio 1:1, and the carboxyl group of the guests were incorporated into the cavity of CD.38
Papp values were determined using the PAMPA model for the application simulating permeability through the gastrointestinal membranes and the BBB. The increase in the TA solubility induced a rise in permeability through the gastrointestinal membranes (Papp for TA-MS-M-β-CD 2.057 x 10−5 cm s−1 and for TA-MS-HP-β-CD 2.091 x 10−5 cm s−1 vs for TA+MS mixture 1.825 x 10−5 cm s−1). Papp values allow TA systems to be classified as well penetrating through the walls of the GIT.33 Permeability through membranes simulating the BBB of TA systems was significantly maximized – Papp for TA-MS-M-β-CD 3.658 x 10−5 cm s−1 and for TA-MS-HP-β-CD 3.629 x 10−5 cm s−1 comparing to the TA mixture with MS Papp 3.014 x 10−5 cm s−1. It is assumed that the majority of drugs transport is based on the diffusion process, however active transport is also present.39 Bearing that in mind, the next stage was the assessment of the increase of pharmacological activity in the in vivo model.
In the in vivo part of the present research, we used a mouse model of migraine-like pain, namely the NTG model, and two behavioral tests for the assessment of the effect of the TA forms on tactile allodynia and heat hyperalgesia induced by NTG administration. This migraine model is based on using NTG, a nitric oxide donor, as a tool to provoke migraine-like pain in rodents. In mice and rats, intraperitoneal NTG has been widely used to develop a model to study sensory hypersensitivity typical for migraine. The infusion of NTG in mice caused thermal and mechanical allodynia, and these symptoms were reversed by sumatriptan.23 Moreover, some studies showed that NTG-treated mice developed aversion to light and increased meningeal blood flow and this mouse model was also used to study the progression of the disorder from an acute to a chronic condition. Taken together, this NTG model may be considered as a “gold standard” and a reliable screening tool in the search for novel therapies in migraine treatment.2 Although analgesic properties of TA can be assessed in several screening pain tests for NSAIDs, including a mouse writhing test, formalin test or the rat Randal-Selitto test, in this research we focused on the NTG model.40–42 Firstly because analgesic properties of TA in other animal pain and inflammation models were described previously, and secondly, our study was the first-in-animal experiment that assessed novel TA forms and it was aimed to be focused on their potential use in migraine.43,44 Therefore we selected a pain model specific for migraine. Thirdly, TA is widely used in humans suffering from migraine, so the confirmation of the effect of TA and novel TA forms in a mouse model of migraine-like pain would be important from the translational point of view.14 Importantly, although migraine is more common in women, for this present research we chose male mice. Both male and female mice are widely tested in the NTG rodent model of migraine-like pain but available literature data indicate that male mice may be more resistant to this model.29,45,46 Therefore, by using male subjects we wanted to assess, whether the pharmacological activity of the tested TA forms can be observed even under these drug-resistant conditions.
The von Frey test enables to assess mechanical (tactile) allodynia in mice. Cutaneous tactile allodynia (ie, perception of pain following non-painful stimuli) occurs in more than 70% of migraineurs, so sensory hypersensitivity to mechanical stimulation is a serious problem for these patients.47 In the present experiment we showed that tactile allodynia developed in all NTG-treated groups. The comparison of the antiallodynic efficacy of all three TA forms revealed that the dose 25 mg/kg of TA-MS-HP-β-CD system was almost equally effective as that of TA-MS-M-β-CD system. Importantly, the dose of 25 mg/kg of these forms of TA, in particular that of TA-MS-M-β-CD system, was only slightly less effective than the dose of 50 mg/kg of pure TA. Therefore, for ethical reasons we did not test the dose of 25 mg/kg of pure TA which was expected to have marginal analgesic effect. Significantly, both TA-MS-HP-β-CD and TA-MS-M-β-CD systems used at 50 mg/kg were more effective than the same dose of pure TA in the von Frey test in NTG-treated mice. TA-MS-M-β-CD system at 50 mg/kg was more effective than the corresponding dose of TA-MS-HP-β-CD system.
nTG also induced heat hyperalgesia in mice. Interestingly, none of the tested TA forms was able to attenuate these symptoms of sensory (heat) hypersensitivity measured in the hot plate test. A possible explanation for this lack of efficacy of TA forms is that in the hot plate assay the characteristic nocifensive response that occurs (jumping, licking of the paws) is of central origin and drugs with antinociceptive properties in the hot plate test act primarily in the spinal medulla and/or higher CNS levels. In this test, peripherally acting analgesic drugs (eg, TA forms tested) are generally not active.48 It has to be noted that in the hot plate test, the post-drug latencies were even reduced as compared to the pre-drug ones. A potential explanation for this observation has been demonstrated in previous studies showing that the hot plate test latency might decrease with repeated testing and this effect might be due to the learning abilities of mice.49 It should also be noted that no adverse effects or signs of toxicity were observed in NTG and all forms of TA-treated mice.
The increase in pharmacological activity for API with a different mechanism of action after their introduction into CD cavities, confirmed by the results of in vivo studies were reported, eg, for antimicrobial activity cefuroxime axetil, for the anti-inflammatory activity of naringenin and for reduction of arterial pressure by valsartan.50–52 In our previous studies, we also confirmed that an increase in the analgesic effect for sumatriptan after introduction into CD structures was possible.23 Other studies confirming the beneficial impact of CD systems on the analgesic effect were also obtained in the case of ketoprofen, where antipyretic, analgesic and anti-inflammatory effects increased, and in the mice study for meloxicam, where the elevated API serum levels maximized its analgesic activity.53,54
The strengths of the studies performed include increasing the solubility of TA with a method that can be easily used in the preparation of a drug form for patients. Prepared systems with CDs and MS that allow the implementation of microenvironmental pH modification are easy to prepare. No specialized equipment is required on a laboratory scale. In addition, only water and small amounts of alcohol were used for their preparation, which is consistent with green chemistry principles. There are also some limitations of the study. Scaling up the repeatable preparation of the systems might be challenging and time-consuming, which can delay implementing the prepared systems for use in patients. Analgesic effectiveness of novel TA forms compared to pure TA has been shown only in one animal model, and in males only. In addition, the efficacy of TA systems was assessed only at one time point of testing and in this first-in-animal study the duration of the analgesic effect was not assessed. Hence, at present we cannot assess if the duration of the analgesic effect might be modified due to changes in physicochemical properties of TA. Also, we did not observe antihyperalgesic properties of the test compounds in a test based on thermal stimulation. Hence, further studies using other than the NTG model are necessary to confirm anti-migraine properties of TA derivatives.2,55 Also, the use of female mice and testing TA forms at additional time points and in other than mice animal species with migraine-like pain symptoms are highly warranted.
Conclusions
The apparent solubility of undetectable TA was significantly increased according to pH-modification and efficient inclusion into the CDs. The maximized obtained value was 8.66 x 10−3 mg/mL for TA-MS-HP-β-CD. As a consequence, the permeability through GIT and BBB membranes was enlarged. The in vivo study confirmed the influence of TA improved physicochemical properties on the increase in analgesic activity in a mouse migraine-like pain model.
Abbreviations
API, active pharmaceutical ingredient; BBB, blood-brain barrier; BCS, Biopharmaceutics Classification System; CD, cyclodextrin; CGRP, calcitonin gene-related peptide; CNS, central nervous system; COX, cyclooxygenase; DFT, Density Functional Theory; FT-IR, Fourier transform infrared spectroscopy; GIT, gastrointestinal tract; HP-β-CD, 2-hydroxypropyl-β-cyclodextrin; MD, molecular dynamics; MS, magnesium stearate; M-β-CD, methyl-β-cyclodextrin; NSAID, non-steroidal anti-inflammatory drug; NTG, nitroglycerin; PAMPA, Parallel Artificial Membrane Permeability Assay; Papp, permeability coefficient; PG, prostaglandin; SEM, standard error of measurement; TA, tolfenamic acid; XRPD, X-ray powder diffraction.
Acknowledgments
The scientific work was performed within the project “Uniwersalna platforma modułowa do przeprowadzania badań uwalniania symulujących warunki fizjologiczne dla doustnych postaci leku” supported by The National Centre for Research and Development in the program EUROSTARS-2.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Edvinsson L, Haanes KA, Warfvinge K. Does inflammation have a role in migraine? Nat Rev Neurol. 2019;15(8):483–490. doi:10.1038/s41582-019-0216-y
2. Tardiolo G, Bramanti P, Mazzon E. Migraine: experimental models and novel therapeutic approaches. Int J Mol Sci. 2019;20(12):2932. doi:10.3390/ijms20122932
3. Antonova M, Wienecke T, Olesen J, Ashina M. Prostaglandins in migraine: update. Curr Opin Neurol. 2013;26(3):269–275. doi:10.1097/WCO.0b013e328360864b
4. Ramachandran R. Neurogenic inflammation and its role in migraine. SeminImmunopathol. 2018; 40:301–314. doi:10.1007/s00281-018-0676-y
5. Vapaatalo H. Tolfenamic acid and migraine‐aspects on prostaglandins and leukotrienes. Pharmacol Toxicol. 1994;75:76–80. doi:10.1111/j.1600-0773.1994.tb02004.x
6. Ong JJY, De Felice M. Migraine treatment: current acute medications and their potential mechanisms of action. Neurotherapeutics. 2018;15(2):274–290. doi:10.1007/s13311-017-0592-1
7. Biswas G, Kim W, Kim K-T, et al. Synthesis of ibuprofen conjugated molecular transporter capable of enhanced brain penetration. J Chem. 2017;2017. doi:10.1155/2017/4746158
8. Stab J, Zlatev I, Raudszus B, et al. Flurbiprofen-loaded nanoparticles can cross a primary porcine in vitro blood-brain barrier model to reduce amyloid-β 42 burden. J Nanomed Biother Discov. 2017;6:140.
9. Ahmed S, Sheraz MA, Ahmad I. Tolfenamic Acid. Brittain H, In: Profiles of Drug Substances, Excipients and Related Methodology. Vol. 43. Elsevier; 2018:255–319.
10. Nyfos L. A comparative clinical study of a new antirheumatic agent, tolfenamic acid (Clotam), and Phenylbutazone in rheumatoid arthritis. Scand J Rheumatol. 1979;8(sup24):5–7. doi:10.1080/03009742.1979.12088618
11. Rejholec V, Vapaatalo H, Tokola O, Gothoni G. Tolfenamic acid in the treatment of rheumatoid arthritis. Scand J Rheumatol. 1979;8(sup24):9–12. doi:10.1080/03009742.1979.12088619
12. Rejholec V, Vapaatalo H, Tokola O, Gothoni G. Tolfenamic acid in ankylosing spondylarthritis: a double-blind comparison to indomethacin. Scand J Rheumatol Suppl. 1980;36:1–7.
13. Schlapp G, Goyeneche L, Fernández G, Menchaca A, Crispo M. Administration of the nonsteroidal anti-inflammatory drug tolfenamic acid at embryo transfer improves maintenance of pregnancy and embryo survival in recipient mice. J Assist Reprod Genet. 2015;32(2):271–275. doi:10.1007/s10815-014-0378-x
14. Feldman D, Leahy E, Lee S-H. Chemopreventive properties of tolfenamic acid: a mechanistic review. Curr Med Chem. 2018;25(14):1598–1608. doi:10.2174/0929867324666170414155107
15. Subaiea GM, Adwan LI, Ahmed AH, Stevens KE, Zawia NH. Short-term treatment with tolfenamic acid improves cognitive functions in Alzheimer’s disease mice. Neurobiol Aging. 2013;34(10):2421–2430. doi:10.1016/j.neurobiolaging.2013.04.002
16. Liu P, Li Y, Liu D, et al. Tolfenamic acid attenuates 3-nitropropionic acid-induced biochemical alteration in mice. Neurochem Res. 2018;43(10):1938–1946. doi:10.1007/s11064-018-2615-7
17. Tang W, Sima AD, Gong J, Wang J, Li T. Kinetic difference between concomitant polymorphism and solvent-mediated phase transformation: a case of tolfenamic acid. Cryst Growth Des. 2020;20(3):1779–1788. doi:10.1021/acs.cgd.9b01503
18. Wang X, Xu S, Jia L, et al. Drug–drug salts of mefenamic acid\tolfenamic acid and piperazine to improve physicochemical properties for potential veterinary use. Cryst Eng Comm. 2019;21(35):5284–5291. doi:10.1039/C9CE00781D
19. Rozou S, Michaleas S, Antoniadou‐Vyza E. Supramolecular interactions between tolfenamic acid and various cyclodextrins: effects of complexation on physicochemical and spectroscopic data. Pharm Pharmacol Commun. 1999;5(2):79–84. doi:10.1211/146080899128734497
20. Vavia PR, Adhage NA. Freeze-dried inclusion complexes of tolfenamic acid with β-cyclodextrins. Pharm Dev Technol. 2000;5(4):571–574. doi:10.1081/PDT-100102040
21. Eddine ZK, Fatiha M, Amal Z, Leila N, Rachid M. Investigation of the inclusion complex of tolfenamic acid with β-cyclodextrin: geometry and NBO analysis. Comptes Rendus Chim. 2015;18(2):193–198. doi:10.1016/j.crci.2014.04.012
22. Subaiea GM, Ahmed AH, Adwan LI, Zawia NH. Reduction of amyloid-β deposition and attenuation of memory deficits by tolfenamic acid. J Alzheimer’s Dis. 2015;43(2):425–433. doi:10.3233/JAD-132726
23. Paczkowska M, Mizera M, Sałat K, et al. Enhanced pharmacological efficacy of sumatriptan due to modification of its physicochemical properties by inclusion in selected cyclodextrins. Sci Rep. 2018;8(1):1–13. doi:10.1038/s41598-018-34554-w
24. The ARRIVE guidelines [on the Internet]. Animal research: reporting of in vivo experiments. Available from: https://arriveguidelines.org/.
25. DIFFRAC.EVA Version 4.2.2. Bruker AXS GmbH Karlsruhe. Germany; 2016.
26. Paczkowska M, Chanaj-Kaczmarek J, Romaniuk-Drapała A, et al. Mucoadhesive chitosan delivery system with Chelidonii herba lyophilized extract as a promising strategy for vaginitis treatment. J Clin Med. 2020;9(4):1208. doi:10.3390/jcm9041208
27. Kaproń B, Łuszczki JJ, Siwek A, et al. Preclinical evaluation of 1, 2, 4-triazole-based compounds targeting voltage-gated sodium channels (VGSCs) as promising anticonvulsant drug candidates. Bioorg Chem. 2020;94:103355. doi:10.1016/j.bioorg.2019.103355
28. Svobodova B, Mezeiova E, Hepnarova V, et al. Exploring structure-activity relationship in tacrine-squaramide derivatives as potent cholinesterase inhibitors. Biomolecules. 2019;9(8):379. doi:10.3390/biom9080379
29. Bates EA, Nikai T, Brennan KC, et al. Sumatriptan alleviates nitroglycerin-induced mechanical and thermal allodynia in mice. Cephalalgia. 2010;30(2):170–178. doi:10.1111/j.1468-2982.2009.01864.x
30. Niopas I, Georgarakis M, Sidi-Frangandrea V, Chrisanthopoulos C, Liara E. Pharmacokinetics of tolfenamic acid in pediatric patients after single oral dose. Eur J Drug Metab Pharmacokinet. 1995;20(4):293–296. doi:10.1007/BF03190247
31. Sałat K, Gawlik K, Witalis J, et al. Evaluation of antinociceptive and antioxidant properties of 3-[4-(3-trifluoromethyl-phenyl)-piperazin-1-yl]-dihydrofuran-2-one in mice. Naunyn Schmiedebergs Arch Pharmacol. 2013;386(6):493–505. doi:10.1007/s00210-013-0847-2
32. Sałat K, Podkowa A, Kowalczyk P, et al. Anticonvulsant active inhibitor of GABA transporter subtype 1, tiagabine, with activity in mouse models of anxiety, pain and depression. Pharmacol Rep. 2015;67(3):465–472. doi:10.1016/j.pharep.2014.11.003
33. Fischer H, Kansy M, Avdeef A, Senner F. Permeation of permanently positive charged molecules through artificial membranes—influence of physico-chemical properties. Eur J Pharm Sci. 2007;31(1):32–42. doi:10.1016/j.ejps.2007.02.001
34. Di L, Kerns EH, Fan K, McConnell OJ, Carter GT. High throughput artificial membrane permeability assay for blood–brain barrier. Eur J Med Chem. 2003;38(3):223–232. doi:10.1016/S0223-5234(03)00012-6
35. Abhijeet K, Namita D. Studies on solubility enhancement of poorly soluble NSAID using dual approach of micro-environmental pH modulation and melt granulation. Curr Drug Deliv. 2017;14(8):1201–1212. doi:10.2174/1567201814666170413120513
36. Gupta P, Bansal AK. Spray drying for generation of a ternary amorphous system of celecoxib, PVP, and meglumine. Pharm Dev Technol. 2005;10(2):273–281. doi:10.1081/PDT-54460
37. Wallace JL. Mechanisms, prevention and clinical implications of nonsteroidal anti-inflammatory drug-enteropathy. World J Gastroenterol WJG. 2013;19(12):1861. doi:10.3748/wjg.v19.i12.1861
38. Huang Y, Quan P, Wang Y, et al. Host-guest interaction of β-cyclodextrin with isomeric ursolic acid and oleanolic acid: physicochemical characterization and molecular modeling study. J Biomed Res. 2017;31(5):395. doi:10.7555/JBR.31.20160073
39. Yang Y, Wen W, Luo Y, et al. Vanillin enhances the passive transport rate and absorption of drugs with moderate oral bioavailability in vitro and in vivo by affecting the membrane structure. Food Funct. 2020;11(1):700–710. doi:10.1039/C9FO02846C
40. Miranda HF, Lopez J, Sierralta F, Correa A, Pinardi G. NSAID antinociception measured in a chemical and a thermal assay in mice. Pain Res Manag. 2001;6(4):190–196. doi:10.1155/2001/701427
41. Miranda HF, Noriega V, Sierralta F, Poblete P, Aranda N, Prieto JC. Non-steroidal anti-inflammatory drugs in tonic, phasic and inflammatory mouse models. Drug Res (Stuttg). 2019;69(10):572–578. doi:10.1055/a-0956-673
42. Takesue EI, Schaefer W, Jukniewicz E. Modification of the Randall-Selitto analgesic apparatus. J Pharm Pharmacol. 1969;21(11):788–789. doi:10.1111/j.2042-7158.1969.tb08175.x
43. Kovala‐Demertzi D, Hadjipavlou‐Litina D, Primikiri A, Staninska M, Kotoglou C, Demertzis MA. Anti‐Inflammatory, antiproliferative, and radical‐scavenging activities of tolfenamic acid and its metal complexes. Chem Biodivers. 2009;6(6):948–960. doi:10.1002/cbdv.200800120
44. Paliwal M, Jain S. Synthesis and biological evaluation of mutual prodrugs of carboxylic group containing some non-steroidal anti-inflammatory drugs and propyphenazone. Curr Drug Deliv. 2017;14(8):1213–1224. doi:10.2174/1567201814666170213153509
45. Pradhan AA, Smith ML, McGuire B, Tarash I, Evans CJ, Charles A. Characterization of a novel model of chronic migraine. PAIN®. 2014;155(2):269–274. doi:10.1016/j.pain.2013.10.004
46. Tipton AF, Tarash I, McGuire B, Charles A, Pradhan AA. The effects of acute and preventive migraine therapies in a mouse model of chronic migraine. Cephalalgia. 2016;36(11):1048–1056. doi:10.1177/0333102415623070
47. Scuteri D, Adornetto A, Rombolà L, et al. New trends in migraine pharmacology: targeting calcitonin gene–related peptide (CGRP) with monoclonal antibodies. Front Pharmacol. 2019;10:363. doi:10.3389/fphar.2019.00363
48. Vogel HG. Drug Discovery and Evaluation: Pharmacological Assays. Springer Science & Business Media; 2002.
49. Gebhart GF, Sherman AD, Mitchell CL. The influence of learning on morphine analgesia and tolerance development in rats tested on the hot plate. Psychopharmacologia. 1971;22(3):295–304. doi:10.1007/BF00401791
50. Mizera M, Szymanowska D, Stasiłowicz A, et al. Computer-aided design of cefuroxime axetil/cyclodextrin system with enhanced solubility and antimicrobial activity. Biomolecules. 2020;10(1):24. doi:10.3390/biom10010024
51. Gratieri T, Pinho LAG, Oliveira MA, et al. Hydroxypropyl-β-cyclodextrin-complexed naringenin by solvent change precipitation for improving anti-inflammatory effect in vivo. Carbohydr Polym. 2020;231:115769. doi:10.1016/j.carbpol.2019.115769
52. de Matos Jensen CE, Dos Santos RAS, Denadai AML, Santos CFF, Braga ANG, Sinisterra RD. Pharmaceutical composition of valsartan: β-cyclodextrin: physico–chemical characterization and anti-hypertensive evaluation. Molecules. 2010;15(6):4067–4084. doi:10.3390/molecules15064067
53. Lu W-L, Zhang Q, Zheng L, et al. Antipyretic, analgesic and anti-inflammatory activities of ketoprofen β-cyclodextrin inclusion complexes in animals. Biol Pharm Bull. 2004;27(10):1515–1520. doi:10.1248/bpb.27.1515
54. Janovský M, Doležal T, Procházková M, Slíva J, Kršiak M. Influence on analgesic activity and serum levels after meloxicam complexation with beta-cyclodextrin in mice and rats. Arzneimittelforschung. 2010;60(06):320–323. doi:10.1055/s-0031-1296294
55. Harriott AM, Strother LC, Vila-Pueyo M, Holland PR. Animal models of migraine and experimental techniques used to examine trigeminal sensory processing. J Headache Pain. 2019;20(1):1–15. doi:10.1186/s10194-019-1043-7
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.