")
Back to Journals » Drug Design, Development and Therapy » Volume 18
The Impact of Baohuoside I on the Metabolism of Tofacitinib in Rats
Authors Shi Y, Lu Z, Song W, Wang Y, Zhou Q, Geng P, Zhou Y, Wang S , Han A
Received 22 August 2023
Accepted for publication 13 March 2024
Published 25 March 2024 Volume 2024:18 Pages 931—939
DOI https://doi.org/10.2147/DDDT.S436549
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Georgios Panos
Yaru Shi,* Zebei Lu,* Wei Song, Yu Wang, Quan Zhou, Peiwu Geng, Yunfang Zhou, Shuanghu Wang, Aixia Han
Key Laboratory of Joint Diagnosis and Treatment of Chronic Liver Disease and Liver Cancer of Lishui, The Sixth Affiliated Hospital of Wenzhou Medical University, Lishui People’s Hospital, Lishui, Zhejiang, 323000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Aixia Han; Shuanghu Wang, Key Laboratory of Joint Diagnosis and Treatment of Chronic Liver Disease and Liver Cancer of Lishui, The Sixth Affiliated Hospital of Wenzhou Medical University, Lishui People’s Hospital, Lishui, Zhejiang, 323000, People’s Republic of China, Tel +86 5782780081, Email [email protected]; [email protected]
Purpose: To study the potential drug–drug interactions between tofacitinib and baohuoside I and to provide the scientific basis for rational use of them in clinical practice.
Methods: A total of eighteen Sprague-Dawley rats were randomly divided into three groups: control group, single-dose group (receiving a single dose of 20 mg/kg of baohuoside I), and multi-dose group (receiving multiple doses of baohuoside I for 7 days). On the seventh day, each rat was orally administered with 10 mg/kg of tofacitinib 30 minutes after giving baohuoside I or vehicle. Blood samples were collected and determined using UPLC-MS/MS. In vitro effects of baohuoside I on tofacitinib was investigated in rat liver microsomes (RLMs), as well as the underlying mechanism of inhibition. The semi-inhibitory concentration value (IC50) of baohuoside I was subsequently determined and its inhibitory mechanism against tofacitinib was analyzed. Furthermore, the interactions between baohuoside I, tofacitinib and CYP3A4 were explored using Pymol molecular docking simulation.
Results: The administration of baohuoside I orally has been observed to enhance the area under the concentration-time curve (AUC) of tofacitinib and decrease the clearance (CL). The observed disparity between the single-dose and multi-dose groups was statistically significant. Furthermore, our findings suggest that the impact of baohuoside I on tofacitinib metabolism may be a mixture of non-competitive and competitive inhibition. Baohuoside I exhibit an interaction with arginine (ARG) at position 106 of the CYP3A4 enzyme through hydrogen bonding, positioning itself closer to the site of action compared to tofacitinib.
Conclusion: Our study has demonstrated the presence of drug–drug interactions between baohuoside I and tofacitinib, which may arise upon pre-administration of tofacitinib. Altogether, our data indicated that an interaction existed between tofacitinib and baohuoside I and additional cares might be taken when they were co-administrated in clinic.
Keywords: drug–drug interaction, pharmacokinetics, tofacitinib, baohuoside I, cytochrome P450
Introduction
Tofacitinib, a pharmacologically active compound possessing a pyrrole [2,3-d] pyrimidine framework, has been sanctioned to treat rheumatoid arthritis by the Food and Drug Administration (FDA) in 2012.1,2 Furthermore, it has recently received FDA approval in 2018 for the long-term therapy of ulcerative colitis.3 This inhibitor primarily targets Janus kinase (JAK)1 and 3, while also exhibiting a lesser degree of inhibition towards JAK2.
Following the administration of tofacitinib via oral route to individuals without any medical conditions, the pharmacokinetics parameters showed that the apparent volume of distribution (Vz/F) was 1.24 L/kg, elimination half-life (t1/2) was 3.2 hours and about 40% of tofacitinib exhibited plasma protein binding.4,5 Tofacitinib is predominantly eliminated from the body through liver metabolism (70%) and renal excretion (30%), with the enzymes CYP3A4 and CYP2C19, particularly CYP3A4, being primarily involved in its metabolism.6 The primary metabolic pathways of tofacitinib involve oxidation and N-demethylation on the pyrrole-pyrimidine ring and piperidine ring, which subsequently undergo further metabolism to form glucuronate conjugates.4 Tofacitinib exhibits no substantial impact on other drugs, including those metabolized by the CYP450 isoenzyme or excreted by the kidneys. Research findings indicate that the administration of 30 mg tofacitinib twice daily for a continuous period of 6 days in healthy individuals did not result in any alterations in midazolam pharmacokinetics, which was widely used as a benchmark for assessing drug-CYP isoenzyme interactions.7 Tofacitinib exhibits interactions when co-administered with inhibitors or inducers of CYP3A4. For instance, in patients concurrently using CYP3A4 inhibitors (such as ketoconazole) or a combination of CYP3A4 and CYP2C19 inhibitors (like fluconazole), it is recommended to reduce the daily dosage of tofacitinib by half. Conversely, the concomitant use of CYP3A4 inducers (such as rifampicin) substantially diminishes the area under the curve (AUC) of tofacitinib, resulting in a decrease in efficacy.7 It is postulated that the co-administration of tofacitinib with drugs capable of modifying the metabolic activity of CYP3A4 or CYP2C19 may result in alterations in its metabolism.
Epimedium, a traditional Chinese herbal medicine extensively employed in clinical and pharmacological studies in China, has been demonstrated to possess diverse pharmacological effects, particularly in the areas of hormonal regulation, anti-osteoporosis, immune function regulation, antioxidant and anti-tumor properties, anti-aging effects, anti-atherosclerosis activity, and antidepressant activity.8 Epimedium has been developed into a wide range of traditional Chinese medicine preparations as a common clinical medicine in orthopedics and traumatology in China. Many traditional Chinese medicinal preparations have been developed from epimedium, including the Xianling Gubao capsules and ZhuangguGuanjie pills, which are still in use today. Icariin, a flavonoside compound extracted from the dried stems and leaves of Epimedium, was administered orally to rats, resulting in the identification of seven metabolites in plasma except for the prototype drug icariin. Baohuoside I was discovered as the primary metabolite extracted from Epimedium. In addition, baohuoside I has various pharmacological activities, such as anti-tumor and anti-osteoporosis properties.9,10
Baohuoside I has been reported as an inhibitor of CXC chemokine receptor 4 (CXCR4) exhibiting apoptotic-inducing properties and demonstrating antitumor activity.11 Furthermore, it has been observed that baohuoside I could effectively inhibit the metastasis of nasopharyngeal cancer cells. Consequently, it can be considered a promising therapeutic agent for the clinical management of nasopharyngeal carcinoma.12 Notably, baohuoside I has been found to possess a higher inhibitory effect on osteoclast formation in RAW 264.7 cells compared to icariin, as evidenced by various studies.13,14 The potential interaction between the coadministration of baohuoside I and tofacitinib warrants further investigation, as the impact of baohuoside I on the pharmacokinetics of tofacitinib has not been definitively established. Furthermore, the absence of any documented clinical studies examining the combined use of baohuoside I and tofacitinib necessitates additional research in this area.
The objective of this study was to investigate the impact of baohuoside I on tofacitinib both in rat liver microsomes (RLMs) and in rats. In addition, the metabolic stability of tofacitinib in the presence of baohuoside I was assessed using RLMs, and the effect of baohuoside I on tofacitinib was examined in vitro by ultra high-performance liquid chromatography-tandem mass spectrometry (UPLC/MS-MS). What’s more, the plasma samples of rats were also determined by UPLC/MS-MS and pharmacokinetic parameters of tofacitinib in rats were analyzed. Our data could demonstrate the presence of drug–drug interactions between baohuoside I and tofacitinib, which may provide the scientific basis for rational use of them in clinical practice.
Materials and Methods
Chemicals and Biologicals
Tofacitinib (purity > 98%) and baohuoside I (purity > 98%) were purchased from the Guangzhou Zero One Biotechnology Co. Ltd. (Guangzhou, China). Midazolam (internal standard, IS; purity > 98%) was purchased from the Tianjin KingYork Pharmaceutical Co. Ltd. (Tianjin, China). Acetonitrile and methanol were purchased from Merck Co. Ltd. (Darmstadt, Germany). Ultrapure water was obtained by using a Milli-Q water purification system (Millipore, Billerica, MA, USA). RLMs were prepared in our laboratory.
Animals and Treatment
Male Sprague-Dawley (SD) rats were purchased from the Experimental Animal Center of Wenzhou Medical University. Rats were housed in a breeding room maintained at a temperature of 22.5±2.5°C with a humidity level of 60 ± 5% and a 12-hour light-dark cycle. They were provided with tap water and common feed. Prior to commencing the animal experiments, the rats were acclimatized to the above conditions for a duration of two weeks. All animal experiments were approved by the Animal Care and Use Committee of Wenzhou Medical University (No. xmsq2021-0409). We followed the GB/T35892 Guidelines for Ethical Review of Animal Welfare and we also follow the rules of 3R laboratory animal welfare principles.
Instruments and Operation Conditions
The concentration of tofacitinib were determined by UPLC-MS/MS, comprising an ACQUITY I Class UPLC and a XEVO TQD triple quadrupole mass spectrometer (Waters Corp., Milford, MA, USA). The tofacitinib, tofacitinib M8, and midazolam were separated on an ACQUITY UPLC HSS T3 Column (2.1 mm×100 mm, 1.8 μm), with the column temperature of 40°C. The mobile phase A was acetonitrile, while the mobile phase B was water with 0.1% formic acid and 5 mM ammonium formate. The elution process of mobile phase follows a linear gradient in which the concentration of acetonitrile increased from 10% to 30% over a period of 0 to 0.5 minutes, rapidly increased from 30% to 95% between 0.5 to 1.0 minutes, and remained constant at 95% from 1.0 to 2.0 minutes. Subsequently, the concentration decreased to 10% between 2.0 to 2.3 minutes. The flow rate of mobile phase employed was 0.4 mL/min, and the entire process lasted for a duration of 3 minutes.
The mass scan mode utilized in this study was positive ionization and multiple reaction monitoring mode with an electrospray ionization source. The precursor ion and product ion were observed at m/z 313.180→149.030 for tofacitinib, m/z 299.1620→98.0969 for tofacitinib M8, and m/z 325.9800→291.0700 for midazolam. The optimal MS parameters for mass detection of tofacitinib, tofacitinib M8 and midazolam were: 40, 40, and 50 V for Cone voltages and 30, 30, and 26 V for collision energies, respectively.
Pharmacokinetic Interactions Between Baohuoside I and Tofacitinib in Rats
Eighteen SD rats were randomly divided into 3 groups with 6 rats in each group: control group, single-dose group and multi-dose group. The experiment lasted for 7 days. The control group was only fed normal saline for 7 days; the single-dose group was fed normal saline for the first 6 days and took 20mg/kg of baohuoside orally once on the seventh day. The multi-dose group was given 20mg/kg of baohuoside I orally once a day for seven days. On day 7, Eighteen rats were received oral saline (control group) or baohuoside I (single dose group, multiple-dose group), after 30 minutes, then received 10mg/kg of tofacitinib orally once. 50 μL blood samples were collected from the tail veins of the rats using heparinized glass capillary tubes at various time intervals, including 5, 15, and 30 minutes, as well as 1, 2, 3, 4, 6, 8, 12, and 24 hours. In the present study, 100 μL acetonitrile (containing IS) were added in a 1.5 mL Eppendorf tube. The resulting mixture was subjected to vortexing for a duration of 30 seconds, followed by centrifugation at a speed of 12,000 rpm for a duration of 10 minutes. Finally, the supernatant was transferred into a separate sample bottle.10 microliters of this supernatant were immediately analyzed using a sensitive and reliable UPLC-MS/MS method.
Inhibitory Effects of Baohuoside I on Tofacitinib in RLMs
Based on the evidence from in vivo experiments demonstrating that baohuoside I inhibits tofacitinib metabolism, we aimed to investigate the inhibitory effects of baohuoside I in vitro. RLMs were prepared and the inhibitory effect of baohuoside I on tofacitinib metabolism was assessed using methods similar to those previously described.15 The volume of incubation system was 200 µL, containing 100 mM pH 7.4 potassium phosphate buffer, 0.5 mg/mL RLMs, and the varying concentration of tofacitinib (1, 2.5, 5, 10, 25, 50, 100, 200 and 400 μM). The system was preincubated at 37 °C for 5 min. The reaction was then initiated by the addition of reduced nicotinamide adenine dinucleotide phosphate (NADPH) at a final concentration of 1 mM, and the incubation was continued for 30 min. To determine the half-maximal inhibitory concentration (IC50) of baohuoside I, various concentrations of baohuoside I (100, 50, 10, 5, 1, 0.1, 0.01, and 0 μM) were selected along with 40 µM of tofacitinib, which is close to its Km value. In order to investigate the inhibitory mechanism of baohuoside I on tofacitinib, the two drugs were subjected to incubation at various concentration gradients, including 1/4 Km, 1/2 Km, Km, and 2Km of tofacitinib, as well as 0, 1/4 IC50, 1/2 IC50, IC50, and 2IC50 of baohuoside I. The incubation procedure remained consistent with the afore mentioned method.
Molecular Docking Simulations
The interaction between tofacitinib and baohuoside I was studied using the Auto Dock 4.2 docking program. Initially, crystal structure PDB files of the CYP3A4 protein were obtained from the PDB database website (https://www.rcsb.org/). Subsequently, the 3D molecular structures of baohuoside I and tofacitinib were acquired in SDF file formats from the Pubchem website (https://pubchem.ncbi.nlm.nih.gov) and converted into PDB files via OpenBabel 3.1.1 to PDB files. Finally, proteins and small molecules underwent pretreatment using Autodock Tools 1.5.7 to remove water and introduce hydrogen atoms, and then molecular docking was performed with Autodock Vina.
Statistical Analysis
The IC50 value, Lineweaver-Burk plot and plasma concentration–time curves were obtained by GraphPad prism software (version 9.0, San Diego, CA, USA). Drug and Statistics (DAS) 3.0 software (Version 3.2.8; Lishui People’s Hospital, China) in a non-atrial model was selected to calculate the pharmacokinetic parameters. All data were assessed distribution by using QQ plots. The relevant data was statistically analyzed using SPSS software (version 27.0), including Welch ANOVA test with LSD post-hoc test, with a significance threshold of P < 0.05 to for determine significant variations in the rubric. Kruskal–Wallis with Dunn’s post-hoc test were used for data that were not normally distributed. The sample size was determined according to the resource equation method outlined in the guidelines for the website. The “resource equation method” specifies between 10 and 20 acceptable error degrees of freedom in one-way ANOVA. The samples for our animal experiments were divided into three groups and based on the formula 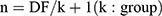 , a sample size of 5 to 7 individuals was recommended for each group. The sample size was set at six rats per group.
, a sample size of 5 to 7 individuals was recommended for each group. The sample size was set at six rats per group.
Results
Effects of Baohuoside I on the Pharmacokinetics of Tofacitinib in vivo
Figure 1 exhibited the mean plasma concentration–time curves of tofacitinib in three groups. And the pharmacokinetic parameters were determined and presented in Table 1. All data were assessed distribution by using QQ plots as show in Figure S1. The control group was Vz/F, 1 single-dose group was Tmax, Vz/F, and multi-dose group were Vz/F which did not conform to the normal distribution. Kruskal–Wallis non-parametric tests are used for data that is not normally distributed (Tmax, Vz/F), the result is shown in Figures S2–S4. Pretreatment with single- and multi-dose baohuoside I resulted in significantly increased AUC(0-t), AUC(0-∞), MRT (0-t), MRT(0-∞), and Tmax values of tofacitinib compared to the control group. Conversely, the CLz/F values were significantly lower in the baohuoside I pretreatment groups. However, there were no significant alterations observed in the t1/2, Cmax and Vz/F values of tofacitinib in the multi-dose group following baohuoside I pretreatment. Furthermore, subsequent to baohuoside I pretreatment, there was a marginal increase observed in the values of t1/2 and Vz/F with the single-dose group. Conversely, a slight decrease was observed in the Cmax value, although no statistically significant difference was found (P>0.05). These results indicate that the administration of baohuoside I, whether in single or multiple doses, hinders the metabolic process of tofacitinib in rats.
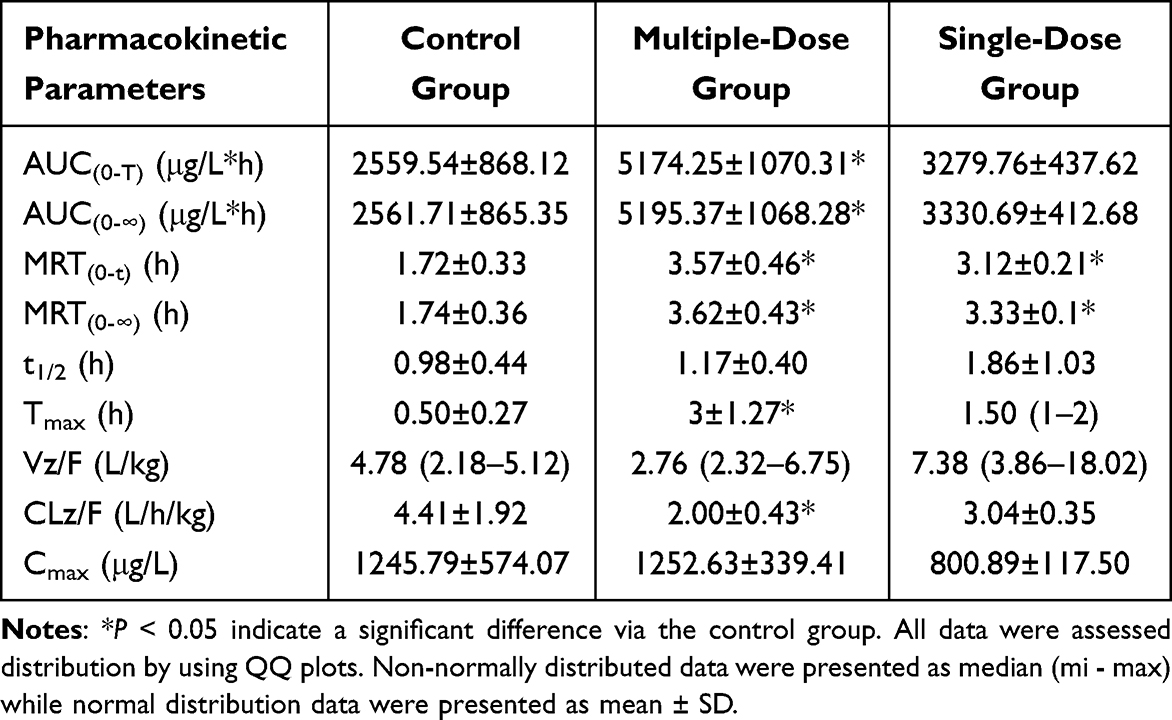 |
Table 1 Main Pharmacokinetic Parameters of Tofacitinib in Rats. (n = 6, Mean ± SD) |
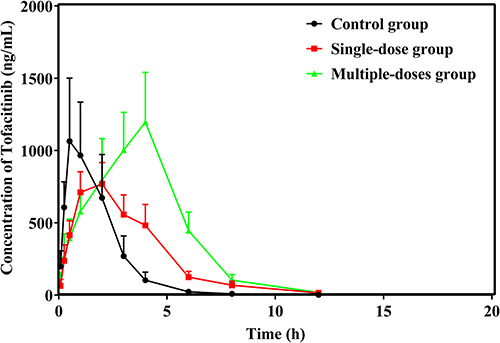 |
Figure 1 Mean plasma concentration–time curves of tofacitinib in control group, single-dose group, and multiple-dose group. |
Evaluation of the Influence of Baohuoside I on Tofacitinib Metabolism in vitro
To investigate the inhibitory effect of baohuoside I on tofacitinib in RLMs. The IC50 value for tofacitinib inhibition was determined by detecting tofacitinib in RLMs and baohuoside I, as shown in Figure 2. The Vmax, Km, and IC50 values were found to be 4.2824 ng/min/mg, 39.87 μM, and 3.234 μM, respectively. The inhibition mechanism of baohuoside I on tofacitinib in RLMs was elucidated in Figure 3. In addition, the Ki and αKi values of tofacitinib in RLM are 5.854 μM, 6.107 μM, respectively. This result indicates that baohuoside I has a clear in vitro inhibitory effect on tofacitinib, and its inhibition mechanism is a mixture of non-competitive and competitive inhibition.
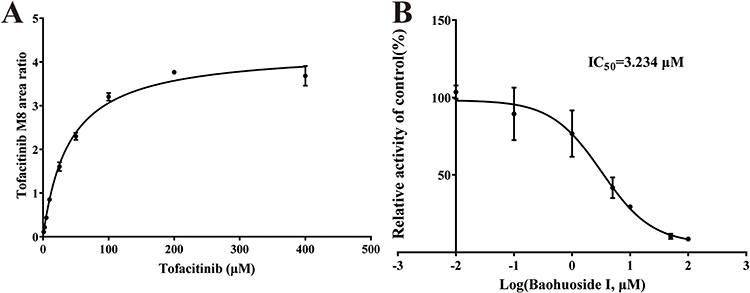 |
Figure 2 Michaelis–Menten kinetics (A) and the IC50 value (B) of tofacitinib in RLMs. |
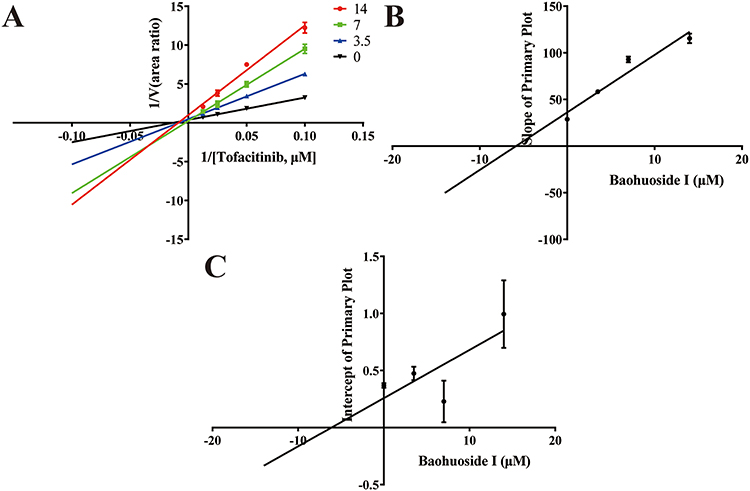 |
Figure 3 Lineweaver–Burk plot (A) and the secondary plot for Ki (B) and αKi (C) in the inhibition of tofacitinib metabolism by various concentrations of baohuoside I in RLMs. |
Molecular Docking of Baohuoside I and Tofacitinib
To gain a deeper learning of the interaction mechanism between baohuoside I and tofacitinib, molecular docking analysis was conducted using established methods. The results of the simulation performed Pymol revealed that baohuoside I formed a hydrogen bond with arginine (ARG) and Glutamate (GLU) at position CYP3A4 106 with a distance of 3.1 A and 3.3 A. Similarly, tofacitinib also exhibited interaction with CYP3A4 106 arginine (ARG) with a distance of 3.4 A, as shown in Figure 4. This spatial proximity between the two drugs may contribute to their facile interaction.
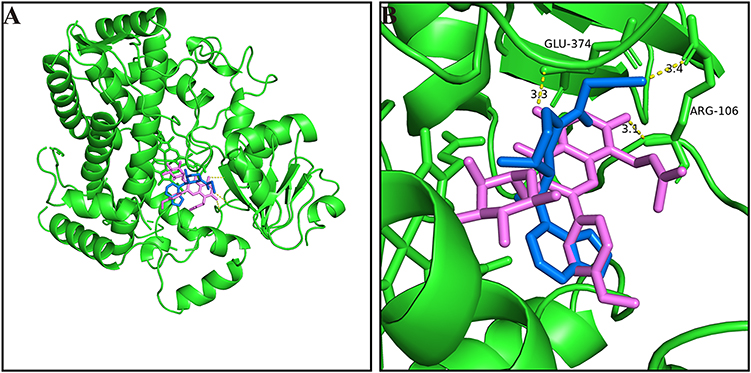 |
Figure 4 (A) Molecular docking scheme of baohuoside I and tofacitinib; (B) Action site between baohuoside I, tofacitinib and CYP3A4 via hydrogen bonding. |
Discussion
The objective of this study is to investigate the alterations in the pharmacokinetics of tofacitinib in rats subsequent to the administration of baohuoside I. Numerous recent studies have examined the pharmacokinetics of tofacitinib in various diseases, including liver damage,16 kidney failure,17 psoriasis,18 as well as inflammatory bowel disease.19 However, these studies have primarily focused on elucidating the correlation between drug concentration and therapeutic efficacy without providing a comprehensive explanation of how plasma concentrations differ across diseases in relation to the basis of pharmacokinetics.
In the metabolism of tofacitinib, CYP3A4 and CYP2C19 play a significant role in humans, with CYP3A4 being the primary enzyme involved.4 Conversely, rats primarily rely on CYP3A1(23)/2 and CYP2C11 for drug metabolism.20,21 It is worth noting that there is a high degree of similarity between CYP2C19 and CYP2C11 for human and rat, while 73% homology were exhibited between CYP3A4 and CYP3A1(23). The study revealed that CYP3A1(23) and CYP2C11 were the main CYPs in rats.22 Furthermore, following oral administration, tofacitinib was swiftly absorbed by the rats, resulting in the attainment of peak plasma concentration at approximately 1.1 h. In addition, this investigation demonstrated rapid elimination of tofacitinib from plasma with a t1/2 elimination of 0.98±0.44 h. It is worth noting that drug–drug interactions (DDIs) represent a prevalent cause of adverse drug reactions or compromised therapeutic efficacy.23 Clinical practitioners must be aware of possible DDIs when treating patients. Drug metabolism can be affected by irrelevant, synergistic, additive, or antagonistic outcomes. It is critical to be aware of potential DDIs in treating patients in clinical practice to adjust drug dosage appropriately.
As the introduction part, Baohuoside I is extracted and isolated from a traditional Chinese herb called Epimedium. Epimedium. In China, epimedium, as a commonly used clinical medicine in orthopedics and traumatology, has been developed into hundreds of traditional Chinese medicine preparations, such as compound preparations Xianling Gubao Capsules and Zhuanggu Guanjie Pills are still in use. However, few studies have been conducted on DDI when tofacitinib is administered in combination with traditional Chinese medicine. Therefore, it is of great significance to study the interaction between baohuoside I and tofacitinib.
When rats in control group were pretreated with tofacitinib, the AUC(0-∞) value of tofacitinib ranged from 2561.71±865.35, the MRT(0-∞) value of tofacitinib ranged from 1.74±0.36, and the Tmax value of tofacitinib ranged from 0.50±0.27. In contrast, when rats were pretreated with baohuoside I, the AUC(0-∞) value of tofacitinib ranged from 3330.69 ± 412.68 (single dose) to 5195.37 ± 1068.28 (multiple dose); the MRT(0-∞) value of tofacitinib each ranged from 3.62 ± 0.43 (multiple dose) to 3.33 ± 0.1 (single dose); and the Tmax value of tofacitinib ranged from 3 ± 1.27 (multiple dose) to 1.50 (1-2) (single dose).
The aforementioned findings indicate that the administration of baohuoside I hinders the metabolism of tofacitinib. In comparison to the control group, the CLz/F value of tofacitinib decreased by 2.2-fold and 1.45-fold in groups pretreated with baohuoside I. These results demonstrate a substantial impact of baohuoside I on the pharmacokinetic parameters of tofacitinib in rats. In addition, the multi-dose group exhibited more notable alterations in the pharmacokinetic parameters of tofacitinib when compared to the single-dose group, suggesting that the inhibitory effect of baohuoside I could potentially be influenced by the frequency and timing of its administration.
It is well established that tofacitinib undergoes hepatic metabolism (70%) and renal elimination (30%), primarily mediated by the enzymes CYP3A4 and CYP2C19, with CYP3A4 being the predominant enzyme involved.4 Therefore, it is plausible that baohuoside I inhibit the metabolism of tofacitinib by exerting an inhibitory effect on the CYP3A4 and CYP2C19 enzymes. Based on the outcomes of in vitro RLM incubation, it was observed that baohuoside I significantly impeded the metabolism of tofacitinib in RLM. Furthermore, the application of Michael’s equation to analyze the interaction between tofacitinib and baohuoside I revealed the following values: Vmax (4.2824 ng/min/mg), Km (39.87 μM), and IC50 (3.234 μM), denoting the extent of inhibition exerted by baohuoside I on tofacitinib in an in vitro setting, with its inhibition mechanism being a combination of non-competitive and competitive inhibition (as indicated by the inequality of Ki and αKi).
Numerous drug interactions are influenced by the chemical composition of the drug and the structure of proteins. Research has demonstrated that the relocation of drugs from their protein-binding sites in vitro may potentially alter the pharmacological effects of displaced drugs, either enhancing or diminishing their efficacy.24 To assess the molecular docking status of baohuoside I, tofacitinib and CYP3A4 enzymes, Pymol was employed for simulation. The results showed that baohuoside I interacted with arginine (ARG) at position CYP3A4106 through hydrogen bonding at a distance of 3.1A. Similarly, tofacitinib exhibited interaction with CYP3A4 106 arginine (ARG) at a distance of 3.4A.
These results are of great significance for future clinical research on the interaction between tofacitinib and baohuoside I. Extrapolating the present outcomes from rat models to humans suggests that the administration of tofacitinib should be modified. However, it is imperative to conduct additional clinical studies to validate the aforementioned findings.
Conclusions
In summary, the findings of this study provide clear evidence that baohuoside I has a significant impact on the pharmacokinetic parameters of tofacitinib. Specifically, oral administration of baohuoside I leads to an increase in the AUC of tofacitinib, a prolongation of the Tmax, and a decrease in the CL of the drug. Notably, the observed differences between the single-dose and multi-dose groups were found to be statistically significant. Furthermore, our results suggest that baohuoside I may exert a mixed inhibitory effect on the metabolism of tofacitinib. In addition, it was observed that baohuoside I interacts with the 106-position arginine (ARG) of CYP3A4 via hydrogen bonding and this interaction occurs in closer proximity to the site of action compared to tofacitinib. Therefore, when taking tofacitinib for rheumatoid arthritis, people should pay attention to the intake of food or herbs containing Baohuoside I, and may adjust the dose of tofacitinib.
Acknowledgments
This work was supported by the Public Welfare Technology Research Funding Project of Zhejiang (LTGY24H100002 and LGF21H310001), the Public Welfare Technology Research Funding Project of Lishui (2021SJZC022, 2021SJZC023, 2020GYX23, 2022SJZC079, 2023GYX04, 2022GYX24 and 2023GYX18), the Key Research and Development Project of Lishui (2021ZDYF13, 2021ZDYF15, and 2023zdyf15), Medical and health science and technology plan project Zhejiang province (2022RC302, 2022RC303 and 2023RC312).
Disclosure
The authors have no conflicts of interest and no disclosures to declare.
References
1. Flanagan ME, Blumenkopf TA, Brissette WH, et al. Discovery of CP-690,550: a potent and selective Janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection. J Med Chem. 2010;53(24):8468–8484. doi:10.1021/jm1004286
2. Fraenkel L, Bathon JM, England BR, et al. 2021 American College of Rheumatology Guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol. 2021;73(7):1108–1123. doi:10.1002/art.41752
3. Palasik BN, Wang H. Tofacitinib, the first oral janus kinase inhibitor approved for adult ulcerative colitis. J Pharm Pract. 2021;34(6):913–921. doi:10.1177/0897190020953019
4. Dowty ME, Lin J, Ryder TF, et al. The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a janus kinase inhibitor, in humans. Drug Metab Dispos. 2014;42(4):759–773. doi:10.1124/dmd.113.054940
5. Scott LJ. Tofacitinib: a review of its use in adult patients with rheumatoid arthritis. Drugs. 2013;73(8):857–874. doi:10.1007/s40265-013-0065-8
6. Lee JS, Kim SH. Dose-dependent pharmacokinetics of tofacitinib in rats: influence of hepatic and intestinal first-pass metabolism. Pharmaceutics. 2019;11(7):318. doi:10.3390/pharmaceutics11070318
7. Gupta P, Alvey C, Wang R, et al. Lack of effect of tofacitinib (CP-690,550) on the pharmacokinetics of the CYP3A4 substrate midazolam in healthy volunteers: confirmation of in vitro data. Br J Clin Pharmacol. 2012;74(1):109–115. doi:10.1111/j.1365-2125.2012.04168.x
8. Ma H, He X, Yang Y, et al. The genus Epimedium: an ethnopharmacological and phytochemical review. J Ethnopharmacol. 2011;134(3):519–541. doi:10.1016/j.jep.2011.01.001
9. Sun Y, Pang B, Wang Y, et al. Baohuoside I inhibits the proliferation of hepatocellular carcinoma cells via apoptosis signaling and NF-kB pathway. Chem Biodivers. 2021;18(6):e2100063. doi:10.1002/cbdv.202100063
10. Xi Y, Jiang T, Yu J, et al. Preliminary studies on the anti-osteoporosis activity of Baohuoside I. Biomed Pharmacother. 2019;115:108850. doi:10.1016/j.biopha.2019.108850
11. Kim B, Park B. Baohuoside I suppresses invasion of cervical and breast cancer cells through the downregulation of CXCR4 chemokine receptor expression. Biochemistry. 2014;53(48):7562–7569. doi:10.1021/bi5011927
12. Wang Q, Jiang S, Wang W, et al. Effects of baohuoside-I on epithelial-mesenchymal transition and metastasis in nasopharyngeal carcinoma. Hum Exp Toxicol. 2021;40(4):566–576. doi:10.1177/0960327120960765
13. Ma M, Fan AY, Liu Z, et al. Baohuoside I inhibits osteoclastogenesis and protects against ovariectomy-induced bone loss. Front Pharmacol. 2022;13:874952. doi:10.3389/fphar.2022.874952
14. Zhang LB, Yan Y, He J, et al. Epimedii Herba: an ancient Chinese herbal medicine in the prevention and treatment of rheumatoid arthritis. Front Chem. 2022;10:1023779. doi:10.3389/fchem.2022.1023779
15. Wang B, Shen J, Zhou Q, et al. Effects of naringenin on the pharmacokinetics of tofacitinib in rats. Pharm Biol. 2020;58(1):225–230. doi:10.1080/13880209.2020.1738504
16. Lawendy N, Lamba M, Chan G, et al. The effect of mild and moderate hepatic impairment on the pharmacokinetics of tofacitinib, an orally active Janus kinase inhibitor. Clin Pharmacol Drug Dev. 2014;3(6):421–427. doi:10.1002/cpdd.143
17. Bae SH, Chang SY, Kim SH. Slower elimination of tofacitinib in acute renal failure rat models: contribution of hepatic metabolism and renal excretion. Pharmaceutics. 2020;12(8):714. doi:10.3390/pharmaceutics12080714
18. Ighani A, Georgakopoulos JR, Yeung J. Tofacitinib for the treatment of psoriasis and psoriatic arthritis. G Ital Dermatol Venereol. 2020;155(4):400–410. doi:10.23736/S0392-0488.20.06643-2
19. Ma C, Battat R, Jairath V, et al. Advances in therapeutic drug monitoring for small-molecule and biologic therapies in inflammatory bowel disease. Curr Treat Options Gastroenterol. 2019;17(1):127–145. doi:10.1007/s11938-019-00222-9
20. Bae SH, Choi HG, Park SY, et al. Effects of isosakuranetin on pharmacokinetic changes of tofacitinib in rats with N-dimethylnitrosamine-induced liver cirrhosis. Pharmaceutics. 2022;14(12):2684. doi:10.3390/pharmaceutics14122684
21. Won JM, Choi HG, Park SY, et al. Effects of hyperlipidemia on the pharmacokinetics of tofacitinib, a JAK 1/3 inhibitor, in rats. Pharmaceutics. 2023;15(9):2195. doi:10.3390/pharmaceutics15092195
22. Gwak EH, Yoo HY, Kim SH. Effects of diabetes mellitus on the disposition of tofacitinib, a janus kinase inhibitor, in rats. Biomol Ther. 2020;28(4):361–369. doi:10.4062/biomolther.2020.006
23. Bettonte S, Berton M, Marzolini C. Magnitude of drug-drug interactions in special populations. Pharmaceutics. 2022;14(4):789. doi:10.3390/pharmaceutics14040789
24. Kristensen MB. Drug interactions and clinical pharmacokinetics. Clin Pharmacokinet. 1976;1(5):351–372. doi:10.2165/00003088-197601050-00003
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.