Back to Archived Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 10
The identification of protein domains that mediate functional interactions between Rab-GTPases and RabGAPs using 3D protein modeling
Received 1 September 2016
Accepted for publication 23 November 2016
Published 10 April 2017 Volume 2017:10 Pages 47—56
DOI https://doi.org/10.2147/AABC.S121245
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Juan Fernandez-Recio
Jeremiah J Davie, Silviu L Faitar
Department of Biology and Mathematics, School of Arts, Sciences, and Education, D’Youville College, Buffalo, NY, USA
Abstract: Currently, time-consuming serial in vitro experimentation involving immunocytochemistry or radiolabeled materials is required to identify which of the numerous Rab-GTPases (Rab) and Rab-GTPase activating proteins (RabGAP) are capable of functional interactions. These interactions are essential for numerous cellular functions, and in silico methods of reducing in vitro trial and error would accelerate the pace of research in cell biology. We have utilized a combination of three-dimensional protein modeling and protein bioinformatics to identify domains present in Rab proteins that are predictive of their functional interaction with a specific RabGAP. The RabF2 and RabSF1 domains appear to play functional roles in mediating the interaction between Rabs and RabGAPs. Moreover, the RabSF1 domain can be used to make in silico predictions of functional Rab/RabGAP pairs. This method is expected to be a broadly applicable tool for predicting protein–protein interactions where existing crystal structures for homologs of the proteins of interest are available.
Keywords: GTP hydrolysis, Rab proteins, RabGAPs, protein–protein interactions, structural informatics, computational biology, Evi5, Evi5L
Introduction
Rab proteins represent the largest subset of small GTPase proteins in the Ras superfamily and are critical components for the functioning of eukaryotic cells. The hydrolysis of guanosine triphosphate (GTP) by these proteins permits them to oversee processes as diverse as vesicular trafficking, organelle formation, cilium formation, and cytokinesis.1–3 As seen in other eukaryotic systems, additional proteins modulate control of Rab activity. Rab proteins are active only when bound to GTP and can be deactivated by the action of Rab-GTPase activating proteins (RabGAPs). RabGAP proteins trigger Rab proteins to hydrolyze the bound GTP, leaving the protein inert until the guanosine diphosphate (GDP) can be swapped for GTP by the action of a second protein interaction partner, a guanine nucleotide exchange factor (GEF). Finally, GDP dissociation inhibitors (GDIs) bind to GDP-bound Rabs to prevent their reactivation.3
One of the current challenges of investigating the Rab-family of GTPases is the difficulty in determining the capacity for a functional interaction between a given Rab and RabGAP. These studies have historically involved a substantial amount of time and laboratory work, as there are >40 recognized Rabs and >40 RabGAPs.2 Considering that some RabGAPs are capable of interacting with more than one Rab (and vice versa), there are >1,600 possible interactions to be evaluated. Although studies have successfully utilized a serial testing methodology to identify functional Rab–RabGAP interactions,4 advancement in the field of cell biology would be accelerated if a mechanism existed to limit the experiment to only those Rabs with an elevated possibility of physical interaction.
Previously, Periera-Leal and Seabra5 identified a series of conserved protein domains common to Rab proteins, which they classified as the Rab family motif and Rab-family submotifs. However, the high degree of amino acid conservation typical to these domains among Rabs has made ascribing a functional role to them challenging. This task has been further complicated by the capacity for overlapping interactions between certain Rabs and RabGAPs. In the 16 years since the identification of these domains, advances in structural informatics and computational biology have made it possible to rapidly predict the three-dimensional (3D) structure of proteins with homology to other proteins with solved crystal structures. One highly useful method is found on the Phyre2 webserver,6 which is able to produce a 3D model of the protein of interest, and is connected to the PhyreInvestigator platform. PhyreInvestigator acts as a structural informatics portal capable of integrating information from the Protein Data Bank (PDB)7 the Conserved Domain Database (CDD),8 and a suite of structural informatics programs to provide a robust description of the most probable function, spatial location, and functional domains found within the predicted protein structure.9–12
To facilitate the future functional characterization of Rab–RabGAP interaction pairs, we sought to undertake a bioinformatics-based approach that would use these advances in protein structural informatics to identify homologous or nearly homologous regions of Rab proteins that are known to interact with the same RabGAP. By identifying these regions, we may increase the likelihood of predicting in advance what Rab–RabGAP pairings may functionally interact, both in vitro and in vivo. Here, we describe the initial implementation of these complementary bioinformatics methods by identifying domains present in the Rab11 and Rab23 proteins that are likely to mediate interactions with their known partner RabGAPs, Evi5, and Evi5L, respectively.13,14
Materials and methods
3D structure prediction and analysis
Amino acid sequences for Rab proteins were downloaded from the GenBank accession numbers listed in Table 1. These sequences were submitted to the Phyre2 webserver (www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi) for a homology-dependent prediction of the 3D structure for each protein.6 Only 3D models with 100% confidence in 3D prediction were considered for submission to the associated PhyreInvestigator platform for additional analyses.6 Prediction of protein–protein interaction sites present in each model was accomplished by the PI-Site9 and ProtinDb programs (http://protindb.cs.iastate.edu). Similarly, prediction of structural sensitivity to mutation and binding-pocket detection were made by the SUSPect12 and fpocket211 programs, respectively. All programs were utilized as implemented by PhyreInvestigator using default settings.
 | Table 1 Accession numbers for protein sequences used in this study |
Domain homology analysis
Predicted functional domains were identified using InterPro (v48.0; https://www.ebi.ac.uk/interpro/).10 Multiple sequence alignments were prepared using the ClustalW program as implemented in the MacVector software package (v13.0.2, MacVector, Inc., Cary, NC, USA) using the BLOSUM matrix with default parameters. Domains of interest identified by the various programs described above were compared as described in the Results section. The phylogenetic analysis of Rab proteins and their protein domains was accomplished through the implementation of the unweighted pair group method with arithmetic mean algorithm in the MacVector software package, using the following settings: Best Tree with random tiebreaking and distance calculated using an uncorrected “p” with gaps distributed proportionally.
Visualization of 3D structures
Visualization of predicted 3D protein structures was accomplished using the MacPyMol program (v1.8.2.3; Schrödinger, LLC, NY, USA) or the JSmol program as used in PhyreInvestigator. Crystal structures of Rab11A (PDB_ID: 1YZK) and Rab23 (PDB_ID: 1Z22) were downloaded from the PDB for visualization and structural alignment using MacPyMol. Details regarding the crystallization of Rab11A and Rab23 can be found in the study by Eathiraj et al.15
Results
Prediction of Rab 3D structures and comparison to existing crystal structures
The recent release of the Phyre2 protein modeling and associated PhyreInvestigator structural bioinformatics platform has made analysis of protein tertiary structure more efficient and straightforward.6 The Phyre2 program employs a homology-based approach that identifies the closest homologs with solved crystal structures to the protein of interest.6 These crystal structures are then used for one-to-one protein threading modeling to create a series of 3D structure predictions of varying confidence levels. To develop an in silico method of identifying structural features that could be used to predict successful Rab-RabGAP interaction partners, we selected two Rab proteins, Rab11 and Rab23, whose crystal structures had previously been solved and are known to interact with the RabGAPs Evi5 and Evi5L, respectively, in a mutually exclusive fashion. A broad outline of the bioinformatics workflow used in this study is presented in Figure 1. As expected, Phyre2 generated a number of predicted structures for each submitted amino acid sequence. The model with the highest overall ranking at the time of the study was selected for further analysis; details regarding the coverage and confidence of each model can be found in Table 2. In general, model predictions of Rab proteins possessed excellent confidence and each model covered >80% of the experimentally derived crystal structure that was used as the template for protein-threading modeling. The comparison of the Phyre2-generated models to the previously solved crystal structures reveals that the modeled ones are nearly identical to the solved structures, with the primary difference being the positioning of certain loop regions (Figure 2). Given the high quality of structure prediction and the need to assess the use of this method on computationally derived structures, the remainder of the structural analyses in this work was performed on the in silico-generated models.
 | Figure 1 Flow diagram of bioinformatics analyses utilized in the current study. Abbreviations: CDD, Conserved Domain Database; UPGMA, unweighted pair group method with arithmetic mean. |
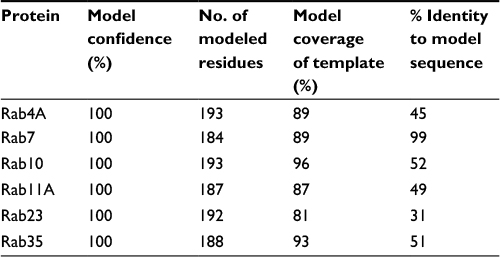 | Table 2 Description of structural predictions of proteins used in this study |
 | Figure 2 Phyre2 structure predictions closely align to experimentally solved Rab crystal structures. Notes: Phyre2 homology-based 3D structural predictions of Rab11A (A) and Rab23 (D). (B and E) Experimentally-derived 3-D structures solved previously by Eathiraj et al15 as visualized from publically available PDB co-ordinate files for Rab11A (Panel B; PDB_ID: 1YZK) and Rab23 (Panel E; PDB_ID: 1Z22). The superimposition of each predicted structure over the solved crystal structure for Rab11A and Rab23 (C and F, respectively) demonstrates the high degree of accuracy of modeling Rab proteins using this method. Abbreviations: 3D, three dimensional; PDB, Protein Data Bank. |
Analysis of 3D structure and identification of functional domains
The PhyreInvestigator platform represents a single-entry portal for the application of a suite of bioinformatics programs.6 The highest ranked structure identified by Phyre2 was submitted to PhyreInvestigator, and a series of domain predictions was performed. Binding-pocket locations were predicted using the fpocket2 program, whereas the prediction of protein–protein interaction sites was achieved using both PI-Site and ProtinDb; these data are summarized in Table 3. The fpocket2 program identified a GTP-binding pocket in both Rab11A and Rab23, consistent with their known role as GTP-binding proteins. Similarly, both the PI-Site and ProtinDb programs identified multiple residues predicted to be involved in protein–protein interactions, with PI-Site including more residues in its analysis than the more conservative ProtinDb. As both PI-Site and fpocket2 predicted similar numbers of residues to form GTP-binding pockets and protein–protein interaction sites, respectively, we investigated the possibility that these two domains could coordinate in 3D space. Mapping of these residues onto the 3D model confirmed that these two predicted regions coordinated for both Rab11A and Rab23 (Figure 3A–F). The in silico prediction of the catalytic residues of Rab11A and Rab23 was accomplished via comparison to the Catalytic Site Atlas (as implemented in PhyreInvestigator) and identified two catalytic residues each. Catalytic glutamine residues were identified at positions 70 and 68, in Rab11A and Rab23, respectively. Whereas the catalytic site of Rab23 was identified to include an alanine residue at position 19, the nonglutamine catalytic residue in Rab11A is a glycine located at position 21.
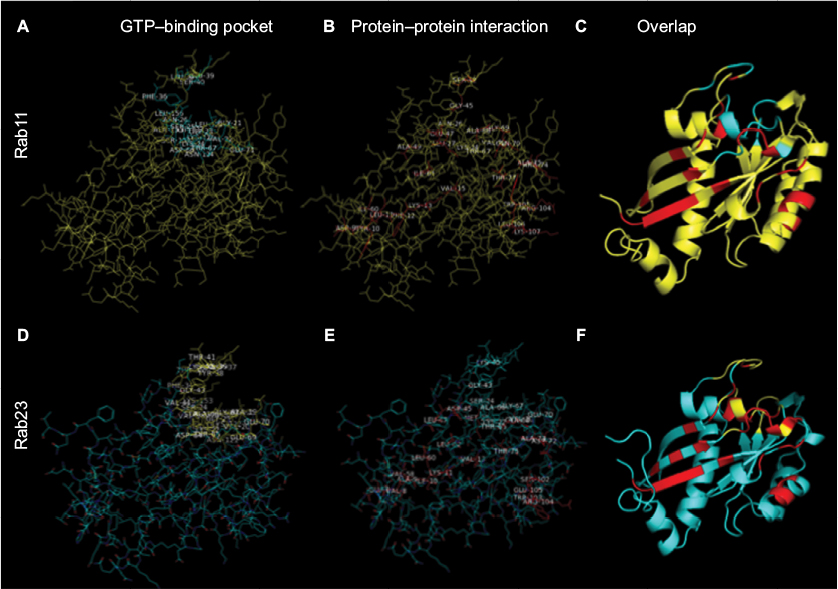 | Figure 3 Critical Rab residues coordinate in 3D space. Notes: The predicted 3D structure of Rab11 (top panels) and Rab23 (bottom panels) were obtained using the Phyre2 webserver and analyzed. (A, D) The GTP-binding pocket location – amino acid residues predicted by the fpocket2 algorithm to be a part of the Rab GTP-binding pocket are highlighted in cyan (Rab11A) or in yellow (Rab23). (B, E) Amino acid residues predicted by the PI-Site algorithm to be a part of the Rab protein interaction interface are highlighted in red (both). Residues predicted exclusively by ProtinDb are not shown. (C, F) Amino acids highlighted in A and B or D and E were highlighted simultaneously on the modeled structure. All panels: visualization achieved using MacPyMol. Abbreviations: 3D, three dimensional; GTP, guanosine triphosphate. |
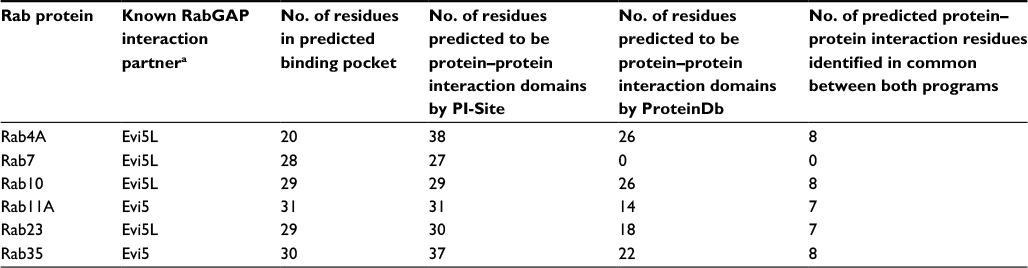 | Table 3 Binding pocket and protein–protein interaction site predictions among Rab proteins known to interact with Evi5 or Evi5L Note: aExperimentally validated pairs of Rab and RabGAP proteins are reviewed in Frasa et al.2 |
Rab proteins possess a well-described domain signature.5,16 To further validate our predicted structures, the primary amino acid sequences of Rab11 and Rab23 were submitted to InterPro database for protein domain identification. These analyses revealed that the putative GTP-binding domains of Rab11 and Rab23 would correspond to amino acids 10–168 and 10–165, respectively. The InterPro analysis was consistent with the fpocket2 analysis, which identified the binding pockets of Rab11 and Rab23 to be formed by a subset of amino acids present in the regions between amino acids 21–156 and 19–153, respectively.
In an attempt to confirm the above-mentioned results, Rab11 and Rab23 were queried for the presence of any other recognized functional domains known to the CDD (as implemented in PhyreInvestigator). This analysis identified 20 structural motifs or function regions present in each protein (Table 4). Of the features mentioned in Table 4, only nine are characteristic of Rab proteins.5,16 Although the expected Rab domains were identified, their existence has not yet been correlated with a specific function. Multiple sequence alignments of the Rabs revealed the presence of amino acid substitutions within the Rab-specific domain signatures (Figure 4). These variations occurred in patterns that mimicked the ability of the Rab proteins in question to bind to Evi5 or Evi5L. In particular, among Rabs capable of interacting with the same RabGAP, binding appeared to be associated with a high degree of homology between the RabF1, RabF2, RabF3, RabF4, RabF5, RabSF1, and RabSF4 domains. When comparing Rabs that cannot interact with the same RabGAP, RabF2, RabF5, RabSF1, and RabSF2 show diminished homology. Among these two groups of Rab-family domains, the RabSF1 and RabF2 domains are both highly similar between Rabs interacting with the same RabGAP and dissimilar between Rabs interacting with the different RabGAPs. This line of inquiry was expanded by exploring whether the RabSF1 and/or RabF2 domains could be used to infer RabGAP-binding preferences. Alignments of whole protein or the RabF2 and RabSF1 domains revealed that, unlike alignments of the whole Rab proteins, alignments of the RabSF1 domain could be used to reconstruct a tree of Rab proteins that could interact with the same RabGAP (Figure 5).
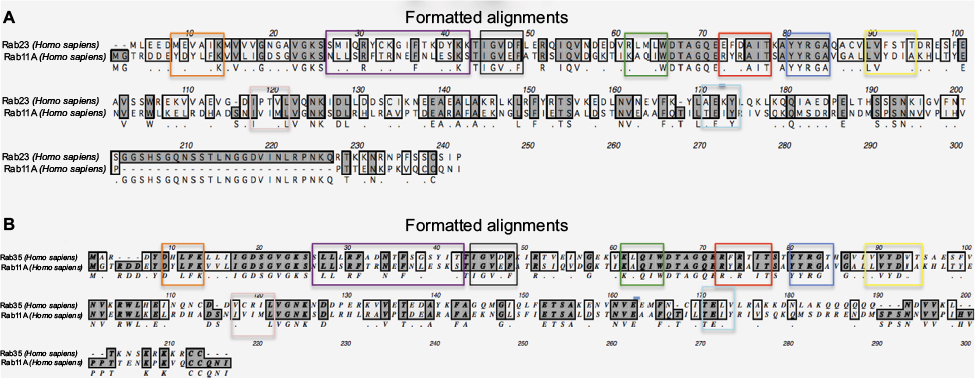 | Figure 4 Rab proteins capable of functional interaction with the same Rab-GTPase activating proteins share greater homology at specific Rab (RabF) and Rab subfamily (RabSF) domains. Notes: ClustalW alignments of (A) Rab11 (interacts with Evi5) and Rab23 (cannot interact with Evi5) reveal lesser homology, whereas (B) Rab proteins both capable of interaction with Evi5 show greater homology, particularly in domains RabF2 (green box) and RabSF2 (purple box). Other domains shown: RabF1 (black box), RabF3 (red box), RabF4 (blue box), RabF5 (yellow box), RabSF1 (orange box), RabSF3 (pink box), and Rab SF4 (light blue box). |
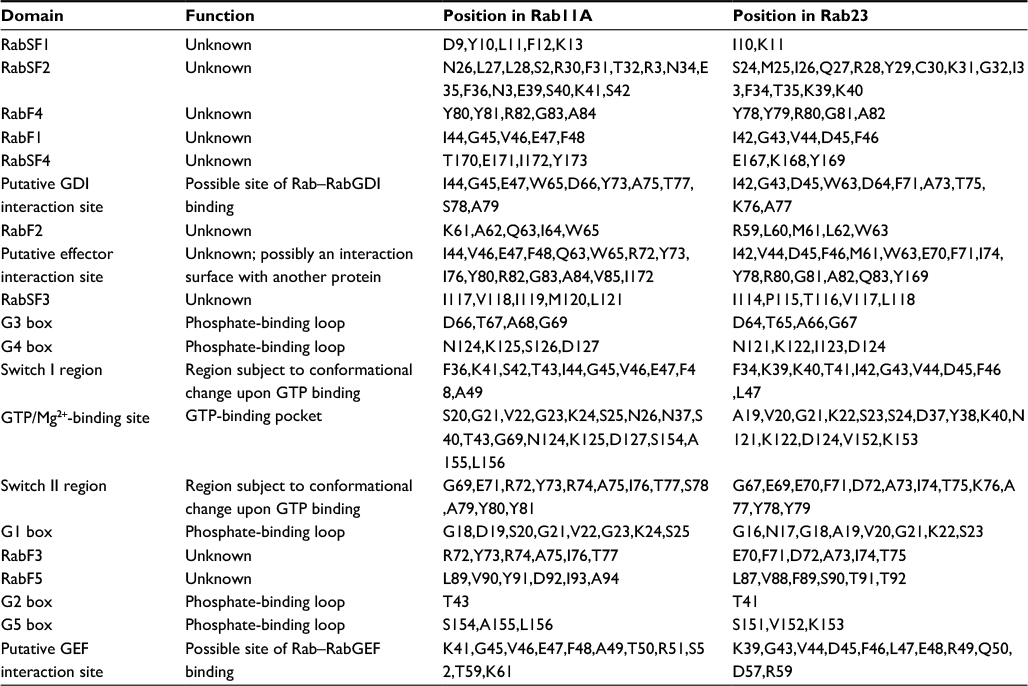 | Table 4 Structural and functional motifs present in Rab11A and Rab23 Note: “RabF” denotes Rab family motifs, while “RabSF” denotes Rab family submotifs as identified by Pereira-Leal and Seabra.5 Abbreviations: GDI, guanosine diphosphate dissociation inhibitor; GEF, guanine nucleotide exchange factor; GTP, guanosine triphosphate. |
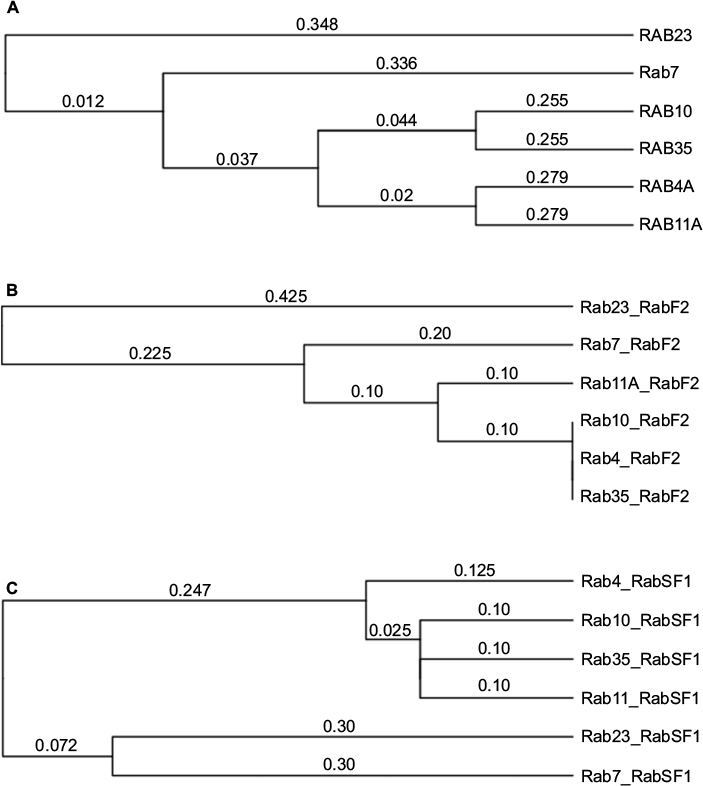 | Figure 5 Hierarchical clustering analyses demonstrate that RabSF1 domain sequence variations correlate with known Rab-Rab-GTPase activating proteins interaction pairs. Notes: Hierarchical clustering analyses were performed using the unweighted pair group method with arithmetic mean algorithm using (A) whole Rab protein sequence, (B) RabF2 domain only, or the (C) RabSF1 domain only. The RabSF1 domains from Rabs capable of interacting with the same RabGAP were consistently placed on the same branches. Hierarchical clustering analyses performed using the Neighbor-Joining method produced similar results (data not shown). |
In order for RabF2 and RabSF1 to mediate interactions between their respective Rab and RabGAP partner, these same domains would be expected to be in close physical proximity to one another, as well as with the residues predicted to be involved in protein–protein interactions. Visualization of both the Rab11 and Rab23 models revealed that RabF2 and RabSF1 domains are adjacent to one another on the same beta-pleated sheet (Figure 6A, B). Furthermore, the amino acid residues predicted to mediate protein–protein interactions are also present in this beta-pleated sheet. To further explore the significance of these domains, we next considered the effect of mutation on the predicted structures. If the previously identified domains were associated with the interaction with a RabGAP, then mutation of some or all of these residues would result in a phenotypic or functional change. To assess this in silico, we used the SUSPect algorithm implemented within PhyreInvestigator (Figure 6A–D). This algorithm predicted that the amino acids corresponding to the RabF2 domain, but not the RabSF1 domain, are highly sensitive to mutation. These results suggested that the RabF2 domain might stabilize the physical location of the RabSF1 domain in a manner that permits the interaction with one or multiple RabGAP proteins (Figure 6C). Additional analysis revealed that the residues of both domains exhibit moderate scores with respect to relative amino acid conservation (Figure 6D).
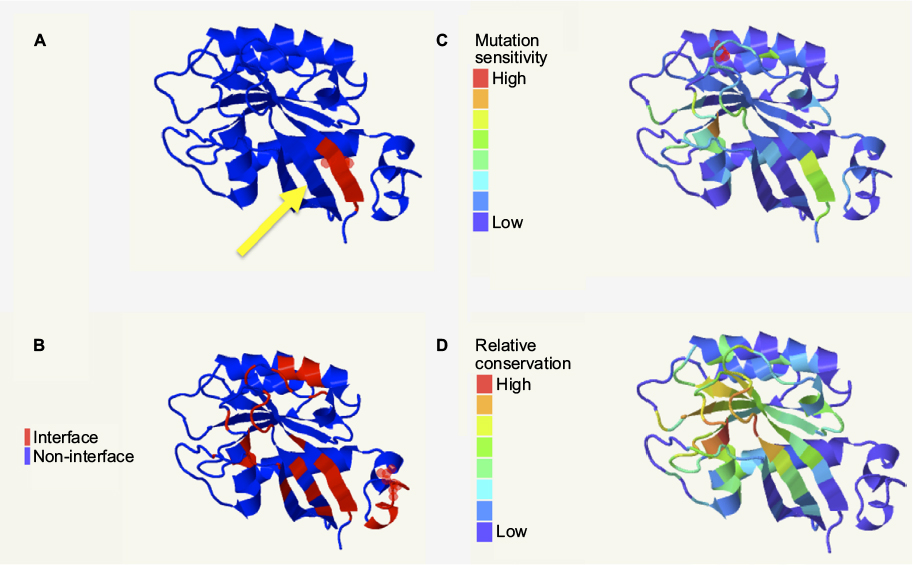 | Figure 6 RabF2 and RabSF1 domains correspond to predicted protein–protein interaction surfaces and are expected to be subject to mutational and conservational pressure. Notes: (A) The RabSF1 (red) and RabF2 (yellow arrow) are predicted to be adjacent to one another in the same β-pleated sheet. (B) This β-pleated sheet is also predicted as part of the protein–protein interaction surface. Only the RabSF1 domain is predicted to be sensitive to mutation (C), whereas both domains are functionally conserved (D) when analyzed using Jensen–Shannon divergence as implemented by PhyreInvestigator. All panels: visualization achieved using JSmol. |
Discussion
One of the primary uses of computational biology is to guide experimental design prior to in vitro experimentation. In cell biology, the development of in silico methods for predicting the interaction of Rab and RabGAP proteins could become a valuable tool for accelerating the pace of Rab–RabGAP interaction mapping. In this study, we have demonstrated that the RabSF1 and RabF2 domains of Rab11 and Rab23 are colocated in 3D space and comprise many of the residues predicted to be involved in protein–protein binding. The residues in these domains were found to be highly conserved with respect to their function. The RabSF1 domain is further predicted to be highly sensitive to mutation, suggesting that alteration to this domain may result in a loss of RabGAP binding and sub- sequent disregulation of Rab-specific activities. Collectively, these results suggest that these domains serve together as the site of Rab–RabGAP interaction. Furthermore, we have established an in silico framework for the identification of possible interactions between Rab and RabGAP proteins based on experimentally verified pairs of interacting Rabs and RabGAPs, where the Rab protein has a solved crystal structure. It is our expectation that this method should aid in limiting the number of wet-lab experiments necessary to demonstrate a functional interaction between a Rab and a RabGAP protein regardless of the existence of prior X-ray crystallography data. In practice, this method should be easy to use, requiring the investigator to align the RabSF1 domain of the Rab(s) of interest to that of a Rab known to interact with the RabGAP of interest and a control Rab known not to interact with that same RabGAP. In addition, applications of this method may prove useful in identifying the interaction surfaces for Rabs and their nonregulatory protein interaction partners. Several other domains (RabF1, RabF2–5, and RabSF2–4) are currently without a confirmed function; a comparison of Rabs known to interact with similar or identical cellular partners using variations of this method may suggest roles for these domains as well. Similarly, this technique could be applied more broadly to other sets of interacting proteins with homology to solved crystal structures; while the domains putatively specifying interaction may differ, methods for combining structural predictions with scanning for domain-specific amino acid signatures are expected to be useful.
The use of two different computer programs for predicting amino acid residues involved in protein–protein interactions resulted in the identification of an overlapping, but nonidentical set of amino acids (Table 3). In each of the Rab structures analyzed, with the exception of Rab7, seven to eight common amino acids were found when using both methods. These amino acids represent the residues most likely to mediate physical interactions between Rabs and other proteins. However, as both the ProtinDb and PI-Site databases are composed of protein-interaction residues identified from solved crystal structures deposited in the PDB, it is possible that both methods, while computationally different, may identify biologically significant residues among the nonoverlapping residue collection.9,17 However, in order to confirm which, if any, of these predicted amino acids are involved in Rab–protein interactions, future in vitro experimentation will be needed.
The method of structure determination used by Phyre2 is highly accurate and provides several possible structures. Each one of them is subsequently ranked on the basis of the accuracy that the underlying algorithms used.6 Assessment of model quality was made on the basis of comparing each one to existing crystal structures of the same Rab proteins. All modeled structures were predicted with high confidence (>80% confidence value in all cases) and the generated structures for Rab11 and Rab23 were nearly identical to their experimentally solved crystal structures. Any possible dissimilarities are likely the result of the assembly of the final molecular model based on the individual Phyre2 algorithm components. Further support of the structural modeling’s accuracy can be found in the location of predicted catalytic residues. A comparison to the Catalytic Site Database has identified residues in Rab11 (G21 and Q70) and Rab23 (A19 and Q68) that are expected to have catalytic activity. This is consistent with the position of the GTP-binding pocket predictions for Rab11 (positions 21–71, 124–128, and 154–156) and Rab23 (positions 19–70, 121–125, and 151–153), as well as with the position of these residues within the solved crystal structures.15 Similarly, the GTP-binding pocket and predicted protein interaction surfaces colocate, further supporting the validity of these models (Figure 3A–F). Moreover, although limited details are available regarding the accuracy of the ProtinDb program, the PI-Site program draws upon the library of solved 3D protein structures to infer residues in the sequence of interest that are similar to those known to be involved in protein–protein interactions.9,17 The PI-Site algorithm was designed to be conservative, requiring >90% sequence identity among the protein chains being compared for the presence of similar binding sites. These observations support the approach described in this work, and the authors expect that this method will be broadly applicable in cases where no solved structure exists for the molecule being analyzed.
With this in mind, the authors consider that further work in refining this method may prove valuable. For example, the data in Figure 5 indicate that the use of the RabSF1 domain alone may result in false-positive identifications, namely the placing of the Evi5L-interacting Rab4A and Rab10 on the same branch as the Evi5-interacting Rab11 and Rab35. However, it should be noted that in the study by Itoh et al, the evidence of interaction of Evi5L with both Rab4A and Rab10 was described by the authors as weak but statistically significant.4 A later study by Yoshimura et al showed extremely little GTP hydrolysis by Rab4A and none by Rab10, in the presence of Evi5L.18 It is also equally important to take into account the effect of environmental conditions on Rab–RabGAP interactions. The GTPase activity of Rab proteins in the presence and/or absence of RabGAPs could be significantly affected by environmental factors during both in vivo and in vitro experiments. The use of bioinformatics tools similar to the method described, for predicting the optimal conditions necessary for a functional protein–protein interaction, would further enhance the practical application of this method. In addition, Evi5 and Evi5L represent members of the classical Group I (RQ catalytic residues) RabGAPs.2 Expansion of this method to the Rab–RabGAP interactions involving members of Groups II–VI has not been attempted as most of these RabGAPs have no known Rab partner.2 Future studies also need to include the identification of Rab-GDI and Rab-GEF-binding domains, and the swapping of RabGAP-binding domains between Rabs, followed by the testing of their target RabGAP specificities. The authors recognize that further in vitro research is warranted on these topics, and efforts in these areas are planned.
One promising avenue of future research regards the RabGAP proteins Evi5 and Evi5L. Presently, efforts for the development of a high-confidence model for Evi5 and Evi5L are ongoing. While the current iteration of these models have high confidence for only select regions of each protein (data not shown), we plan to develop more complete models allowing us to better predict their interaction with partner Rabs. Such efforts would make it possible to compare them to solved crystal structures of Rab–RabGAP interaction pairs. In X-ray crystallography studies where this type of experiment has been attempted, the Rab partner has an alpha-helical domain that interacts with the RabGAP in a manner that is consistent with our in silico prediction of the surfaces of Rab11A and Rab23 that potentially interact with Evi5 and Evi5L, respectively.19,20 In silico confirmation of such interactions would further enhance the accuracy and performance of the method described herein for the study of other Rab/RabGAP pairs.
Acknowledgments
The authors thank the D’Youville College Department of Biology and Mathematics, as well as the Research Committee of the D’Youville College Faculty Council, for supporting this project.
Authors’ contributions
JJD and SLF designed the study. JJD performed the analyses and prepared the manuscript. SLF critically reviewed the manuscript. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Barr FA, Gruneberg U. Cytokinesis: placing and making the final cut. Cell. 2007;131(5):847–860. | ||
Frasa MAM, Koessmeier KT, Ahmadian MR, Braga VMM. Illuminating the functional and structural repertoire of human TBC/RABGAPs. Nat Rev Mol Cell Biol. 2012;13(2):67–73. | ||
Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129(5):865–877. | ||
Itoh T, Satoh M, Kanno E, Fukuda M. Screening for target Rabs of TBC (Tre-2/Bub2/Cdc16) domain-containing proteins based on their Rab-binding activity. Genes Cells. 2006;11(9):1023–1037. | ||
Pereira-Leal JB, Seabra MC. The mammalian Rab family of small GTPases: definition of family and subfamily sequence motifs suggests a mechanism for functional specificity in the Ras superfamily. J Mol Biol. 2000;301(4):1077–1087. | ||
Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10(6):845–858. | ||
Rose PW, Prlic´ A, Bi C, et al. The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015;43:D345–D356. | ||
Marchler-Bauer A, Derbyshire MK, Gonzales NR, et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015;43:D222–D226. | ||
Higurashi M, Ishida T, Kinoshita K. PiSite: a database of protein interaction sites using multiple binding states in the PDB. Nucleic Acids Res. 2009;37:D360–D364. | ||
Mitchell A, Chang HY, Daugherty L, et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 2015;43:D213–D221. | ||
Schmidtke P, Le Guilloux V, Maupetit J, Tuffery P. fpocket: online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010;38:W582–W589. | ||
Yates CM, Filippis I, Kelley LA, Sternberg MJE. SuSPect: Enhanced prediction of single amino acid variant (SAV) phenotype using network features. J Mol Biol. 2014;426(14):2692–2701. | ||
Dabbeekeh JTS, Faitar SL, Dufresne CP, Cowell JK. The EVI5 TBC domain provides the GTPase-activating protein motif for RAB11. Oncogene. 2007;26(19):2804–2808. | ||
Lim YS, Tang BL. The Evi5 family in cellular physiology and pathology. FEBS Lett. 2013;587(12):1703–1710. | ||
Eathiraj S, Pan X, Ritacco C, Lambright DG. Structural basis of family-wide Rab GTPase recognition by Rabenosyn-5. Nature. 2005;436(7049):415–419. | ||
Pereira-Leal JB, Seabra MC. Evolution of the Rab family of small GTP-binding proteins. J Mol Biol. 2001;313(4):889–901. | ||
Jordan R, Wu F, Dobbs D, Honavar V. ProtinDb: a data base of protein-protein interface residues. In preparation. Available from: http://ailab1.ist.psu.edu/protInDb/index.py. Accessed August 01, 2016. | ||
Yoshimura S-I, Egerer J, Fuchs E, Haas AK, Barr FA. Functional dissection of Rab GTPases involved in primary cilium formation. J Cell Biol. 2007;178(3):363–369. | ||
Gavriljuk K, Gazdag E-M, Itzen A, Kötting C, Goody RS, Gerwert K. Catalytic mechanism of a mammalian Rab·RabGAP complex in atomic detail. Proc Natl Acad Sci. 2012;109(52):21348–21353. | ||
Pan X, Eathiraj S, Munson M, Lambright DG. TBC-domain GAPs for Rab GTPases accelerate GTP hydrolysis by a dual-finger mechanism. Nature. 2006;442(7100):303–306. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.