")
Back to Journals » Infection and Drug Resistance » Volume 16
The Homologous Gene of Chromosomal Virulence D (chvD) Presents High Resolution as a Novel Biomarker in Mycobacterium Species Identification
Authors Yu X, He Y, Gu Y, Zhang T, Huo F, Liang Q, Wu J, Hu Y, Wang X, Tang W, Huang H , Liu G
Received 11 June 2023
Accepted for publication 17 August 2023
Published 11 September 2023 Volume 2023:16 Pages 6039—6052
DOI https://doi.org/10.2147/IDR.S422191
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Xia Yu,1,* Yingxia He,2,* Yuzhen Gu,1,* Tingting Zhang,1 Fengmin Huo,1 Qian Liang,1 Jing Wu,1 Yan Hu,2 Xuan Wang,2 Wei Tang,2 Hairong Huang,1 Guan Liu2
1National Clinical Laboratory on Tuberculosis, Beijing Key Laboratory on Drug-Resistant Tuberculosis, Beijing Chest Hospital, Capital Medical University, Beijing, 101149, People’s Republic of China; 2Wuhan Pulmonary Hospital, Wuhan Institution of Tuberculosis Control, Wuhan, 430030, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hairong Huang; Guan Liu, Email [email protected]; [email protected]
Objective: To evaluate the resolution of chromosomal virulence D (chvD) as a novel marker for mycobacterial species identification.
Methods: A segment of chvD (652 bp) was amplified by PCR from 63 mycobacterial reference strains, 163 nontuberculous mycobacterial clinical isolates, and 16 M. tuberculosis complex (MTBC) clinical isolates. A phylogenetic tree based on the reference strains was constructed by the neighbor-joining and IQ-tree methods. Comparative sequence analysis of the homologous chvD gene efficiently differentiated the species within the genus Mycobacterium. Slowly growing Mycobacterium (SGM) and rapidly growing Mycobacterium (RGM) were separated in the phylogenetic tree based on the chvD gene.
Results: The sequence discrepancies were obvious between M. kansasii and M. gastri, M. chelonae and M. abscessus, and M. avium and M. intracellulare, none of which could be achieved by 16S ribosomal RNA (rRNA) homologous gene alignment. Furthermore, chvD manifested larger intraspecies diversity among members of M. intracellulare subspecies. A total of 174 of the 179 (97.21%) clinical isolates, consisting of 12 mycobacterial species, were identified correctly by chvD blast. Four M. abscessus subsp. abscessus were identified as M. abscessus subsp. bolletii by chvD. MTBC isolates were indistinguishable, because they showed 99.84%– 100% homology.
Conclusion: Homologous chvD is a promising gene marker for identifying mycobacterial species, and could be used for highly accurate species identification among mycobacteria.
Keywords: species identification, Mycobacterium, chromosomal virulence D, phylogenetic tree
Introduction
The clinical symptoms and signs of pulmonary disease caused by nontuberculous mycobacteria (NTM) are very similar to tuberculosis, so differentiating the two becomes extremely challenging. Both diseases manifest cough, expectoration, hemoptysis, chest pain, and other respiratory symptoms, as well as systemic symptoms, such as fatigue and anorexia.1–3 Furthermore, NTM pulmonary disease cannot be easily differentiated from tuberculosis by radiographic images either, even though nodular or cavitary, or multifocal bronchiectasis with multiple small nodules fit best with diseases caused by Mycobacterium avium complex (MAC), M. kansasii and M. abscessus.4 Currently, more than 200 species of NTM are known, but most of them are nonpathogenic and mainly found in water and soil.5 However, a very small minority of the NTM species can cause human diseases.6 During the last few decades, NTM infections have increased globally.7,8 In the absence of species identification, NTM diseases are often misdiagnosed as tuberculosis. Because of the evident differences in drug-susceptibility profiles of different mycobacterial species, accurate species identification is tremendously important before commencing treatment.4
Mycobacterial species identification was once dependent on the conventional biochemical method, which relies on a series of biochemical experiments, pigmentation, and growth characteristics. These had inherent drawbacks, such as slow turnaround time and limited accuracy, due to similar phenotypes of different mycobacterial species in biochemical experiments that brought great challenges to the interpretation of results.2 Then, from the end of last century, molecular tests based on homologous gene/sequence comparison have become a major tool to identify NTM at the species level. The 16S rRNA-encoding gene (16s rRNA) is highly conserved, but exhibits obvious nucleotide variations in different organisms, which makes it an ideal sequence maker for bacterial species identification, including Mycobacterium. Although 16s rRNA can clearly differentiate most of the NTM species, it fails to separate some of the most frequently isolated species, such as M. abscessus and M. chelonae, M. avium, and M. intracellulare, as well as M. kansasii and M. gastric.4,9 This failure is typically attributed to the presence of almost identical 16s rRNA sequences in some mycobacterial species. The hsp65 gene exhibits a homology tendency similar to that of rpoB. The homology among M. intracellulare, M. chimaera is greater than 99.6%, making them indistinguishable.10 In addition, 16S–23S rRNA gene ITS is not a useful method for identification at the species level, although it may be a method for intraspecific differentiation.11 Therefore, alternative or complementary phylogenetic markers for the 16S rRNA gene are needed to increase the resolution power of species identification.
Chromosomal virulence D (chvD, Rv2477c) encodes a probable macrolide-transport ATP-binding protein ABC transporter in M. tuberculosis (https://mycobrowser.epfl.ch). As one of the efflux-pump genes, chvD was thought to be involved in the active transportation of drugs across the membrane, which has been observed during ofloxacin stress in M. tuberculosis.12 In addition, chvD is expected to be in 144 mycobacteria species (reported Mycobacterium species from LPSN (http://www.bacterio.net/mycobacterium.html), and Mycobacterium has a single copy of chvD in the genome (Table S1). Our preliminary data analysis found that the sequence similarity among different mycobacterial species with known chvD gene sequences was 86.05%–100%. These outcomes imply that chvD can be valuable in phylogenetic studies of the genus Mycobacterium.13 In this study, we report the resolution and reliability of the homologous chvD gene as a novel biomarker for mycobacterial species identification.
Methods
Ethics
All the mycobacterial reference and clinical isolates were stored in the Biobank at Beijing Chest Hospital (Beijing, China). The study was approved by the Ethics Committee of the Beijing Chest Hospital, Capital Medical University (2021–32-01).
Mycobacterial Reference Strains and Clinical Isolates
A total of 63 reference Mycobacterium strains (Table 1), 163 clinical NTM isolates, and 16 M. tuberculosis complex (MTBC) clinical isolates were investigated. The reference strains were obtained either from the American Type Culture Collection (ATCC) or the German Collection of Microorganisms (DSM). The clinical isolates of Mycobacterium were obtained from the Biobank at the National Clinical Laboratory on Tuberculosis, Beijing Chest Hospital in northeast China (n=149) and Wuhan Pulmonary Hospital in central China (n=30). All the clinical NTM isolates were classified as NTM, preliminarily by p-nitrobenzoic acid-containing Löwenstein–Jensen medium (500 µg/mL) and were subsequently identified at species level by sequence alignment of 16S rRNA, hsp65, rpoB, and 16–23S rRNA internal transcribed spacer (ITS) sequences.14
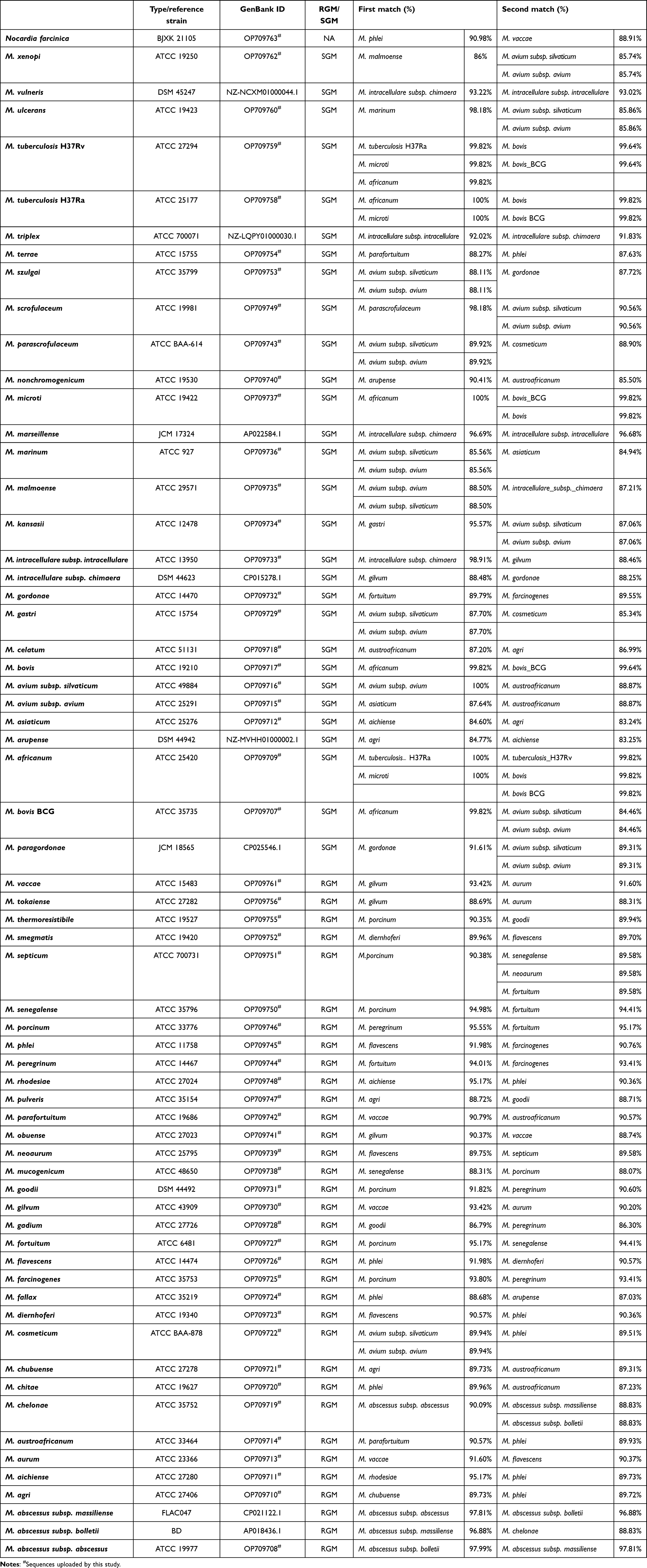 |
Table 1 chvD sequence matches among types/reference strains |
chvD Gene Amplification and Sequencing
DNA was extracted from cultured mycobacteria by boiling them in TE buffer for 10 min. After centrifugation, the supernatant was used as a template for PCR amplification. The forward primer, 5’-TGCCCTCGAACCAGAACC−3’, corresponded to positions 96–113 in the chvD gene of M. tuberculosis (GenBank accession number NC000962.3). The reverse primer, 5’-CTGCAGCGCTACGAGGAG-3’, corresponded to positions 799–816 in the chvD gene of M. tuberculosis (GenBank accession number NC000962.3). PCR amplification conditions were 5 min at 95°C, followed by 30 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 45 seconds, with a final extension step at 72°C for 10 min. The amplified product, 721 bp in length, was sequenced by Rui Biotech (Beijing, China).
Sequence Analysis and Phylogenetic Tree Constructions
In addition to the sequences of 56 reference strains obtained in this study, the chvD sequence of seven other Mycobacterium species were retrieved from GenBank, including M. abscessus subsp. massiliense (GenBank accession number CP021122.1), M. abscessus subsp. bolletii (GenBank accession number AP018436.1), M. arupense (GenBank accession number NZ_MVHH01000002.1), M. intracellulare subsp. chimaera (GenBank accession number CP015278.1), M. marseillense (GenBank accession number AP022584.1), M. paragordonae (GenBank accession number CP025546.1), and M. vulneris (GenBank accession number NZ_NCXM01000044.1). Then, a 652 bp region (excluding the terminal nucleotides at both ends that represented the primer binding site) of sequencing was phylogenetically analyzed using both neighbor joining and IQ-tree. The sequence of Nocardia farcinica (Genebank accession number OP709763) was used as the outgroup to construct a rooted tree. In addition, hypervariable regions of 16S rRNA was widely used for Mycobacterium species identification, and an ~873 bp region of 16S rRNA of the above 64 reference strains were retrieved from GenBank.
Species Identification of the Clinical Isolates
To evaluate the performance of chvD in species identification of mycobacteria, clinical isolates were obtained and analyzed. The strain inclusion criteria were as follows. For frequently isolated species, like M. abscessus complex, MAC, M. kansasii, M. fortuitum, and MTBC, one or a few dozen strains were included. For the less frequently isolated species, all the strains available were recruited. Sequences minus the known PCR primer sequences were assembled using SeqMan (version 7.1.0; DNAstar, Madison, WI). Isolates were identified by comparing sequences using a FASTA BLASTn search with MegAlign (version 10.1.0; DNAstar) to an in-house database of sequences consisting of type and reference strains from external culture collections.
Results
chvD Sequence Alignment of the Reference Strains
The 63 tested reference strains and one additional mycobacterial species (whose sequences were obtained from the GenBank database) demonstrated 86.05%–100% sequence identity (Table 1). Among the 29 reference strains of the slowly growing mycobacteria (SGM), besides themselves, 13 strains had >97% homology with the other first-matched mycobacterial species, including MTBC members and subspecies of M. avium and M. intracellulare, M. ulcerans and M. marinum. Among the 34 reference strains of rapidly growing Mycobacterium (RGM), only three had sequence identity >97% with the other first-matched species (Table 1), ie, M. parafortuitum and M. triviale shared identical chvD sequences, whereas the intrasubspecies of M. abscessus complex demonstrated 97%–98% sequence identity. Furthermore, except for the five MTBC member strains, all involved strains were well separated from the second-matched strains (homology was lower than 97%). Notably, the pathogenic M. kansasii was easily differentiated from the nonpathogenic M. gastri (with 96.03% homology). Those two species were not distinguishable by the 16S rRNA sequence alignment. For some other species, for which 16S RNA provides inadequate separation, chvD also demonstrated very distinct sequence variation: 88.80% sequence identity between M. chelonae and M. abscessus, 88.26% between M. avium and M. intracellulare, and 84.66% sequence identity between M. szulgai and M. malmoense were observed. In contrast to 16S rRNA, chvD also easily differentiated the subspecies of M. abscessus complex, ie, homology of 96.74% between M. bolletii and M. massiliense, 97.78% between M. bolletii and M. abscessus, and 97.61% between M. massiliense and M. abscessus were observed. However, the sequence similarity between MTBC members was high, 99.84%–100%, which indicated incapacity in differentiating members of MTBC (Table S2).
Phylogenetic Tree Construction
A phylogenetic tree that provided the basis for species differentiation in the genus Mycobacterium was constructed (Figure 1). The reliability of the phylogenetic tree was verified by the bootstrap method, using Nocardia farcinica as the outgroup. All 63 tested species showed good separation. The phylogenetic tree built upon the 63 reference strains was robust and discriminatory. Four SGM species — M. celatum, M. terrae, M. trivial and M. nonchromogenicum — were incorrectly placed among the RGM strains. Otherwise, the SGM and RGM were well separated. The sequence discrepancy was obvious between M. kansasii and M. gastri, M. chelonae and M. abscessus, M. avium and M. intracellulare, and M. szulgai and M. malmoense, which could not be achieved by 16S rRNA gene comparison (Figure 1). Notably, chvD also showed higher discrimination within the intraspecies of M. intracellulare complex and M. abscessus complex.
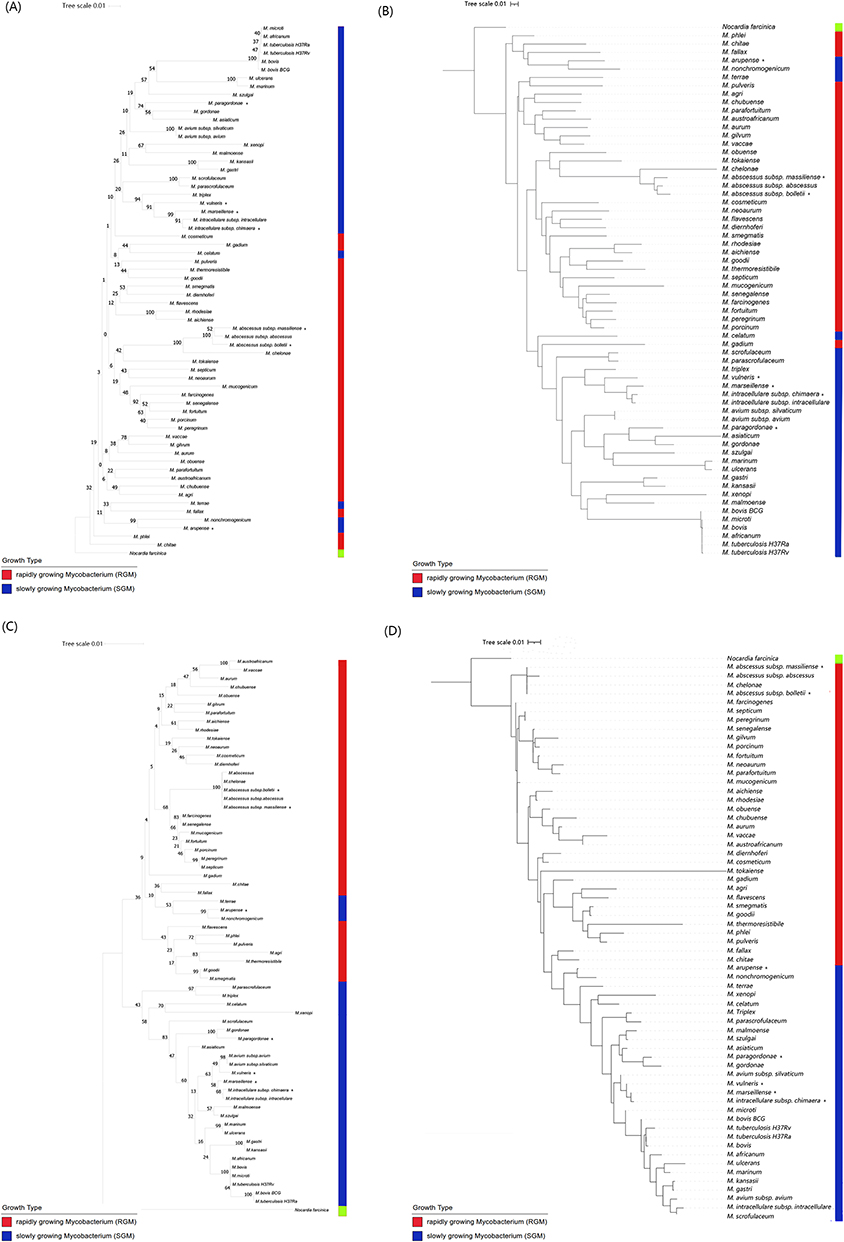 |
Figure 1 Phylogenetic tree based on chvD and 16S rRNA gene sequences shows the relationship of the 63 type strains of mycobacteria and one outgroup strain. Bootstrap values (percentages) are shown next to the nodes. (A) Tree of 652 bp region of chvD reconstructed with neighbor joining. (B) Tree of 652 bp region of chvD reconstructed with IQ tree. (C) Tree of 873 bp hypervariable regions of 16S rRNA reconstructed with neighbor joining. (D) Tree of 873 bp hypervariable regions of 16S rRNA reconstructed with IQ tree. Seven other Mycobacterium spp. retrieved from GenBank marked with an asterisk at the end of their names. |
Species-Identification Outcomes of Clinical Isolates
To evaluate the performance of chvD in identifying mycobacteria, 179 clinical isolates were tested, including 11 mycobacterial species, three subspecies, and clinical strains of M. tuberculosis (Table 2). chvD sequence identity in intraspecies was very high, generally 97%–100% (Table S2). Only three clinical strains of M. gordonae demonstrated significant variation when compared with the chvD sequence of the M. gordonae reference strain. Using the first-matched species’ sequence similarity (ie, >97%) as a cutoff value, chvD correctly identified 174 of the 179 (97.21%) clinical isolates (Table 2). All clinical strains of the frequently isolated pathogenic species, including M. intracellulare, M. avium, M. abscessus and MTBC, had been accurately identified. Misidentification was encountered in one of the six tested M. gordonae clinical isolates, which was identified as M. paragordonae. In addition, four (10.26%) M. abscessus subspecies abscessus of the 39 M. abscessus complex isolates were incorrectly identified as M. abscessus subspecies bolletii (Table 2). Owing to the high similarity between MTBC members (99.84–100%), chvD can not differentiate between members of MTBC taking 97% as a cutoff value.
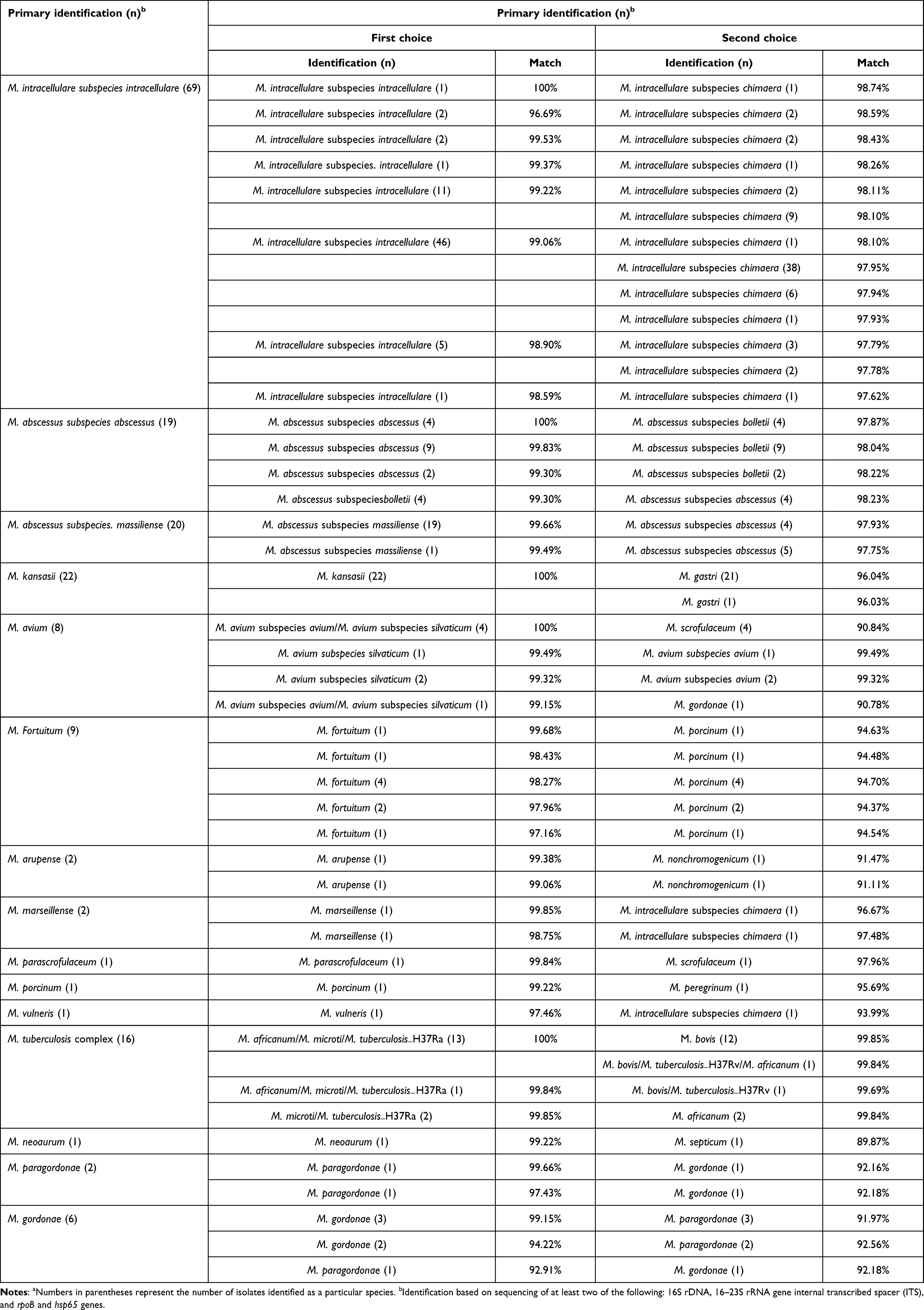 |
Table 2 Comparison of mycobacteria identified by 16S rRNA/ITS/hsp65/rpoB gene sequencing and chvD sequencing among the 179 clinical isolatesa |
Discussion
While 16S rRNA has good resolution in identifying most NTM species, many species that have close phylogenic relatedness often have indistinguishable 16S rRNA gene sequences. For example, the difference between M. abscessus and M. chelonae was only four nucleotides across the several-hundred bp lengths of the genes.15 A previous study showed that about a third of the included clinical strains were wrongly categorized when using 16S rRNA alone, due to high similarity of the sequences.16 Additional/supplementary DNA markers are needed for species that cannot be identified confidently by 16S rRNA.17 rpoB was reported to be an effective DNA marker for the species identification of mycobacteria.18 Addition of rpoB increased the resolution to 84% in contrast to 48% achieved using only the 16S rRNA gene.19 However, inconsistent outcomes between these two markers are frequently encountered (24.4%, 42 of 172).9 As a common genomic locus used in species identification, the rate of species-level identification of ITS was 81.55%.2 Hence, more makers are needed to resolve this inconstancy and to further improve species identification.
To be a qualified marker for species differentiation, the target gene should be a single-copy gene in the genome and should be conserved among species, but with enough random sequence variations. Therefore, extremely conserved genes or highly variable genes are not eligible. As a single-copy gene in 144 Mycobacterium genomes (Table S1), chvD works well in discriminating Mycobacterium species without ambiguous identification. Compared to chvD, 16S rRNA had higher homology within our tested mycobacteria (ie, 94.3%–100% compared to 86.05%–100%). According to our findings, chvD presented excellent potential as a supplementary marker to 16S rRNA in identification of Mycobacterium species. In this study, the 64 recruited reference strains resulted in 2016 paired comparisons (2016=[na1+n(n-1)d/2]; a1=1, n=63, d=1), and chvD successfully differentiated 99.01% (1996 of 2016) of them when using 97% sequence similarity as the cutoff value. Furthermore, the mean sequence identity of the chvD gene was 93.67%, which is significantly lower than the 16S rRNA genes (96.6%), indicating a higher discriminatory power of chvD.20 Phylogenetic analysis and tree construction further increased the resolution of chvD compared to the cutoff value method. Among the 179 clinical isolates, 97.21% (174 of 179) were identified correctly by chvD gene blast. One M. gordonae clinical strain was incorrectly identified as M. paragordonae at species level, which may have been caused by the high degree of intraspecies variability in M. gordonae.21 Additionally, at the subspecies level, four (10.26%) M. abscessus subspecies abscessus of 39 M. abscessus complexes were incorrectly identified as M. abscessus subspecies bolletii by chvD blast. M. abscessus subspecies bolletii was described as a new member of M. abscessus complex in 2009,22 and often infects patients with cystic fibrosis.23 In China, M. abscessus subspecies bolletii is rarely isolated,24 and none was detected in the 1755 NTM isolates from the year 2014 to 2021 in our laboratory. Therefore, these four isolates, identified as M. abscessus subspecies bolletii by chvD, need to be reconfirmed by other more powerful approaches, such as whole-genome sequencing and MALDI-TOF.
In this study, the homologous chvD gene demonstrated robust capacity in identification of RGM and SGM species. Five SGM species — M. celatum, M. terrae, M. arupense and M. nonchromogenicum — were allocated to the RGM group in the phylogenetic tree. The general rationale showed that RGM have two rRNA gene operons, while SGM have only one.25 Surprisingly, SGM species (M. terrae and M. celatum) have been reported to contain two rRNA genes, which suggests that these two species could be intermediate transition species between SGM and RGM. In addition, M. trivial and M. nonchromogenicum belong to the M. terrae complex. This complex is often placed into the group of RGM based on hsp65, dnaK and secA1,21,26 as well as rpsA, which we previously reported as a novel potential marker for Mycobacterium species identification.27 Consistently, the M. terrae complex (M. terrae, M. arupense, and M. nonchromogenicum) was placed between RGM and SGM based on tmRNA sequences in phylogenetic tree28 and allocated to the RGM group in the phylogenetic tree by 16S rRNA. Overall the M. terrae complex may phylogenetically be an intermediate transition species between SGM and RGM, according to this and other studies.
All the included 64 reference strains were distinguished by the chvD phylogenetic tree. Some paired species, such as M. senegalense and M. thermoresistibile, M. austroafricanum and M. terrae, are known to be not properly separated from each other by other markers alone, such as 16S rRNA, ITS, rpoB and hsp65.27 The chvD sequence identity between the above species was 88.37% and 87.99%, respectively. Thus, the data suggest that chvD had an advantage in resolving certain species over 16S rRNA, ITS, rpoB and hsp65 when used as sole marker.
Even with the suboptimal resolution, the 16S rRNA gene is still frequently firstly selected because it has been well recognized and its sequence dataset is highly abundant. To increase the resolution, at least one additional maker, such as ITS, rpoB, hsp65, rpsA (previously reported by us) and chvD (in this study) are recommended to be used as a supplementary maker. Integration of different loci would be helpful to avoid conflicting or dubious outcome yields. Furthermore, other results of species identification, including biochemical tests, high-performance liquid chromatography (HPLC), and matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry, could be used for increasing the precision of diagnoses.
There were some limitations in our study. Firstly, the analyses were from culture-positive isolates and not DNA extracted directly from clinical samples. The performance of the homologous chvD gene in species identification should be further evaluated in clinical samples. Secondly, some uncommon yet clinically relevant species like M. chimaera have not been clearly evaluated owning to no such isolates being able to be collected in our hospital. Thirdly, similar to current DNA markers like 16S rRNA, chvD was unable to differentiate members of MTBC owing to the high similarity between MTBC members.
In conclusion, the homologous chvD gene is a valuable DNA marker for mycobacterial species identification. For certain specific species, chvD manifested better discrimination power than other frequently used DNA markers, which suggests its utility in increasing the resolution of Mycobacterium species identification.
Funding
This work was supported by the Tongzhou Science and Technology Project (KJ2022CX041) and Tongzhou Yunhe Plan (YH201905), the Beijing Hospitals Authority Youth Programme (QML20211602), and the Wuhan Municipal Health Commission Scientific research Project (WX17Q33).
Disclosure
Xia Yu, Yingxia He and Yuzhen Gu share first authorship. The authors declare no conflicts of interest.
References
1. He Y, Wang JL, Zhang YA, Wang MS. Prevalence of culture-confirmed tuberculosis among patients with nontuberculous mycobacterial disease. Infect Drug Resist. 2022;15:3097–3101. doi:10.2147/IDR.S363765
2. Kim SH, Shin JH. Identification of nontuberculous mycobacteria using multilocous sequence analysis of 16S rRNA, hsp65, and rpoB. J Clin Lab Anal. 2018;32(1):e22184. doi:10.1002/jcla.22184
3. Wassilew N, Hoffmann H, Andrejak C, Lange C. Pulmonary disease caused by non-tuberculous mycobacteria. Respiration. 2016;91(5):386–402. doi:10.1159/000445906
4. Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175(4):367–416. doi:10.1164/rccm.200604-571ST
5. Tortoli E, Fedrizzi T, Meehan CJ, et al. The new phylogeny of the genus Mycobacterium: the old and the news. Infect Genet Evol. 2017;56:19–25. doi:10.1016/j.meegid.2017.10.013
6. Dávalos AF, Garcia PK, Montoya-Pachongo C, et al. Identification of Nontuberculous Mycobacteria in Drinking Water in Cali, Colombia. Int J Environ Res Public Health. 2021;18(16):8451. doi:10.3390/ijerph18168451
7. Baldwin SL, Larsen SE, Ordway D, Cassell G, Coler RN. The complexities and challenges of preventing and treating nontuberculous mycobacterial diseases. PLoS Negl Trop Dis. 2019;13(2):e0007083. doi:10.1371/journal.pntd.0007083
8. Brode SK, Marchand-Austin A, Jamieson FB, Marras TK. Pulmonary versus Nonpulmonary Nontuberculous Mycobacteria, Ontario, Canada. Emerg Infect Dis. 2017;23(11):1898–1901. doi:10.3201/eid2311.170959
9. Kazumi Y, Mitarai S. The evaluation of an identification algorithm for Mycobacterium species using the 16S rRNA coding gene and rpoB. Int J Mycobacteriol. 2012;1(1):21–28. doi:10.1016/j.ijmyco.2012.01.004
10. Kim MJ, Kim KM, Shin JI, et al. Identification of Nontuberculous Mycobacteria in Patients with Pulmonary Diseases in Gyeongnam, Korea, Using Multiplex PCR and Multigene Sequence-Based Analysis. Can J Infect Dis Med Microbiol. 2021;2021:8844306. doi:10.1155/2021/8844306
11. Trček J, Barja F. Updates on quick identification of acetic acid bacteria with a focus on the 16S-23S rRNA gene internal transcribed spacer and the analysis of cell proteins by MALDI-TOF mass spectrometry. Int J Food Microbiol. 2015;196:137–144. doi:10.1016/j.ijfoodmicro.2014.12.003
12. Gupta AK, Katoch VM, Chauhan DS, et al. Microarray analysis of efflux pump genes in multidrug-resistant mycobacterium tuberculosis during stress induced by common anti-tuberculous drugs. Microb Drug Resist. 2010;16(1):21–28. doi:10.1089/mdr.2009.0054
13. Zhou L, Ma C, Xiao T, et al. A New Single Gene Differential Biomarker for Mycobacterium tuberculosis Complex and Non-tuberculosis Mycobacteria. Front Microbiol. 2019;10:1887. doi:10.3389/fmicb.2019.01887
14. Lee JC, Whang KS. Mycobacterium aquiterrae sp. nov., ion of acetic acid bacteria with a focus on the 16S-23S rRNA a rapidly growing bacterium isolated from groundwater. Int J Syst Evol Microbiol. 2017;67(10):4104–4110. doi:10.1099/ijsem.0.002261
15. Hall L, Doerr KA, Wohlfiel SL, Roberts GD. Evaluation of the MicroSeq system for identification of mycobacteria by 16S ribosomal DNA sequencing and its integration into a routine clinical mycobacteriology laboratory. J Clin Microbiol. 2003;41(4):1447–1453. doi:10.1128/JCM.41.4.1447-1453.2003
16. Pauls RJ, Turenne CY, Wolfe JN, Kabani A. A high proportion of novel mycobacteria species identified by 16S rDNA analysis among slowly growing AccuProbe-negative strains in a clinical setting. Am J Clin Pathol. 2003;120(4):560–566. doi:10.1309/VF401U7H7DHE0FRE
17. Woo PC, Lau SK, Teng JL, Tse H, Yuen KY. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 2008;14(10):908–934. doi:10.1111/j.1469-0691.2008.02070.x
18. Meghdadi H, Khosravi AD, Hashemzadeh M, Tabandeh MR. New design of multilocus sequence analysis of rpoB, ssrA, tuf, atpE, ku, and dnaK for identification of Mycobacterium species. Mol Biol Rep. 2022;49(8):7967–7977. doi:10.1007/s11033-022-07638-0
19. Simmon KE, Kommedal S, Karlsen B, Petti CA. Simultaneous Sequence Analysis of the 16S rRNA and rpoB Genes by Use of RipSeq Software To Identify Mycobacterium Species. J Clin Microbiol. 2010;48(9):3231–3235. doi:10.1128/JCM.00362-10
20. Yamada-Noda M, Ohkusu K, Hata H, et al. Mycobacterium species identification--A new approach via dnaJ gene sequencing. Syst Appl Microbiol. 2007;30:453–462. doi:10.1016/j.syapm.2007.06.003
21. Zelazny AM, Calhoun LB, Li L, Shea YR, Fischer SH. Identification of Mycobacterium Species by secA1 Sequences. J Clin Microbiol. 2005;43(3):1051–1058. doi:10.1128/JCM.43.3.1051-1058.2005
22. Adékambi T, Drancourt M. Mycobacterium bolletii Respiratory Infections. Emerg Infect Dis. 2009;15(2):302–305. doi:10.3201/eid1502.080837
23. Griffith DE, Daley CL. Treatment of Mycobacterium abscessus Pulmonary Disease. Chest. 2022;161(1):64–75. doi:10.1016/j.chest.2021.07.035
24. Sun Q, Yan J, Liao X, et al. Trends and Species Diversity of Non-tuberculous Mycobacteria Isolated From Respiratory Samples in Northern China, 2014-2021. Front Public Health. 2022;10:923968. doi:10.3389/fpubh.2022.923968
25. Bercovier H, Kafri O, Sela S. Mycobacteria possess a surprisingly small number of ribosomal RNA genes in relation to the size of their genome. Biochem Biophys Res Commun. 1986;136(3):1136–1141. doi:10.1016/0006-291X(86)90452-3
26. Dai J, Chen Y, Dean S, Morris JG, Salfinger M, Johnson JA. Multiple-Genome Comparison Reveals New Loci for Mycobacterium Species Identification. J Clin Microbiol. 2011;49(1):144–153. doi:10.1128/JCM.00957-10
27. Duan H, Liu G, Wang X, et al. Evaluation of the Ribosomal Protein S1 Gene (rpsA) as a Novel Biomarker for Mycobacterium Species Identification. Biomed Res Int. 2015;2015:271728. doi:10.1155/2015/271728
28. Mignard S, Flandrois JP. Identification of Mycobacterium using the EF-Tu encoding (tuf) gene and the tmRNA encoding (ssrA) gene. J Med Microbiol. 2007;56(8):1033–1041. doi:10.1099/jmm.0.47105-0
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.