")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 5
The hereditary basis of bicuspid aortic valve disease: a role for screening?
Authors Gharibeh L, Nemer M
Received 10 October 2014
Accepted for publication 11 November 2014
Published 23 December 2014 Volume 2015:5 Pages 11—17
DOI https://doi.org/10.2147/AGG.S59832
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr John Martignetti
Lara Gharibeh, Mona Nemer
Molecular Genetics and Cardiac Regeneration Laboratory, Department of Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, ON, Canada
Abstract: Over the past years, human and molecular genetic studies have provided new understanding of valve development and the molecular pathogenesis of bicuspid aortic valve (BAV) disease. BAV is an autosomal dominant disease with incomplete penetrance and is found to affect 1%–2% of the population. It can occur in isolation or coexists with other congenital heart diseases such as ventricular septal defect and tetralogy of fallot. BAV is a risk factor for premature cardiovascular disease and can lead to severe complications affecting the aorta and the valves. To date, NOTCH1 and GATA5 are the only genes linked to human BAV, and the genetic basis for most BAVs remains unidentified. Large-scale screening as well as whole exome sequencing studies hold promise for uncovering BAV-causing genes. Similarly, molecular analysis of valve development in animal models is needed for better insight of normal and pathologic valve formation. Together, these approaches will undoubtedly accelerate discovery of disease-causing genes opening the way for early diagnosis of BAV and prevention of valve degeneration and cardiovascular complications.
Keywords: congenital heart disease, valvulogenesis, genetic screening
General overview
The formation of the heart is a very complex process that begins at early stages of fetal development and is orchestrated by complex regulatory networks ensuring the formation of a fully mature four-chambered heart. The mammalian heart contains three layers: an inner endothelial cell lining called the endocardium, surrounded by a muscular component forming the majority of the heart called the myocardium, and an outer protective layer that envelops the myocardium called the pericardium. The unidirectional flow of blood into the whole body is ensured by the presence of thin structures, composed of leaflets opening and closing with each heart contraction. The valve leaflets originate from mesenchymal outgrowths known as the cardiac cushions. These cardiac cushions result from the transformation of endothelial cells into mesenchymal cells through a process called EMT (epithelial to mesenchymal transformation), where endothelial cells invade the cardiac jelly separating the outer myocardium from the inner endothelium, at the base of the outflow tract (OFT). The cushions containing mesenchymal-derived cells then elongate and undergo extensive remodeling to gradually mature and form thin leaflets known as valves. Mature valves are composed of a highly organized extracellular matrix (ECM) consisting of three distinct layers made of collagens (fibrosa), proteoglycans (spongiosa), and elastin (ventricularis).1,2 The heart valves comprise the semilunar valves (aortic and pulmonary valves), controlling the systemic flow of blood; and the atrioventricular valves (mitral and tricuspid valves), controlling the flow of blood between the heart compartments. The semilunar valves arise from two types of cushions: the conotruncal and the intercalated cushions situated at the OFT. The mature aortic valve (AoV) is composed of three symmetrical leaflets (also called tricuspid aortic valve [TAV]):the right and left leaflets originate from the conotruncal cushions, and the posterior aortic leaflet is formed from the right-posterior intercalated cushion, adjacent to the conotruncal cushion.3–5 The proper formation of the heart valves ensures the unidirectional flow of blood into the systemic circulation.
Semilunar valve malformations are very common and affect 2%–3% of the population causing a variety of valvular complications including valve stenosis and/or regurgitation.6 There are two types of valve diseases: 1) congenital valve diseases that are present at birth such as bicuspid aortic valve (BAV), mitral valve prolapse, and tricuspid atresia and 2) acquired valve diseases that develop later in life such as valve calcification and dysfunction.7 BAV is the most common congenital abnormality affecting 1%–2% of the population with a higher (3:1) male prevalence.8 It results in the presence of two asymmetrical cusps or leaflets instead of three symmetrical ones with the presence or absence of a raphe (connecting ridge) on the larger leaflet marking the failure of separation between leaflets (Figure 1). Familial clustering studies have shown that BAV is a heritable trait with a prevalence of 9% among first-degree relatives and up to 24% in families where more than one member is affected. BAV follows an autosomal dominant mode of inheritance with incomplete penetrance suggestive of gene–gene and gene–environment interactions.9–11 Leaflets orientation varies widely among BAV patients resulting in different types of BAVs classified according to the leaflets position relative to the right and left coronary arteries (Figure 1). The most frequent type in human is the RL-type BAV resulting from fusion of the right and left coronary cusps and representing 59% of all BAV cases. The RN-type results from fusion of the right and noncoronary cusps and is less frequent accounting for 37% of BAV cases.12 A less common type, the LN BAV, results from a failure of separation between the left and noncoronary leaflets. Recent studies on animal models suggest that RL- and RN-type BAVs have distinct etiologies: RL-type BAV results from defective septation during valvulogenesis, while RN BAVs are caused by a defect in the formation of the OFT cushions.13 Despite its importance, the underlying pathways leading to the different types of BAV remain undetermined.
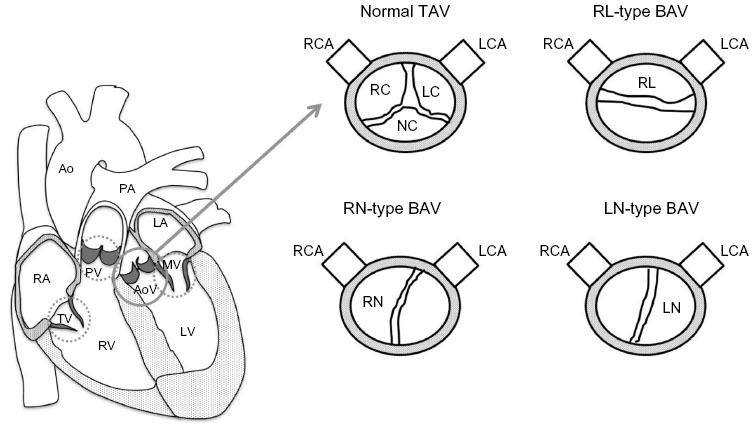 | Figure 1 Representation of a normal TAV and different types of BAV. |
BAV is a risk factor for premature valvulo-vascular disease
BAV occurs in isolation or coexists with other congenital heart diseases such as coarctation of the aorta, interruption of the aortic arch, and ventricular septal defects. Individuals with BAV are often asymptomatic but are at an increased risk of several life threatening events ranging from aortic stenosis and regurgitation, aneurisms, and dilation and rupture of the aorta to infective endocarditis.12,14 So it is clear that BAV is a risk factor for many valvulo-vascular complications. Increased shear stress (effect of blood velocity and viscosity) is thought to play a crucial role in the pathogenesis of BAV-associated disease. The presence of BAV might be associated with pressure overload that could in part be due to the presence of malfunctioning valves; both conditions are risk factors for aortic dissection and rupture.15 The most common phenotypic pattern in BAV disease is mid ascending aortic dilatation and is correlated with older age, whereas the other pattern, aortic root dilatation, is more associated with younger age and male sex.16 The normal aortic wall consists of an internal intimal endothelial layer (intima), a medial layer (media) composed of smooth muscle cells with an ECM (rich in elastic and collagen fibers), and a covering protective layer rich in collagen fibers called the adventia.17 BAV patients tend to develop aortic dilation, which is thought to be due to a disruption of the ECM by itself, caused by the lack of Fibrillin-1 and the presence of a cystic medial necrosis. Fibrillin-1 is a glycoprotein that ensures the strong interaction between vascular smooth muscle cells and elastin and collagen-rich matrix as well as maintaining the structural integrity of the aortic wall.18 Studies have shown that regardless of the AoV function, BAV patients have a defect in the structure of the aortic wall that will eventually result in aortic disease.19 BAV was found to be a pathological event since it was associated with accelerated degeneration of the medial layer of the aorta.20 In the aorta of BAV patients, less elastic tissue loss and fragmentation, and less changes in the intimal layer were observed when compared to aortas from TAV patients.21 Examination of specimens taken from BAV patients with aortic dissection showed a thinner medial layer due to elastic fiber necrosis (less vascular smooth muscle cells) leaving a greater distance between elastin fibers in those patients, which was not the case in TAV patients.22 On the other hand, an elevation of matrix metalloproteinases, endogenous enzymes that degrade matrix components, has been reported to play an important role in the formation of aortic aneurysm in the aorta of patients with BAVs.20 Beyond cardiovascular diseases, an increased risk of development of intracranial aneurysms was also found to correlate with the presence of BAV in a study done by Schievink et al. The study was done on a small population (61 BAV patients), so additional investigations are required before drawing any conclusions.23 Together, these data raise the possibility that other subclinical genetic abnormalities in vascular endothelial or smooth muscle cells coexist with BAV and contribute to aortic disease. The development of animal models where BAV and TAV are present on a similar genetic background will help answer this important question.
With regard to the AoVs and their function, BAV is found to lead to severe valvular complications at late onset. The valve’s ECM is regulated by valve interstitial cells (VICs) that are fibroblast like and quiescent in normal conditions. Changes in the hemodynamic environment are controlled by valve endothelial cells, which form a protective layer around the leaflets and communicate with the VICs through different molecular signals. In the presence of BAV, disruption of normal valve morphogenesis is accompanied by changes in the ECM composition, endothelial cell disruption, and VIC activation, eventually causing valvular dysfunction and calcification.2,24 Valve disease is most commonly diagnosed as regurgitation, a leaking of the AoV that causes blood to flow in the reverse direction, or stenosis, a disease of the AoV in which the opening of the valves is narrowed. Both disease presentations are caused by disruptions in the structure and function of the valves. A study of 542 surgical BAV patients showed that 75% of the BAV cases had aortic stenosis; the same study showed that only 13% had aortic regurgitation. In fact, the regurgitation group had a higher rate of annular dilatation and a higher male to female ratio when compared to the stenosis group that had a higher prevalence of aortic calcification.25 Studies have also shown that the presentation of valve disease varies among different BAV subtypes. RL-type BAVs are found to have more severe stenosis when compared to RN-type BAVs that develop more severe regurgitation.26 In congenital bicuspid valve stenosis, aortic stenosis, which is secondary to the BAV, is found to lead to the development of calcification, occurring usually within the cusp’s spongiosa.27 This calcification in BAVs is due to the presence of an abnormal distribution of stress across the cusps in the deformed valves, leading to calcification earlier than in the case of TAV.28 It can also be the result of mechanical disruptions (elastin and collagen fragments) that lead to the formation of nucleation sites for calcification.29 In the case of calcified aortic valve disease, activation of the VICs is found to lead to the expression of bone development genes such as osteopontin, Runx2, and osteocalcin.30 Once the valves are calcified, a surgical intervention is recommended in the case of severe valve dysfunction or the presence of comorbidities, which could further affect cardiac function and lead ultimately to heart failure.
Genetic basis of BAV: what we know from human and animal models
Even though the heritability of BAV is well documented, BAV causing genes remain largely unknown. Recent studies suggest that BAV is a complex process that can be caused by several genes, each with a distinct mode of inheritance, which might explain why identifying the genetics of BAV through classical human genetic studies has been challenging.10 To date, mutations in only two genes in human, NOTCH1 (member of the Notch signaling pathway) and GATA5 (member of the GATA family of transcription factors), were found to be linked to BAV. Based on DNA-sequencing of all coding exons and adjacent splice consensus sequences of the NOTCH1 gene, missense mutations (p.T596M and p.P1797H) leading to amino acid substitution in a well-conserved region in NOTCH1 were identified in patients with sporadic BAV, which were absent in 327 healthy individuals.31 In another study, two novel mutations, p.P284L and p.Y1619X, a missense and a nonsense mutation, were found in the NOTCH1 gene in two unrelated Italian families where the mutations segregated with the disease in both families.32 Garg et al also identified two mutations in NOTCH1: the first mutation (p.R1108X) in a European-American five generation family showed linkage for BAV to a single locus on chromosome 9q34-35, whereas the second mutation (p.H1505del) cosegregated with the disease in three affected family members.33 Consistent with a role for the BMP signaling in AoV formation, mutations in SMAD6, a component of the BMP intracellular signal transduction pathway, were reported in two out of 24 BAV patients with aortic stenosis and coarctation. Whether the mutations are causally linked with BAV remains uncertain since inactivation of SMAD6 in mice did not cause BAV; instead, hyperplastic thickening of the valves and aortic ossification were observed.34
In order to further understand the complex process of valve morphogenesis, animal models lacking important regulators of valve formation were generated. Inactivation of key players during this process led to a wide variety of valvular malformations including BAV. One of the first models of BAV was reported in mice lacking NOS3 (endothelial nitric oxide synthase) where BAV was reported in five out of 12 mature NOS3-deficient mice (41.6%), all sharing the RN type with no BAV observed in any of the 26 wild-type littermates.35 Another study by Bosse et al supports the important role of NOS3 in AoV formation by regulating NOTCH1 signaling and AoV disease prognosis. NOS3−/− NOTCH1+/− mice display 100% penetrance of BAV compared to the partial penetrance in the NOS3−/− (26%) and in the NOTCH1+/− (8%) single knockouts.36 These results confirm a crucial role for the Notch pathway in AoV development and suggest that any deficiency in one or more of its key regulators could lead to valve defects including BAV.
In 2011, our group reported that targeted deletion of GATA5 in mice leads to partially penetrant BAV (26%). GATA5, a member of the GATA family of transcription factors crucial for heart morphogenesis, is restricted to endocardial cells during early embryonic development. Whether GATA5 is deleted in all cells or only in endocardial cells, partial penetrance of RN-type BAV is consistently observed. Mechanistically, GATA5 was found to regulate important pathways associated with endocardial cushion differentiation such as NOS3, NOTCH, BMP4, and TBX20.37 GATA5 mutations were later reported in several patients with BAV. A genetic analysis in 100 BAV cases identified four rare nonsynonymous GATA5 variants (Gln3Arg, Ser19Trp, Tyr142His, and Gly166Ser) in GATA5 transactivational domain, pointing out a possible role for GATA5 in the pathogenesis of BAV. These mutations were in highly conserved and functional regions, which would affect protein function.38 In another study, 78 BAV patients were analyzed using Sanger sequencing to screen for these GATA5 variants. Two rare nonsynonymous heterozygote mutations, p.Gln3Arg and p.Leu233Pro, with a frequency of 2.6% (2/78), were found in patients with aortic coarctation and RN- or RL-type BAV.39
Even though no human mutations linked to BAV in genes other than NOTCH1 and GATA5 were reported, potential candidate genes that could underlie the formation of BAV were discovered by examination of the AoVs of genetically engineered mice. Activin type I receptor (ALK2) is expressed in endocardial cells, and its role in endothelial EMT in the atrioventricular cushions is well established.40 Deletion of ALK2 from the cushion mesenchyme after EMT using GATA5-Cre results in AoV defect (BAV) and development of aortic insufficiency and/or stenosis in some of the mutant mice. These results indicate an important role of ALK2 in AoV development and suggest that its absence can lead to AoV diseases.41 No human mutations in HOXA1 gene are observed in BAV patients, but its inactivation in mice was found to result in 24% penetrance of BAV, among other cardiac malformations. HOXA1 null mice show defects such as interrupted aortic arch, tetralogy of fallot, and abnormal subclavian artery, pointing to an important role of HOXA1 in patterning of the OFT of the heart.42 On the other hand, studies done on NKX2-5 heterozygous mice showed an increased frequency of 8.2% of stenotic BAV with three out of 35 NKX2-5 heterozygous mutant mice showing an increase in the aortic blood flow velocity and BAV with mild thickening upon autopsy. The presence of BAV in these mice was dependent on the genetic background confirming the presence of genetic modifiers as suggested in human genetic studies.43
All the mice models of BAV mentioned above displayed the RN-type BAV. A study by Sans-Coma et al in 1995 reported the first RL BAV model in inbred Syrian hamsters. In all, 16 out of 29 embryos had a fusion between the left and right cushions resulting in two abnormal valve cushions (a ventral and a dorsal).44 To date, no genetically modified mouse model of RL BAV, the most common type of BAV observed in human, has been reported.
Given the lack of identification of BAV causative genes, studies have aimed at determining genetic loci associated with familial BAV. Early studies have identified genomic regions 17q29.3 (KCJN2 gene) and 9q34.3 (NOTCH1) to be responsible for BAV in a small proportion of cases. A mutation in KCJN2 gene, encoding the inward-rectifying potassium current, KIR2.1, was reported in patients with Anderson syndrome having sex-specific cardiac (BAV) and skeletal muscle phenotypes.32,45 Genome-wide scan studies have identified three chromosomal regions, 18q, 5q, and 13q, associated with BAV, but the genes therein remain to be identified.46 BAV is also manifested in some human syndromes such as Loeys–Dietz and Marfan. Loeys–Dietz syndrome is caused by a defect in the TGFB signaling pathway (mutations in TGFBR1 [TGFB receptor 1] and TGFBR2) and is characterized by the presence of congenital heart disease and mental retardation among other abnormalities. Marfan syndrome is a connective tissue disorder characterized by cardiovascular, skeletal, and ocular manifestations. It is caused by a defect in the fibrillin-1 (FBN1) gene (essential for the formation of elastic fibers found in connective tissues) localized on chromosome 15q21.1 and is inherited in an autosomal dominant manner.47,48 Mutations in HOXA1 gene were also found in Bosley-Salih-Alorainy as well as in Athabaskan brainstem dysgenesis syndromes, which are characterized by a wide range of heart defects including ventricular septal defects, tetralogy of fallot, and BAV.49 A high prevalence of BAV is also observed in patients with Turner syndrome, caused by a deletion of the short arm of the X chromosome. This suggests that abnormal AoV and aortic arch development can also be caused by happloinsufficiency in Xp gene (Table 1).50
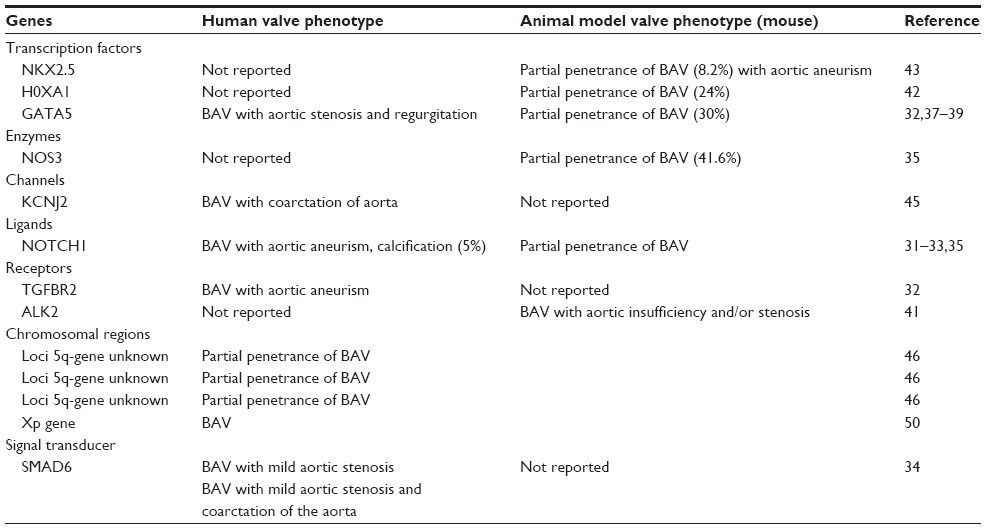 | Table 1 Genes associated with human and animal (mouse) BAV phenotypes |
Is it important to screen?
Asymptomatic individuals with BAV may go undetected for a lifetime and many will be disease free. However, a significant proportion will develop cardiovascular events that could be life threatening, requiring surgical intervention in some cases. A recent study showed that 25% of BAV subjects had to undergo premature valvular or aortic surgeries.51 Whether the BAV is the direct cause of aortic events or whether subtle defects in vascular cells – caused by the same genetic alterations – are responsible for BAV-associated cardiovascular complications remain to be determined. Be it as it may, BAV is a cardiovascular risk factor as also evidenced in the ASTRONOMER study.52 It is now well established that early detection of affected individuals allows for better monitoring and preventative care. Echocardiography and magnetic resonance imaging have been reliable techniques so far to detect BAVs and dilated aortas, but the infrastructure and cost of these tests make them impractical for large-scale population screening. Moreover, valvular problems and dilated aortas caused by BAV are not detected at childhood, and the late identification of BAV often occurs when AoV disease has become irreversible.
Given the established familial contribution to BAV, the use of next-generation technologies such as whole exome sequencing (WES) and whole genome sequencing should be considered. Given the high cost of whole genome sequencing, WES is currently a more viable approach to determine the protein-coding sequence variations. In previous studies, WES has proven to be useful in revealing genetic variants in individuals affected with rare forms of heart diseases such as BAG3 gene in familial dilated cardiomyopathy.53 Other genes causing severe hypercholesterolemia (ABCG) and familial combined hypolipidemia (ANGPTL3) could not have been identified without the availability of this technique.54,55 In a not so distant future, WES and eventually WEG might well become routine techniques for sequencing due to accelerated developments that are resulting in rapid drop in cost. Investments in those technologies will accelerate discovery of BAV causing genes, ultimately enhancing patient management and treatment.56 Recent studies have shown that WES is useful for disease diagnosis, which informs patient treatment. Since most BAVs diagnosed to date usually have a familial history, it is important to screen first-degree relatives of a patient with BAV. WES has helped in the diagnosis of Crohn’s disease due to a mutation in the X-linked inhibitor of apoptosis gene in a 15-month old boy. The patient successfully underwent a hematopoietic progenitor cell transplant.57 Discovery of BAV causing genes will also help develop genotype–phenotype correlations and open the way to personalized treatment. For example, knowledge of which BAV genotype might be predictive of valvulo-vascular complications will decrease unnecessary interventions such as unnecessary aortic aneurism repair surgeries while pointing to individuals who require closer cardiovascular monitoring. Therefore, it is important to accelerate the search for new causative genes that will allow for genetic screening as well as a better understanding of the molecular basis of BAV formation and degeneration. This holds great promise for secondary prevention and might open new therapeutic avenues.
Acknowledgment
Work in our lab is supported by the Canadian Institutes of Health Research, the Heart and Stroke Foundation of Canada, and the McCain Foundation.
Disclosure
The authors report no conflicts of interest in this work.
References
Tam PP, Parameswaran M, Kinder SJ, Weinberger RP. The allocation of epiblast cells to the embryonic heart and other mesodermal lineages: the role of ingression and tissue movement during gastrulation. Development. 1997;124(9):1631–1642. | |
Hinton RB Jr, Lincoln J, Deutsch GH, et al. Extracellular matrix remodeling and organization in developing and diseased aortic valves. Circ Res. 2006;98(11):1431–1438. | |
Restivo A, Piacentini G, Placidi S, Saffirio C, Marino B. Cardiac outflow tract: a review of some embryogenetic aspects of the conotruncal region of the heart. Anat Rec A Discov Mol Cell Evol Biol. 2006;288(9):936–943. | |
Okamoto N, Akimoto N, Hidaka N, Shoji S, Sumida H. Formal genesis of the outflow tracts of the heart revisited: previous works in the light of recent observations. Congenit Anom (Kyoto). 2010;50(3):141–158. | |
Anderson RH, Webb S, Brown NA, Lamers W, Moorman A. Development of the heart: (2) septation of the atriums and ventricles. Heart. 2003;89(8):949–958. | |
Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. First of two parts. N Engl J Med. 2000;342(5):334–342. | |
LaHaye S, Lincoln J, Garg V. Genetics of valvular heart disease. Curr Cardiol Rep. 2014;16(6):487. | |
Ward C. Clinical significance of the bicuspid aortic valve. Heart. 2000;83(1):81–85. | |
Padang R, Bannon PG, Jeremy R, Semsarian C, Richmond DR, Vallely M. The genetic and molecular basis of bicuspid aortic valve associated thoracic aortopathy: a link to phenotype heterogeneity. Ann Cardiothorac Surg. 2013;2(1):83–91. | |
Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. J Am Coll Cardiol. 2004;44(1):138–143. | |
Siu SC, Silversides CK. Bicuspid aortic valve disease. J Am Coll Cardiol. 2010;55(25):2789–2800. | |
Friedman T, Mani A, Elefteriades JA. Bicuspid aortic valve: clinical approach and scientific review of a common clinical entity. Expert Rev Cardiovasc Ther. 2008;6(2):235–248. | |
Fernández B, Durán AC, Fernández-Gallego T, et al. Bicuspid aortic valves with different spatial orientations of the leaflets are distinct etiological entities. J Am Coll Cardiol. 2009;54(24):2312–2318. | |
Yuan S, Jing H. The bicuspid aortic valve and related disorders review article. Sao Paulo Med J. 2010;128(5):296–301. | |
Homme JL, Aubry M-C, Edwards WD, et al. Surgical pathology of the ascending aorta: a clinicopathologic study of 513 cases. Am J Surg Pathol. 2006;30(9):1159–1168. | |
Della Corte A, Bancone C, Quarto C, et al. Predictors of ascending aortic dilatation with bicuspid aortic valve: a wide spectrum of disease expression. Eur J Cardiothorac Surg. 2007;31(3):397–404; discussion 404–405. | |
Choudhury N, Bouchot O, Rouleau L, et al. Local mechanical and structural properties of healthy and diseased human ascending aorta tissue. Cardiovasc Pathol. 2009;18(2):83–91. | |
Aboulhosn J, Child JS. Left ventricular outflow obstruction: subaortic stenosis, bicuspid aortic valve, supravalvar aortic stenosis, and coarctation of the aorta. Circulation. 2006;114(22):2412–2422. | |
Hahn RT, Roman MJ, Mogtader AH, Devereux RB. Association of aortic dilation with regurgitant, stenotic and functionally normal bicuspid aortic valves. J Am Coll Cardiol. 1992;19(2):283–288. | |
Fedak PW, de Sa MP, Verma S, et al. Vascular matrix remodeling in patients with bicuspid aortic valve malformations: implications for aortic dilatation. J Thorac Cardiovasc Surg. 2003;126(3):797–806. | |
Collins MJ, Dev V, Strauss BH, Fedak PW, Butany J. Variation of the histopathological features of patients with ascending aortic aneurysms: a study of 111 surgically excised cases. J Clin Pathol. 2008;61(4):519–523. | |
Bauer M, Pasic M, Meyer R, et al. Morphometric analysis of aortic media in patients with bicuspid and tricuspid aortic valve. Ann Thorac Surg. 2002;74(1):58–62. | |
Schievink WI, Raissi SS, Maya MM, Velebir A. Screening for intracranial aneurysms in patients with bicuspid aortic valve. Neurology. 2010; 74(18):1430–1433. | |
Tao G, Kotick JD, Lincoln J. Heart valve development, maintenance, and disease: the role of endothelial cells. Curr Top Dev Biol. 2012;100: 203–232. | |
Sabet HY, Edwards WD, Tazelaar HD, Daly RC. Congenitally bicuspid aortic valves: a surgical pathology study of 542 cases (1991 through 1996) and a literature review of 2,715 additional cases. Mayo Clin Proc. 1999;74(1):14–26. | |
Sonoda M, Takenaka K, Uno K, Ebihara A, Nagai R. A larger aortic annulus causes aortic regurgitation and a smaller aortic annulus causes aortic stenosis in bicuspid aortic valve. Echocardiography. 2008;25(3):242–248. | |
Isner JM, Chokshi SK, DeFranco A, Braimen J, Slovenkai GA. Contrasting histoarchitecture of calcified leaflets from stenotic bicuspid versus stenotic tricuspid aortic valves. J Am Coll Cardiol. 1990;15(5):1104–1108. | |
Ladich E, Nakano M, Carter-Monroe N, Virmani R. Pathology of calcific aortic stenosis. Future Cardiol. 2011;7(5):629–642. | |
Bailey MT, Pillarisetti S, Xiao H, Vyavahare NR. Role of elastin in pathologic calcification of xenograft heart valves. J Biomed Mater Res A. 2003;66(1):93–102. | |
Rajamannan NM, Evans FJ, Aikawa E, et al. Calcific aortic valve disease: not simply a degenerative process: a review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: calcific aortic valve disease-2011 update. Circulation. 2011;124(16):1783–1791. | |
Mohamed SA, Aherrahrou Z, Liptau H, et al. Novel missense mutations (p.T596M and p.P1797H) in NOTCH1 in patients with bicuspid aortic valve. Biochem Biophys Res Commun. 2006;345(4):1460–1465. | |
Foffa I, Ait Alì L, Panesi P, et al. Sequencing of NOTCH1, GATA5, TGFBR1 and TGFBR2 genes in familial cases of bicuspid aortic valve. BMC Med Genet. 2013;14:44. | |
Garg V, Muth AN, Ransom JF, et al. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437(7056):270–274. | |
Tan HL, Glen E, Töpf A, et al. Nonsynonymous variants in the SMAD6 gene predispose to congenital cardiovascular malformation. Hum Mutat. 2012;33(4):720–727. | |
Lee TC, Zhao YD, Coutman DW, Stewart DJ. Abnormal aortic valve development in mice lacking endothelial nitric oxide synthase. Circulation. 2000;101(20):2345–2348. | |
Bosse K, Hans CP, Zhao N, et al. Endothelial nitric oxide synthase regulates Notch1 in aortic valve disease. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J Mol Cell Cardiol. 2013;60:27–35. | |
Laforest B, Andelfinger G, Nemer M. Loss of Gata5 in mice leads to bicuspid aortic valve. J Clin Invest. 2011;121(7):2876–2887. | |
Padang R, Bagnall RD, Richmond DR, Bannon PG, Semsarian C. Rare non-synonymous variations in the transcriptional activation domains of GATA5 in bicuspid aortic valve disease. J Mol Cell Cardiol. 2012; 53(2):277–281. | |
Bonachea EM, Chang SW, Zender G, et al. Rare GATA5 sequence variants identified in individuals with bicuspid aortic valve. Pediatr Res. 2014;76(2):211–216. | |
Wang J, Sridurongrit S, Dudas M, et al. Atrioventricular cushion transformation is mediated by ALK2 in the developing mouse heart. Dev Biol. 2005;286(1):299–310. | |
Thomas PS, Sridurongrit S, Ruiz-Lozano P, Kaartinen V. Deficient signaling via Alk2 (Acvr1) leads to bicuspid aortic valve development. PLoS One. 2012;7(4):e35539. | |
Makki N, Capecchi MR. Cardiovascular defects in a mouse model of HOXA1 syndrome. Hum Mol Genet. 2012;21(1):26–31. | |
Biben C, Weber R, Kesteven S, et al. Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2-5. Circ Res. 2000;87(10):888–895. | |
Sans-Coma V, Fernández B, Durán AC, et al. Fusion of valve cushions as a key factor in the formation of congenital bicuspid aortic valves in Syrian Hamsters. Anat Rec. 1996;244(4):490–498. | |
Andelfinger G, Tapper AR, Welch RC, Vanoye CG, George AL Jr, Benson DW. KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am J Hum Genet. 2002;71(3):663–668. | |
Martin LJ, Ramachandran V, Cripe LH, et al. Evidence in favor of linkage to human chromosomal regions 18q, 5q and 13q for bicuspid aortic valve and associated cardiovascular malformations. Hum Genet. 2007;121(2):275–284. | |
Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, cranofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37(3):275–281. | |
Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337–339. | |
Tischfield MA, Bosley TM, Salih MA, et al. Homozygous HOXA1 mutations disrupt human brainstream, inner ear, cardiovascular and cognitive development. Nat Genet. 2005;37(10):1035–1037. | |
Bondy C, Bakalov VK, Cheng C, Olivieri L, Rosing DR, Arai AE. Bicuspid aortic valve and aortic coarctation are linked to deletion of the X chromosome short arm in Turner syndrome. J Med Genet. 2013; 50(10):662–665. | |
Michelena HI, Desjardins VA, Avierinos JF, et al. Natural history of asymptomatic patients with normally functioning or minimally dysfunctional bicuspid aortic valve in the community. Circulation. 2008;117(21):2776–2784. | |
Chan KL, Teo K, Dumesnil JG, Ni A, Tam J, ASTRONOMER Investigators. Effect of Lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation. 2010;121(2):306–314. | |
Norton N, Li D, Rieder MJ, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011;88: 273–282. | |
Musunuru K, Pirruccello JP, Do R, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010;363:2220–2227. | |
Rios J, Stein E, Shendure J, Hobbs HH, Cohen JC. Identification by whole genome resequencing of gene defect responsible for severe hypercholesterolemia. Hum Mol Genet. 2011;19:4313–4318. | |
Rabbani B, Tekin M, Mahdieh N. The promise of whole-exome sequencing in medical genetics. J Hum Genet. 2014;59(1):5–15. | |
Worthey EA, Mayer AN, Syverson GD, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011;13:255–262. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.