")
Back to Journals » Advances and Applications in Bioinformatics and Chemistry » Volume 7
The hemagglutinin of the influenza A(H1N1)pdm09 is mutating towards stability
Authors Castelán-Vega JA, Magaña-Hernández A, Jiménez-Alberto A, Ribas-Aparicio RM
Received 6 June 2014
Accepted for publication 1 July 2014
Published 3 October 2014 Volume 2014:7 Pages 37—44
DOI https://doi.org/10.2147/AABC.S68934
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Juan A Castelán-Vega, Anastasia Magaña-Hernández, Alicia Jiménez-Alberto, Rosa María Ribas-Aparicio
Departamento de Microbiología, Escuela Nacional de Ciencias Biológicas del Instituto Politécnico Nacional, Mexico City, Mexico
Abstract: The last influenza A pandemic provided an excellent opportunity to study the adaptation of the influenza A(H1N1)pdm09 virus to the human host. Particularly, due to the availability of sequences taken from isolates since the beginning of the pandemic until date, we could monitor amino acid changes that occurred in the hemagglutinin (HA) as the virus spread worldwide and became the dominant H1N1 strain. HA is crucial to viral infection because it binds to sialidated cell-receptors and mediates fusion of cell and viral membranes; because antibodies that bind to HA may block virus entry to the cell, this protein is subjected to high selective pressure. Multiple alignment analysis of sequences of the HA from isolates taken since 2009 to date allowed us to find amino acid changes that were positively selected as the pandemic progressed. We found nine changes that became prevalent: HA1 subunits D104N, K166Q, S188T, S206T, A259T, and K285E; and HA2 subunits E47K, S124N, and E172K. Most of these changes were located in areas involved in inter- and intrachain interactions, while only two (K166Q and S188T) were located in known antigenic sites. We conclude that selective pressure on HA was aimed to improve its functionality and hence virus fitness, rather than at avoidance of immune recognition.
Keywords: influenza A, hemagglutinin evolution, virus fitness
Introduction
A novel influenza A(H1N1) virus of swine origin emerged in Mexico and in the United States in March of 2009. The outbreak and spread of the first influenza pandemic of the 21st century challenged licensed vaccine manufacturers to rapidly mobilize and generate a prophylactic vaccine.1 Influenza pandemics occur when an influenza virus with a hemagglutinin (HA), against which there was little or no existing immunity, emerges in the human population and efficiently transmits from human to human.2
The HA protein is a trimer, with each monomer composed of a heavy (HA1) (~40 kDa) and a light (HA2) (~20 kDa) chain cleaved from a single precursor (HA0).3,4 A single disulfide bridge and noncovalent interactions hold HA1 and HA2 together. HA1 forms a more variable globular head region, which contains the receptor binding sites, while the membrane anchoring HA2 contains a relatively conserved fusion peptide. Many of the HA-specific neutralizing antibodies target any one of the antigenic sites (Sa, Sb, Ca1, Ca2, and Cb) that are located at the globular head of HA1.5 HA activity is crucial during the initial stages of cell infection because it is in charge of sialic acid receptor binding and, after virus uptake into endosomes, of fusion of viral and cell membranes.4,6 Modifications in the HA glycoprotein of the influenza A virus are believed to be a catalyst for previous world pandemics. These includes reassortment between cocirculating animal and human viruses and mutations in the HA, which grant better transmissibility between hosts. The pandemic influenza A virus from 2009, A(H1N1)pdm09, originated from reassortment among three cocirculating swine-like and avian-like viruses; hence, its HA comes from a swine virus origin.7 Additionally, HA can undergo rapid evolution, due to its plasticity and to the error-prone nature of the virus’ RNA-dependent RNA polymerase, which leads to a gradual change in genetic sequences by point mutations (antigenic drift), and can result in various phenotypic changes, including changes in antigenicity, receptor preference (and thereby cell tropism), altered fusion functionality, and virulence. This rapid and large change can give rise to new strains very different from any virus encountered previously, with the potential to cause a pandemic.8
Since the spring of 2009, the influenza virus A(H1N1)pdm09 has demonstrated many diverse adaptive mutations in the HA protein, which have been reported in the gene databases. An example of one such mutation that has been studied is the D222G mutation, which has been shown to alter receptor specificity and tropism, and to increase virulence.9–11 In contrast, for many mutations that have arisen, there exists little data reported other than epidemiology, phylogeny, and some limited antigenicity studies. Little is known about the effects these mutations have on the structure and function of the HA molecule.
In a previous study, we found that HA variants that proliferated as the pandemic progressed showed changes in antigenic sites but no significant alterations in ligand binding, while variants that declined had an increased ligand affinity. These findings reveal some of the pathways that this pandemic strain used to disseminate in the human population as a response to selective pressure, such as receptor recognition, increased infectivity, and evasion of the immune response.8
Viral isolates from 2009 to 2012 did not reveal any considerable changes in the antigenic properties of influenza A(H1N1)pdm09 viruses. Single amino acid substitutions displaying opposite effects on receptor binding (increased binding by S186P and S188T, and decreased binding by A137T and A200T) occurred in combinations (S186P or S188T together with A137T or A200T, respectively) that had no net effect on receptor binding. Thus, during the first 3 years of the pandemic influenza A(H1N1), HA mutations that increased receptor binding avidity, followed by the selection of compensatory mutations that restored binding to the original levels were presented.12
Recent surveillance data has revealed the emergence of a prominent mutation, E47K (HA2 numbering), in the HA2 stalk region of influenza A(H1N1)pdm09 isolates.13 This mutation determines the threshold pH of fusion and affects the thermal stability of HA due to intermonomer interactions between HA2 and HA1 in the stalk region of HA. Furthermore, variants with E47K are more infectious in ferrets, underscoring the importance of this mutant for viral host adaptation and fitness.14
In this study we found that the HA is steadily evolving and that rather than antigenic drift, mutations are being introduced in regions that confer structural stability. Moreover, we describe two mutations in antigenic sites that may cause antigenic drift in the virus as more mutations are incorporated in these sites. This makes monitoring of HA variants an imperative measure, to be able to modify vaccine composition according to current circulating strains. In addition, mutations that increase stability but have no effect in antigenicity are valuable to improve the development and manufacture of vaccines and diagnostic reagents for influenza.
Material and methods
HA influenza sequences
A total of 7,479 full-length amino acid sequences of the HA from the influenza A(H1N1)pdm09 virus were downloaded from the OpenFlu database (http://openflu.vital-it.ch/browse.php).15 To determine amino acid changes that occurred as the influenza pandemic evolved, sequences were grouped semiannually, from 2009 to the first semester of 2014.
Multiple-sequence alignments
Each group of sequences was aligned with MAFFT version 7 (http://mafft.cbrc.jp/alignment/software/), using the progressive method (fast Fourier transform – normalized similarity matrix [FFT-NS-1]) for a fast analysis.16 The consensus sequence for each alignment was calculated with BioEdit 7.1.9. (http://www.mbio.ncsu.edu/bioedit/bioedit.html).17 Positional frequencies were calculated, and a consensus was considered when at least 51% of the amino acids were identical at a given position. The resulting consensus sequences (nine sequences) were then aligned with MAFFT.
Effect of mutation on protein stability
To determine the effect of the selected mutation on protein stability we used the iStable server (http://predictor.nchu.edu.tw/iStable/). iStable integrates the results from several stability prediction programs, and with the support of a support vector machine, calculates the effect of point mutations on protein stability.18 Stability calculations were done with the HA structure (Protein Data Bank [PDB] code 3LZG) at 37°C and pH =7.0.
Visualization of mutations
Selected mutations were mapped in the structure of the HA from the influenza A/California/4/2009 H1N1 virus (PDB: 3LZG) with PyMOL version 1.5.0.4 (http://www.pymol.org).19 Figures were prepared with the “Mutagenesis” wizard and the ray trace renderer of Pymol.
Results
Evolution of amino acid sequences in the HA of the influenza A(H1N1)pdm09 virus
The identification of the amino acids that have been positively selected from the origin of the influenza pandemics of 2009 to present was done in a two-stage sequence alignment procedure. A total of 7,479 sequences were grouped by semester (Table 1); each group was then aligned, and the consensus sequence was calculated. Finally, all consensus sequences were aligned, using the sequence from early 2009 as a reference to detect the mutations as time proceeded (Figure 1). More than half of the sequences were from samples collected in 2009, given the urgency to characterize the virus at the beginning of the pandemic. In comparison with other groups, there were very few sequences from late 2011 and 2012; as can be seen in Figure 1, although the imbalance in the number of sequences did not alter the mutation tendencies. A total of nine mutations have become prevalent since 2009 and currently are the dominant variants (Table 2). The mutations located in HA1 (globular portion of HA) are D104N, K166Q, S188T, S206T, A259T, and K285E, while the mutations located in HA2 (stem portion of HA) are E47K, S124N, and E172K (Figure 2).
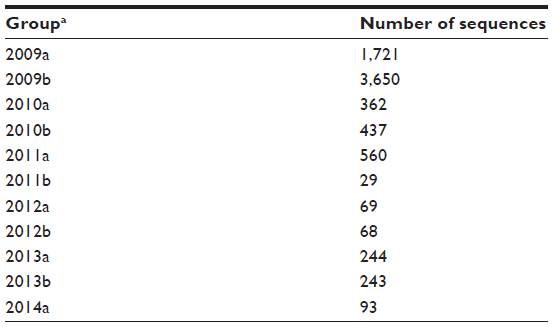 | Table 1 Amino acid sequences of the influenza A(H1N1)pdm09 hemagglutinin from 2009 until 2014 |
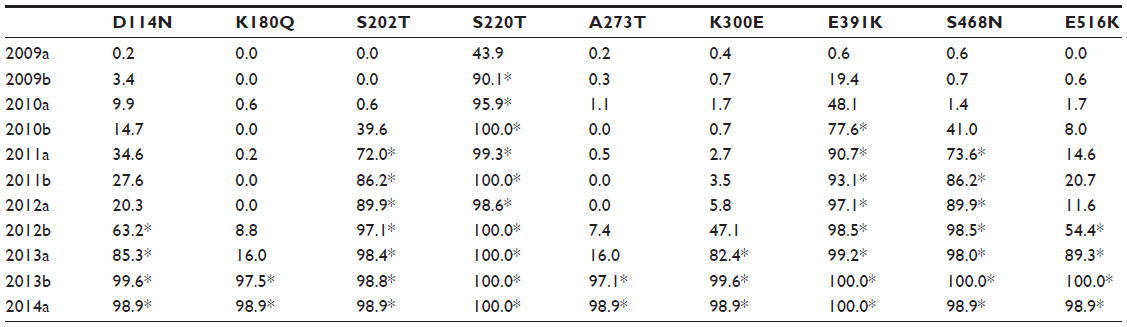 | Table 2 Frequency of appearance of the selected mutants according to the time of isolation |
 | Figure 1 Evolution of the amino acid sequence of the HA from the influenza A(H1N1)pdm09 virus. |
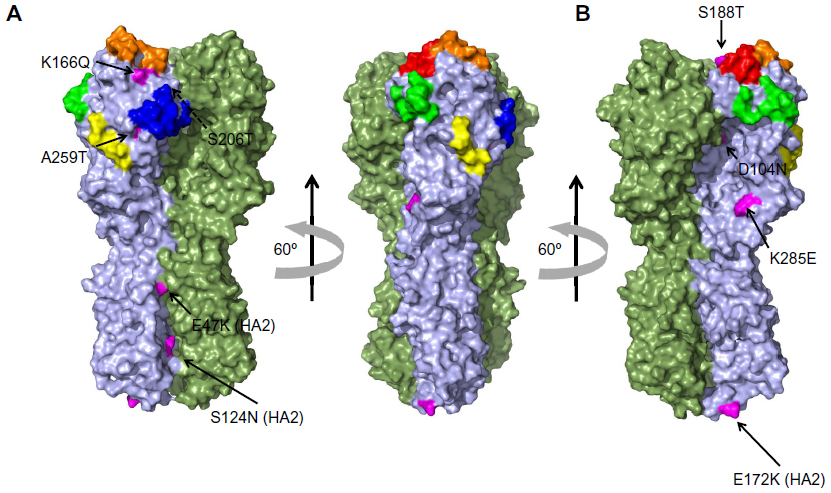 | Figure 2 Location of the mutants in the structure of the HA from the A/California/4/2009 H1N1 strain. |
Mutations K166Q and S188T are likely the result of antigenic drift because they are located in known antigenic sites (Sa and Sb, respectively) (Figure 2) and their side chains are surface-exposed (Figure 3). Both mutations are located near the receptor binding site. Additionally, the functional group of glutamine in K166Q can interact with the backbone of S126 (Figure 3A); S188T has its side chain exposed to the solvent and does not interact with other amino acids (Figure 3B). Mutations D104N, S206T, and A259T are located in the globular part of HA. The location of these mutations causes us to assume they have a role in the structural stability of HA. The change D104N generates interchain interactions with the backbone of L73 (Figure 4A), while mutations S206T and A259T introduce a bulkier functional group within the hydrophobic pocket where they are located (Figure 4B and C). Free-energy analysis showed that D104N and A259T have a structural stabilizing effect, whereas S206T is slightly destabilizing (Table 3).
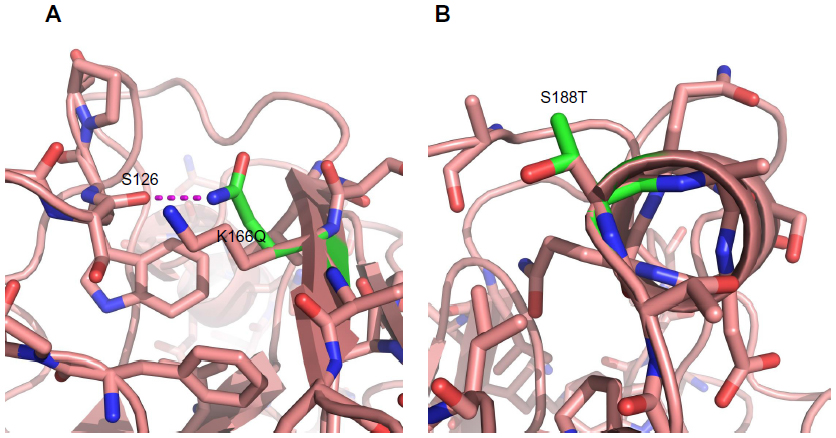 | Figure 3 Mutations K166Q and S188T can lead to antigenic drift. (A) Amino acids neighboring mutation K166Q; (B) amino acids neighboring mutation K166Q. Notes: Wild-type amino acids are shown in pink; mutations are shown in green. Potential interactions are shown as a dashed magenta line. Positions 166 and 188 are located in antigenic sites Sa and Sb, respectively, and are exposed to the solvent. |
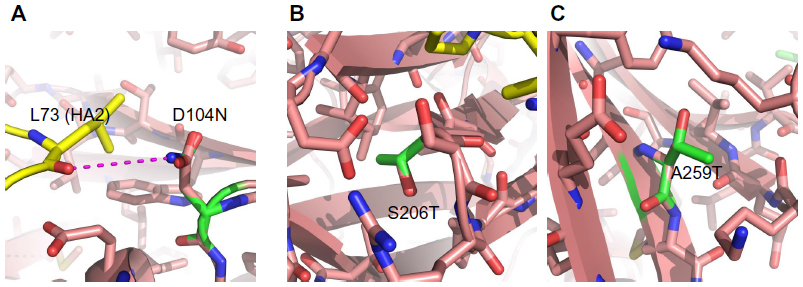 | Figure 4 Mutations D104N, S206T, and A259T impact the stability of HA. (A) Amino acids neighboring mutation D104N; (B) amino acids neighboring mutation S206T; (C) amino acids neighboring mutation A259T. |
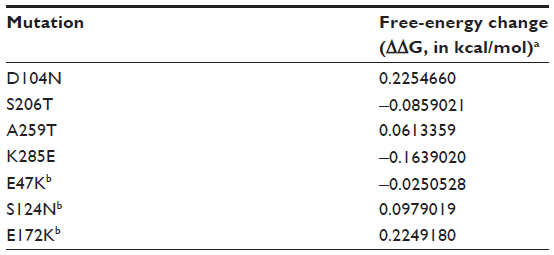 | Table 3 Folding free-energy changes for selected mutations |
Position 285 is located in a basic patch in the stalk region of HA. K285 interacts with the backbone of I297; in E285, this interaction is lost, but instead, the glutamic acid can interact with H47 and H298 (Figure 5A); loss of this interaction has a destabilizing effect on HA according to the free-energy analysis (Table 3). Mutation E47K (HA2) resides near the fusion peptide. In the wild-type structure, E47 interacts with Y110 (HA2); however, in E47K the mutation abolishes interactions with Y110 and creates interactions with E31 (HA1), which is located near the fusion peptide (Figure 5B). According to the free-energy analysis, this mutation is destabilizing (Table 3); however, interactions with E31 can still stabilize the structure by hampering the transition of HA to the fusogenic state.
 | Figure 5 Stabilizing effect of mutations K285E and E47K. (A) Amino acids neighboring mutation K285E; (B) amino acids neighboring mutation E47K. |
Mutation S124N creates intrachain interactions with a nearby arginine (R123) and interchain interactions with E132 (Figure 6A). Finally, mutation E172K is located at the base of the HA (Figure 6B). K172 makes contact with N169, which could stabilize the bottom of the HA stem. Both mutations are stabilizing according to the free-energy analysis (Table 3).
 | Figure 6 Stabilizing effect of mutations S124N and E172K. These mutations are located in the HA2 subunit. (A) Amino acids neighboring mutation S124N; (B) amino acids neighboring mutation E172K. |
Discussion
Surface proteins of influenza viruses are subjected to a high selective pressure because they have to avoid recognition by the host’s immune system as well as improve virus fitness by modulating their activity according to the nature of the host. One of the main mechanisms of variation is antigenic drift, which is the introduction of mutations in known antigenic sites as a response to recognition by the immune system.20,21 The influenza A(H1N1)pdm09 virus has been an excellent model to study antigenic drift because since its appearance in 2009, laboratories worldwide have contributed to better knowledge of this virus by releasing a huge amount of sequence data from isolates all around the world. In this study, we analyzed this data to detect amino acid changes in the HA from the influenza A(H1N1)pdm09 virus that could have an impact in future prevention and diagnostic strategies against influenza. The mutations that became prevalent as the pandemic advanced can be grouped as mutations that can cause antigenic drift (K166Q and S188T), and mutations that can increase HA stability through generation of favorable inter- and intramonomer interactions (of D104N and A259T, in HA1; and of HA2: E47K, S124N, and E172K, in K285E).
Because of its recent appearance, K166Q is a mutation that requires close examination, particularly regarding vaccine efficacy. This mutant was first described in isolates from Peru, and our analysis showed that its location within the antigenic site Sa could allow the virus to escape from the immune response elicited by current vaccines.22 Moreover, this change has been described in escape mutants of the HA from 1918.23 Interestingly, despite that mutant S188T is located in antigenic site Sb and has become prevalent since 2009,24 it appears to have had little impact in antigenic drift because no evidence has been reported to date of reduced vaccine efficacy caused by this mutant. This mutant increases receptor-binding avidity of HA,12 and its increased prevalence reflects the continuous adaptation of the influenza virus. Evidently, the presence of several mutations in the antigenic sites can lead to a rapid decrease in vaccine efficacy if changes in vaccine formulated are not introduced on time.
Since the first manufacture of lots of vaccines, it has been reported that the HA from the influenza A(H1N1)pdm09 virus is intrinsically unstable.25 This instability leads to increased proteolytic cleavage,1 increased pH of fusion, and dissociation into monomers.26 At the beginning of the pandemic, this instability was advantageous to the virus, but as the pandemic progressed, more stable variants originated. Our results suggest that many mutations arising since 2009 have likely stabilized the structure of HA by introducing favorable interactions within and between monomers. Single point mutations seem to have little impact on HA stability,26 but recruitment of several mutations could have a great impact on the overall stability of HA. The only single mutation that has been shown to increase HA stability is E47K. Despite that our free-energy analysis suggested a destabilizing effect, the contrary has been corroborated experimentally.14 We suggest that interactions that are lost with the mutation are compensated by interactions with other amino acids, leading to a more stable HA. In the case of mutant K285E, the replacement of a basic amino acid by an acidic one masks the basic patch located in this area. As in the case of E47K, while the free-energy analysis suggests a destabilizing effect, interaction with nearby histidine residues could obstruct the transition of HA to the fusogenic state when the virus is endocytosed, in this way increasing the stability of HA.
Conclusion
The HA from influenza A(H1N1)pdm09 virus is steadily evolving towards a more stable and antigenically distinct protein. This trend indicates that current influenza vaccines are likely to have a decreased efficacy if the same strain is used in future vaccine formulations. Some of the mutations described here can be considered for inclusion, to improve vaccine manufacturing, given the stabilizing effect that they have on the influenza HA.
Author contributions
JAC-V and RMR-A contributed equally to the original concept and design described in the manuscript and supervised the overall project, providing essential guidance. AM-H and AJ-A participated in the data collection, and analysis and interpretation of the data. All of the authors participated in drafting of the manuscript and approved the final version.
Acknowledgments
Authors are grateful for support of this work from the Instituto Politécnico Nacional, in the form of grants (numbers SIP-20143977, SIP-20141506, and SIP-20144063) and fellowships (Comisión de Operación y Fomento de Actividades Académicas del Instituto Politécnico Nacional [COFAA], Estímulo al Desempeño de los Investigadores [EDI], Estímulo al Desempeño Docente [EDD]).
Disclosure
The author reports no conflicts of interest in this work.
References
Feshchenko E, Rhodes DG, Felberbaum R, et al. Pandemic influenza vaccine: characterization of A/California/07/2009 (H1N1) recombinant hemagglutinin protein and insights into H1N1 antigen stability. BMC Biotechnol. 2012;12:77. | |
Garten RJ, Davis CT, Russell CA, et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science. 2009;325(5937):197–201. | |
Ambroggio XI, Dommer J, Gopalan V, Dunham EJ. HASP server: a database and structural visualization platform for comparative models of influenza A hemagglutinin proteins. BMC Bioinformatics. 2013;14:197. | |
Harris AK, Meyerson JR, Matsuoka Y, et al. Structure and accessibility of HA trimers on intact 2009 H1N1 pandemic influenza virus to stem region-specific neutralizing antibodies. Proc Natl Acad Sci U S A. 2013;110(12):4592–4597. | |
Li OT, Poon LL. One step closer to universal influenza epitopes. Expert Rev Anti Infect Ther. 2009;7(6):687–690. | |
Russell RJ, Kerry PS, Stevens DJ, et al. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc Natl Acad Sci U S A. 2008;105(46):17736–17741. | |
Smith GJ, Vijaykrishna D, Bahl J, et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature. 2009;459(7250):1122–1125. | |
Jiménez-Alberto A, Alvarado-Facundo E, Ribas-Aparicio RM, Castelán-Vega JA. Analysis of adaptation mutants in the hemagglutinin of the influenza A(H1N1)pdm09 virus. PLoS One. 2013;8(7):e70005. | |
Chutinimitkul S, Herfst S, Steel J, et al. Virulence-associated substitution D222G in the hemagglutinin of 2009 pandemic influenza A(H1N1) virus affects receptor binding. J Virol. 2010;84(22):11802–11813. | |
Wedde M, Wählisch S, Wolff T, Schweiger B. Predominance of HA-222D/G polymorphism in influenza A(H1N1)pdm09 viruses associated with fatal and severe outcomes recently circulating in Germany. PLoS One. 2013;8(2):e57059. | |
Vazquez-Perez JA, Isa P, Kobasa D, et al. A (H1N1) pdm09 HA D222 variants associated with severity and mortality in patients during a second wave in Mexico. Virol J. 2013;10:41. | |
de Vries RP, de Vries E, Martínez-Romero C, et al. Evolution of the hemagglutinin protein of the new pandemic H1N1 influenza virus: maintaining optimal receptor binding by compensatory substitutions. J Virol. 2013;87(24):13868–13877. | |
Maurer-Stroh S, Lee RT, Eisenhaber F, Cui L, Phuah SP, Lin RT. A new common mutation in the hemagglutinin of the 2009 (H1N1) influenza A virus. PLoS Curr. 2010;2:RRN1162. | |
Cotter CR, Jin H, Chen Z. A single amino acid in the stalk region of the H1N1pdm influenza virus HA protein affects viral fusion, stability and infectivity. PLoS Pathog. 2014;10(1):e1003831. | |
Liechti R, Gleizes A, Kuznetsov D, et al. OpenFluDB, a database for human and animal influenza virus. Database (Oxford). 2010;2010:baq004. | |
Katoh K, Toh H. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 2008;9(4):286–298. | |
Hall T. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series. 1999;41:95–98. | |
Chen CW, Lin J, Chu YW. iStable: off-the-shelf predictor integration for predicting protein stability changes. BMC Bioinformatics. 2013;14(Suppl 2):S5. | |
Schrödinger LLC. The PyMOL Molecular Graphics System, Version 1.5.0.4. Available from: http://www.pymol.org/citing. Accessed September 24, 2014. | |
Klein EY, Serohijos AW, Choi JM, Shakhnovich EI, Pekosz A. Influenza A H1N1 pandemic strain evolution – divergence and the potential for antigenic drift variants. PLoS One. 2014;9(4):e93632. | |
García-Barreno B, Delgado T, Benito S, et al. Characterization of an enhanced antigenic change in the pandemic 2009 H1N1 influenza virus haemagglutinin. J Gen Virol. 2014;95(Pt 5):1033–1042. | |
Padilla C, Condori F, Huaringa M, et al. Full genome analysis of influenza A(H1N1)pdm09 virus isolated from Peru, 2013. Genome Announc. 2014;2(2). | |
Krause JC, Tsibane T, Tumpey TM, et al. Epitope-specific human influenza antibody repertoires diversify by B cell intraclonal sequence divergence and interclonal convergence. J Immunol. 2011;187(7):3704–3711. | |
El Moussi A, Ben Hadj Kacem MA, Pozo F, et al. Genetic diversity of HA1 domain of heammaglutinin gene of influenza A(H1N1)pdm09 in Tunisia. Virol J. 2013;10:150. | |
Farnsworth A, Cyr TD, Li C, Wang J, Li X. Antigenic stability of H1N1 pandemic vaccines correlates with vaccine strain. Vaccine. 2011;29(8):1529–1533. | |
Yang H, Chang JC, Guo Z, et al. Structural stability of influenza A(H1N1)pdm09 virus hemagglutinins. J Virol. 2014;88(9):4828–4838. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.