")
Back to Journals » The Application of Clinical Genetics » Volume 8
The genetic basis of familial adenomatous polyposis and its implications for clinical practice and risk management
Authors Leoz M, Carballal S, Moreira L, Ocaña T, Balaguer F
Received 23 January 2015
Accepted for publication 27 February 2015
Published 16 April 2015 Volume 2015:8 Pages 95—107
DOI https://doi.org/10.2147/TACG.S51484
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Maria Liz Leoz, Sabela Carballal, Leticia Moreira, Teresa Ocaña, Francesc Balaguer
Department of Gastroenterology, Hospital Clínic, Centro de Investigación Biomédica en Red en Enfermedades Hepáticas y Digestivas (CIBERehd), Institut d'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Catalonia, Spain
Abstract: Familial adenomatous polyposis (FAP) is an inherited disorder that represents the most common gastrointestinal polyposis syndrome. Germline mutations in the APC gene were initially identified as responsible for FAP, and later, several studies have also implicated the MUTYH gene as responsible for this disease, usually referred to as MUTYH-associated polyposis (MAP). FAP and MAP are characterized by the early onset of multiple adenomatous colorectal polyps, a high lifetime risk of colorectal cancer (CRC), and in some patients the development of extracolonic manifestations. The goal of colorectal management in these patients is to prevent CRC mortality through endoscopic and surgical approaches. Individuals with FAP and their relatives should receive appropriate genetic counseling and join surveillance programs when indicated. This review is focused on the description of the main clinical and genetic aspects of FAP associated with germline APC mutations and MAP.
Keywords: colorectal cancer, familial adenomatous polyposis, MAP, APC, MUTYH
Introduction
Familial adenomatous polyposis (FAP) refers to an inherited syndrome characterized by the development of multiple adenomas in the colorectum, a high risk of colorectal cancer (CRC), and the existence of extracolonic manifestations. Germline adenomatous polyposis coli (APC) mutations were firstly described in 19911,2 as causing FAP with an autosomal dominant pattern of inheritance. Since then, a great body of evidence has been generated, including pathophysiology, genetics, clinical phenotype, and prevention. In 2002, another polyposis gene was identified, the mutY homolog (MUTYH) gene, in which biallelic mutations cause an autosomal recessive pattern of inheritance, usually referred to as MUTYH-associated polyposis (MAP).3 This review is focused on the description of the key clinical and genetics aspects of FAP associated with germline APC mutations and MAP.
APC-associated FAP
APC-FAP (OMIM #175100) is an autosomal-dominant inherited disease characterized by the development of multiple adenomas throughout the colorectum.4 It represents less than 1% of all CRC cases, and is the most common gastrointestinal polyposis syndrome, with an incidence of one case per 10,000 subjects.5
APC gene
APC is a tumor-suppressor gene that is located on chromosome 5q21-q22. The gene has 15 exons, with exon 15 individually representing >75% of the coding sequence and the most common target of both germline and somatic mutations. The APC gene encodes a protein of 2,843 aminoacids (310 kDa) that plays an outright role in the Wnt signaling pathway.6 This multifunctional protein occurs in several isoforms within cells, containing several aminoacid motifs and domains allowing it to oligomerize, as well as interact with numerous other molecules (Figure 1A).7,8 The APC protein functions as a tumor suppressor by negatively regulating the β-catenin oncoprotein. The APC protein leads to ubiquitination and degradation of β-catenin; so in the absence of it, β-catenin accumulates in the nucleus and interacts with factors that up-regulate the transcription of genes involved in cell cycle entry, proliferation, differentiation, migration, apoptosis, and progression.9 In addition, APC stabilizes microtubules, leading to chromosomal stability.10 Inactivation of APC can lead to defective chromosome segregation and aberrant mitosis.
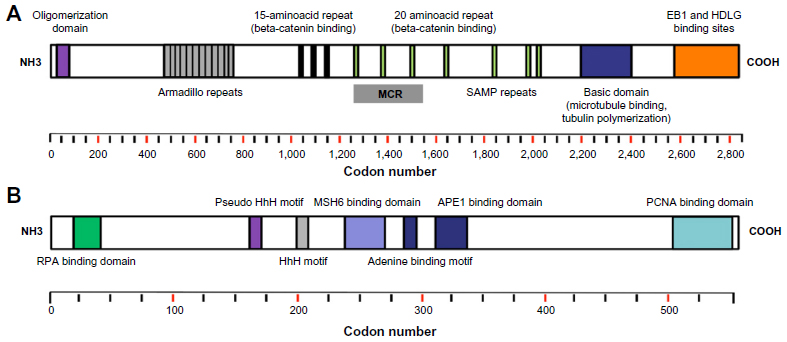 | Figure 1 The functional domains of the APC and MUTYH proteins. |
Germline mutations in the APC gene are responsible for most cases of FAP. Individuals with a germline APC mutation develop multiple adenomas as a result of inactivation of the remaining allele in the colorectum by additional somatic APC mutations or loss of heterozygosity (LOH) at this locus.
In a recent large cross-sectional study, APC mutations were found in 80% (95% CI 71%–87%) of individuals with more than 1,000 adenomas, 56% (95% CI 54%–59%) in those with 100–999 adenomas, 10% (95% CI 9%–11%) in those with 20–99 adenomas, and 5% (95% CI 4%–7%) in those with 10–19 adenomas.11 Inheritance of FAP is autosomal dominant although up to 25% of FAP patients carry de novo germline mutations.12,13
Mutational landscape
Since the identification of the APC gene, more than 1,100 unique likely pathogenic germline mutations have been reported (http://www.lovd.nl/apc). The majority of them represent truncating mutations, being nonsense mutations (28%), small insertions (10%) or small deletions (46%) that lead to a truncated protein.14 Although infrequent, there are also missense mutations (3%) and gross alterations (ie, single or multiexon deletions and duplications) (13%).14
- Truncation mutations: As mentioned earlier, the great majority of FAP-associated APC mutations lead to a truncated protein, mainly due to a frameshift or nonsense variant. The most common nonsense changes are C>T mutations,15 and the majority of germline mutations in APC occur in the 5′ half of the gene, leading to the elimination of most, if not all, of the 20-aminoacid repeats involved in regulating β-catenin levels and SAMP repeats involved in axin binding (Figure 1A).16,17
- Missense mutations: Although rare, more than 60 different missense variants of APC have been described in the literature as potentially pathogenic. The two missense variants most frequently reported are I1307K and E1317Q. The I1307K variant, present in 6% of all individuals of Ashkenazi Jewish ancestry, does not lead to a polyposis phenotype, but it carries an increased (10%–20%) lifetime risk of developing CRC.16 The E1317Q missense variant has also been associated with a moderate risk for colorectal adenomas and CRC.18
- Splicing mutations and gross alterations: Splice altering mutations and large deletions/duplications have also been described, and recent data suggest that especially gross alterations affecting the promoter of coding regions may have been underreported, accounting for up to 20% of FAP families.19–21
- De novo mutations and germline mosaicism: Most of the APC germline mutations are inherited, but can also occur de novo in a patient with no family history of the disease, representing between 11% and 25% of all FAP cases.22 The estimated rate of APC new mutations is between 4 and 9×106 mutations/gametes/generation,23 and equal susceptibility for mutagenesis during spermatogenesis and oogenesis has been described. A significant percentage of the de novo cases arise in the mosaic form, affecting only a subset of cells in the affected individual (it is estimated that one-fifth of the de novo cases of FAP are mosaic).24,25
- Mutational hotspots: The mutational hotspots in the APC gene are located in the 5′ part of exon 15 at codons 1,309 and 1,061, accounting for approximately 17% and 11% of all germline APC mutations, respectively. Because of the accumulation of mutations from codon 1,250 to 1,464, this region is termed the mutation cluster region (MCR) (Figure 1A).26
The type of germline mutation in the APC gene determines the nature of the second hit. If the germline mutation occurs between codons 1,194 and 1,392, then there is a strong association with allelic loss of APC as the second hit. If the germline mutation occurs outside this region, the second hit is most likely to induce a truncating mutation in the MCR.27
Phenotypes: classic and attenuated FAP (AFAP)
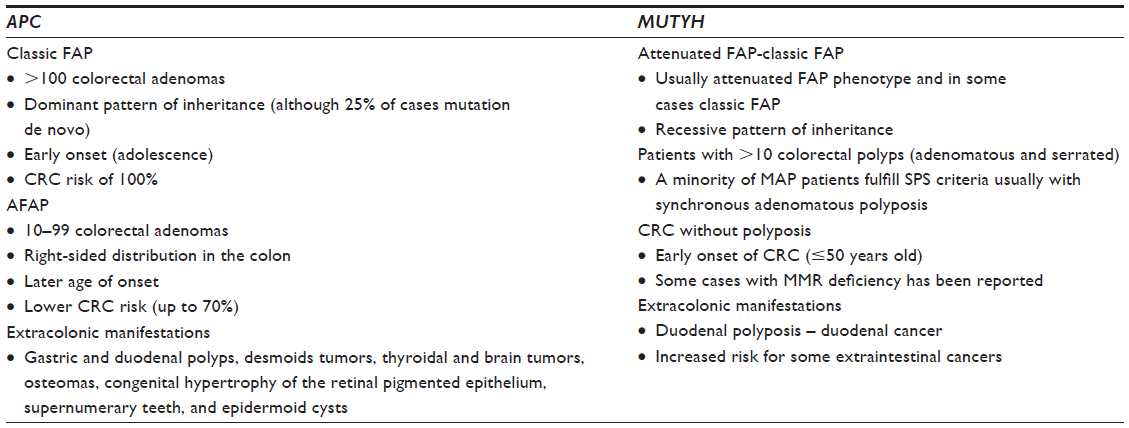
According to the number of polyps and the age of onset, two major phenotypes have been described for FAP (Table 1):
- Classic FAP: It is characterized by the presence of hundreds to thousands of adenomatous polyps throughout the colon and the rectum. At the time of adolescence, the polyps are usually identified in the recto-sigmoid as small polyps, and thereafter, increase in size and number. About half of FAP patients develop adenomas by 15 years of age and 95% by 35 years.28,29 CRC inevitably occurs at an earlier age than sporadic CRC (average age of 35 years), but only rarely before the age of 20 years.
- AFAP: It is a variant of FAP with a mild disease course, characterized by a reduced number of polyps (10–100), later age of onset, frequently right-sided distribution of polyps, and lower CRC risk (up to 70%). The clinical definition of AFAP is controversial and should be considered in any patient with 10–99 adenomas, although a precise diagnosis is often difficult in a single patient.11 APC-associated AFAP can mimic MAP or even sporadic polyp development. Examination of multiple family members can often determine the phenotype.
 | Table 1 Clinical phenotypes of APC-associated FAP and MUTYH-associated polyposis |
Extracolonic manifestations
In many FAP patients, extracolonic manifestations are present, including gastric and duodenal polyps, desmoid tumors (DT) thyroidal and brain tumors, osteomas, congenital hypertrophy of the retinal pigmented epithelium, supernumerary teeth, and epidermoid cysts.
Gastroduodenal polyps
The most common extracolonic manifestations in FAP patients are upper gastrointestinal polyps. They are located in the stomach, duodenum, and the periampullary region. Gastric polyps are usually benign fundic gland polyps (FGP), and occur in 20%–84% of FAP patients.30 Although FAP-associated FGPs have been traditionally thought of as nonneoplastic and usually do not require intervention, cases of high-grade dysplasia and gastric carcinoma arising from FGPs in FAP have been reported.31 Gastric adenomatous polyps represent around 10% of gastric polyps, and when they occur, they are most commonly located in the antrum. Despite the malignant potential of gastric dysplastic FGP and adenomas, gastric carcinoma is very rare in FAP patients (incidence <1%).31
After the colorectum, the duodenum is the second most common site of polyps in FAP patients. Duodenal adenomas arise in most patients with FAP with a lifetime risk of almost 100%.32 These polyps have a predilection for the second and third portion of the duodenum, especially the periampullary region. This pattern probably reflects the exposure of duodenal mucosa to bile acids, suggesting a role for these compounds in duodenal carcinogenesis. Duodenal cancer is the second cause of cancer death in FAP patients, with a cumulative lifetime risk of 5%.33
Extraintestinal manifestations
Both benign and malignant extracolonic manifestations are common in FAP patients. Congenital hypertrophy of the retinal pigment epithelium (CHRPE) is the most common extracolonic manifestation of FAP (70%–80%). It appears as gray-brown to black round or oval lesions in the retina, and is not known to cause any clinical problems. Epidermoid cysts (50%) and fibromas (25%–50%) are considered subcutaneous lesions, and may cause cosmetic problems. Other benign manifestations include dental abnormalities (79%–90%), osteomas (50%–90%), and desmoid tumors (DT) (10%–15%).34,35
DTs are slow-growing mesenchymal neoplasms characterized by the lack of metastatic potential but an aggressive local behavior due to their infiltrative growth and a high local recurrence rate following complete resection. Compared to the general population, FAP patients are at approximately 1,000-fold increased risk of developing DT.36 Most DTs in FAP patients arise in the abdomen, most frequently in the abdominal wall or intra-abdominal. Risk factors for DT development include abdominal surgery, a positive family history for desmoids, and as explained below, the site of the mutation in the APC gene. Although benign histologically, they represent one of the main causes of death in patients with FAP.37
Extracolonic malignancies include thyroid cancer (2%–3%), pancreatic mucinous adenocarcinomas (1%), hepatoblastoma (1%), and brain tumors (ie, medulloblastoma; <1%).
Papillary thyroid carcinoma is the third most common malignancy associated with FAP (after CRC and duodenal cancer). The lifetime risk of developing thyroid cancer is low, and estimated to be 2%–3% with a rate of approximately 160 times that of the general population.37 There is a remarkable female preponderance (female to male ratio 17:1), and the average age at diagnosis is 27 years.35 Although thyroid cancer in FAP patients can be multifocal and regional lymph node involvement may occur, prognosis is usually very favorable.
Hepatoblastoma is an embryonal neoplasm that predominantly occurs in children between 6 months and 3 years of age, but the age at diagnosis can range from prenatal stages to 16 years. Although the combination of chemotherapy and surgery is very successful, an estimated 25% of all patients do not survive this disease.
The combination of colorectal and extracolonic manifestations is known as Gardner’s syndrome, whereas the association between colorectal polyposis and brain tumors corresponds to Turcot’s syndrome.
Genotype–phenotype correlation
The existence of a spectrum of polyposis caused by mutations located in different regions of the APC gene was suggested by Leppert et al in 1990.38 Since then, several studies have observed an association between the clinical manifestation and the location of the germline mutation. Broadly, the classic phenotype of more than 100 adenomas is associated with mutations between codons 178 and 309, and between codons 409 and 1,580, corresponding to exons 5–8, 9–14 and the first half of the large final exon 15.6 Based upon the genotype–phenotype correlation, FAP can be classified into three categories. Profuse or aggressive polyposis (characterized by an earlier onset and greater number of polyps) has been associated with mutations from codons 1,250 to 1,464, mainly in codon 1,309. AFAP is usually associated with mutations at the extreme 5′ (before codon 157) and 3′ (after codon 1,595) ends of the APC gene, and in the alternatively spliced region of exon 9 (codons 213–412). Finally, in classic FAP, the intermediate phenotype, mutations are located in the remainder of the APC gene, in particular the 5′ end between codon 157 and 1,595 excluding codon 1,309.6,26,39,40 (Figure 2).
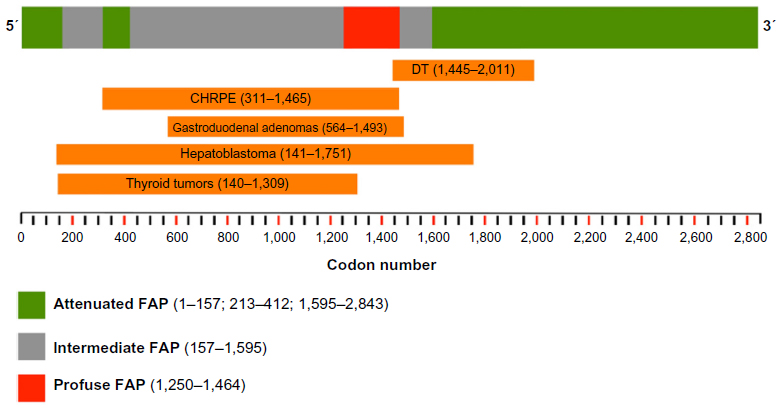 | Figure 2 Genotype–phenotype correlation for the APC gene. |
Extracolonic manifestations have been also associated with specific APC mutations, especially those located beyond codon 1,400. CHRPE is linked to mutations located between codon 311 and codon 1,465, and the presence of DTs is related to mutations at the 3′ end of the APC gene, in general downstream codon 1,400 (1,445–2,011). The presence of gastric and duodenal polyps have been related to mutations at the 3′ end, before codon 1,395, but also exon 4 and codons 564–1,493.26 Other genotype–phenotype correlations have been observed with limited evidence.29 Almost 95% of the mutations in hepatoblastoma patients are located on the 5′ to mid region of the APC gene between codons 141 and 1,751. Thyroid tumors have been related to mutations between codons 140 and 1,309 (Figure 2).26,35
Although genotype–phenotype association is observed, there is a considerable variability among individuals, even among family members, suggesting the influence of environmental factors and/or the effect of modifier genes.41,42
Genetic testing algorithm
Prior to gene testing, affected individuals must receive genetic counseling, so that they understand the pros and cons of cancer genetic testing. Patients must be able to determine whether such testing is acceptable emotionally and take into account other potential issues (such as confidentiality). Once genetic counseling is properly done, if the causative mutation is detected, then presymptomatic diagnosis can be offered to at-risk relatives of the index case. Mid-adolescence is the right time to perform genetic testing, when the diagnosis begins to gain clinical importance in terms of cancer prevention. If germline pathogenic mutation is not found, gene testing cannot be offered to family members, and clinical diagnosis and surveillance is mandatory for all first-degree relatives.
Several methods for APC gene testing have been used. Direct sequencing of all 15 coding exons of the APC gene is considered the gold standard for mutation detection. However, other approaches have been used. In the past, several clinical laboratories used the RNA-based protein truncation test (PTT). The method is based on the size analysis of products resulting from in vitro transcription and translation, which has a sensitivity ranging from 70% to 90%. However, the PTT approach has disadvantages, including assay artifacts and an inability to detect nontruncating mutations. Other methods include scanning methods (such as conformation strand gel electrophoresis), followed by sequencing of aberrant fragments. However, none of these methods has the detection sensitivity of direct sequencing, which is a standard method in most clinical laboratories for detecting point mutations and small insertions or deletions, which account for >85% of the APC mutations. The remaining 10%–15% of mutations are gross deletions and duplications, which can be detected by multiplex ligation-dependent probe amplification (MLPA), Southern blot, or real-time quantitative PCR analysis.43–45
Current guidelines recommend that FAP testing should be performed using full sequencing of the APC gene, and if no mutation is detected, then testing for large gene rearrangements should be completed.14
Clinical management
Colorectal adenomas and CRC
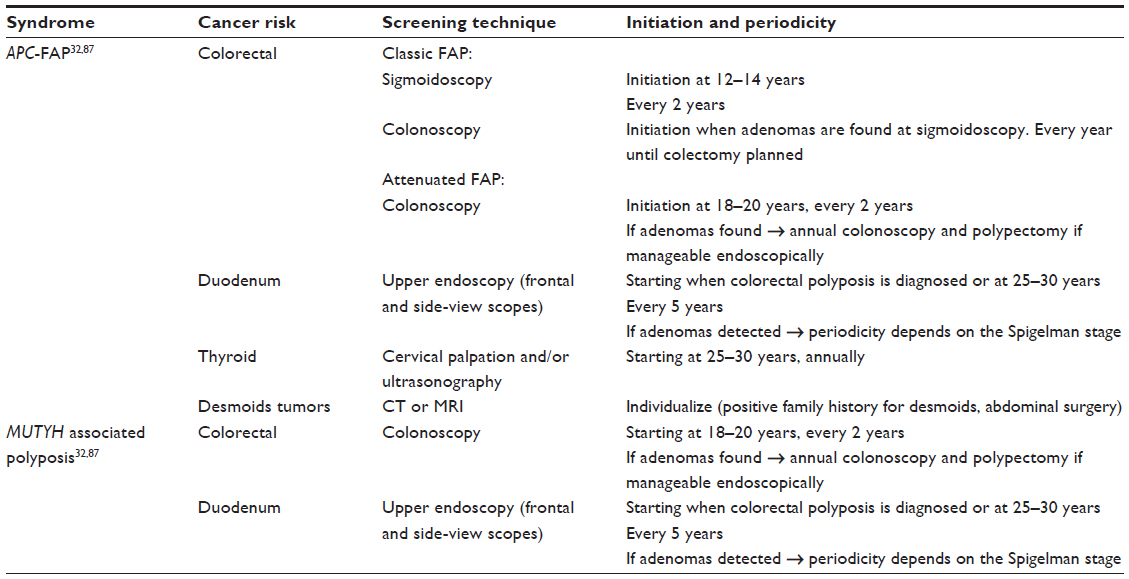
The goal of colorectal neoplasia management in FAP patients is to prevent CRC.13 This management includes both endoscopic polypectomy and surgery. In families with classic FAP, flexible sigmoidoscopy is an adequate technique because of the almost universal distribution of adenomas, including the rectum. The age at which screening should start depends on the risk of malignant transformation of the colorectal adenomas. In FAP patients, the risk of developing CRC before age 20 is very low; however, up to 1.5% of CRC occur between 11 and 20 years of age.34 Accordingly, sigmoidoscopy screening should be carried out every 2 years, starting at age 12–14 years, and be continued lifelong in mutations carriers. Once adenomas are detected, total colonoscopy should be carried out annually until colectomy is planned (Table 2).34
 | Table 2 Management recommendations for FAP |
In AFAP cases, since adenomas can be localized in the right colon, colonoscopy is recommended instead of sigmoidoscopy. In this setting, screening should be carried out every 2 years until polyposis is diagnosed, starting at the age of 18–20 years. Once adenomas are detected, colonoscopy should be carried out annually.34
Surgical removal of the colon at a premalignant stage is a key to prevent the morbidity and mortality associated with advanced CRC. In classic FAP, prophylactic colectomy is usually recommended, when polyposis is profuse or “worrisome” polyps are identified (ie, >1 cm, ulcerated, high grade dysplasia). Most patients with classic FAP undergo surgery between the age of 15 and 25 years. The treatment of AFAP is commonly endoscopic, and only if this is not possible, surgery is performed in a similar manner as in classical FAP.
Surgical options include both proctocolectomy with ileal pouch-anal anastomosis (IPAA) and total colectomy with ileorectal anastomosis (IRA). Compared with IPAA, IRA is relatively simple, with a lower complication rate and usually good bowel function after surgery. For IPAA, more extensive surgery is needed (including pelvic dissection), causing reduction of fertility and worse bowel function.46 The choice of the surgical technique mainly depends on the age at diagnosis, desmoids, fertility, and the severity of rectal polyposis (>15–20 polyps), as well as patient decision after receiving comprehensive information on the benefits and risks of each approach.34 Some authors have proposed to use the evidence of genotype–phenotype association in guiding the surgical treatment of patients with a relatively spared rectum.26,34,47 An IPAA may be recommended in patients with a severe genotype because such patients are at increased risk of developing severe rectal polyposis that will require a secondary proctectomy if IRA is performed.
After surgery, endoscopic follow-up is recommended for those patients with rectal remnant, due to the risk of developing rectal cancer (up to 30% of the cases). Many studies have shown that adenomas and occasionally even adenocarcinomas have been found in the ileo-anal pouch after restorative proctocolectomy.13,34 Therefore, the surveillance of the pouch and the transitional anal zone is essential.
Gastroduodenal polyps
In general, screening for extracolonic manifestations should start when colorectal polyposis is diagnosed or at the age of 25–30 years. Gastroduodenal endoscopy using both front and side-view scopes (in order to correctly visualize the Vater’s ampulla) should be performed every 5 years until adenomas are detected.34
In current practice, given the relative rarity of gastric adenocarcinoma, upper gastrointestinal endoscopic surveillance is driven by the greater risk of duodenal cancer. The stomach is visualized as part of this surveillance, but biopsy or polypectomy is undertaken only for large or unusual looking lesions, especially in the antrum.30,31
To standardize the management of duodenal polyps in FAP, Spigelman et al developed a classification system based on four prognostic variables (Table 3): number of polyps, size, histology, and degree of dysplasia.32,48 Stage I (4 points) indicates mild disease, whereas stages III–IV (>6 points) imply severe duodenal polyposis, with a significant risk of duodenal cancer (7%–36%).49 Approximately 80% of the patients have stage I–III disease, and 10%–20% have stage IV disease. Current evidence indicates that duodenal inspection with chromoendoscopy or narrow-band imaging increase the detection of duodenal adenomas but without a considerable change in Spigelman stages.50,51
 | Table 3 Spigelman classification |
The management of patients with multiple larger adenomas (Spigelman stage III or greater) is challenging, and should be centralized in expert centers. The recurrence rate of adenoma development after endoscopic treatment is high (>50%), and treatment is associated with a high complication rate (perforation, hemorrhage, and pancreatitis).52 Because it is not feasible to remove all adenomas, the usual approach is to remove only large adenomas (>1 cm) or adenomas with high-grade dysplasia, with the aim of delaying/avoiding surgery. In Spigelman IV cases, surgery is often necessary, including duodenotomy with polypectomy, pancreas-sparing duodenectomy, and duodenal-pancreatectomy.33,52
Management of other tumors
Given the increased risk for thyroid cancer, there is expert consensus that thyroid palpation and/or annual cervical ultrasonography should be performed, starting at the age of 25–30 years.34
The development of DT is mainly related to a positive family history, abdominal surgery, and the site of the mutation, and can occur inside the abdomen or in the abdominal wall. DT can be diagnosed by computed tomography (CT) scanning or magnetic resonance imaging (MRI). The options for treatment include pharmacological treatment (nonsteroidal anti-inflammatory drugs [NSAIDs] and/or antiestrogens), chemotherapy, surgical excision, or radiotherapy.34 The evidence for the efficacy of these treatments is poor and is based on small studies. However, because of the high recurrence rate of DT, surgical resection of intra-abdominal tumors should be delayed unless complications appear. Experts recommend as the first line of treatment in patients with large or growing DT sulindac with tamoxifen, and when patients with intra-abdominal tumors do not respond to this treatment, chemotherapy or radiation is indicated. Abdominal wall DT and mesenteric DT should be considered differently. For the treatment for abdominal wall DT, surgery is usually used as a first-line treatment. However, for mesenteric DT, the optimal management strategy should be individualized, taking into consideration the extent of disease, morbidity, and potential benefit versus risk of the different treatment modalities.34
The incidence of other less common extraintestinal malignancies (pancreatic, brain, adrenal, among others) is so low that the use of additional costly imaging studies is not currently recommended. However, surveillance tests should be considered in patients with a strong family history of any of these specific extraintestinal manifestations and in those presenting with symptoms that could be attributed to these tumors.
Chemoprevention
Chemoprevention using NSAIDs has been proposed in FAP patients. The first drug that was shown to be effective in FAP was sulindac.53 Long-term use of this drug reduced the number of colorectal adenomas by >50% in the colon as well as in the rectum of patients after colectomy, but not in duodenal polyposis. However, sulindac does not prevent the development of adenomas in FAP.34 Selective COX-2 inhibitor (cyclooxygenase-2) celecoxib, associated with fewer gastrointestinal side effects than sulindac, was found to reduce the number of colorectal adenomas by 28%,54 and also reduced the number of duodenal adenomas.52,55 However, cardiovascular effects (myocardial infarction or stroke) have been described in long-term users of another selective COX-2 inhibitor (rofecoxib), and therefore the role of these drugs remain controversial, and should be considered only in selected patients without cardiovascular risk factors.34
Although NSAIDs (sulindac, celecoxib) do not replace surgical treatment for colonic FAP, they may play a role in postponing surgery in patients with colonic polyposis or patients with rectal polyposis after colectomy. Regarding duodenal polyposis, the use of celecoxib might be justifiable for patients with severe duodenal polyposis (Spigelman stage III or IV), because the endoscopic and surgical treatment options in such cases are associated with significant complications. Though celecoxib is registered for the treatment of FAP in several countries, some specialists are reluctant to prescribe it because of the cardiovascular effects, and hence this drug is rarely used.34
MAP
MAP (OMIM #608456) is an autosomal-recessive inherited disorder caused by biallelic germline mutations in the MUTYH gene. It was first described in 2002 by Al-Tassan et al in a British family with three affected members and recessive inheritance of multiple colorectal adenomas and carcinoma.3 Patients with MAP show a great variability in clinical features, but usually present with an attenuated polyposis phenotype, showing fewer than 100 adenomas. Some patients develop extracolonic manifestations indistinguishable from that of FAP patients.
MUTYH gene
The MUTYH gene is located on chromosome 1p34.3–1p32.1, and contains 16 exons that encode a 535 aminoacid protein (Figure 1B).56 The MUTYH gene encodes a member of the base excision repair (BER) system. This system is composed of three enzymes (MUTYH, OGG1, and MTH1) that contribute to protect cells against the mutagenic effects of aerobic metabolism, specifically the oxidation of a guanine, leading to the formation of 8-oxo-7, 8-dihydro-2′-deoxyguanosine (8-oxoG). MUTYH acts together with MTH1 and OGG1 to prevent somatic mutations induced by 8-oxoG and its high affinity for adenine (A) instead of cytosine. Specifically, MUTYH is responsible for the removal of adenines mispaired with 8-oxoG.57–59 In the absence of a functional copy of MUTYH due to biallelic mutations, when an oxo-G:A mismatch is present in the DNA template, a G:C to T:A transversion occurs in the subsequent round of replication. For this reason, somatic G:C to T:A transversions in genes such as APC or KRAS frequently occur in MUTYH-associated adenomas and tumors.3,60 One such transversion in the KRAS gene (c.34G>T in codon 12) is frequently encountered (64%) in patients with MAP CRC.61 Therefore, the analysis of somatic KRAS has been recommended as a pre-screening test to identify CRC patients eligible for MUTYH germline molecular genetic testing. Since MAP patients can present with conventional adenomas as well as serrated polyps (hyperplastic polyps, sessile serrated adenomas), the existence of two distinct pathways has been suggested, one leading to conventional adenomas with APC and/or KRAS mutations, and one separate non-APC route leading to hyperplastic polyps and sessile serrated adenomas with KRAS mutations.62,63
Mutational landscape
More than 300 unique sequence variants have been identified in this gene, including more than 80 pathogenic mutations (http://www.lovd.nl/mutyh).64 The majority of them are missense substitutions followed by a minority of splice site or truncation mutations.39 Although infrequent, gross genomic deletions, frameshift, and nonsense mutations have also been reported.14,57,60 Mutations have been described in almost all exons (except exons 1 and 2). The predominance of missense mutations is based mainly on the two hotspot mutations, p.Y179C (c.536A>G;p.Tyr179Cys, previously known as p.Y165C) in exon 7 and p.G396D (c.1187G>A;p.Gly396Asp, previously known as p.G382D) in exon 13, that represent around 70%–80% of all mutations in populations of European origin (Table 4).65
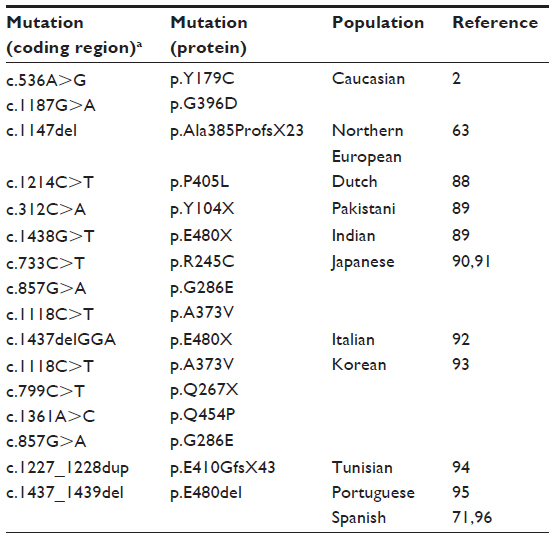 | Table 4 MUTYH germline mutations |
An increasing number of publications on MUTYH have unraveled the ethnic and geographic differences in the mutation frequency of this gene. As mentioned earlier, among European populations, the two missense mutations p.Y179C and p.G396D are by far the most common disease-causing variants. Moreover, different variants have a larger role in other populations, pointing to ethnic and geographic differences, such as p.Y104X in Pakistani patients and p.E480X in Indian patients.66 Other specific mutations have been reported in the Japanese and Southern Europe populations (Table 4).
Several studies have examined the frequency of the two common missense mutations (p.Y179C and p.G396D), and approximately 1%–2% of the general population (of European origin) is predicted to be a carrier.67–72
Phenotypes
The clinical spectrum of MUTYH germline mutations is heterogeneous and can include a wide range of phenotypes (Table 1).
Attenuated and classic adenomatous polyposis
Most biallelic MUTYH mutation carriers have between ten and a few hundred polyps, and very few patients develop more than 500 polyps. Biallelic mutations were initially identified in nearly one-third of cases with APC-negative AFAP with more than 15 adenomas and in about 10% of cases with APC-negative classical FAP, especially in those cases where there is an evident recessive pattern of inheritance.73 A recent study found biallelic MUTYH mutations in 2/119 (2%, 95% CI 0.2%–6%) patients with >1,000 adenomas, 94/1,338 (7%, 95% CI 6%–8%) patients with 100–999 adenomas, 233/3,253 (7%, 95% CI 6%–8%) patients with 20–99 adenomas, and 37/970 (4%, 95% CI 3%–5%) patients with 10–19 adenomas.74
Serrated polyposis syndrome and patients with multiples polyps
Serrated polyps and adenomas are a common finding in patients with MAP. A minority of MAP patients fulfill the WHO criteria for serrated polyposis syndrome (defined when a patient fulfills at least one of the following criteria: 1) at least five serrated polyps proximal to the sigmoid colon, two of which are greater than 10 mm in diameter; 2) any number of serrated polyps occurring proximal to the sigmoid colon in an individual who has a first-degree relative with serrated polyposis; and 3) more than 20 serrated polyps of any size distributed throughout the colon),75 usually in the setting of a synchronous adenomatous polyposis.62,76 In a recent study centered on patients diagnosed with at least ten polyps of any histology (including adenomatous and serrated polyps), 6.7% (27/405) patients were found to be MUTYH biallelic carriers.76
CRC without polyposis
In population-based CRC studies, where patients were recruited on the basis of the diagnosis of CRC, biallelic MUTYH mutations were found in 0.3%–2.0% of the population.60,71 In several population-based studies, up to a third of proven biallelic MUTYH mutation carriers did not have a polyposis phenotype.67–72 Accordingly, biallelic MUTYH mutations are not always associated with a polyposis phenotype; therefore, biallelic MUTYH mutations should be considered also in early onset CRC patients (≤50 years old), especially if the Lynch syndrome has been ruled out. However, atypical phenotypes have been described.77 In a recent study that analyzed the MUTYH gene in 85 cases with suspected Lynch syndrome with tumors showing mismatch repair deficiency without detectable germline mutation, one biallelic (p.Y179C) carrier was identified that developed CRC, urothelial carcinoma, and a sebaceous gland carcinoma.78 In this particular patient, sequencing of the tumor revealed two somatic mutations in the mismatch repair system, thus explaining the phenotype.
Heterozygous MUTYH mutations and CRC risk
The risk of CRC in individuals with monoallelic (heterozygous) mutations has been the focus of an intense debate. Several meta-analyses have been performed, with ORs (odds ratios) or RRs (relative ratios) between 1.11 and 1.27, most of them showing no statistical differences.79–84 The most recent systematic review has shown that monoallelic mutation carrier frequency was greater for cases ascertained due to a family history (3.3%) than for controls (1.4%; P=0.02), suggesting that monoallelic carriers with a family history of CRC may be at a greater risk.
Extracolonic manifestations
Since oxidative stress is present in different tissues, it can be expected that a defective MUTYH gene leads to neoplasms in other organs. Vogt et al recently described the spectrum of extracolonic features of a large cohort of MAP patients (276 patients with MAP). In this study, duodenal polyposis occurs in 17% of cases with a lifetime risk of duodenal cancer of 4% and a 38% lifetime risk of any extraintestinal cancer.85 The incidence of extraintestinal malignancies among cases was almost twice that of the general population with a significant increase in the incidence of ovarian, bladder, and skin cancers and a trend of increased risk of breast cancer.85 The median age at diagnosis for the different extraintestinal malignancies varied between 51 and 61 years.
Other manifestations also seen in FAP patients have been reported in a small number of MAP patients: gastric FGP, lipomas, CHRPE, epidermoid cyst, DT, and thyroid carcinoma.60 Overall, the incidence of FAP-related manifestations in MAP patients is lower than in FAP patients.
Genotype–phenotype correlation
As in the APC gene, genotype–phenotype associations have been described for the common MUTYH mutations. Some studies have observed that in accordance with the functionality assays that show a greater reduction in MUTYH glycosylase activity for p.Y179C as compared to p.G396D, patients with biallelic Y179C mutations are associated with a more aggressive phenotype (ie, earlier onset and a greater number of polyps) compared to biallelic G396D mutations.65,71,76,86 A recent study reported that the p.G396D variant is associated with the development of serrated polyps in MAP patients.76
Genetic testing algorithm
Biallelic MUTYH mutations should be suspected in patients with an attenuated form of adenomatous polyposis or classical FAP with a recessive pattern of inheritance. It should also be considered in CRC patients diagnosed before the age of 50 years, and in patients with multiple colonic polyps (>10, including both adenomatous and serrated ones) (Table 1).
Once an individual is found to carry biallelic MUTYH mutations, presymptomatic testing can be offered to first-degree relatives, especially to siblings, who have a 25% risk of carrying biallelic mutations. The risk of MAP in the children of MAP patients depends on the status of their reproductive partner.
Germline analysis usually comprises the two most common mutations (p.G396D and p.Y179C).14 Full-gene sequencing is recommended in: 1) individuals who are found to carry one of the two common mutations; 2) individuals whose ethnic ancestry is not Caucasian; and 3) individuals with Caucasian ancestry with a suggestive clinical presentation and a family history found to be negative for the common mutations. Since large deletions or duplications have been exceptionally reported in the MUTYH gene, there is no need for applying methods to analyze these mutations.14,87
Clinical management
The suggested surveillance protocol for MAP patients is similar to that for patients with AFAP (Table 2).34 Individuals should undergo total colonoscopy every 2 years, starting at age 18–20 years and continuing lifelong. Colorectal polyps management is similar to that proposed for patients with AFAP. Because of the usual attenuated phenotype, in some patients it is possible to remove these polyps endoscopically. If surgery is required, decisions should be made as in AFAP.
Upper endoscopy starting at age of 25–30 years is recommended, following the same strategy described for AFAP. There is no evidence of the usefulness of any chemopreventive measure in this condition.
Currently, there is no evidence that justifies the screening for extraintestinal manifestations in patients with MAP.
Although still under debate, CRC screening in monoallelic mutation carriers is recommended for first-degree relatives of a patient with sporadic CRC.29
Acknowledgments
This work was supported by grants from the Plan Nacional de I+D+I y cofinanciado por el ISCIII-Subdirección General de Evaluación y el Fondo Europeo de Desarrollo Regional (FEDER) (PI10/00384; PI13/00719), and a grant from the Spanish Gastroenterology Association (Beca Gonzalo Miño). CIBERehd is funded by the Instituto de Salud Carlos III.
Disclosure
The authors report no conflicts of interest in this work.
References
Nishisho I, Nakamura Y, Miyoshi Y, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253(5020):665–669. | |
Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66(3):589–600. | |
Al-Tassan N, Chmiel NH, Maynard J, et al. Inherited variants of MYH associated with somatic G:C-->T: a mutations in colorectal tumors. Nat Genet. 2002;30(2):227–232. | |
Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919–932. | |
Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol. 2006;101(2):385–398. | |
Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Hum Mol Genet. 2001;10(7):721–733. | |
Lipton L, Tomlinson I. The genetics of FAP and FAP-like syndromes. Fam Cancer. 2006;5(3):221–226. | |
Sulekova Z, Reina-Sanchez J, Ballhausen WG. Multiple APC messenger RNA isoforms encoding exon 15 short open reading frames are expressed in the context of a novel exon 10A-derived sequence. Int J Cancer. 1995;63(3):435–441. | |
Sieber OM, Tomlinson IP, Lamlum H. The adenomatous polyposis coli (APC) tumour suppressor – genetics, function and disease. Mol Med Today. 2000;6(12):462–469. | |
Zumbrunn J, Kinoshita K, Hyman AA, Nathke IS. Binding of the adenomatous polyposis coli protein to microtubules increases microtubule stability and is regulated by GSK3 beta phosphorylation. Curr Biol. 2001;11(1):44–49. | |
Nielsen M, Hes FJ, Nagengast FM, et al. Germline mutations in APC and MUTYH are responsible for the majority of families with attenuated familial adenomatous polyposis. Clin Genet. 2007;71(5):427–433. | |
Bisgaard ML, Fenger K, Bulow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat. 1994;3(2):121–125. | |
Balmana J, Balaguer F, Cervantes A, Arnold D, Group EGW. Familial risk-colorectal cancer: ESMO Clinical Practice Guidelines. Ann Oncol. 2013;24(Suppl 6):vi73–vi80. | |
Hegde M, Ferber M, Mao R, Samowitz W, Ganguly A; Working Group of the American College of Medical Genetics and Genomics (ACMG) Laboratory Quality Assurance Committee. ACMG technical standards and guidelines for genetic testing for inherited colorectal cancer (Lynch syndrome, familial adenomatous polyposis, and MYH-associated polyposis). Genet Med. 2014;16(1):101–116. | |
Jais P, Laurent-Puig P, Olschwang S. [APC protein: protein interactions and cellular functions]. Gastroenterol Clin Biol. 1998;22(12):1071–1080. French. | |
Heinen CD. Genotype to phenotype: analyzing the effects of inherited mutations in colorectal cancer families. Mutat Res. 2010;693(1–2):32–45. | |
Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25(57):7531–7537. | |
Popat S, Stone J, Coleman G, et al. Prevalence of the APC E1317Q variant in colorectal cancer patients. Cancer Lett. 2000;149(1–2):203–206. | |
Aretz S, Uhlhaas S, Sun Y, et al. Familial adenomatous polyposis: aberrant splicing due to missense or silent mutations in the APC gene. Hum Mutat. 2004;24(5):370–380. | |
Montera M, Piaggio F, Marchese C, et al. A silent mutation in exon 14 of the APC gene is associated with exon skipping in a FAP family. J Med Genet. 2001;38(12):863–867. | |
Snow AK, Tuohy TM, Sargent NR, Smith LJ, Burt RW, Neklason DW. APC promoter 1B deletion in seven American families with familial adenomatous polyposis. Clin Genet. Epub 2014 Sep 19. | |
Aretz S, Uhlhaas S, Caspari R, et al. Frequency and parental origin of de novo APC mutations in familial adenomatous polyposis. Eur J Hum Genet. 2004;12(1):52–58. | |
Ripa R, Bisgaard ML, Bulow S, Nielsen FC. De novo mutations in familial adenomatous polyposis (FAP). Eur J Hum Genet. 2002;10(10):631–637. | |
Hes FJ, Nielsen M, Bik EC, et al. Somatic APC mosaicism: an underestimated cause of polyposis coli. Gut. 2008;57(1):71–76. | |
Necker J, Kovac M, Attenhofer M, Reichlin B, Heinimann K. Detection of APC germ line mosaicism in patients with de novo familial adenomatous polyposis: a plea for the protein truncation test. J Med Genet. 2011;48(8):526–529. | |
Nieuwenhuis MH, Vasen HF. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Crit Rev Oncol Hematol. 2007;61(2):153–161. | |
Lamlum H, Ilyas M, Rowan A, et al. The type of somatic mutation at APC in familial adenomatous polyposis is determined by the site of the germline mutation: a new facet to Knudson’s ‘two-hit’ hypothesis. Nat Med. 1999;5(9):1071–1075. | |
Petersen GM, Slack J, Nakamura Y. Screening guidelines and premorbid diagnosis of familial adenomatous polyposis using linkage. Gastroenterology. 1991;100(6):1658–1664. | |
Carballal S, Leoz M-L, Moreira L, Ocan~a T, Balaguer F. Hereditary colorectal cancer syndromes. Colorectal Cancer. 2014;3(1):1–20. | |
Sarre RG, Frost AG, Jagelman DG, Petras RE, Sivak MV, McGannon E. Gastric and duodenal polyps in familial adenomatous polyposis: a prospective study of the nature and prevalence of upper gastrointestinal polyps. Gut. 1987;28(3):306–314. | |
Bianchi LK, Burke CA, Bennett AE, Lopez R, Hasson H, Church JM. Fundic gland polyp dysplasia is common in familial adenomatous polyposis. Clin Gastroenterol Hepatol. 2008;6(2):180–185. | |
Bulow S, Bjork J, Christensen IJ, et al; the DAF Study Group. Duodenal adenomatosis in familial adenomatous polyposis. Gut. 2004;53(3):381–386. | |
Biasco G, Pantaleo MA, Di Febo G, Calabrese C, Brandi G, Bulow S. Risk of duodenal cancer in patients with familial adenomatous polyposis. Gut. 2004;53(10):1547. [author reply 1547]. | |
Vasen HF, Möslein G, Alonso A, et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut. 2008;57(5):704–713. | |
Groen EJ, Roos A, Muntinghe FL, et al. Extra-intestinal manifestations of familial adenomatous polyposis. Ann Surg Oncol. 2008;15(9):2439–2450. | |
Gurbuz AK, Giardiello FM, Petersen GM, et al. Desmoid tumours in familial adenomatous polyposis. Gut. 1994;35(3):377–381. | |
Sturt NJ, Clark SK. Current ideas in desmoid tumours. Fam Cancer. 2006;5(3):275–285. [discussion 287–278]. | |
Leppert M, Burt R, Hughes JP, et al. Genetic analysis of an inherited predisposition to colon cancer in a family with a variable number of adenomatous polyps. N Engl J Med. 1990;322(13):904–908. | |
Torrezan GT, da Silva FC, Santos EM, et al. Mutational spectrum of the APC and MUTYH genes and genotype-phenotype correlations in Brazilian FAP, AFAP, and MAP patients. Orphanet J Rare Dis. 2013;8:54. | |
Newton KF, Mallinson EK, Bowen J, et al. Genotype-phenotype correlation in colorectal polyposis. Clin Genet. 2012;81(6):521–531. | |
Grandval P, Blayau M, Buisine MP, et al. The UMD-APC database, a model of nation-wide knowledge base: update with data from 3,581 variations. Hum Mutat. 2014;35(5):532–536. | |
Crabtree MD, Fletcher C, Churchman M, et al. Analysis of candidate modifier loci for the severity of colonic familial adenomatous polyposis, with evidence for the importance of the N-acetyl transferases. Gut. 2004;53(2):271–276. | |
Aihara H, Kumar N, Thompson CC. Diagnosis, surveillance, and treatment strategies for familial adenomatous polyposis: rationale and update. Eur J Gastroenterol Hepatol. 2014;26(3):255–262. | |
Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis. 2009;4:22. | |
Renkonen ET, Nieminen P, Abdel-Rahman WM, et al. Adenomatous polyposis families that screen APC mutation-negative by conventional methods are genetically heterogeneous. J Clin Oncol. 2005;23(24):5651–5659. | |
Fazio VW, Kiran RP, Remzi FH, et al. Ileal pouch anal anastomosis: analysis of outcome and quality of life in 3707 patients. Ann Surg. 2013;257(4):679–685. | |
Vasen HF, van der Luijt RB, Slors JF, et al. Molecular genetic tests as a guide to surgical management of familial adenomatous polyposis. Lancet. 1996;348(9025):433–435. | |
Spigelman AD, Williams CB, Talbot IC, Domizio P, Phillips RK. Upper gastrointestinal cancer in patients with familial adenomatous polyposis. Lancet. 1989;2(8666):783–785. | |
Groves CJ, Saunders BP, Spigelman AD, Phillips RK. Duodenal cancer in patients with familial adenomatous polyposis (FAP): results of a 10 year prospective study. Gut. 2002;50(5):636–641. | |
Lopez-Ceron M, van den Broek FJ, Mathus-Vliegen EM, et al. The role of high-resolution endoscopy and narrow-band imaging in the evaluation of upper GI neoplasia in familial adenomatous polyposis. Gastrointest Endosc. 2013;77(4):542–550. | |
Dekker E, Boparai KS, Poley JW, et al. High resolution endoscopy and the additional value of chromoendoscopy in the evaluation of duodenal adenomatosis in patients with familial adenomatous polyposis. Endoscopy. 2009;41(8):666–669. | |
Brosens LA, Keller JJ, Offerhaus GJ, Goggins M, Giardiello FM. Prevention and management of duodenal polyps in familial adenomatous polyposis. Gut. 2005;54(7):1034–1043. | |
Cruz-Correa M, Hylind LM, Romans KE, Booker SV, Giardiello FM. Long-term treatment with sulindac in familial adenomatous polyposis: a prospective cohort study. Gastroenterology. 2002;122(3):641–645. | |
Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342(26):1946–1952. | |
Phillips RK, Wallace MH, Lynch PM, et al; FAP Study Group. A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut. 2002;50(6):857–860. | |
Slupska MM, Baikalov C, Luther WM, Chiang JH, Wei YF, Miller JH. Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J Bacteriol. 1996;178(13):3885–3892. | |
Yamaguchi S, Ogata H, Katsumata D, et al. MUTYH-associated colorectal cancer and adenomatous polyposis. Surg Today. 2014;44(4):593–600. | |
Ohtsubo T, Nishioka K, Imaiso Y, et al. Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res. 2000;28(6):1355–1364. | |
Takao M, Zhang QM, Yonei S, Yasui A. Differential subcellular localization of human MutY homolog (hMYH) and the functional activity of adenine:8-oxoguanine DNA glycosylase. Nucleic Acids Res. 1999; 27(18):3638–3644. | |
Nielsen M, Morreau H, Vasen HF, Hes FJ. MUTYH-associated polyposis (MAP). Crit Rev Oncol Hematol. 2011;79(1):1–16. | |
Lipton L, Halford SE, Johnson V, et al. Carcinogenesis in MYH-associated polyposis follows a distinct genetic pathway. Cancer Res. 2003;63(22):7595–7599. | |
Boparai KS, Dekker E, Van Eeden S, et al. Hyperplastic polyps and sessile serrated adenomas as a phenotypic expression of MYH-associated polyposis. Gastroenterology. 2008;135(6):2014–2018. | |
Castells A. MYH-associated polyposis: adenomas and hyperplastic polyps, partners in crime? Gastroenterology. 2008;135(6):1857–1859. | |
Venesio T, Balsamo A, D’Agostino VG, Ranzani GN. MUTYH-associated polyposis (MAP), the syndrome implicating base excision repair in inherited predisposition to colorectal tumors. Front Oncol. 2012;2:83. | |
Nielsen M, Joerink-van de Beld MC, Jones N, et al. Analysis of MUTYH genotypes and colorectal phenotypes in patients With MUTYH-associated polyposis. Gastroenterology. 2009;136(2):471–476. | |
Out AA, Tops CM, Nielsen M, et al. Leiden Open Variation Database of the MUTYH gene. Hum Mutat. 2010;31(11):1205–1215. | |
Jenkins MA, Croitoru ME, Monga N, et al. Risk of colorectal cancer in monoallelic and biallelic carriers of MYH mutations: a population-based case-family study. Cancer Epidemiol Biomarkers Prev. 2006;15(2):312–314. | |
Croitoru ME, Cleary SP, Di Nicola N, et al. Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. J Natl Cancer Inst. 2004;96(21):1631–1634. | |
Peterlongo P, Mitra N, Chuai S, et al. Colorectal cancer risk in individuals with biallelic or monoallelic mutations of MYH. Int J Cancer. 2005;114(3):505–507. | |
Enholm S, Hienonen T, Suomalainen A, et al. Proportion and phenotype of MYH-associated colorectal neoplasia in a population-based series of Finnish colorectal cancer patients. Am J Pathol. 2003;163(3):827–832. | |
Balaguer F, Castellví-Bel S, Castells A, et al; Gastrointestinal Oncology Group of the Spanish Gastroenterological Association. Identification of MYH mutation carriers in colorectal cancer: a multicenter, case-control, population-based study. Clin Gastroenterol Hepatol. 2007;5(3):379–387. | |
Farrington SM, Tenesa A, Barnetson R, et al. Germline susceptibility to colorectal cancer due to base-excision repair gene defects. Am J Hum Genet. 2005;77(1):112–119. | |
Sieber OM, Lipton L, Crabtree M, et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med. 2003;348(9):791–799. | |
Grover S, Kastrinos F, Steyerberg EW, et al. Prevalence and phenotypes of APC and MUTYH mutations in patients with multiple colorectal adenomas. JAMA. 2012;308(5):485–492. | |
Snover DC, Ahnen DJ, Burt RW, Odze RD. WHO Classification of Tumours of the Digestive System. Serrated Polyps of the Colon and Rectum and Serrated (“Hyperplastic”) Polyposis. Berlin, Germany: Springer-Verlag; 2010. | |
Guarinos C, Juárez M, Egoavil C, et al. Prevalence and characteristics of MUTYH-associated polyposis in patients with multiple adenomatous and serrated polyps. Clin Cancer Res. 2014;20(5):1158–1168. | |
Segui N, Navarro M, Pineda M, et al. Exome sequencing identifies MUTYH mutations in a family with colorectal cancer and an atypical phenotype. Gut. 2015;64(2):355–356. | |
Morak M, Heidenreich B, Keller G, et al. Biallelic MUTYH mutations can mimic Lynch syndrome. Eur J Hum Genet. 2014;22(11):1334–1337. | |
Avezzù A, Agostini M, Pucciarelli S, et al. The role of MYH gene in genetic predisposition to colorectal cancer: another piece of the puzzle. Cancer Lett. 2008;268(2):308–313. | |
Webb EL, Rudd MF, Houlston RS. Colorectal cancer risk in monoallelic carriers of MYH variants. Am J Hum Genet. 2006;79(4):768–771. [author reply 771–762]. | |
Win AK, Hopper JL, Jenkins MA. Association between monoallelic MUTYH mutation and colorectal cancer risk: a meta-regression analysis. Fam Cancer. 2011;10(1):1–9. | |
Tenesa A, Campbell H, Barnetson R, Porteous M, Dunlop M, Farrington SM. Association of MUTYH and colorectal cancer. Br J Cancer. 2006; 95(2):239–242. | |
Cleary SP, Cotterchio M, Jenkins MA, et al. Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology. 2009;136(4):1251–1260. | |
Jones N, Vogt S, Nielsen M, et al. Increased colorectal cancer incidence in obligate carriers of heterozygous mutations in MUTYH. Gastroenterology. 2009;137(2):489–494, 494e481. [quiz 725–486]. | |
Vogt S, Jones N, Christian D, et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology. 2009;137(6):e1910–e1971. | |
Lubbe SJ, Di Bernardo MC, Chandler IP, Houlston RS. Clinical implications of the colorectal cancer risk associated with MUTYH mutation. J Clin Oncol. 2009;27(24):3975–3980. | |
Torrezan GT, da Silva FC, Krepischi AC, et al. Breakpoint characterization of a novel large intragenic deletion of MUTYH detected in a MAP patient: case report. BMC Med Genet. 2011;12:128. | |
Nielsen M, Franken PF, Reinards TH, et al. Multiplicity in polyp count and extracolonic manifestations in 40 Dutch patients with MYH associated polyposis coli (MAP). J Med Genet. 2005;42(9):e54. | |
Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362(9377):39–41. | |
Miyaki M, Iijima T, Yamaguchi T, et al. Germline mutations of the MYH gene in Japanese patients with multiple colorectal adenomas. Mutat Res. 2005;578(1-2):430–433. | |
Yanaru-Fujisawa R, Matsumoto T, Ushijima Y, et al. Genomic and functional analyses of MUTYH in Japanese patients with adenomatous polyposis. Clin Genet. 2008;73(6):545–553. | |
Gismondi V, Meta M, Bonelli L, et al. Prevalence of the Y165C, G382D and 1395delGGA germline mutations of the MYH gene in Italian patients with adenomatous polyposis coli and colorectal adenomas. Int J Cancer. 2004;109(5):680–684. | |
Kim DW, Kim IJ, Kang HC, et al. Germline mutations of the MYH gene in Korean patients with multiple colorectal adenomas. Int J Colorectal Dis. 2007;22(10):1173–1178. | |
Abdelmaksoud-Dammak R, Miladi-Abdennadher I, Amouri A, et al. High prevalence of the c.1227_1228dup (p.Glu410GlyfsX43) mutation in Tunisian families affected with MUTYH-associated-polyposis. Fam Cancer. 2012;11(3):503–508. | |
Isidro G, Laranjeira F, Pires A, et al. Germline MUTYH (MYH) mutations in Portuguese individuals with multiple colorectal adenomas. Hum Mutat. 2004;24(4):353–354. | |
Gomez-Fernandez N, Castellvi-Bel S, Fernandez-Rozadilla C, et al. Molecular analysis of the APC and MUTYH genes in Galician and Catalonian FAP families: a different spectrum of mutations? BMC Med Genet. 2009;10:57. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.