Back to Journals » Cancer Management and Research » Volume 13
The Functional Hallmarks of Cancer Predisposition Genes
Authors Capellini A, Williams M, Onel K, Huang KL
Received 20 March 2021
Accepted for publication 8 May 2021
Published 1 June 2021 Volume 2021:13 Pages 4351—4357
DOI https://doi.org/10.2147/CMAR.S311548
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmet Emre Eşkazan
Alexandra Capellini,1,* Matthew Williams,1,* Kenan Onel,1– 3 Kuan-Lin Huang1– 3
1Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, USA, New York, NY, 10029, USA; 2Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, USA, New York, NY, 10029, USA; 3Icahn Institute for Data Science and Genomic Technology, Icahn School of Medicine at Mount Sinai, USA, New York, NY, 10029, USA
*These authors contributed equally to this work
Correspondence: Kenan Onel; Kuan-Lin Huang
Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, Box 1498, New York, NY, 10029, USA
Tel +1 212-824-6134
Email [email protected]; [email protected]
Abstract: The canonical model for hereditary cancer predisposition is a cancer predisposition gene (CPG) that drives either one or both of two fundamental hallmarks of cancer, defective genomic integrity and deregulated cell proliferation, ultimately resulting in the accumulation of mutations within cells. Thus, the genes most commonly associated with cancer-predisposing genetic syndromes are tumor suppressor genes that regulate DNA repair (eg, BRCA1, BRCA2, MMR genes) and/or cell cycle (eg, APC, RB1). In recent years, however, the spectrum of high-penetrance CPGs has expanded considerably to include genes in non-canonical pathways such as oncogenic signaling, metabolism, and protein translation. We propose here that, given the variety of pathways that may ultimately affect genome integrity and cell proliferation, the model of cancer genetic predisposition needs to be expanded to account for diverse mechanisms. This synthesis calls for modeling and multi-omic studies applying novel experimental and computational approaches to understand cancer genetic predisposition.
Keywords: cancer, genetics, predisposition, multi-omics, genomics, oncogenesis
Introduction
Cancer arises from accumulated mutations, which are the products of DNA surveillance and repair errors and the number of cellular divisions.1 The presence of a diverse mutational landscape in healthy tissue under environmental assault is an important precursor in the micro-evolutionary process of carcinogenesis. Recent reports that sequenced biopsies of physiologically normal tissues, including sun-exposed eyelid epidermis, Barrett’s esophagus, colorectal epithelial cells, and liver, demonstrated a diverse array of “driver” mutations associated with tumors with varying degrees of clonal expansion among these non-cancerous biopsy specimens, highlighting a precancerous landscape for clonal selection that begins with mutation acquisition.2–4
It can thus be reasoned that individuals with elevated mutation rates will have a higher risk of developing cancer. Aligned with this rationale, many of the known hereditary cancer predisposition genes (CPGs)—genes harboring inherited germline variants that confer high or moderate risks—are critical components of DNA damage repair pathways, such as BRCA1 and BRCA2 in homologous recombination. Additionally, increased cellular proliferation directly contributes to the number of cellular divisions. An established CPG that directly regulates cellular proliferation is RB1, whose gene products halt DNA replication and mediates cell survival, apoptosis, and differentiation. Using the differential risks of retinoblastoma in germline RB1 carriers versus non-carriers, Alfred Knudson statistically demonstrated the two-hit model of cancer predisposition in 1972.5
Advances in sequencing technology engendered the discovery of CPGs in pedigrees enriched for familial cases and large sets of non-familial cancer cases. In these large sample sets, it was found that approximately 5–15% of cancer cases are carriers of pathogenic variation within CPGs.6–8 However, many of the newly identified CPGs do not have a primary function in these two canonical predisposition processes. These CPGs affect diverse pathways including metabolism, protein translation, and rRNA processing. How they result in higher cancer risks remains unclear. Herein, we highlight this crucial knowledge gap and discuss a few possible mechanistic contributions of emerging cancer predisposition pathways to stress-induced somatic mutation acquisition and cellular proliferation, which may ultimately lead to clonal diversification (Figure 1). This synthesis is not meant to be comprehensive, but rather, it illustrates a conceptual model and describes through representative examples how non-canonical cancer predisposition pathways may mediate cancer risks.
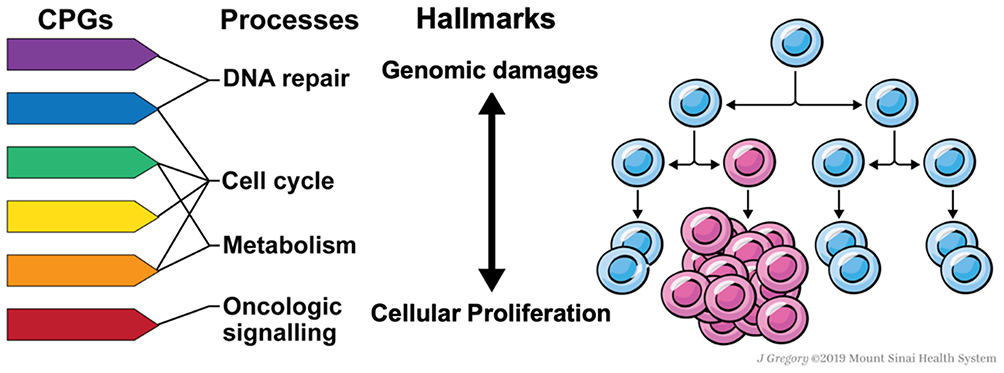 |
Figure 1 Proposed model of cancer genetic predisposition through a micro-evolutionary perspective. Pathogenic germline variants of cancer predisposition genes (CPGs) affecting a host of cellular pathways including DNA repair, oncogenic signaling, metabolism, and cell cycle, among others, can predispose to human cancer on a spectrum from increasing mutation acquisition and cellular proliferation. While the cancer predisposition field has traditionally focused on dysfunctional DNA repair as a mechanism of carcinogenesis largely through increasing clonal diversity, we propose that additional pathways that might also confer clonal fitness advantages that are important to consider in a cancer predisposition model. |
Genomic Integrity
The majority of well-known cancer predisposition pathways are directly implicated in the maintenance of genomic stability. DNA damage repair pathways including homologous recombination (ex: BRCA1, BRCA2, PALB2), DNA damage sensing (ex: ATM, ATR, CHEK2), mismatch repair (ex: MSH2, MLH1, MSH6, PMS1/2), nucleotide excision repair (ex: XPC, ERCC2) and Fanconi anemia (ex: FANCA, FANCD2, FANCJ/BRIP1) are associated with a variety of cancer predisposition syndromes.9,10 Loss of helicase function (ex: BLM) also enhances mutation acquisition via the inability to rescue stalled replication forks, dissolve double Holliday Junctions, and disentangle topologically constrained molecules.11 These CPGs typically harbor loss-of-function germline variants that conceivably give rise to elevated somatic mutation rates. This hypothesis is supported by experimental evidence, including recent multiplex assays of variant effect (MAVEs) that scan thousands of possible mutations in a CPG (eg, BRCA1) and show that the known pathogenic alleles correspond almost perfectly to those that showed loss-of-function in the associated DNA damage repair (eg, homologous directed repair).12
Notably, many CPGs implicated in DNA damage repair pathways are known to show genetic pleiotropy and may also contribute to cellular proliferation. For example, BRCA1/2 are involved in a wide range of functions beyond DNA repair, including regulation of cell-cycle progression and apoptosis via p53 and ubiquitylation.13 Additionally, the so-called “Guardian of the Genome” TP53 has essential roles in maintaining genomic stability through a broad range of mechanisms. In addition to activating nucleotide excision repair, base excision repair, and mismatch repair pathways, p53 is important for cell cycle arrest at the G1/S phase and G2/M phase transitions and inducing apoptosis.14 Cell cycle checkpoints and apoptosis can be important for preventing the persistence of somatic mutations in subsequent lineages. When TP53’s cell cycle checkpoint and apoptotic functions are lost, there is a persistence of somatic mutations to progeny and a “cut-the-break” phenomenon promoting dysregulated proliferation. Comprehensive investigation of genetic pleiotropy is required to fully understand cancer predisposition at the level of an individual CPG and integration among broader pathways.
Cellular Proliferation and Oncogenic Signaling
The other canonical cancer predisposition process is cellular proliferation. Errors in cell cycle checkpoints can lead to dysregulated cell cycle progression (ex: RB1, TP53). Additionally, constitutive activation of pro-proliferative pathways that enhance cell division and can predispose to cancer development include WNT/B-Catenin (ex: APC), receptor tyrosine kinase (ex: RET, MET) and MAPK (ex: TMEM127, NF1) signaling pathways. Pathogenic variants in RET and MET disrupt the RTK signaling pathway with downstream effects leading to aberrant MAPK and PI3K/AKT signaling, potentially altering the rate of cellular proliferation in predisposed individuals.15,16 Pathogenic variants in APC reduce antagonism of the WNT/B-Catenin pathway, which promotes cellular proliferation through beta-catenin accumulation and WNT-triggered gene expression. Pathogenic variants in APC likely increase cancer risk by shifting the WNT pathway toward an on state that promotes continuous cellular proliferation.17 Additionally, pathogenic variants in TMEM127 may promote oncogenesis through upregulation of the mammalian target of rapamycin (mTOR) signaling pathway. The mTOR pathway controls cellular proliferation by integrating growth factor-activated signals with nutrients and energy sufficient for cell growth and survival;18 upregulation of mTOR can therefore lead to dysregulated cellular proliferation that increases the TMEM127 carriers’ risk for cancer.19 Finally, inactivating NF1 variants can cause prolonged activation of the RAS/RAF/MAPK pathway and result in increased cell proliferation and loss of cell growth control. Dysregulated cellular signaling by somatic mutation or epigenetic alteration is well-known in carcinogenesis,20 and pathogenic germline variants affecting the same pathways present a notable overlap between heritable germline and acquired somatic alterations.
Metabolism
Deregulated cellular energetics is also a hallmark known for the somatic genome of cancer.20 Pathogenic germline variants in metabolic genes that predispose individuals to cancer include HFE, VHL, FH, FAH, and SDH A/B/C/D. While these genes are implicated in a variety of metabolic processes, mutations in FH, SDH A/B/C/D, VHL, and MITF have all been reported to contribute to oncogenesis through the HIF-1alpha pathway. Activation of the HIF-1alpha pathway produces hypoxia-stimulated consequences. Over-expression of hypoxia-inducible gene products such as VEGF and PDGF promotes angiogenesis to support cellular proliferation. However, the inefficiencies of this vascular development stress cellular machinery, often resulting in mutation acquisition.21 The downstream effects of HIF-1alpha pathway activation therefore can contribute to both cellular proliferation and accumulation of somatic mutations. FAH dysfunction can increase replication error rates via mitotic spindle disruption. It may also lead to dysregulated cellular proliferation through the activation of the MAPK pathway.22 Aberrant HFE function results in disrupted iron metabolism. The iron-induced oxidative stress can cause DNA damage, resulting in a cooperative process whereby cellular proliferation and elevated replication rate potentiate each other.23 While dysfunction in many metabolic CPGs may promote oncogenesis by synergistic effects that promote mutation acquisition and clonal diversification via elevated mutation rates and enhanced replicative capacity, these genes are highly pleiotropic and additional mechanisms need to be considered. For example, VHL has been implicated in a range of cancer-promoting molecular pathways including microtubule stabilization,24 ciliary maintenance,25 and fibronectin matrix assembly,26 among others.
Protein Translation and Ribosomal Processing
Some CPGs affect ribosomal proteins and co-translational protein-targeting pathways. Pathogenic germline variants in 19 different ribosomal proteins can predispose individuals to develop Diamond-Blackfan Anemia (DBA), a syndrome of bone marrow failure. DBA is characterized by a disruption in ribosomal protein function responsible for nucleolar stress signaling, p53 activation and ultimately, cell cycle arrest or apoptosis. This disruption places selective pressure on clonal populations that have the propensity for accelerated clonal diversification and enhanced cellular proliferation. At least two hypotheses can explain how DBA affects dysregulated cellular proliferation. First, a DBA patient may develop a somatic mutation in the p53 pathway that would confer a proliferation advantage, resulting in the outgrowth of selected clones carrying p53 mutations. Second, a pathogenic variant that enhances ribosomal protein synthesis to reduce nucleolar stress can lead to oncogenic gene activation, favoring proliferative clonal selection. An over-expressed c-Myc oncogene (eg, c-MYC) can further upregulate protein translational machinery and facilitate the growth of clonal populations with a survival advantage that predisposes to cancer.27 Cases of breast and colon cancer have been documented among young adults living with DBA.28
Pathogenic germline variants in a variety of genes involved in ribosomal and ribosomal RNA (rRNA) processing can contribute to oncogenesis and are collectively referred to as ribosomopathies.29 The ribosome maturation factor SBDS has been implicated in the maintenance of genomic stability and the stromal microenvironment. Loss of SBDS function may contribute to dysregulated cellular proliferation via its effects on the microenvironment through increased expression of osteoprotegerin and VEGF-A.30 However, the absence of wild-type SBDS may also contribute to somatic mutations due to its known function in mitotic spindle formation and MTOC assembly in neutrophils.30 Ribosomopathies may also be linked downstream to cellular stress and metabolic derangements that promote cancer development.29 The molecular mechanisms whereby ribosomal and rRNA processing dysfunctions lead to cancer remains an important question in the field, but it appears to involve multiple pathways including genetic integrity, cellular proliferation, and metabolism that may ultimately converge onto enhanced clonal diversification.
Predisposition Processes and Tissue-Specificity in Cancer Risk
Although mutagenesis is driven by elevated replication error rate and cellular proliferation, the development of cancer occurs in a tissue-microenvironment where both cell-autonomous and non-cell-autonomous factors need to be carefully considered. The mechanism by which CPGs predispose to cancer types in a highly tissue-specific manner remains a long-standing question in cancer genetics. For example, pathogenic variation in BRCA1/2 is predominantly associated with breast and ovarian cancers, and, to a lesser degree, prostate cancer, pancreatic cancer, and melanoma. There is a differential risk of cancer for each tissue type between the two genes, and even within genes, the penetrance of pathogenic variants differs. The cumulative breast cancer risk to age 80 is about 70% for BRCA1/2 carriers, while the cumulative ovarian cancer risk for BRCA1 and BRCA2 is only 44% and 17%, respectively.31 It is possible that the unique stromal and immune cell interactions as well as hormonal sensitivities, in a microenvironment, affect an individual’s cancer risk. For example, while replicative errors may result in oncogenic mutations that confer precancerous cells a selective growth advantage, these errors may also create neoantigens that can be recognized by the immune system. Recent studies have demonstrated that cancer development occurs through a microevolutionary pressure whereby mutations are significantly biased towards peptides that are not presented by the cancer patient’s specific MHC-I and MHC-II complexes.32,33 In an immune-excluded tissue microenvironment, neoantigens associated with replication error-inducing CPGs may not be detected by the immune system, providing these precancerous cells more opportunities for mutagenic proliferation. Given the different immune accessibility of each tissue, clonal populations likely survive through various pressures of immune-editing, resulting in tissue-specific cancer risks.
Given our synthesis of diverse pathways amounting to cancer risks, how cellular processes such as proliferation signaling, metabolism, and protein translation differ across tissues may also be considered. For example, uncontrolled cellular proliferation is thought to induce hypoxic stress in the tumor microenvironment without a matched functional vasculature.34 As a result, the HIF-1alpha pathway is frequently activated by tumors. This results in the stimulation of angiogenic pathways to improve tumor oxygenation, an increased propensity for anaerobic metabolism via the Warburg Effect, as well as activation of pro-proliferative and pro-survival pathways.35 Mutations in many of the metabolic CPGs, such as FH, SDH A/B/C/D, and VHL, have been reported to activate the HIF-1alpha pathway. Pathogenic variants of these metabolic genes may result in the premature activation of the HIF-1alpha pathway, causing an enhanced propensity for dysregulated cellular proliferation and tissue invasion. The degree of hypoxic stress likely varies across microenvironments, contributing to distinct cancer risks across tissue sites. The underlying mechanisms of tissue specificity remain unclear. For example, SDH A/B/C/D mutations predispose to the development of pheochromocytoma/paraganglioma (PCPG). SDH A/B/C/D- and VHL- mutated PCPGs share a pseudo-hypoxic phenotype that likely differs in etiology. VHL-mutated, compared to SDH B-mutated, adrenal medulla tissue expresses higher levels of HIF1-alpha target gene mRNA36 and increased rates of glycolysis.37 These distinctions suggest an alternate pseudo-hypoxic transcriptome that drives oncogenesis via SDH mutations38 in the highly vascularized neuroendocrine tissue where PCPGs arise.
In addition to a predilection for tissue specificity, certain cancer predisposition syndromes demonstrate cell-type specificity. For example, patients with Fanconi Anemia are prone to developing squamous cell carcinomas in a variety of organs including head, neck, and gynecologic tissues.39 It remains to be investigated how specific CPGs pertain to the cell-specific pathway wiring or responses to environmental insults, thus inducing cell-type specific cancer risk.
Altogether, different selective pressures arising from the interactions between CPG-associated pathways and unique tissue-microenvironments may jointly shape the process of cancer development, providing plausible explanations of tissue-specificity of cancer risk in CPG carriers.
Future Directions
Growing evidence demonstrates that genetic predisposition to cancer involves diverse biological functions. Here, we examined how diverse CPGs and pathways may converge onto the cancer hallmarks of elevated somatic mutations and cellular proliferation, both of which can lead to cancer-inducing mutagenesis. These observations amount to a new hypothesis of cancer predisposition that needs to be carefully addressed through innovative methodologies. Several of the novel cancer predisposition pathways discussed herein may contribute to stress-induced somatic mutation acquisition, cellular proliferation and ultimately clonal diversification. Meanwhile, CPGs could also possess unknown roles outside of their primary pathways or known function, so further gene ontology and functional investigation will be imperative to improve our understanding of the genetic pleiotropy and crosslinks that may exist between pathways.
Albeit out of the scope of this review, it may be worth investigating whether predisposition genes nominated through genome-wide association studies (GWAS) may also converge onto pathways affecting somatic mutations and cellular proliferation. Given that the majority of the GWAS-identified variants are found in non-coding regions, improved mapping of genetic variants to their regulatory functions integrating epigenetic, transcriptomic, and chromatin interaction data are required to dissect their underlying genes and pathways. For example, a recent study applying targeted RNA-Seq revealed that breast cancer GWAS risk variants may exert their functional effect through long non-coding RNAs.40
Rapidly-expanding patient cohorts with matched germline/somatic genomes, epigenomes and transcriptomes can clarify the relationship between germline CPGs and somatic cancer hallmarks. For example, studies using these cohorts can reveal the somatic mutation rates and specific mutation types accumulated in different CPG carriers. The mechanisms underlying epigenome and transcriptional alterations in cancer remain in the early stages of understanding. Although hereditary epigenetic deregulations in cancer have been identified, further study of the genome-epigenome interplay is required.41 Further, distinct epigenomic regulation across tissue types may present as another plausible cause of differential cancer risk across tissues.
Genetic perturbation of precancerous experimental models can also help reveal how diverse CPGs affect somatic mutations and cellular proliferation in the early stages of oncogenesis. Recent innovations in genome-editing technologies and large-scale parallel assays allow for genetic engineering and variant assessment, which can reveal the functions of a large array of genes and pathogenic variants of interest.42 Lastly, advances in the ability to simulate the tumor microenvironment in new experimental models43 hold promise for more accurate characterization of CPGs in tissue-specific microenvironmental contexts. Integrating evidence from diverse approaches, including multi-omic integration using patient cohorts and mechanistic investigations using new functional models, will be required to further establish the mechanistic links between CPGs, functional hallmarks, and cancer risk.
Acknowledgments
The authors thank J. Gregory for making the figure and members of the Mount Sinai Computational Omics lab for discussions.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The authors declare no financial support related to this review.
Disclosure
The authors declare no potential conflicts of interest.
References
1. Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017;355(6331):1330–1334. doi:10.1126/science.aaf9011
2. Martincorena I, Roshan A, Gerstung M, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348(6237):880–886. doi:10.1126/science.aaa6806
3. Lee-Six H, Olafsson S, Ellis P, et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature. 2019;574(7779):532–537. doi:10.1038/s41586-019-1672-7
4. Brunner SF, Roberts ND, Wylie LA, et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature. 2019;574(7779):538–542. doi:10.1038/s41586-019-1670-9
5. Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68(4):820–823. doi:10.1073/pnas.68.4.820
6. Huang KL, Mashl RJ, Wu Y, et al. Pathogenic germline variants in 10,389 adult cancers. Cell. 2018;173(2):355–370.e314.
7. Rahman N. Realizing the promise of cancer predisposition genes. Nature. 2014;505(7483):302–308. doi:10.1038/nature12981
8. Lu C, Xie M, Wendl MC, et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat Commun. 2015;6(1):10086. doi:10.1038/ncomms10086
9. Garber JE, Offit K. Hereditary cancer predisposition syndromes. J Clin Oncol. 2005;23(2):276–292. doi:10.1200/JCO.2005.10.042
10. Chae YK, Anker JF, Carneiro BA, et al. Genomic landscape of DNA repair genes in cancer. Oncotarget. 2016;7(17):23312–23321. doi:10.18632/oncotarget.8196
11. Cunniff C, Bassetti JA, Ellis NA. Bloom’s syndrome: clinical spectrum, molecular pathogenesis, and cancer predisposition. Mol Syndromol. 2017;8(1):4–23. doi:10.1159/000452082
12. Findlay GM, Daza RM, Martin B, et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature. 2018;562(7726):217–222. doi:10.1038/s41586-018-0461-z
13. Friedenson B. BRCA1 and BRCA2 pathways and the risk of cancers other than breast or ovarian. MedGenMed. 2005;7(2):60.
14. Williams AB, Schumacher B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med. 2016;6(5):a026070. doi:10.1101/cshperspect.a026070
15. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11(12):834–848. doi:10.1038/nrm3012
16. Arighi E, Borrello MG, Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 2005;16(4–5):441–467.
17. Saito-Diaz K, Chen TW, Wang X, et al. The way Wnt works: components and mechanism. Growth Factors. 2013;31(1):1–31.
18. Ballou LM, Lin RZ. Rapamycin and mTOR kinase inhibitors. J Chem Biol. 2008;1(1–4):27–36. doi:10.1007/s12154-008-0003-5
19. Trovó-Marqui AB, Tajara EH. Neurofibromin: a general outlook. Clin Genet. 2006;70(1):1–13. doi:10.1111/j.1399-0004.2006.00639.x
20. Hanahan D, Weinberg Robert A. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674.
21. Baluk P, Hashizume H, McDonald DM. Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev. 2005;15(1):102–111. doi:10.1016/j.gde.2004.12.005
22. Jorquera R, Tanguay RM. Fumarylacetoacetate, the metabolite accumulating in hereditary tyrosinemia, activates the ERK pathway and induces mitotic abnormalities and genomic instability. Hum Mol Genet. 2001;10(17):1741–1752. doi:10.1093/hmg/10.17.1741
23. Toyokuni S. Role of iron in carcinogenesis: cancer as a ferrotoxic disease. Cancer Sci. 2009;100(1):9–16. doi:10.1111/j.1349-7006.2008.01001.x
24. Hergovich A, Lisztwan J, Barry R, Ballschmieter P, Krek W. Regulation of microtubule stability by the von Hippel-Lindau tumour suppressor protein pVHL. Nat Cell Biol. 2003;5(1):64–70. doi:10.1038/ncb899
25. Lolkema MP, Mans DA, Ulfman LH, Volpi S, Voest EE, Giles RH. Allele-specific regulation of primary cilia function by the von Hippel-Lindau tumor suppressor. Eur J Hum Genet. 2008;16(1):73–78. doi:10.1038/sj.ejhg.5201930
26. Ohh M, Yauch RL, Lonergan KM, et al. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell. 1998;1(7):959–968. doi:10.1016/S1097-2765(00)80096-9
27. Lipton JM, Ellis SR. Diamond-Blackfan anemia: diagnosis, treatment, and molecular pathogenesis. Hematol Oncol Clin North Am. 2009;23(2):261–282. doi:10.1016/j.hoc.2009.01.004
28. Willig TN, Niemeyer CM, Leblanc T, et al. Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 Diamond-Blackfan anemia patients. DBA group of Société d’Hématologie et d’Immunologie Pédiatrique (SHIP), Gesellshaft für Pädiatrische Onkologie und Hämatologie (GPOH), and the European Society for Pediatric Hematology and Immunology (ESPHI). Pediatr Res. 1999;46(5):553–561. doi:10.1203/00006450-199911000-00011
29. Sulima SO, Kampen KR, De Keersmaecker K. Cancer Biogenesis in Ribosomopathies. Cells. 2019;8(3):3. doi:10.3390/cells8030229
30. Myers KC, Davies SM, Shimamura A. Clinical and molecular pathophysiology of Shwachman-Diamond syndrome: an update. Hematol Oncol Clin North Am. 2013;27(1):117–128, ix. doi:10.1016/j.hoc.2012.10.003
31. Kuchenbaecker KB, Hopper JL, Barnes DR, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. 2017;317(23):2402–2416. doi:10.1001/jama.2017.7112
32. Marty Pyke R, Thompson WK, Salem RM, Font-Burgada J, Zanetti M, Carter H. Evolutionary pressure against MHC class II binding cancer mutations. Cell. 2018;175(7):1991. doi:10.1016/j.cell.2018.11.050
33. Marty R, Kaabinejadian S, Rossell D, et al. MHC-I genotype restricts the oncogenic mutational landscape. Cell. 2017;171(6):1272–1283.e1215. doi:10.1016/j.cell.2017.09.050
34. Eales KL, Hollinshead KE, Tennant DA. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis. 2016;5(1):e190. doi:10.1038/oncsis.2015.50
35. Masoud GN, Li W. HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharmaceutica Sinica B. 2015;5(5):378–389. doi:10.1016/j.apsb.2015.05.007
36. Fliedner SM, Kaludercic N, Jiang XS, et al. Warburg effect’s manifestation in aggressive pheochromocytomas and paragangliomas: insights from a mouse cell model applied to human tumor tissue. PLoS One. 2012;7(7):e40949. doi:10.1371/journal.pone.0040949
37. Favier J, Brière JJ, Burnichon N, et al. The Warburg effect is genetically determined in inherited pheochromocytomas. PLoS One. 2009;4(9):e7094. doi:10.1371/journal.pone.0007094
38. Kluckova K, Tennant DA. Metabolic implications of hypoxia and pseudohypoxia in pheochromocytoma and paraganglioma. Cell Tissue Res. 2018;372(2):367–378. doi:10.1007/s00441-018-2801-6
39. Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010;24(3):101–122. doi:10.1016/j.blre.2010.03.002
40. Moradi Marjaneh M, Beesley J, O’Mara TA, et al. Non-coding RNAs underlie genetic predisposition to breast cancer. Genome Biol. 2020;21(1):7. doi:10.1186/s13059-019-1876-z
41. Nebbioso A, Tambaro FP, Dell’Aversana C, Altucci L. Cancer epigenetics: moving forward. PLoS Genet. 2018;14(6):e1007362. doi:10.1371/journal.pgen.1007362
42. Sanchez-Rivera FJ, Jacks T. Applications of the CRISPR-Cas9 system in cancer biology. Nat Rev Cancer. 2015;15(7):387–395. doi:10.1038/nrc3950
43. Neal JT, Li X, Zhu J, et al. Organoid modeling of the tumor immune microenvironment. Cell. 2018;175(7):1972–1988.e1916. doi:10.1016/j.cell.2018.11.021
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.