")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
The Expression of GLAST and GLT1 in a Transient Cerebral Ischemia Mongolian Gerbil Model
Authors Shen Y, Lu H, Xu R , Tian H, Xia X, Zhou FH, Wang L , Dong J , Sun L
Received 13 November 2019
Accepted for publication 10 March 2020
Published 23 March 2020 Volume 2020:16 Pages 789—800
DOI https://doi.org/10.2147/NDT.S238455
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Yuping Ning
Yanling Shen,1,2 Huiling Lu,1 Runnan Xu,1 Haibo Tian,1 Xuewei Xia,1 Fiona H Zhou,3 Liping Wang,3 Jianghui Dong,4 Liyuan Sun1
1Department of Guangxi Key Laboratory of Brain and Cognitive Neuroscience, Guilin Medical University, Guilin, Guangxi 541004, People’s Republic of China; 2Department of Pathology, Affiliated Chenggong Hospital, Xiamen University, Xiamen, Fujian 361000, People’s Republic of China; 3School of Pharmacy and Medical Sciences, University of South Australia, Cancer Research Institute, University of South Australia, Adelaide, SA 5001, Australia; 4College of Pharmacy, Guilin Medical University, Guilin, Guangxi 541199, People’s Republic of China
Correspondence: Jianghui Dong
College of Pharmacy, Guilin Medical University, Guangxi, Guilin 541199, People’s Republic of China
Tel +86-773-5891498
Fax +86-773-5891498
Email [email protected]
Liyuan Sun
Department of Guangxi Key Laboratory of Brain and Cognitive Neuroscience, Guilin Medical University, Guilin 541004, Guangxi, People’s Republic of China
Tel +86-773-5893516
Fax +86-773-5898939
Email [email protected]
Purpose: Excitatory amino acid transporters (EAATs) have an indispensable function in the reuptake of extracellular glutamate. To investigate the relationship and the expression of neuronal and astrocytic markers after brain ischemia, the temporal profile of glial EAATs in both peripheral and core regions of the cortex was examined.
Methods: Transient common carotid artery occlusion was used to induce unilateral transient forebrain ischemia of Mongolian gerbils, and post-ischemic brains (6 h to 2 w) were collected and prepared for immunohistochemical and Western blotting analysis of glutamine synthetase (GS), GLT-1, GLAST, S100β, and NeuN, and for Alizarin red staining of calcium deposits.
Results: The expression of GLAST and GLT-1 were significantly escalated at 6 h both in the core and periphery regions, while reduced from 12 h to 2 w in the core region post-ischemia. GS-positive cells increased at 6 h both in the core and periphery regions, while the density of Alizarin red-positive cells increased and peaked at 12 h in the ischemic cortex. The density of S100β-positive cells decreased in the ischemic core and increased in the periphery region. Immunofluorescence staining showed that S100β and TUNEL double-positive cells increased at 12 h in the core region.
Conclusion: The results of GLT-1 and GLAST expression in the cortex indicate that their up-regulation was time-dependent and occurred in the acute post-ischemia period, whereas their down-regulation was region-dependent and it is involved in the pathological progress of nerve cell and glial cell death, and has a series of cascade reactions.
Keywords: glutamate transporter, forebrain ischemia, astrocyte, neuronal death, Mongolian gerbil
Introduction
Focal ischemic stroke is the most common form of stroke which is a leading cause of morbidity and mortality. Currently, regenerative therapies are limited and symptoms are managed with medication. The study on the changes of molecular events with time after ischemia is of great significance.1 Influx of calcium resulted from ATP-depletion and depolarization induced by brain ischemia is an upstream event of the neuronal injury pathway. In the central nervous system (CNS), the uptake activity of excitatory amino acid transporters (EAATs) primarily controls extracellular glutamate homeostasis. Transporters of glutamate thus play a significance role in the pathophysiology cascade of neuronal injury.2,3 The inhibition or attenuation of glutamate excitotoxicity is considered a potential therapy in neuronal protection after ischemia.
EAAT1 (GLAST), EAAT2 (GLT-1), EAAT3 (EAAC1), EAAT4 and EAAT5 have been identified as a sodium-dependent and distinct high-affinity EAATs. EAAT1 and EAAT2 are mainly localized in astrocytes,2,4-6 while EAAT3, EAAT4 and EAAT5 are widely distributed in neurons.5,7-9 Generally, it is accepted that glutamate uptake of the normal brain mainly depends on the specific glutamate transporters GLAST and GLT-1 in astrocytes, though both astrocytes and neurons contain EAATs.5,10 GLT-1 makes up 1% of the total brain protein in the CNS, is responsible for 95% of glutamate’s transport activity and plays a predominant role in glutamate clearance in the brain.11–15 After being transported into astrocytes, glutamate is transformed into glutamine by glutamine synthetase (GS),16 which is localized to astrocytes and is a critical enzyme in the glutamate-glutamine pathway of the brain.17
Previous studies have reported the expression GLT-1 is altered after ischemia: the level of GLT-1 protein decreased in the cortex on the same side of the ischemic area after transient focal cerebral ischemia or in the CA1 region of the brain after transient global ischemia, while the expression of GLAST was not affected.18,19 Another study showed both GLT-1 and GLAST protein levels were reduced in the ischemic core and were not changed in the ischemic boundary region.20 In contrast, thrombotic focal ischemia increased GLAST expression in the penumbra region after 72 h.21 These studies indicate that the variation in the animal model, ischemic region and the duration of time after the ischemic insults can affect the expression of EAATs differently.22–24
Different to mice, in gerbils, there are no posterior communicating arteries, and nearly a third of the proximal part of the bilateral anterior cerebral arteries have no connecting vessels, and the blood supply of the two sides of the brain is relatively independent.25 A forebrain focal ischemia model for stroke can be established with ease by clipping one common carotid artery. Mongolian gerbil’s ischemia model has high success rate, high survival rate, small damage to animals, reduced additional brain damage and experimental interference caused by animal stress. It is widely used in various kinds of research on neurological dysfunction and injury mechanism of brain ischemia injury.26–28 Unilateral forebrain ischemia mode of Mongolian gerbils, as a highly reproducible late death model, there is a slow maturation process in the infarct after 4 d to 1 w, it is often used to study the progression of nerve injury after ischemia.29–31 Since little is known about the EAAT expression profile in this model of cerebral ischemic, the aims of the current study are to: 1) exam expression of GLAST, GLT-1 and GS in a forebrain focal ischemia model; and 2) to investigate the temporal relationship between astrocyte death and neuron damage.
Materials and Methods
The current work was approved by Guilin Medical University Animal Committee. Male Mongolian gerbils (60–72 g, 16–20 w old, from Sankyo Laboratory Animal Center, Tokyo, Japan) were used and the animal experiments were carried out according to NIH guidelines for the care and use of animals.32
Surgical Procedures
To induce ischemia, 10% chloral hydrate was used to anesthetize the animals, and microvascular clamps were used to occlude the left common carotid artery for 10 min.23 A stroke index (SI) was used to evaluate stroke symptoms from the initial carotid artery occlusion. Brain infarction is developed in animals if the SI value is more than 10 points.33 In this study, animals with SI values between 14 and 17 points were chosen as “post-ischemic animals” to minimize the differences in the degree of ischemic injury. Due to the low mortality rate of this procedure,23 a second 10-min ischemia was implemented in the post-ischemic animals 5 h after the first ischemic procedure. Similar operations were conducted in sham animals (n = 4), however without the occlusion in the left carotid artery. At 6 h, 12 h, 1 d, 4 d, 7 d, and 14 d following ischemia, (n = 4 at each time point) brain tissues were collected from mice for three preparations (n = 28, included sham animals at 1 d): 1) paraffin tissue, 2) frozen tissue and 3) Western blot samples.
Histological Preparation
For paraffin embedding of the brain, animals were anesthetized with diethyl ether, and transcardiac perfused with 4% paraformaldehyde in PBS (PFA). Then, the fixed brain slices used for paraffin embedding were prepared by coronally cutting at superior colliculus level (3 mm posterior to the bregma), chiasmal level (0.5 mm anterior to the bregma), and infundibular level (1.5 mm posterior to the bregma). The paraffin-embedded brain slices were section at 4 µm and mounted on glass slides. For frozen OCT embedding, animals were perfused with 4% PFA, the brains were processed into slices as mentioned above followed by dehydration using different concentrations of sucrose (low to high). Next, brain slices were embedded in OCT compound and 16 µm sections were mounted on to glass slides.
Regions of Interest and Morphometric Analysis
In accordance with the previous study,22,31 the regions of interest (ROIs) anterior to bregma for 0.5 mm in the coronal plane were defined for evaluating the expression of GLAST, GLT-1, GS and neuronal and astrocytic markers.31 The cortical columns with the width of 0.5 mm (ie, the ROIs, Figure 1) were perpendicularly placed toward the cortical surface that corresponded to 5.7 mm at the left side of the brain midline, and 2.5 mm to the left side of the brain midline, where the selective neuronal death at 14 d after ischemia will be examined.22,31
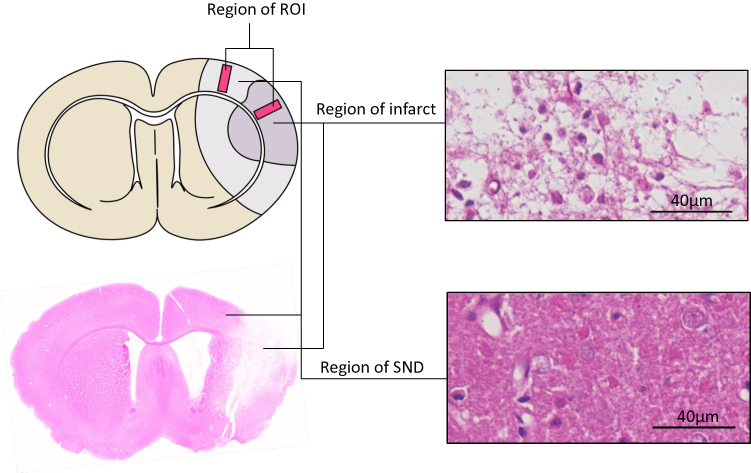 |
Figure 1 Define regions of interest (ROIs). Schematic drawing of infarction and SND area, pink sliver indicate ROIs, the ROIs – cortical columns 0.5 mm in width – were placed perpendicular to the cortical surface which correspond to 5.7 mm left of the midline, where normal tissue developed infarction (right, above), and 2.5 mm left of the midline, where selective neuronal death (right, below) was consistently observed 2 w after ischemia. Bar scale=40 µm. |
A microscope at a magnification of x200 with a drawing tube (Nikon, Tokyo, Japan) was used to count the numbers of immuno-positive and Alizarin red-positive cells in all cortical layers of each ROI (0.85 mm2).
Histological Assessment
Hematoxylin & Eosinophil (HE) staining was used for histological analysis. The samples were collected at 6 h, 12 h, 4 d and 2 w, and the sham group was analyzed, respectively. The paraffin embedding tissues were dewaxed 3 times in xylene for 10 min followed by routine washes of gradient alcohol and distilled water to rehydrate the sections. Routine HE staining was performed and morphological changes were examined and captured under a light microscope. The cells with intact nuclear structure and no red staining in the cytoplasm are living cells. The changes of ischemic cells showed homogeneous red staining of cytoplasm and pyknosis of dead neurons.
In order to detect the formation of calcium deposits in the ischemic brain, 1% Alizarin red (AMR, Solon, Ohio, USA) was used to stain the sections for 10 min as described in our previous work.31
Immunohistochemistry
The main antibodies in this study are listed: guinea pig anti-glutamate transporter GLAST (Chemicon, Temecula, CA, USA; 1:500 dilution); guinea pig anti-glutamate transporter GLT-1 (Chemicon, Temecula, CA, USA; 1:500); anti-glutamine synthetase of mouse (Chemicon, Temecula, CA, USA; 1:1000) for examining glutamine conversion; mouse anti-NeuN (Chemicon, Temecula, CA, USA; 1:2000), a neuronal marker; and mouse anti-S100β (β-subunit) (Sigma, Temecula, CA, USA; 1:500), an astrocyte marker.
After dewaxing the paraffin embedding sections as described above in the histology assessment, antigen retrieval was performed. The sections were placed in sodium citrate buffer (0.01 M, pH 6.0) and heated to 95°C in a water bath, for 10–15 mins. To reduce endogenous peroxidase activity, brain sections were incubated in 3% H2O2 for 30 mins, and rinsed with PBS, before blocking in 10% normal goat serum for 30 min at room temperature. Brain sections (frozen) were incubated in various diluted primary antibodies in goat serum at 4°C overnight listed above. After rinsing in PBS, the sections were further incubated in biotinylated goat anti-guinea mouse (or pig) IgG secondary antibody for 60 min. The immuno-stained sections were incubated in the avidin-biotinylated peroxidase complex (ABC) for 60 min, before adding the diaminobenzidine (DAB) substrate to detect the specific antibodies bound. Finally, IHC pictures were obtained by a bright light microscope (BX53, Olympus Corporation, Japan).
Immunofluorescence Staining
After incubation with a mouse monoclonal antibody against S100β (dilution in 1:500 for overnight at 4°C) and anti-mouse Ig-rhodamine (dilution in 1:200 at 37°C for 1 h), the sections were rinsed with PBS. Then, the sections were incubated with TUNEL (terminal deoxynucleotidyl transferase-mediated deoxyuridinetri-phosphate nick-end labeling) reaction mixture (in situ cell death detection kit, POD Roche, GER) at 37°C for 1 h, which included TUNEL label, fluorescein and TUNEL enzyme. The negative control sections for TUNEL staining were treated with the labelling solution rather than the TUNEL reaction mixture. Double-positive cells were observed by a Zeiss M510 laser scanning confocal imaging system.
Western Blotting Analysis
For isolation of total protein, animals were anesthetized with diethyl ether, perfused with PBS, and the cortex tissues in regions of interest (Figure 1) were dissected and homogenized. In order to accurately examine the protein expression of GLT-1 and GLAST, each dissected tissue sample was homogenized in 25 mM Tris-HCL (pH 6.8) buffer plus protease inhibitors (benzenesulfonyl fluoride, bestatin, leupeptin and aprotinin) and 2 mM EDTA on ice. The homogenate was centrifuged at 11,000 g at 4°C for 300 s, and the non-soluble protein pellet was resuspended in fresh buffer (sodium dodecyl sulfate 2%, glycerol 25%, Tris 60 mM and h-mercaptoethanol 14.4 mM), then was further centrifuged at 20,000 g for 30 min.34 The concentration of protein in the supernatant was examined by protein assay using Coomassie blue. We electrophoresed the proteins on polyacrylamide gels, transferred proteins on to nitrocellulose membranes and probed with polyclonal affinity-purified antibodies, such as anti-GLAST and anti-GLT-1, then the HRP-conjugated goat anti-guinea pig IgG. Subsequently, immunoreactive proteins were visualized using the ECL Western blotting kit (Amersham Pharmacia Biotech, Piscataway, NJ). Immunoblot intensities were standardized by reprobing the same blot with β-tubulin antibody. The density of protein bands detected was quantified using the NIH Image program.
Statistical Analysis
The data were analyzed using the repeated-measures analysis of variance (ANOVA) and day of experiment was regarded as the argument in the analysis. The Bonferroni post hoc test was applied for multiple tests among the different groups. All values were presented as mean ± SEM. The level of statistical significance was set at P < 0.05.
Results
Immunostaining showed that both GLAST and GLT-1 were activated at 6 h post-ischemia both in the core of ischemia and periphery followed by a distinct decrease in the ischemic core region (Figure 2). Western blot analysis (Figure 3A–D) showed that the protein bands of GLAST and GLT-1 were 58.1 ± 0.2% and 48.2 ± 0.1%, respectively, in the sham animals and peaked at 6 h (88.5 ± 8.4% and 74.8 ± 15%), and then decreased from 12 h to 4 d in the ischemic core (Figure 3A and C). The level of GLAST and GLT-1 also showed overexpression at 6 h in the ischemic periphery (Figure 3B and D).
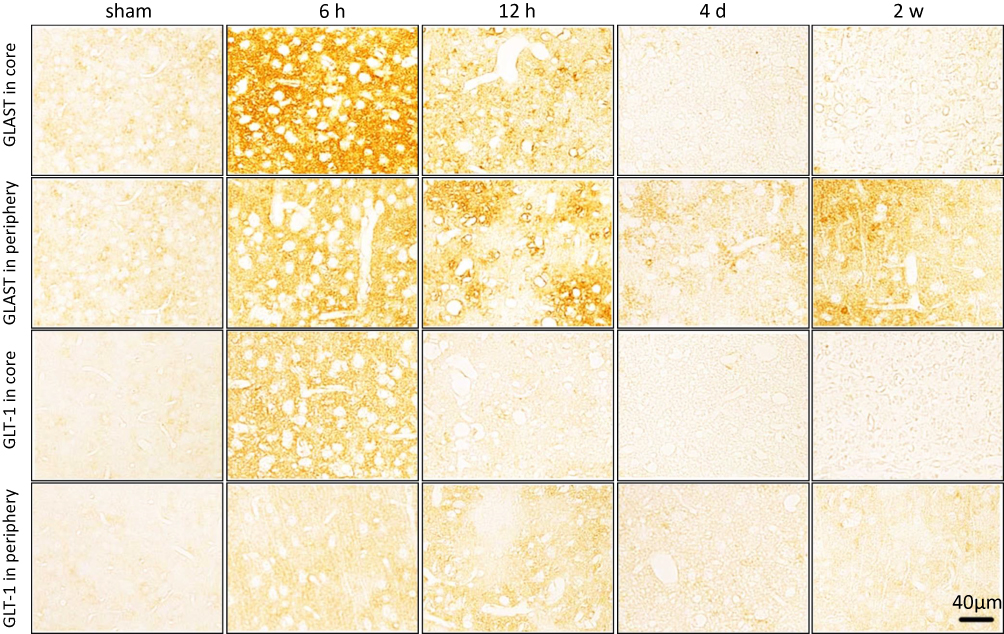 |
Figure 2 Representative images of immunohistochemical staining of GLAST and GLT-1 glutamate transporters in the ischemic core and periphery regions of the cortex. Both GLAST and GLT-1 were activated at 6 h post-ischemia following a distinct decrease from 12 h in the ischemic core region while GLT-1 levels remained increased at 12 h in the ischemic periphery region. Bar scale= 40 µm. |
 |
Figure 3 Western blot analysis of GLAST and GLT-1 in the ischemic core (A) and periphery (B) regions of the cortex. Upper rows show representative protein bands for GLAST and GLT-1, and lower rows show the re-probed β-tubulin protein bands. Quantitative analysis of the intensity level of GLAST and GLT-1 protein normalized to β-tubulin peaking at 6 h and decreasing from 12 h to 4 d in the ischemic core (C) and periphery (D) regions of the brain. **P < 0.01, *P < 0.05, compared to sham; #P < 0.05, compared to 6 h. |
A similar pattern was observed in the expression of GS. Compared to the sham animals where the density of GS-positive glial cells, the density of GS-positive cells significantly increased at 6 hand decreased from 12 h to 2 w (Figure 4).
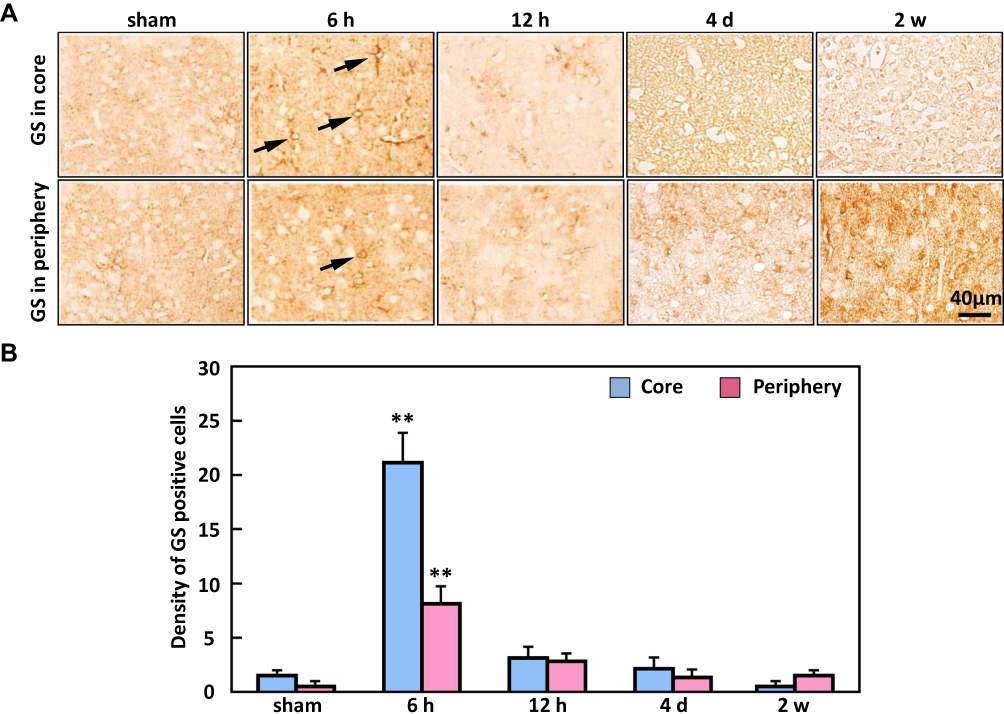 |
Figure 4 The expression of glutamine synthetase (GS) in the ischemic core and periphery regions of the cortex. (A) Representative immunostaining showed that GS-positive cells (arrows) were significantly increased at 6 h in both the core and periphery. (B) The density of glutamine synthetase peaked at 6 h in the ischemic core and periphery regions. Bar scale = 40 µm. n=20, **P < 0.01, compared to sham. |
To detect calcium deposits in neuronal cells as a result of ischemia brain injury, Alizarin red staining indicated that the colour density significantly increased from 12 h (50 ± 9% and 236.3 ± 35.5%) in the ischemic periphery region and core region, respectively, compared to the minimal levels determined in the non-ischemic periphery region and the core region of the sham animals (Figure 5). In contrast to the raise in Alizarin red-positive cells at 12 h, the immunostaining density of NeuN-positive cells was markedly reduced in the ischemic core region from 12 h (357.6 ± 77%) compared to sham, while the reduction in the periphery region occurred from 4 d (Figure 6).
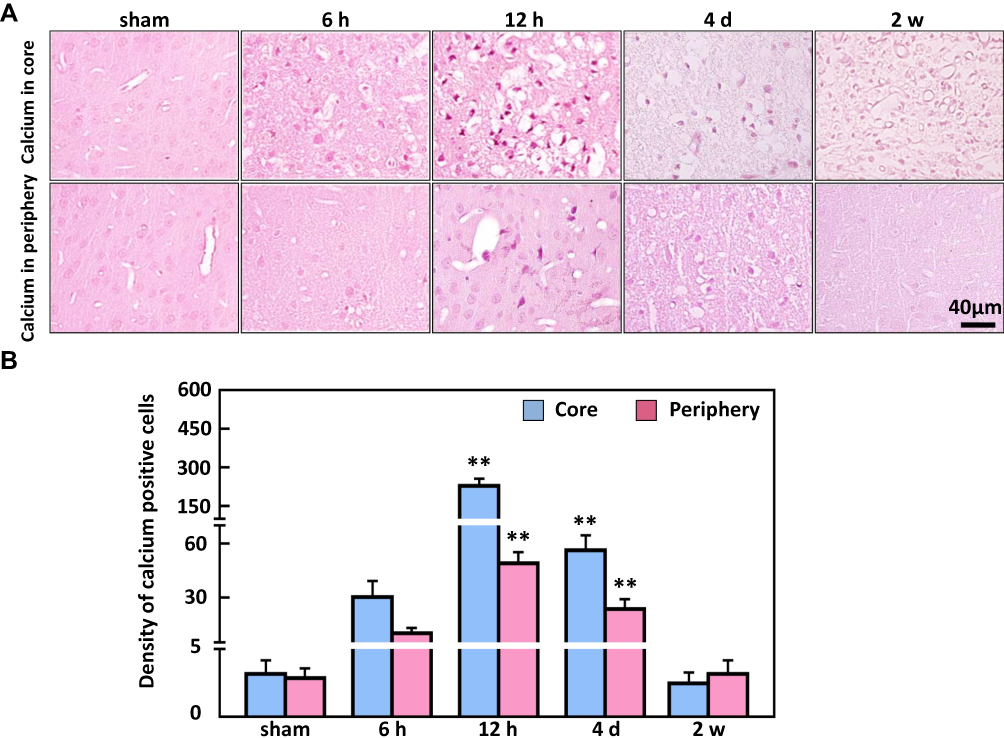 |
Figure 5 The analysis of Alizarin red-positive cells in the ischemic core and periphery regions of the cortex. (A) Representative images of Alizarin red staining (B) showing the density of calcium-deposited in cells peaked at 12 h in the ischemic core and periphery regions of the brain. Bar scale= 40 µm. **P < 0.01, compared to sham. |
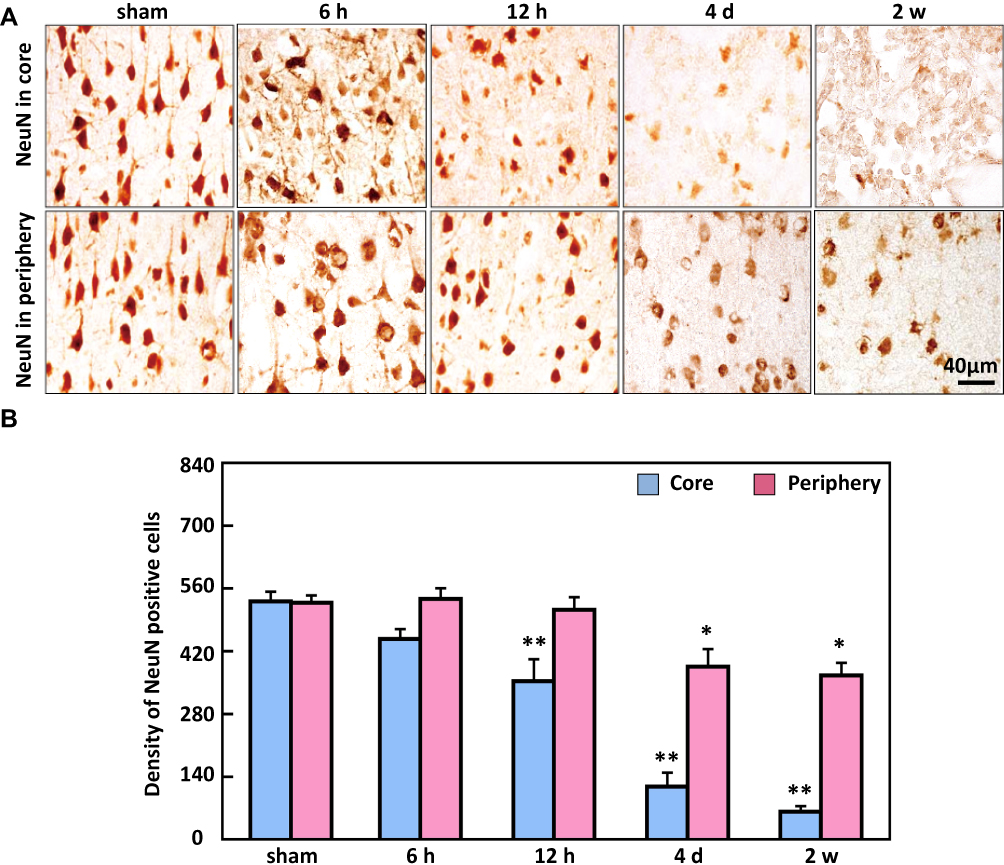 |
Figure 6 The analysis of NeuN-positive cells in the ischemic core and periphery regions of the cortex. (A) Representative images of NeuN immunohistochemical staining. (B) The density of the NeuN-positive cells significantly decreased first in the ischemic core region from 12 h then decreased from 4 d t in the ischemic periphery region. Bar scale= 40 µm. **P < 0.01, *P < 0.05, compared with sham. |
The density of the S100β-positive cells was 81.33 ± 11% in the sham animals, and decreased significantly from 12 h (41 ± 15%) to 2 w in the ischemic core region; the density (112.3 ± 22.5%) was significantly increased at 2 w in the ischemic periphery region (Figure 7).
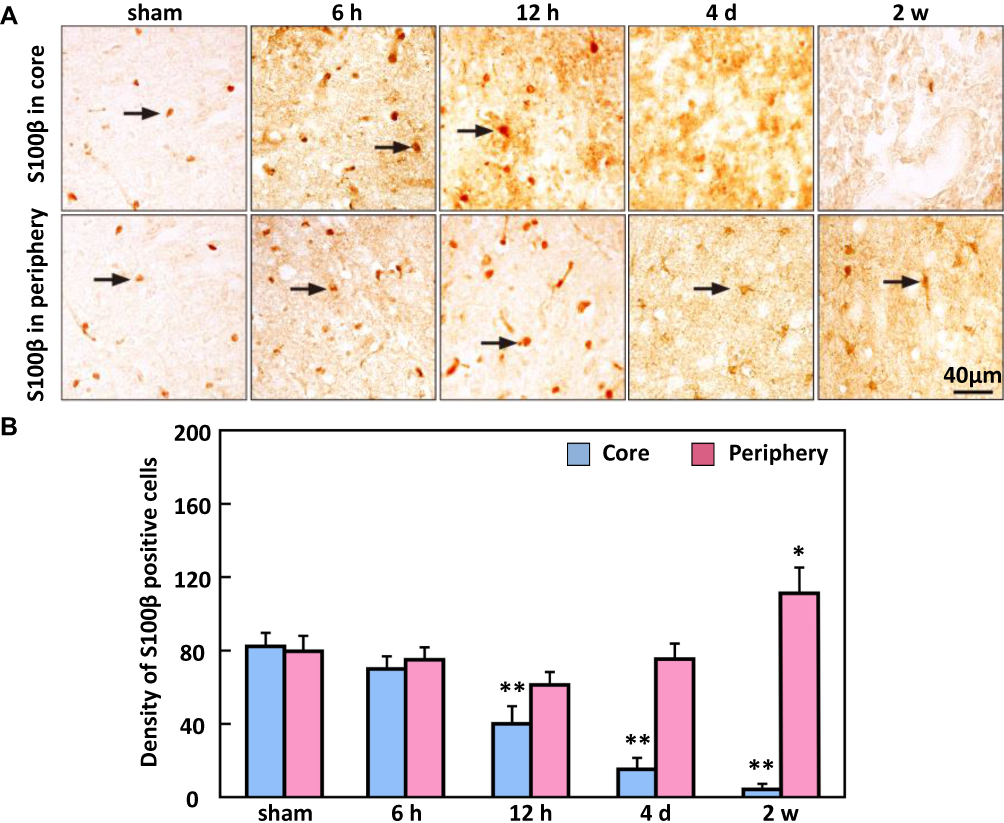 |
Figure 7 The expression of S100β in the ischemic core and periphery regions of the cortex. (A) Representative images of S100β immunohistochemical staining. (B) The density of S100β-positive cells significantly decreased from 12 h in the ischemic core region while in the ischemic periphery regions S100β staining density significantly increased by 2 w. Arrows show that S100β-positive cells. Bar scale=40 µm. **P < 0.01, *P < 0.05, compared to sham. |
Based on the immunofluorescence staining, S100β-TUNEL double-positive cells (Figure 8) were not observed in sham animals. However, they were significantly increased at 12 h (16 ± 3.6%) in the ischemic core of the cortex, and few were observed at 4 d in the ischemic periphery of the cortex (Figure 8).
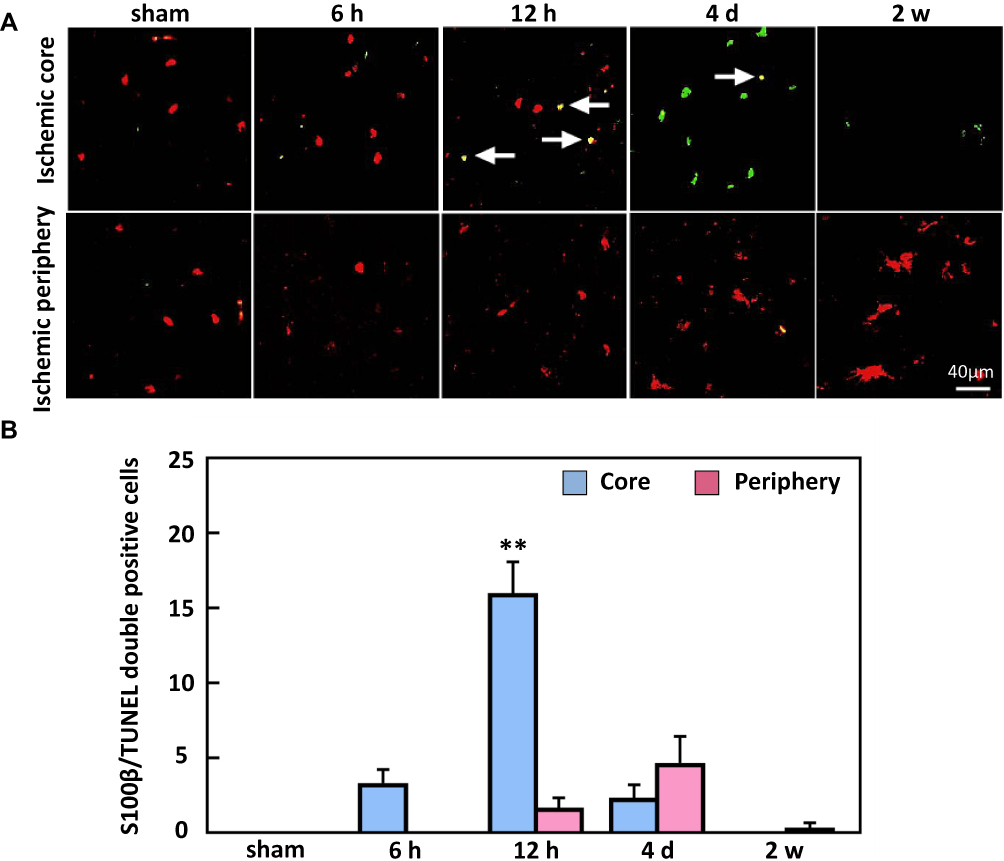 |
Figure 8 Double immunofluorescence staining of S100β and TUNEL. (A) Representative images indicate S100β and TUNEL double-positive cells, and (B) the density of double-positive cells significantly increased by 4 d post-ischemia in the core region of the cortex. Arrows show that double-positive cells. Bar scale=40 µm. **P < 0.01, compared to sham. |
Discussion
The role of EAATs in regulating the Glu concentration of the synaptic gap during cerebral ischemia is still controversial.7 The expression of GLAST and GLT-1 in glial cells is reduced in the early stage of cerebral ischemia,35,36 while increased in these two proteins during chronic cerebral ischemia.37,38 Therefore, the expression of GLAST and GLT-1 is significantly related to the progress of cerebral ischemia injury, but there is little detailed study on the dependence of GLAST and GLT-1, and the correlation between astrocyte death, neuron injury and their expression change. Here, using our previously established mild ischemia model,31 which includes selective neuronal death and cortical infarcts, we examined the expression of GLT-1 and GLAST protein after ischemia. The results obtained indicate that the protein levels of GLT-1 and GLAST were transiently increased in the acute period in the cortex after focal ischemia, and followed by a rapid decrease in the ischemic core that was related to the loss of neurons and astrocytes. The up-regulation situation of GLT-1 and GLAST was time-dependent and occurred in the acute post-ischemia period, whereas the down-regulation was region-dependent occurred in the core, which might be related to the loss of neurons and astrocytes due to the decrease of NeuN and S100b staining.
Glutamate exposure rapidly up-regulates GLAST expression in cultured astrocytes.39 The acute increase in GLAST-positive cells and GLAST protein expression following hypoxia-ischemia suggests that this increase may be a self-compensatory mechanism to protect neurons from further injury.40 Based on the results in this study, it can be speculated that the down-regulation of GLT-1 and GLAST might be involved in the acute neuronal damage in the ischemic cortex.
The clearance of Glu in the physiological state mainly depends on the reuptake of glutamate transporter to terminate its synaptic transmission effect. Under ischemic conditions, glutamate transporters EAATs might be functionally reversed and may release rather than reuptake glutamate in the brain.41,42 GLT-1 absorbs extracellular glutamate to preserve neurons in the early stages of ischemia, and followed its release.43 Here, the increased protein levels of GLT-1 and GLAST were accompanied with increased activated GS-positive cells at 6 h post-ischemia. Since GS localized in astrocytes is a critical enzyme in the glutamate-glutamine pathway in the brain, it is possible that glutamate transported into astrocytes by these EAATs could be converted to glutamine by GS at 6 h post-ischemia. Similarly, the up-regulation of GS expression has been reported in studies of the up-regulation of hippocampal glutamine synthetase in global brain ischemia in rats.16,43
The present study did not examine the levels of extracellular glutamate; however, our results showed that calcium-positive cells were significantly increased from 12 h to 4 d in the ischemic core and the periphery regions of the cortex, which corresponded to the reduced expression of GLT-1 and GLAST. This leads to the accumulation of glutamate in the intercellular space, over activation of glutamate receptors such as NMDAR, calcium overload, mitochondrial dysfunction, activation of calcium-dependent degrading enzyme, promotion of active oxygen generation, destruction of cytoskeleton, and eventually neuronal death. Furthermore, the density of NeuN-positive cells obviously declined in the ischemic core at the same time points which suggests neuronal loss in this stroke model. Several laboratories have noted that the declined expression of glutamate transporters was related to neuronal death following hypoxic-ischemia, forebrain ischemia and traumatic brain injury.36,44,45 Our results are consistent with the finding that the down-regulation in GLT-1 and GLAST expression is related to excitotoxicity and neuronal injury in the ischemic core region. Moreover, there was no obvious decrease in the expression of GLT-1 and GLAST in the ischemic periphery region, when the delayed selective neuronal death occurred from 4 d to 2 w.
Our study found that the decrease in S100β-positive cells and the increase in S100β-TUNEL double-positive cells coincided with the reduction in expression of GLAST and GLT-1. Consistent with our research results, a previous study reported that S100β immunoreactivity was markedly and immediately decreased in the core of the injured area after reperfusion and did not increase again. In the periphery, however, S100β expression increased after transient MCAO.46 Thus, it is possible to consider increasing astrocyte glutamate transporters in order to block the ischemic astrocytic injury pathway.
In the ischemia core region, our results showed a drastic reduction of S100β single-positive cells from 12 h to 4 d, while S100β-TUNEL double-positive cells peaked at 12 h. This suggests that apoptosis of astrocytes may be the reason for the reduction in their numbers. However, TUNEL single-positive cells increased and peaked at 4 d post-ischemia, and these positive cells were mainly eosinophilic neurons as shown in our previous study.31 In the ischemia periphery region, our results showed a few S100β/TUNEL double-positive cells at 4 d post-ischemia, and the numbers of S100β-positive astrocytes increased at 2 w, which corresponds to conventional gliosis. Therefore, the time and regional dependence of GLAST and GLT-1 expression in ischemic tissues, as well as S100 β - TUNEL staining results suggest that their changes or involvement in the pathological process of the nerve cell and glial cell death lead to a series of cascade reactions, which is similar to the previous research conclusions.47,48 However, it was observed that a transient increase in GLT-1 and GLAST in Mongolian gerbils during the acute phase through a transient cerebral ischemia model. In addition, although the glutamate transporter and glutamine enzyme were quantified, the glutamate concentration in the brain was not directly detected. The direct detection of glutamate content can help us to understand the relationship between the expression of glutamate transporter in the acute stage of cerebral ischemia and the damage to neurons and astrocytes.
Conclusion
Using a gerbil focal ischemic stroke model, we showed the up-regulation in GLT-1 and GLAST expressions in the cortex were time-dependent and occurred in the acute post-ischemia period, while the down-regulation was region-dependent and correlated with the loss of neurons and astrocytes in the core region.
Acknowledgments
This work was supported by the funding of the National Natural Science Foundation of China (Grant no. 81260205) and Guangxi Natural Science Foundation (Grant nos. 2016GXNSFCA380028 and 2018GXNSFAA281246).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Wahul AB, Joshi PC, Kumar A, Chakravarty S. Transient global cerebral ischemia differentially affects cortex, striatum and hippocampus in Bilateral Common Carotid Arterial occlusion (BCCAo) mouse model. J Chem Neuroanat. 2018;92:1–15. doi:10.1016/j.jchemneu.2018.04.006
2. Kashem MA, Sultana N, Pow DV, Balcar VJ. GLAST (GLutamate and ASpartate Transporter) in human prefrontal cortex; interactome in healthy brains and the expression of GLAST in brains of chronic alcoholics. Neurochem Int. 2019;125:111–116. doi:10.1016/j.neuint.2019.02.009
3. Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65(1):1–105. doi:10.1016/S0301-0082(00)00067-8
4. Mahmoud S, Gharagozloo M, Simard C, Amrani A, Gris D. NLRX1 enhances glutamate uptake and inhibits glutamate release by astrocytes. Cells. 2019;8(5):400. doi:10.3390/cells8050400
5. Suárez-Pozos E, Thomason EJ, Fuss B. Glutamate Transporters: expression and function in oligodendrocytes. Neurochem Res. 2019;1–10.
6. Martinez-Lozada Z, Guillem AM, Robinson MB. Transcriptional regulation of glutamate transporters: from extracellular signals to transcription factors. Adv Pharmacol. 2016;76:103–145.
7. Zhang L-N, Hao L, Guo Y-S, et al. Are glutamate transporters neuroprotective or neurodegenerative during cerebral ischemia? J Mol Med. 2019;97(3):281–289. doi:10.1007/s00109-019-01745-5
8. De Freitas AP, Ferreira DDP, Fernandes A, et al. Caffeine alters glutamate-aspartate transporter function and expression in rat retina. Neuroscience. 2016;337:285–294. doi:10.1016/j.neuroscience.2016.09.028
9. Young K, Singh G. Biological mechanisms of cancer-induced depression. Front Psychiatry. 2018;9:299. doi:10.3389/fpsyt.2018.00299
10. Pajarillo E, Rizor A, Lee J, Aschner M, Lee E.The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: potential targets for neurotherapeutics. Neuropharmacology.2019;161:107559. doi:10.1016/j.neuropharm.2019.03.002
11. Pregnolato S, Chakkarapani E, Isles AR, Luyt K. Glutamate transport and preterm brain injury. Front Physiol. 2019;10:417. doi:10.3389/fphys.2019.00417
12. Rimmele TS, Rosenberg PA. GLT-1: the elusive presynaptic glutamate transporter. Neurochem Int. 2016;98:19–28. doi:10.1016/j.neuint.2016.04.010
13. Scofield MD, Kalivas PW. Astrocytic dysfunction and addiction: consequences of impaired glutamate homeostasis. Neuroscientist. 2014;20(6):610–622. doi:10.1177/1073858413520347
14. Perisic T, Holsboer F, Rein T, Zschocke J. The CpG island shore of the GLT-1 gene acts as a methylation-sensitive enhancer. Glia. 2012;60(9):1345–1355. doi:10.1002/glia.v60.9
15. Takahashi K, Foster JB, Lin C-LG. Glutamate transporter EAAT2: regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell Mol Life Sci. 2015;72(18):3489–3506. doi:10.1007/s00018-015-1937-8
16. Lemberg A, Fernández MA. Hepatic encephalopathy, ammonia, glutamate, glutamine and oxidative stress. Ann Hepatol. 2009;8(2):95–102. doi:10.1016/S1665-2681(19)31785-5
17. Petito CK, Chung MC, Verkhovsky LM, Cooper AJ. Brain glutamine synthetase increases following cerebral ischemia in the rat. Brain Res. 1992;569(2):275–280. doi:10.1016/0006-8993(92)90639-Q
18. Rao VLR, Bowen KK, Dempsey RJ. Transient focal cerebral ischemia down-regulates glutamate transporters GLT-1 and EAAC1 expression in rat brain. Neurochem Res. 2001;26(5):497–502. doi:10.1023/A:1010956711295
19. Yeh T, Hwang H, Chen J, Wu T, Li A, Wang H. Glutamate transporter function of rat hippocampal astrocytes is impaired following the global ischemia. Neurobiol Dis. 2005;18(3):476–483. doi:10.1016/j.nbd.2004.12.011
20. Fukamachi S, Furuta A, Ikeda T, et al. Altered expressions of glutamate transporter subtypes in rat model of neonatal cerebral hypoxia-ischemia. Dev Brain Res. 2001;132(2):131–139. doi:10.1016/S0165-3806(01)00303-0
21. Yan Y-P, Yin K-J, Sun F-Y. Effect of glutamate transporter on neuronal damage induced by photochemical thrombotic brain ischemia. Neuroreport. 1998;9(3):441–446. doi:10.1097/00001756-199802160-00016
22. Sun L, Kuroiwa T, Ishibashi S, Katsumata N, Endo S, Mizusawa H. Transition of areas of eosinophilic neurons and reactive astrocytes to delayed cortical infarcts after transient unilateral forebrain ischemia in Mongolian gerbils. Acta Neuropathol. 2006;111(1):21–28. doi:10.1007/s00401-005-1081-x
23. Kuroiwa T, Yamada I, Endo S, Hakamata Y, Ito U. 3-Nitropropionic acid preconditioning ameliorates delayed neurological deterioration and infarction after transient focal cerebral ischemia in gerbils. Neurosci Lett. 2000;283(2):145–148. doi:10.1016/S0304-3940(00)00937-X
24. Ouyang Y, Xu L, Liu S, Giffard R. Role of astrocytes in delayed neuronal death: GLT-1 and its novel regulation by MicroRNAs. Adv Neurobiol. 2014;11:171–188.
25. Levy DE, Brierley JB. Communications between vertebro-basilar and carotid arterial circulations in the gerbil. Exp Neurol. 1974;45(3):503–508. doi:10.1016/0014-4886(74)90155-1
26. Ahn JH, Song M, Kim H, et al. Differential regional infarction, neuronal loss and gliosis in the gerbil cerebral hemisphere following 30 min of unilateral common carotid artery occlusion. Metab Brain Dis. 2019;34(1):223–233. doi:10.1007/s11011-018-0345-9
27. Noguchi K, Ali TF, Miyoshi J, et al. Neuroprotective effects of a novel carnosine-hydrazide derivative on hippocampal CA1 damage after transient cerebral ischemia. Eur J Med Chem. 2019;163:207–214. doi:10.1016/j.ejmech.2018.11.060
28. Pegorini S, Braida D, Verzoni C, et al. Capsaicin exhibits neuroprotective effects in a model of transient global cerebral ischemia in Mongolian gerbils. Br J Pharmacol. 2005;144(5):727–735. doi:10.1038/sj.bjp.0706115
29. Laidley DT, Colbourne F, Corbett D. Increased behavioral and histological variability arising from changes in cerebrovascular anatomy of the Mongolian gerbil. Curr Neurovasc Res. 2005;2(5):401–407. doi:10.2174/156720205774962719
30. Hanyu S, Ito U, Hakamata Y, Yoshida M. Repeated unilateral carotid occlusion in Mongolian gerbils: quantitative analysis of cortical neuronal loss. Acta Neuropathol. 1993;86(1):16–20. doi:10.1007/BF00454893
31. Sun L, Kuroiwa T, Ishibashi S, et al. Two region-dependent pathways of eosinophilic neuronal death after transient cerebral ischemia. Neuropathology. 2009;29(1):45–54. doi:10.1111/neu.2008.29.issue-1
32. Council NR. Guide for the Care and Use of Laboratory Animals. National Academies Press; 2010.
33. Ohno K, Ito U, Inaba Y. Regional cerebral blood flow and stroke index after left carotid artery ligation in the conscious gerbil. Brain Res. 1984;297(1):151–157. doi:10.1016/0006-8993(84)90552-3
34. Shen Y, Dai L, Tian H, et al. Treatment of magnesium-L-threonate elevates the magnesium level in the cerebrospinal fluid and attenuates motor deficits and dopamine neuron loss in a mouse model of Parkinson’s disease. Neuropsychiatr Dis Treat. 2019;15:3143–3153.
35. Torp R, Lekieffre D, Levy L, et al. Reduced postischemic expression of a glial glutamate transporter, GLT1, in the rat hippocampus. Exp Brain Res. 1995;103(1):51–58. doi:10.1007/BF00241964
36. Harvey BK, Airavaara M, Hinzman J, et al. Targeted over-expression of glutamate transporter 1 (GLT-1) reduces ischemic brain injury in a rat model of stroke. PLoS One. 2011;6(8):e22135. doi:10.1371/journal.pone.0022135
37. Yatomi Y, Tanaka R, Shimura H, et al. Chronic brain ischemia induces the expression of glial glutamate transporter EAAT2 in subcortical white matter. Neuroscience. 2013;244:113–121. doi:10.1016/j.neuroscience.2013.04.018
38. Inage Y, Itoh M, Wada K, Takashima S. Expression of two glutamate transporters, GLAST and EAAT4, in the human cerebellum: their correlation in development and neonatal hypoxic-ischemic damage. J Neuropathol Exp Neurol. 1998;57(6):554–562. doi:10.1097/00005072-199806000-00003
39. Duan S, Anderson C, Stein B, Swanson R. Glutamate induces rapid upregulation of astrocyte glutamate transport and cell-surface expression of GLAST. J Neurosci. 1999;19(23):10193–10200. doi:10.1523/JNEUROSCI.19-23-10193.1999
40. Mourão FAG, Leite HR, de Carvalho LED, et al. Neuroprotective effect of exercise in rat hippocampal slices submitted to in vitro ischemia is promoted by decrease of glutamate release and pro-apoptotic markers. J Neurochem. 2014;131(1):65–73. doi:10.1111/jnc.12786
41. Roettger V, Lipton P. Mechanism of glutamate release from rat hippocampal slices during in vitro ischemia. Neuroscience. 1996;75(3):677–685. doi:10.1016/0306-4522(96)00314-4
42. Rossi D, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403(6767):316–321. doi:10.1038/35002090
43. Zhang W, Miao Y, Zhou S, Jiang J, Luo Q, Qiu Y. Neuroprotective effects of ischemic postconditioning on global brain ischemia in rats through upregulation of hippocampal glutamine synthetase. J Clin Neurosci. 2011;18(5):685–689. doi:10.1016/j.jocn.2010.08.027
44. Ma X-L, Zhang F, Wang Y-X, et al. Genistein inhibition of OGD-induced brain neuron death correlates with its modulation of apoptosis, voltage-gated potassium and sodium currents and glutamate signal pathway. Chem Biol Interact. 2016;254:73–82. doi:10.1016/j.cbi.2016.05.033
45. Ferrari F, Gorini A, Hoyer S, Villa RF. Glutamate metabolism in cerebral mitochondria after ischemia and post-ischemic recovery during aging: relationships with brain energy metabolism. J Neurochem. 2018;146(4):416–428. doi:10.1111/jnc.14464
46. Li C, Zhang B, Tian S, et al. Early wheel-running promotes functional recovery by improving mitochondria metabolism in olfactory ensheathing cells after ischemic stroke in rats. Behav Brain Res. 2019;361:32–38. doi:10.1016/j.bbr.2018.12.038
47. O’shea R. Roles and regulation of glutamate transporters in the central nervous system. Clin Exp Pharmacol Physiol. 2002;29(11):1018–1023. doi:10.1046/j.1440-1681.2002.03770.x
48. Perego C, Vanoni C, Bossi M, et al. The GLT‐1 and GLAST glutamate transporters are expressed on morphologically distinct astrocytes and regulated by neuronal activity in primary hippocampal cocultures. J Neurochem. 2000;75(3):1076–1084. doi:10.1046/j.1471-4159.2000.0751076.x
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.