")
Back to Archived Journals » Oncolytic Virotherapy » Volume 4
The ex vivo purge of cancer cells using oncolytic viruses: recent advances and clinical implications
Received 24 August 2014
Accepted for publication 17 November 2014
Published 23 January 2015 Volume 2015:4 Pages 13—23
DOI https://doi.org/10.2147/OV.S45525
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Faris Farassati
Jovian J Tsang,1,2 Harold L Atkins2,3
1Department of Biochemistry, University of Ottawa, 2Cancer Therapeutics, Ottawa Hospital Research Institute, 3Blood and Marrow Transplant Program, The Ottawa Hospital, Ottawa, ON, Canada
Abstract: Hematological malignancies are treated with intensive high-dose chemotherapy, with or without radiation. This is followed by hematopoietic stem cell (HSC) transplantation (HSCT) to rescue or reconstitute hematopoiesis damaged by the anticancer therapy. Autologous HSC grafts may contain cancer cells and purging could further improve treatment outcomes. Similarly, allogeneic HSCT may be improved by selectively purging alloreactive effector cells from the graft rather than wholesale immune cell depletion. Viral agents that selectively replicate in specific cell populations are being studied in experimental models of cancer and immunological diseases and have potential applications in the context of HSC graft engineering. This review describes preclinical studies involving oncolytic virus strains of adenovirus, herpes simplex virus type 1, myxoma virus, and reovirus as ex vivo purging agents for HSC grafts, as well as in vitro and in vivo experimental studies using oncolytic coxsackievirus, measles virus, parvovirus, vaccinia virus, and vesicular stomatitis virus to eradicate hematopoietic malignancies. Alternative ex vivo oncolytic virus strategies are also outlined that aim to reduce the risk of relapse following autologous HSCT and mitigate morbidity and mortality due to graft-versus-host disease in allogeneic HSCT.
Keywords: hematopoietic stem cells, oncolytic virus, hematopoietic stem cell transplantation, stem cell graft purging, hematopoietic malignancy, graft vs host disease
Introduction
High-dose chemotherapy, with or without radiation, followed by hematopoietic stem cell transplantation (HSCT) is most commonly used for the treatment of malignancies such as acute leukemia, lymphoma, and myeloma. HSCT can rescue or reconstitute hematopoiesis damaged by anticancer therapy, improving outcomes by allowing larger doses to be given. HSCT has also been used for nonmalignant diseases such as immunodeficiencies, hemoglobinopathies, and autoimmune cytopenias, as well as rheumatological, neurological, and gastrointestinal autoimmune disorders.1 The engrafted hematopoietic stem cells (HSCs) function as a vehicle for providing the corrected gene product (providing metabolic enzymes or replacing cells with a mutant hemoglobin gene, such as in sickle cell disease or thalassemia), or for replacing abnormal immune systems (congenital immunodeficiency and autoimmune diseases).
HSCs can be obtained from the bone marrow, from the circulation after mobilization from the marrow, and from umbilical cord blood.2 The use of mobilized peripheral blood stem cell products has become more prevalent due to donor preference, rapidity of engraftment, and the higher yield of stem cells. Both related and unrelated donors (allogeneic) and the patient themselves (autologous) can serve as sources of HSC graft products. Disease type and donor availability are used to determine the type of HSC graft used.
Autologous HSCT is associated with higher relapse rates than allogeneic HSCT.3 Relapse, at least in part, may come from tumor cells contaminating the autologous peripheral blood product.4 Strategies to eliminate tumor contamination have been attempted, such as positive selection of CD34+ HSC from the graft or ex vivo incubation with cytotoxic agents; however, they have largely remained ineffective in altering the outcome of autologous HSCT.5
Allogeneic HSCT can result in graft-versus-host disease (GvHD), a major complication wherein donor immune cells react against the recipient’s tissue. A significant amount of the morbidity and mortality associated with allogeneic HSCT can be attributed to GvHD effects or complications of its treatment. Immunosuppressive drugs, starting just before graft infusion, are required to mitigate the risk of developing GvHD. Alternatively, the depletion of donor lymphocytes from the HSC graft product, predominantly by immunomagnetic selection, has been used to reduce the subsequent risk of GvHD.6,7
Better purging methods are required to reduce the tumor contamination of the graft in order to improve the outcome associated with autologous HSCT. Similarly, outcomes of allogeneic HSCT may be improved by selectively removing the alloreactive effector cells in a graft rather than wholesale immune cell depletion. Viral agents that selectively replicate in specific cell populations are being studied in experimental models of cancer and immunological diseases, and they have potential applications in the context of HSC graft engineering. In this review, we summarize the progress made using oncolytic viruses (OVs) as ex vivo purging agents for HSC grafts.
Tumor contamination of autologous HSC graft products
Autologous HSC graft products may be contaminated with cancer cells. Clinical experience has shown variable rates of contamination, which have ranged from a few percent to nearly all of the graft products examined in different reports.8 This heterogeneity is undoubtedly related to the extent of the underlying disease, the mobilization and collection procedures used, the detection cut-off, and the method used for tumor cell detection. Tumor cell loads between a few malignant cells per 100,000 nucleated blood cells to several percent of the cells in a graft product have been reported.9,10 Some clinical studies have shown a correlation between reinfusion of grafts containing residual cancer cells and higher rates of relapse.11,12 For instance, in a Phase II clinical trial of autologous bone marrow transplantation for non-Hodgkin’s lymphoma, patients whose grafts were free of lymphoma cells, as determined by polymerase chain reaction (PCR) for the B-cell lymphoma 2 (Bcl2) translocation, had significantly longer relapse-free survival when compared to the survival of patients with detectable lymphoma cells.13 This finding suggests that reinfusion of cancer cells contributes to relapse. Purging unwanted cancer cells from the graft could reduce the risk of relapse by preventing the reintroduction of cancer-initiating cells. The ultimate goal of any purging method is to accomplish tumor cell depletion while leaving normal hematopoietic progenitor cells unharmed.
One ex vivo purging strategy involves isolating hematopoietic progenitor and stem cells away from all other cells, including tumor cells, by positively selecting cells expressing a stem cell antigen, CD34. Briefly, iron-containing beads coated with anti-CD34 antibodies are mixed with the autologous graft product and the antibody-bound cells are isolated magnetically using semiautomated clinical scale devices.14 This method has been tested on autologous graft products obtained from patients with breast cancer, neuroblastoma, myeloma, and lymphoma.15–18 To date, there has only been one randomized controlled clinical trial evaluating the efficacy of an ex vivo purging technology.5 This multicenter Phase III clinical trial tested whether the use of CD34+ immunomagnetic selected purged autologous HSCT conferred a survival benefit over unselected grafts for patients with multiple myeloma. Unfortunately, no significant difference in relapse-free survival or overall survival was observed between the purged and control HSCT groups.5 While this result indicates that the conditioning regimen chemotherapy was unable to completely eliminate all the myeloma-initiating cells, it is possible that the inefficiency of CD34+ selection techniques in purging these tumor cells from the autologous graft product contributed to the lack of demonstrable effect attributable to CD34 selection.
Ex vivo purging has also been performed using a cancer-specific marker that is not expressed on hematopoietic progenitor and stem cells. This technique utilizes antibodies that specifically bind surface markers that are highly expressed on the malignant cells. Antibody-directed effector mechanisms include complement activation or antibody-conjugated toxins. For example, a combination of anti-CD10, anti-CD20 antibodies and rabbit complement have been used to purge B-cell lymphoma cells contaminating autologous bone marrow grafts.13
A third ex vivo purging strategy involves exposing the graft product to cytotoxic pharmacologic agents. Drugs such as 4-hydroperoxycyclophosphamide or mafosfamide have been tested as purging agents. Unfortunately, the narrow therapeutic window of the cytotoxicity produced by these chemicals results in drastically reduced progenitor cell numbers and viability, resulting in prolonged post-transplant pancytopenia.19,20
Current clinically-tested ex vivo purging technologies do not have the capacity to effectively purge cancer cells while maintaining the function of the autologous HSC graft product. OVs infect and kill cancer cells while sparing normal tissue. Research has largely focused on the in vivo applications of these biotherapeutic agents. However, certain OVs may play a role in identifying malignant cells in a graft product. They may be practical tools that can function as ex vivo purging agents, with the ability to eradicate residual cancer cells from autologous graft products.
Viral agents to detect contaminating tumor cells in HSC graft products
A variety of techniques have been used to detect microscopic amounts of cancer, often described as minimal residual disease. Where tumor-specific antigens are known, monoclonal antibodies can be used to identify tumor cells with flow cytometry or immunocytochemistry. However, malignant leukemia cells often express the same array of antigens as normal hematopoietic cells. An alternative, and more sensitive approach, involves detecting tumor-specific mutations or translocations by PCR or fluorescent in situ hybridization.21,22 These methods are most effective for malignancies with characteristic genetic abnormalities, such as the Philadelphia translocation (chronic myelogenous leukemia).
OVs infect and replicate in a cancer-specific manner as they exploit defective antiviral signaling pathways in these cells. By integrating a detectable marker into these viruses, OVs could be used to detect and differentiate malignant cells from normal hematopoietic cells. Bioimaging reporter genes, such as green fluorescent protein (GFP), firefly luciferase, HSV-1 thymidine kinase, or human sodium iodide symporter (NIS), have already been engineered into several OV candidates (Table 1). Furthermore, preclinical studies have shown that many OVs infect malignant leukemia, lymphoma, and myeloma cells. The measles virus (MV) has been engineered to express human NIS (MV-NIS). Expression of the symporter in an infected cell leads to the accumulation of radioiodine (131I) in these cells and ultimately results in their death. They become detectable by positron emission tomography following the accumulation of radioisotopes in these cancer cells. OVs could be used as detection tools for contaminating cancer cells in autologous HSC grafts.
 | Table 1 Oncolytic viruses with reporter genes used for the detection of human cancer |
Viral ex vivo tumor purging agents
OVs kill cancer cells through a number of distinct mechanisms. While tumor lysis after viral infection is the most direct mechanism of killing, following infection, OVs destroy tumor vasculature, induce inflammation in the tumor, and recruit innate and adaptive immune responses. Furthermore, these effector mechanisms are selectively operative in the tumor environment. Preclinical and clinical studies have shown OVs to be safe in normal tissues.39
While the initial clinical experience using replication-competent OVs to treat patients with solid tumors has not resulted in illness associated with viral replication, patients with hematopoietic cancers are generally highly immune suppressed and are possibly at greater risk for toxicity. Patients undergoing HSCT are even more severely immune deficient and at higher risk of untoward complications following systemic treatments with OVs. Viral replication is restricted to cancer cells making OVs suitable for ex vivo graft purging. Ex vivo purging of tumor cells from autologous stem cell grafts could reduce tumor burden while minimizing the exposure of immune-deficient or immune-suppressed patients to potential morbidity from replication-competent OV agents.
To function as an ex vivo purging agent, OVs must directly infect and selectively kill malignant cells as downstream mechanisms, reliant on vasculature or immunity, which are not operative. It is important to note that viral replication may not be required, as high multiplicities of infection (MOIs) could be used. Secondly, viruses should not affect normal hematopoietic elements. Antiviral pathways suppress proliferation and could reduce the engraftment of hematopoietic stem and progenitor cells (ie, interferon suppresses normal hematopoiesis).40 Caution must be taken to monitor antiviral cytokines in the graft following OV treatment. Methods to mitigate the negative effects of these cytokines might include choosing OVs that selectively infect only the malignant cells, or it may involve the use of antibodies or reagents that block inflammatory cytokine signaling.
Harnessing the tumor-selective killing of OVs for ex vivo purging of contaminating tumor cells in a stem cell graft product could be beneficial in the clinical setting. The following discussion will focus on OV candidates that have been tested as ex vivo purging agents for autologous HSC graft products, and the suitability of other OVs that have yet to be applied in this context. Table 2 contains a brief summary of these agents.
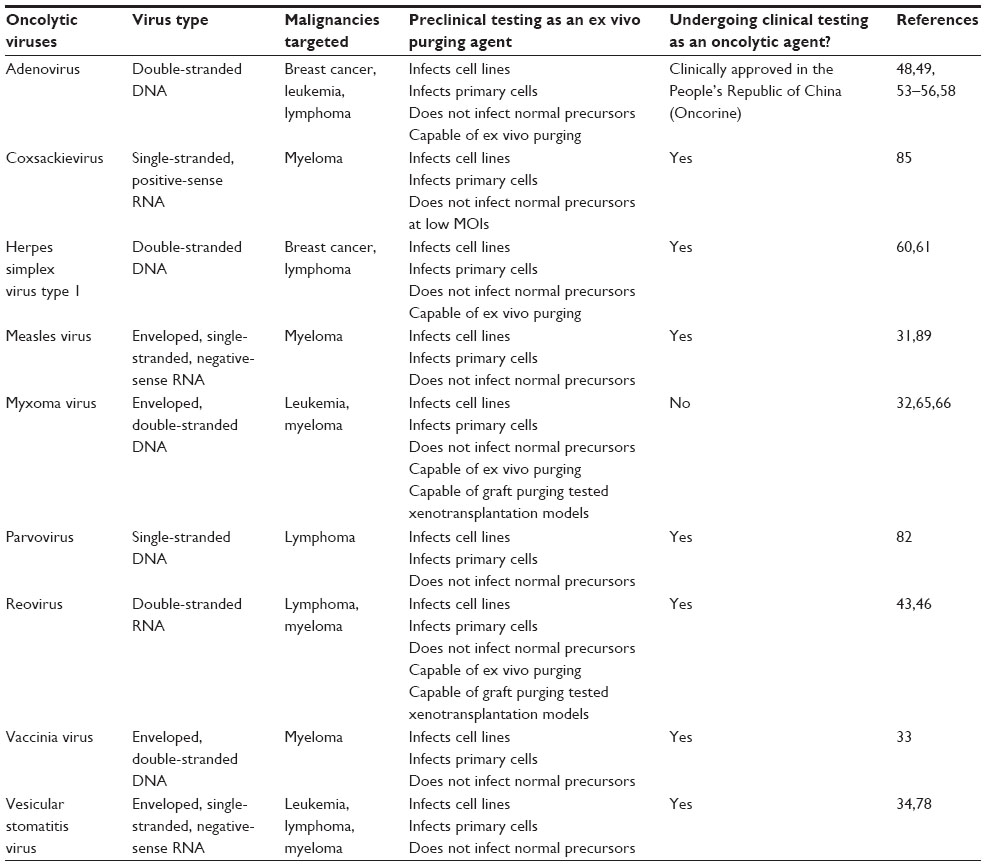 | Table 2 Oncolytic viruses for ex vivo purging of hematopoietic malignancies in autologous bone marrow graft products |
Agents that have been used in preclinical models of graft purging
Reovirus
Reovirus is a nonenveloped double-stranded RNA virus that naturally infects humans, resulting in generally asymptomatic upper respiratory tract infections. Reovirus is internalized by binding to sialic acid receptors on the cell surface. Transformation by the ras oncogene, commonly found in some cancers, results in defective interferon signaling, allowing for selective reovirus replication in tumor cells.41,42
In vitro, reovirus does not affect human progenitor cell viability or colony-forming capacity, yet the virus is able to kill malignant lymphoid cells.43,44 Effective reovirus-induced cytotoxicity has been shown against indolent lymphoid malignancies such as chronic lymphocytic leukemia, and more aggressive lymphoid malignancies such as diffuse large B-cell lymphoma. These studies suggested that reovirus could be utilized as an ex vivo purging agent for hematopoietic malignancies.
Reovirus has demonstrated efficacy in preclinical models of multiple myeloma. Thirukkumaran et al45 found that reovirus infected several human myeloma cell lines and primary myeloma cells isolated from patients. The group also showed that human stem and progenitor cells treated with reovirus were able to engraft sublethally irradiated immune-deficient mice, but these studies did not quantify changes in hematopoietic precursor numbers following OV treatment.
In a separate study, Thirukkumaran et al46 seeded human HSC grafts with human myeloma cell lines, and showed depletion of the myeloma cells following exposure to live reovirus, using flow cytometry and PCR detection strategies. RPMI 8226 contaminated HSC graft product treated with live reovirus successfully engrafted sublethally irradiated mice, which had prolonged survival compared to control mice transplanted with RPMI 8226 spiked grafts treated with killed reovirus.
Currently, reovirus (Reolysin®; Oncolytics Biotech®; Inc., Calgary, AB, Canada) is being tested in a number of single-agent and combination Phase I, II, and III trials for different solid cancers.47 Intriguingly, given its safety profile, Reolysin® is being tested as a systemic agent alone and in combination with chemotherapeutic agents in two Phase I trials for relapsed multiple myeloma. While, reovirus shows promise as an ex vivo purging agent, there are no ongoing clinical trials testing Reolysin® as a stem cell graft purging agent.
Adenovirus
Adenovirus (Ad) is a double-stranded DNA virus that causes upper respiratory tract and enteric infections in humans. There are 57 serotypes (1–57) of the seven Ad species (A–G). Binding to the coxsackie and Ad receptor (CAR) is used by many Ad species to initiate cell entry. Studies using adenoviral gene therapy vectors (Adv) for the modification of CD34+ hematopoietic stem and progenitor cells found that these cells lack CAR and exhibit poor infection efficiency.48,49 These replication-incompetent Adv have been tested as ex vivo purging agents that selectively express prodrug-converting or proapoptotic enzymes in nonhematopoietic malignancies.50,51 An Adv-expressing cytosine deaminase was found to purge as many as six logs of breast cancer cells.52 These agents do not harm HSC engraftment following transplantation into immunodeficient mice.49,50,52 An array of replication-competent Ad strains (Ad5, Ad6, Ad26, and Ad48) selectively killed human CD138+ multiple myeloma cells while sparing CD138− normal bone marrow cells.53 Comparably, a panel of species D Ad types, Ad26, and Ad48 Ad serotypes, exhibited the best killing of human B-cell cancers.54
Conditionally replicative Ad5 (CRAd) with replication mediated by a midkine promoter was tested as an ex vivo purging agent for neuroblastoma and Ewing’s sarcoma. Midkine is a heparin-binding growth factor and its overexpression in these cancer cell lines correlated with enhanced CRAd killing, as determined by MTS viability assays and colony-forming assays.55 In contrast, the colony-forming capacity of bone marrow CD34+ stem cells, which express low levels of midkine, remained unaffected even at a MOI of 1,000.
A challenge for CRAd therapy of leukemia is the lack of CAR expression. ColoAd1 (known as Enadenotucirev; PsiOxus Therapeutics, Ltd., Oxford, UK), a chimeric Ad currently in clinical testing, merits investigation as a HSC graft purging agent. While this virus was isolated from directed evolution on colon cancer cells, it binds to CD46, a cell surface protein that is highly expressed on many hematopoietic malignancies.56,57 Jin et al58 constructed a CRAd expressing chimeric Ad5/35 knob fiber and tumor necrosis factor-related apoptosis-inducing ligand (SG235-TRAIL) to retarget and enhance CAR-independent CRAd killing of leukemia cells. The group found that SG235-TRAIL abrogated the colony-forming ability of several leukemia cell lines and primary acute myeloid leukemia (AML) blasts, while sparing healthy bone marrow mononuclear cells.
An oncolytic Ad with E1B-55k deletion (known as Oncorine®; Shanghai Sunway Biotech Co., LTD., Shanghai, People’s Republic of China) is currently the only clinically approved OV in the world. The Chinese Health Regulatory Body has approved its use for the treatment of head and neck cancer. Although oncolytic Ad has limited experience as an ex vivo purging agent for hematopoietic malignancies, CRAd and retargeted iterations show potential as ex vivo purging agents for hematopoietic malignancies contaminating stem cell graft products.
Herpes simplex virus type 1
Herpes simplex virus type 1 (HSV-1) is a double-stranded DNA virus responsible for recurrent mucosal (oronasal, genital) infections in humans. Occasionally, HSV-1 can cause more serious infections such as encephalitis. Several genetic modifications of HSV-1 have been introduced to create strains with reduced viral pathogenicity and improved oncolytic abilities.59 Hu et al60 showed that the JS-1 strain could selectively kill MDA-MB-231 breast cancer cells when mixed with healthy bone marrow. Additionally, bone marrow samples from breast cancer patients were effectively purged with up to a 95% reduction of contaminating cancer cells, as quantified by cytokeratin-19 expression. Oncolytic HSV-1 can also infect and replicate in T-cell lymphomas and Burkitt’s lymphoma cell lines.61 Currently, talimogene laherparepvec or T-VEC (Amgen Inc., Thousand Oaks, CA, USA), an oncolytic strain of HSV-1 expressing human granulocyte macrophage colony-stimulating factor (GM-CSF), is undergoing Phase III clinical trials for melanoma. The selective killing of malignant cells while maintaining hematopoietic colony-forming capacity highlights the potential role of HSV-1 as a stem cell graft-purging agent.
Myxoma virus
Myxoma virus (MYXV) is a poxvirus, but in contrast to vaccinia virus (VACV), it does not infect humans.62 Pathogenicity is tightly restricted to rabbits, and there have been no reported complications in humans or immune-deficient mice exposed to the virus.63 While it was once thought that this tropism was particular to European rabbits, productive infection occurs in human cancer cells with defective Akt signaling pathways.64
MYXV has been tested as an ex vivo purging agent. Kim et al32 showed that MYXV could infect and kill human primary AML blasts. Normal hematopoietic stem and progenitor cell viability was maintained following exposure to MYXV as measured by colony formation in vitro and semiquantitative in vivo murine xenograft assays. The immunodeficient murine hosts did not develop evidence of myxoma infection. Transplantation of mononuclear cells from patients with AML in a murine xenograft model resulted in engraftment of normal human hematopoiesis. AML cells were detected by PCR of a leukemia marker gene in 10/10 mock-infected samples, but only in one of ten that were treated with MYXV prior to transplantation. A follow-up study by Madlambayan et al65 found that MYXV binding to cells in vitro, rather than viral replication, was sufficient to mediate in vivo killing of leukemic blasts.
MYXV has also been used as an ex vivo purging agent for multiple myeloma. MYXV could infect and kill four human MM cell lines RPMI 8266, MM.1S, HuNS1, and U266.66 A specific reduction in CD138+ myeloma cells, but not CD34+ progenitor cells, was observed in MYXV-infected primary bone marrow samples from myeloma patients. When lethally irradiated immunodeficient mice were transplanted with healthy human bone marrow admixed with U266 cells and infected with MYXV, no MM cells were observed in the bone marrow 6 weeks following transplantation.66
OV graft purging may also prove beneficial in mitigating GvHD following allogeneic stem cell transplantation. Bartee et al67 found that MYXV can inhibit in vitro human leukocyte antigen haplo-mismatched mixed lymphocyte reactions. They showed that ex vivo incubation of an allogeneic stem cell graft with MYXV prior to transplantation limited the in vivo expansion of transplanted CD3+ T-cells and suppressed graft-versus-host alloreactivity, preventing fatal GvHD in a murine model of allogeneic stem cell transplantation. Furthermore, hematopoietic engraftment and alloreactivity against malignant leukemia blasts (graft-versus-leukemia) were not affected.
MYXV has demonstrated promise as an ex vivo purging agent in preclinical models. Its safety profile is truly impressive, with experience in healthy humans, other animals, and immunodeficient animals used for xenograft assays. Furthermore, MYXV treatment of HSC grafts does not affect engraftment. These are advantageous properties for its use as an ex vivo graft purging agent; however, the MYXV killing mechanism may require in vivo effectors that may be prone to biologic variation between individuals.
Agents that have demonstrated activity against hematopoietic cancer
Vaccinia virus
VACV is a large enveloped double-stranded DNA virus belonging to the poxvirus family. Cellular uptake is mediated by the viral particle binding to the cell surface glycosaminoglycans, which initiates macropinocytosis.68 Poxviruses contain an array of genes with immunomodulatory functions, including viral proteins that mimic or bind host cytokines, chemokines, and complement.69 Viral replication in cancer cells can be made more selective by genetic engineering of the virus, eliminating an essential viral product that can be supplied in trans by the cancer cell. This is exemplified by a thymidine kinase-inactivated VACV expressing GM-CSF, known as Pexa-Vec (SillaJen Inc., Busan, South Korea), that is in Phase IIB clinical testing for hepatocellular carcinoma.70
A double-deleted recombinant VACV (deleted genes for thymidine kinase and vaccinia growth factor) has been shown to infect several human multiple myeloma cell lines and patient samples in vitro.33 This attenuated oncolytic strain, however, produced little to no infection of peripheral blood mononuclear cells from healthy donors. Historically, there have been anecdotal reports of documented remission of chronic lymphocytic leukemia following smallpox vaccinations.71,72 Similarly, there was a reduction in myeloma tumor burden in a patient that received the AS strain of VACV.73
There is extensive clinical experience with VACV as a smallpox vaccine. Its safety profile is well understood and there is a very low risk of serious complications in humans, except for those with cellular immune deficiencies including patients with hematological malignancies.74–76 Ex vivo purging may harness the ability of VACV to target hematopoietic malignancies while minimizing the risk of uncontrolled infection.
Vesicular stomatitis virus
Vesicular stomatitis virus (VSV) is a tiny bullet-shaped negative-strand RNA virus with only five genes. Its natural hosts are insects and domestic farm animals. Virus entry into the cells is mediated by binding to low density lipoprotein (LDL) cell surface receptors followed by internalization by endocytosis in clathrin-coated pits.77 The broad expression of the LDL receptor contributes to VSV’s extensive host cell tropism. VSV infection is hindered by type I interferon. Normal cells are able to produce interferon quickly following VSV infection, which shuts down further viral replication. Tumors with defects in interferon signaling are permissive for VSV replication, which results in tumor-specific cytolysis.
VSV could be used as an ex vivo purging agent. Lichty et al34 showed that VSV can effectively kill a number of human leukemia, myeloma, and lymphoma cell lines. Exposing peripheral blood samples from patients with circulating myeloma cells to a single dose of VSV resulted in a significant decrease in CD138+ cells. This selective phenomenon was also observed when Stojdl et al78 took healthy human bone marrow mixed with the human leukemia cell line, OCI/AML3, and infected the samples with VSV. Only normal immune cell colonies were observed in a colony-forming assay 14 days later.78 This study demonstrated that VSV has the potential to eliminate hematologic malignancies contaminating bone marrow and peripheral blood while sparing normal marrow elements.78
When used at very high MOIs, ultraviolet-irradiated, replication-incompetent VSV (nonreplicating rhabdovirus particles [NRRPs]) have been shown to induce cell death in several murine and human leukemia cell lines. The mechanism of killing is not defined. Culturing peripheral blood samples from two patients with chronic myelogenous leukemia blast crisis in the presence of NRRPs resulted in the reduction of CD33+ blasts cells, while parallel short cultures of bone marrow mononuclear cells exposed to NRRPs remained viable.79 Low-dose ultraviolet irradiation could be used as an additional measure to augment safety, reducing the possible risks of infusing live replicating viruses.
There is limited experience using VSV in humans. VSV expressing human interferon-β is currently in Phase I clinical testing as a therapy for hepatocellular carcinoma (NLM identifier: NCT01628640). Preclinical testing of VSV suggests it will be applicable to a range of cancers including those derived from hematopoietic origins. These studies34,78 provide evidence to support the use of VSV as an ex vivo purging agent but, to our knowledge, there are no ongoing clinical trials.
Parvovirus
Oncolytic parvovirus (H-1PV) is a nonenveloped, single-stranded rodent DNA virus. Oncotropism relies on improved replication in transformed cells, while its oncolytic abilities are due to the accumulation of viral nonstructural protein 1, which is selectively toxic to transformed cells.80 H-1PV (clinically known as ParvOryx01; Oryx GmbH & Co. KG, Baldham, Germany) is currently undergoing a Phase I/II clinical trial in patients with glioblastoma multiforme.81 H-1PV has been shown to effectively kill human Burkitt’s lymphoma cell lines while sparing normal B-cells both in vitro and in xenograft preclinical models of lymphoma.82 Other parvoviruses, such as B19 and Aleutian mink disease virus can infect hematopoietic cells, leading to pure red cell aplasia or aplastic anemia.83 Further testing would be required before considering H-1PV as a purging agent.
Coxsackievirus
Oncolytic coxsackievirus A21 (CVA21) is a positive strand RNA virus. Viral entry is facilitated by binding to cell surface intracellular adhesion molecule-1 and decay-accelerating factor.84 Subsequent oncolysis is mediated by viral shutdown of essential cellular processes and by triggering apoptosis. CVA21, clinically known as CAVATAK™ (Viralytics Limited, Sydney, NSW, Australia), is being tested in a Phase IIIC/IV clinical trial for malignant melanoma (NLM identifier: NCT01636882).
CVA21 has been tested on ten bone marrow samples from multiple myeloma patients and was found to produce a dose-dependent reduction of up to 98.7% of the CD138+ cell population.85 Larger viral doses were found to affect the viability of CD138− normal hematopoietic cell populations. A potential concern for using CVA21 as an ex vivo purging agent may be inadvertent virus infection of CD34+ HSC, as these cells express intracellular adhesion molecule and decay-accelerating factor.86,87 Further testing is required to verify whether CVA21 exposure will affect HSC engraftment.
Measles virus
The Edmonton vaccine strain of the MV has proven to be a capable OV. This negative-sense RNA virus binds to cell surface CD46 and signaling lymphocyte activation molecule (SLAM) receptors to initiate cell entry.88 A study by Ong et al31 found a link between the overexpression of CD46 on CD138+ multiple myeloma cell lines, as well as patient samples, and the cytopathic effect of MV therapy. In contrast, normal progenitor cells, with normal CD46 expression, were not harmed, as determined by their intact colony-forming capacity. Oncolytic MV has also been engineered to retarget cancers that do not overexpress its natural receptors; instead, MV can bind CD20, which is overexpressed on lymphoma cells, and gain cell entry.89 A NIS-expressing MV variant has been created and is being tested in a Phase I clinical trial for patients with multiple myeloma (NLM identifier: NCT00450814).
Viral ex vivo tumor-purging agents – summary
OVs are promising biotherapeutic agents for the treatment of cancer. While the majority of clinical applications research examines the role that these viruses play following systemic administration, in some circumstances, OVs may find roles as ex vivo purging agents, eliminating tumor cells that contaminate autologous HSC grafts. Their advantages over other purging agents include the ease of graft manipulation, their selectivity for cancer cells, and their demonstrated ability to kill certain hematopoietic malignancies. In some cases, as will be discussed, infected tumor cells can act as vaccines, inducing immunological responses for an additional therapeutic effect. Preclinical testing has identified oncolytic strains of HSV-1, Ad, reovirus, and MYXV as potentially useful tools for ex vivo graft manipulation, but early-phase clinical trials are now required to select the agents with characteristics that make them feasible to use, at the scale required for purging clinical graft products, before widespread clinical testing for effectiveness can begin.
Alternative ex vivo OV strategies – infected cell vaccines
Relapse continues to be the major concern following autologous stem cell transplantation. Lower relapse rates are seen following allogeneic stem cell transplantation where allograft immune responses – the graft-versus-tumor effect – provide an additional modality for control of any lingering malignant cells. Systemic treatment with OVs induce tumor-specific adaptive immune responses in both preclinical animal models and human clinical trials.39,70,90 Adaptive immune responses can be induced using ex vivo OV-infected tumor cell vaccines (ICVs). The efficacy of ICV has been demonstrated in a number of preclinical models and clinical studies for solid cancers.91–93
ICVs are also effective in hematological malignancies. Mice immunized with MG1 (an oncolytic maraba virus) infected L1210 leukemia cells generate specific anti-L1210 immunity capable of rejecting subsequent challenges with viable L1210 leukemia cells.94 The antileukemia immunity is specific and durable. While it is unlikely that a HSC recipient could mount an immune response in the immediate period following HSCT, the infected leukemia cells in a HSC graft could function as an ICV, although this has not been directly examined.
More likely, future translational efforts would focus on creating a patient-specific ICV at a diagnosis that could be administered after immunological recovery from autologous HSCT in an attempt to induce antileukemia immunity and reduce the risk of relapse.
Conclusion
The role of ex vivo treatment of HSC grafts to eliminate contaminating cancer cells is controversial. The difficulty in demonstrating a role for graft purging is hampered by inefficient purging techniques, toxicity to normal hematopoietic elements in the graft, and incomplete killing of the malignancy by HSCT conditioning regimens. OVs represent a new class of purging agents that have shown promise in preclinical studies and may find their way into translational studies in the future.
Disclosure
HLA was previously a shareholder in Jennerex ULC (a company involved in development of Pexa-Vec since sold to SillaJen, Inc.). JTT reports no conflicts of interest in this work.
References
Passweg JR, Baldomero H, Peters C, et al. Hematopoietic SCT in Europe: data and trends in 2012 with special consideration of pediatric transplantation. Bone Marrow Transplant. 2014;49(6):744–750. | |
Hatzmichael E, Tuthill M. Hematopoietic stem cell transplantation. Stem Cells Cloning. 2010;3:105–117. | |
Pasquini MC, Wang Z. Current use and outcome of hematopoietic stem cell transplantation: CIBMTR Summary Slides, 2013. Available from http://www.cibmtr.org/referencecenter/slidesreports/summaryslides/pages/index.aspx. Accessed August 10, 2014. | |
Brenner MK, Rill DR, Moen RC, et al. Gene-marking to trace origin of relapse after autologous bone-marrow transplantation. Lancet. 1993;341(8837):85–86. | |
Stewart AK, Vescio R, Schiller G, et al. Purging of autologous peripheral-blood stem cells using CD34 selection does not improve overall or progression-free survival after high-dose chemotherapy for multiple myeloma: results of a multicenter randomized controlled trial. J Clin Oncol. 2001;19(17):3771–3779. | |
Giralt S, Hester J, Huh Y, et al. CD8-depleted donor lymphocyte infusion as treatment for relapsed chronic myelogenous leukemia after allogeneic bone marrow transplantation. Blood. 1995;86(11):4337–4343. | |
Ho VT, Soiffer RJ. The history and future of T-cell depletion as graft-versus-host disease prophylaxis for allogenic hematopoietic stem cell transplantation. Blood. 2001;98(12):3192–3204. | |
Shimoni A, Körbling M. Tumor cell contamination in re-infused stem cell autografts: does it have clinical significance? Crit Rev Oncol Hematol. 2002;41(2):241–250. | |
DiPersio JF, Ho AD, Hanrahan J, Hsu FJ, Fruehauf S. Relevance and clinical implications of tumor cell mobilization in the autologous transplant setting. Biol Blood Marrow Transplant. 2011;17(7):943–955. | |
Choi S, Rajan SS, Trivedi MV. The incidence of tumor cell contamination of peripheral blood stem cells: a meta-analysis to evaluate the impact of mobilization regimens and the influence on outcomes in breast cancer patients. Acta Haematol. 2014;131(3):133–140. | |
Sharp JG, Kessinger A, Mann S, et al. Outcome of high-dose therapy and autologous transplantation in non-Hodgkin’s lymphoma based on the presence of tumor in the marrow or infused hematopoietic harvest. J Clin Oncol. 1996;14(1):214–219. | |
Reiffers J, Labopin M, Sanz M, et al. Autologous blood cell vs marrow transplantation for acute myeloid leukemia in complete remission: an EBMT retrospective analysis. Bone Marrow Transplant. 2000;25(11):1115–1119. | |
Gribben JG, Neuberg D, Freedman AS, et al. Detection by polymerase chain reaction of residual cells with the bcl-2 translocation is associated with increased risk of relapse after autologous bone marrow transplantation for B-cell lymphoma. Blood. 1993;81(12):3449–3457. | |
Watts MJ, Somervaille TC, Ings SJ, et al. Variable product purity and functional capacity after CD34 selection: a direct comparison of the CliniMACS (v2.1) and Isolex 300i (v2.5) clinical scale devices. Br J Haematol. 2002;118(1):117–123. | |
Mohr M, Hilgenfeld E, Fietz T, et al. Efficacy and safety of simultaneous immunomagnetic CD34+ cell selection and breast cancer cell purging in peripheral blood progenitor cell samples used for hematopoietic rescue after high-dose therapy. Clin Cancer Res. 1999;5(5):1035–1040. | |
Lode HN, Handgretinger R, Schuermann U, et al. Detection of neuroblastoma cells in CD34+ selected peripheral stem cells using a combination of tyrosine hydroxylase nested RT-PCR and anti-ganglioside GD2 immunocytochemistry. Eur J Cancer. 1997;33(12):2024–2030. | |
Mohr M, Dalmis F, Hilgenfeld E, et al. Simultaneous immunomagnetic CD34+ cell selection and B-cell depletion in peripheral blood progenitor cell samples of patients suffering from B-cell non-Hodgkin’s lymphoma. Clin Cancer Res. 2001;7(1):51–57. | |
Rasmussen T, Björkstrand B, Andersen H, Gaarsdal E, Johnsen HE. Efficacy and safety of CD34-selected and CD19-depleted autografting in multiple myeloma patients: a pilot study. Exp Hematol. 2002;30(1):82–88. | |
Miller CB, Rowlings PA, Zhang MJ, et al. The effect of graft purging with 4-hydroperoxycyclophosphamide in autologous bone marrow transplantation for acute myelogenous leukemia. Exp Hematol. 2001; 29(11):1336–1346. | |
McNiece I, Civin C, Harrington J, et al. Ex vivo expansion of mafosfamide-purged PBPC products. Cytotherapy. 2006;8(5):459–464. | |
Negrin RS, Kiem HP, Schmidt-Wolf IG, Blume KG, Cleary ML. Use of the polymerase chain reaction to monitor the effectiveness of ex vivo tumor cell purging. Blood. 1991;77(3):654–660. | |
Müller P, Weckermann D, Riethmüller G, Schlimok G. Detection of genetic alterations in micrometastatic cells in bone marrow of cancer patients by fluorescence in situ hybridization. Cancer Genet Cytogenet. 1996;88(1):8–16. | |
Hakkarainen T, Rajecki M, Sarparanta M, et al. Targeted radiotherapy for prostate cancer with an oncolytic adenovirus coding for human sodium iodide symporter. Clin Cancer Res. 2009;15(17):5396–5403. | |
Li H, Nakashima H, Decklever TD, Nace RA, Russell SJ. HSV-NIS, an oncolytic herpes simplex virus type 1 encoding human sodium iodide symporter for preclinical prostate cancer radiovirotherapy. Cancer Gene Ther. 2013;20(8):478–485. | |
Dingli D, Peng KW, Harvey ME, et al. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood. 2004;103(5):1641–1646. | |
Haddad D, Chen NG, Zhang Q, et al. Insertion of the human sodium iodide symporter to facilitate deep tissue imaging does not alter oncolytic or replication capability of a novel vaccinia virus. J Transl Med. 2011;9:36. | |
Goel A, Carlson SK, Classic KL, et al. Radioiodide imaging and radiovirotherapy of multiple myeloma using VSV(Delta51)-NIS, an attenuated vesicular stomatitis virus encoding the sodium iodide symporter gene. Blood. 2007;110(7):2342–2350. | |
Abate-Daga D, Andreu N, Camacho-Sánchez J, et al. Oncolytic adenoviruses armed with thymidine kinase can be traced by PET imaging and show potent antitumoural effects by ganciclovir dosing. PLoS One. 2011;6(10):e26142. | |
Bennett JJ, Tjuvajev J, Johnson P, et al. Positron emission tomography imaging for herpes virus infection: Implications for oncolytic viral treatments of cancer. Nat Med. 2001;7(7):859–863. | |
Wang G, Li G, Liu H, et al. E1B 55-kDa deleted, Ad5/F35 fiber chimeric adenovirus, a potential oncolytic agent for B-lymphocytic malignancies. J Gene Med. 2009;11(6):477–485. | |
Ong HT, Timm MM, Greipp PR, et al. Oncolytic measles virus targets high CD46 expression on multiple myeloma cells. Exp Hematol. 2006;34(6):713–720. | |
Kim M, Madlambayan GJ, Rahman MM, et al. Myxoma virus targets primary human leukemic stem and progenitor cells while sparing normal hematopoietic stem and progenitor cells. Leukemia. 2009;23(12):2313–2317. | |
Deng H, Tang N, Stief AE, et al. Oncolytic virotherapy for multiple myeloma using a tumour-specific double-deleted vaccinia virus. Leukemia. 2008;22(12):2261–2264. | |
Lichty BD, Stojdl DF, Taylor RA, et al. Vesicular stomatitis virus: a potential therapeutic virus for the treatment of hematologic malignancy. Hum Gene Ther. 2004;15(9):821–831. | |
Lamfers ML, Fulci G, Gianni D. Cyclophosphamide increases transgene expression mediated by an oncolytic adenovirus in glioma-bearing mice monitored by bioluminescence imaging. Mol Ther. 2006;14(6):779–788. | |
Terada K, Wakimoto H, Tyminski E, Chiocca EA, Saeki Y. Development of a rapid method to generate multiple oncolytic HSV vectors and their in vivo evaluation using syngeneic mouse tumor models. Gene Ther. 2006;13(8):705–714. | |
Kirn DH, Wang Y, Le Boeuf F, Bell J, Thorne SH. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007;4(12):e353. | |
Le Boeuf F, Diallo JS, McCart JA, et al. Synergistic interaction between oncolytic viruses augments tumor killing. Mol Ther. 2010;18(5):888–895. | |
Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14(8):559–567. | |
Raefsky EL, Platanias LC, Zoumbos NC, Young NS. Studies of interferon as a regulator of hematopoietic cell proliferation. J Immunol. 1985;135(4):2507–2512. | |
Shmulevitz M, Pan LZ, Garant K, Pan D, Lee PW. Oncogenic Ras promotes reovirus spread by suppressing IFN-beta production through negative regulation of RIG-I signaling. Cancer Res. 2010;70(12):4912–4921. | |
Norman KL, Lee PW. Not all viruses are bad guys: the case for reovirus in cancer therapy. Drug Discov Today. 2005;10(12):847–855. | |
Thirukkumaran CM, Luider JM, Stewart DA, et al. Reovirus oncolysis as a novel purging strategy for autologous stem cell transplantation. Blood. 2003;102(1):377–387. | |
Alain T, Hirasawa K, Pon KJ, et al. Reovirus therapy of lymphoid malignancies. Blood. 2002;100(12):4146–4153. | |
Thirukkumaran CM, Shi ZQ, Luider J, et al. Reovirus as a viable therapeutic option for the treatment of multiple myeloma. Clin Cancer Res. 2012;18(18):4962–4972. | |
Thirukkumaran CM, Shi ZQ, Luider J, et al. Reovirus as a successful ex vivo purging modality for multiple myeloma. Bone Marrow Transplant. 2014;49(1):80–86. | |
Oncolytics Biotech Inc [webpage on the Internet]. Clinical trials. Calgary, AB: Oncolytics Biotech Inc.; 2014. Available from: http://www.oncolyticsbiotech.com/clinical-trials/default.aspx. Accessed May 2, 2014. | |
Shayakhmetov DM, Papayannopoulou T, Stamatoyannopoulos G, Lieber A. Efficient gene transfer into human CD34(+) cells by a retargeted adenovirus vector. J Virol. 2000;74(6):2567–2583. | |
Hirai M, LaFace D, Robinson S, et al. Ex vivo purging by adenoviral p53 gene therapy does not affect NOD-SCID repopulating activity of human CD34+ cells. Cancer Gene Ther. 2001;8(12):936–947. | |
Wroblewski JM, Lay LT, Van Zant G, et al. Selective elimination (purging) of contaminating malignant cells from hematopoietic stem cell autografts using recombinant adenovirus. Cancer Gene Ther. 1996;3(4):257–264. | |
Hirai M, Kelsey LS, Vaillancourt M, Maneval DC, Watanabe T, Talmadge JE. Purging of human breast cancer cells from stem cell products with an adenovirus containing p53. Cancer Gene Ther. 2000;7(2):197–206. | |
Garcia-Sanchez F, Pizzorno G, Fu SQ, et al. Cytosine deaminase adenoviral vector and 5-fluorocytosine selectively reduce breast cancer cells 1 million-fold when they contaminate hematopoietic cells: a potential purging method for autologous transplantation. Blood. 1998;92(2):672–682. | |
Senac JS, Doronin K, Russell SJ, Jelinek DF, Greipp PR, Barry MA. Infection and killing of multiple myeloma by adenoviruses. Hum Gene Ther. 2010;21(2):179–190. | |
Chen CY, Senac JS, Weaver EA, et al. Species D adenoviruses as oncolytics against B-cell cancers. Clin Cancer Res. 2011;17(21):6712–6722. | |
Adachi Y, Reynolds PN, Yamamoto M, et al. A midkine promoter-based conditionally replicative adenovirus for treatment of pediatric solid tumors and bone marrow tumor purging. Cancer Res. 2001;61(21):7882–7888. | |
Kuhn I, Harden P, Bauzon M, et al. Directed evolution generates a novel oncolytic virus for the treatment of colon cancer. PLoS One. 2008;3(6):e2409. | |
Hara T, Kojima A, Fukuda H, et al. Levels of complement regulatory proteins, CD35 (CR1), CD46 (MCP) and CD55 (DAF) in human haematological malignancies. Br J Haematol. 1992;82(2):368–373. | |
Jin J, Liu H, Yang C, et al. Effective gene-viral therapy of leukemia by a new fiber chimeric oncolytic adenovirus expressing TRAIL: in vitro and in vivo evaluation. Mol Cancer Ther. 2009;8(5):1387–1397. | |
Liu BL, Robinson M, Han ZQ, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10(4):292–303. | |
Hu JC, Booth MJ, Tripuraneni G, et al. A novel HSV-1 virus, JS1/34.5-/47-, purges contaminating breast cancer cells from bone marrow. Clin Cancer Res. 2006;12(22):6853–6862. | |
Zhang X, Zhao L, Hang Z, Guo H, Zhang M. Evaluation of HSV-1 and adenovirus vector-mediated infection, replication and cytotoxicity in lymphoma cell lines. Oncol Rep. 2011;26(3):637–644. | |
Miller S. Virologist helped eradicate smallpox. The Wall Street Journal. November 26, 2010, Available from: http://online.wsj.com/news/articles/SB10001424052748704693104575639520875125424/. Accessed July 23, 2014. | |
Chan WM, Rahman MM, McFadden G. Oncolytic myxoma virus: the path to clinic. Vaccine. 2013;31(39):4252–4258. | |
Stanford MM, McFadden G. Myxoma virus and oncolytic virotherapy: a new biologic weapon in the war against cancer. Expert Opin Biol Ther. 2007;7(9):1415–1425. | |
Madlambayan GJ, Bartee E, Kim M, et al. Acute myeloid leukemia targeting by myxoma virus in vivo depends on cell binding but not permissiveness to infection in vitro. Leuk Res. 2012;36(5):619–624. | |
Bartee E, Chan WM, Moreb JS, Cogle CR, McFadden G. Selective purging of human multiple myeloma cells from autologous stem cell transplantation grafts using oncolytic myxoma virus. Biol Blood Marrow Transplant. 2012;18(10):1540–1551. | |
Bartee E, Meacham A, Wise E, Cogle CR, McFadden G. Virotherapy using myxoma virus prevents lethal graft-versus-host disease following xeno-transplantation with primary human hematopoietic stem cells. PLoS One. 2012;7(8):e43298. | |
Villa NY, Bartee E, Mohamed MR, Rahman MM, Barrett JW, McFadden G. Myxoma and vaccinia viruses exploit different mechanisms to enter and infect human cancer cells. Virology. 2010;401(2):266–279. | |
Johnston JB, McFadden G. Technical knockout: understanding poxvirus pathogenesis by selectively deleting viral immunomodulatory genes. Cell Microbiol. 2004;6(8):695–705. | |
Heo J, Reid T, Ruo L, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19(3):329–336. | |
Hansen RM, Libnoch JA. Remission of chronic lymphocytic leukemia after smallpox vaccination. Arch Intern Med. 1978;138(7):1137–1138. | |
Yettra M. Remission of chronic lymphocytic leukemia after smallpox vaccination. Arch Intern Med. 1979;139(5):603. | |
Kawa A, Arakawa S. The effect of attenuated vaccinia virus AS strain on multiple myeloma; a case report. Jpn J Exp Med. 1987;57(1):79–81. | |
Frey SE, Couch RB, Tacket CO, et al; National Institute of Allergy and Infectious Diseases Smallpox Vaccine Study Group. Clinical responses to undiluted and diluted smallpox vaccine. N Engl J Med. 2002;346(17):1265–1274. | |
Fenner F, Henderson D, Arita I, Ježek Z, Ladnyi I. Development of the global smallbox eradication programme, 1958–1966. In: Smallpox and its Eradication. Geneva, Switzerland: World Health Organization; 1988:365–419. | |
Centers for Disease Control and Prevention [webpage on the Internet]. Frequently asked questions about smallpox vaccine. 2004: Centers for Disease Control and Prevention. Available from: http://www.bt.cdc.gov/agent/smallpox/vaccination/faq.asp. Accessed July 23, 2014. | |
Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci U S A. 2013;110(18):7306–7311. | |
Stojdl DF, Lichty B, Knowles S, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6(7):821–825. | |
Batenchuk C, Le Boeuf F, Stubbert L, et al. Non-replicating rhabdovirus-derived particles (NRRPs) eradicate acute leukemia by direct cytolysis and induction of antitumor immunity. Blood Cancer J. 2013;3:e123. | |
Rommelaere J, Geletneky K, Angelova AL, et al. Oncolytic parvoviruses as cancer therapeutics. Cytokine Growth Factor Rev. 2010; 21(2–3):185–195. | |
Geletneky K, Huesing J, Rommelaere J, et al. Phase I/IIa study of intratumoral/intracerebral or intravenous/intracerebral administration of Parvovirus H-1 (ParvOryx) in patients with progressive primary or recurrent glioblastoma multiforme: ParvOryx01 protocol. BMC Cancer. 2012;12:99. | |
Angelova AL, Aprahamian M, Balboni G, et al. Oncolytic rat parvovirus H-1PV, a candidate for the treatment of human lymphoma: In vitro and in vivo studies. Mol Ther. 2009;17(7):1164–1172. | |
Chen AY, Qiu J. Parvovirus infection-induced cell death and cell cycle arrest. Future Virol. 2010;5(6):731–743. | |
Au GG, Lindberg AM, Barry RD, Shafren DR. Oncolysis of vascular malignant human melanoma tumors by Coxsackievirus A21. Int J Oncol. 2005;26(6):1471–1476. | |
Au GG, Lincz LF, Enno A, Shafren DR. Oncolytic Coxsackievirus A21 as a novel therapy for multiple myeloma. Br J Haematol. 2007;137(2):133–141. | |
Reuss-Borst MA, Bühring HJ, Klein G, Müller CA. Adhesion molecules on CD34+ hematopoietic cells in normal human bone marrow and leukemia. Ann Hematol. 1992;65(4):169–174. | |
Terstappen LW, Nguyen M, Lazarus HM, Medof ME. Expression of the DAF (CD55) and CD59 antigens during normal hematopoietic cell differentiation. J Leukoc Biol. 1992;52(6):652–660. | |
Dhiman N, Jacobson RM, Poland GA. Measles virus receptors: SLAM and CD46. Rev Med Virol. 2004;14(4):217–219. | |
Yaiw KC, Miest TS, Frenzke M, Timm M, Johnston PB, Cattaneo R. CD20-targeted measles virus shows high oncolytic specificity in clinical samples from lymphoma patients independent of prior rituximab therapy. Gene Ther. 2011;18(3):313–317. | |
Naik JD, Twelves CJ, Selby PJ, Vile RG, Chester JD. Immune recruitment and therapeutic synergy: keys to optimizing oncolytic viral therapy? Clin Cancer Res. 2011;17(13):4214–4224. | |
Lemay CG, Rintoul JL, Kus A, et al. Harnessing oncolytic virus-mediated antitumor immunity in an infected cell vaccine. Mol Ther. 2012;20(9):1791–1799. | |
Guillerme JB, Boisgerault N, Roulois D, et al. Measles virus vaccine-infected tumor cells induce tumor antigen cross-presentation by human plasmacytoid dendritic cells. Clin Cancer Res. 2013;19(5):1147–1158. | |
Batliwalla FM, Bateman BA, Serrano D, et al. A 15-year follow-up of AJCC stage III malignant melanoma patients treated postsurgically with Newcastle disease virus (NDV) oncolysate and determination of alterations in the CD8 T cell repertoire. Mol Med. 1998;4(12):783–794. | |
Conrad DP, Tsang J, Maclean M, et al. Leukemia cell-rhabdovirus vaccine: personalized immunotherapy for acute lymphoblastic leukemia. Clin Cancer Res. 2013;19(14):3832–3843. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.