Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 14
The etiologic origins for chronic obstructive pulmonary disease
Authors Huang X, Mu X, Deng L, Fu A, Pu E, Tang T, Kong X ![]()
Received 28 January 2019
Accepted for publication 18 April 2019
Published 27 May 2019 Volume 2019:14 Pages 1139—1158
DOI https://doi.org/10.2147/COPD.S203215
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Xinwei Huang,1,2,* Xi Mu,3,* Li Deng,4 Aili Fu,5 Endong Pu,6 Tao Tang,2 Xiangyang Kong2
1Faculty of Environmental Science and Engineering, Kunming University of Science and Technology, Kunming City, Yunnan Province, People’s Republic of China; 2Medical School, Kunming University of Science and Technology, Kunming City, Yunnan Province, People’s Republic of China; 3Faculty of Life Science and Technology, Kunming University of Science and Technology, Kunming City, Yunnan Province, People’s Republic of China; 4The Pathology Department, First People‘s Hospital of Yunnan Province, Kunming City, Yunnan Province, People’s Republic of China; 5Department of Oncology, Yunfeng Hospital, Xuanwei City, Yunnan Province, People’s Republic of China; 6Department of Thoracic Surgery, Yunfeng Hospital, Xuanwei City, Yunnan Province, People’s Republic of China
*These authors contributed equally to this work
Abstract: COPD, characterized by long-term poorly irreversible airway limitation and persistent respiratory symptoms, has resulted in enormous challenges to human health worldwide, with increasing rates of prevalence, death, and disability. Although its origin was thought to be in the interactions of genetic with environmental factors, the effects of environmental factors on the disease during different life stages remain little known. Without clear mechanisms and radical cure for it, early screening and prevention of COPD seem to be important. In this review, we will discuss the etiologic origins for poor lung function and COPD caused by specific adverse effects during corresponding life stages, as well as try to find new insights and potential prevention strategies for this disease.
Keywords: chronic obstructive pulmonary disease, COPD, early origins, risk factors, air pollution, cigarette smoking
Introduction
COPD, characterized by long-term poorly irreversible airway limitation and persistent respiratory symptoms, is a common and preventable disease.1 According to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) guidelines, three criteria are needed to diagnose the disease: (1) a post-bronchodilator FEV1:FVC ratioof less than 70%, (2) “appropriate symptoms” such as dyspnea, sputum production, chronic cough, or wheezing, (3) “significant exposures to noxious environmental stimuli.2 This disease has at least three phenotypes: emphysema, chronic bronchitis, and small airway remodeling and obstruction,3 and environmental and genetic factors are involved in the pathogenesis and development of the disease. Cigarette smoking is main cause of the disease, whereas only 10–20% of smokers develop COPD,4 and approximately 25–45% of occurrence of COPD is attributed to nonsmoking risk.5 As shown in Table 1 (The Global Burden of Disease study 2017),6 COPD attributed to active smoking, ambient particulate matter pollution, occupational particulate matter/gases/fumes, ambient ozone pollution, household air pollution from solid fuels, secondhand smoke, and lead exposure was responsible for about 3.46 million of global all-age deaths and 79.78 million of disability-adjusted life-years (DALYs) in 2017. Active smoking and ambient particulate matter pollution were the main causes of deaths and DALYs for COPD (Figure 1). Although the global age-standardized death rates and DALY rates for COPD attributing to each of the above risk factors between 2007 and 2017 was reduced, this epidemical tendency forecasting is not optimistic as the growth and aging of population. Nowadays COPD is the fourth leading cause of death worldwide and will become the third leading cause of death by 2030,7 thus it will be an urgent health problem to be solved. Without curative therapies for COPD, palliation of airway obstruction, symptoms and exacerbation are the main clinical managements currently. Therefore, addressing the predisposing factors of COPD and prevent its development seemto be an appropriate intervention strategy for control of the disease in public health. Understanding the effects of risk factors correlated with the development of this disease on population at their different life stages is necessary so that more preventive strategies may be developed. In this review, we will provide a broad overview of etiologic origins for COPD, and try to find some potential preventive strategies and new insights for COPD studies.
 | Table 1 Global all-age attributable deaths and DALYs, and percentage change of age-standardized death rates and DALY rates caused of COPD by environment exposure between 2007 and 2017 |
 | Figure 1 The proportion of all-age deaths and DALYs for COPD attributed to active cigarette smoking, ambient particulate matter pollution, occupational particulate matter/gases/fumes, ambient ozone pollution, household air pollution from solid fuel, and secondhand smoke in 2007 and 2017.Notes: This data comes from part of the article Stanaway JD, Afshin A, Gakidou E, et al. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392(10159):1923–1994.6 R 3.5.2 version was used to plot.Abbreviation: DALYs, disability-adjusted life-years. |
The genetic, epigenetic, and transcriptional origins of COPDand poor lung function
The genetic origins
COPD and poor lung function (FEV1, FEV1/FVC) may already be determined before the birth for some patients. COPD shows independent family aggregation,8–10 and COPD family history shows 18.6% of the population-attributable risk, with more severe diseases, worse quality of life, and more frequent exacerbations.11 In addition, asthma with family aggregation is observed to be the indirect risk of COPD.12 Similarly, inter-individual difference in lung function is partly defined by genetic reasons. The infants with the lowest quartile of functional residual capacity of the FEV1/FVC ratio and FEV1 were lower than that with the highest quartile up to age 22.13 Heritability of FEV1/FVC was higher than that of either FEV1 or FVC, and a significant difference in lung function exists between males and females. Males have higher FEV1 and FVC, while females have higher FEV1/FVC.14 The genome-wide association study (GWAS), whole genome sequencing (WGS), and fine-mapping studies have consistently discovered and well-replicated many COPD associated genes (COPD genes) and loci in various populations. As summarized in Table 2,15–68 a panel of genes associated with COPD and poor lung function, nicotine addiction, and lung injury-repair response are indicated.
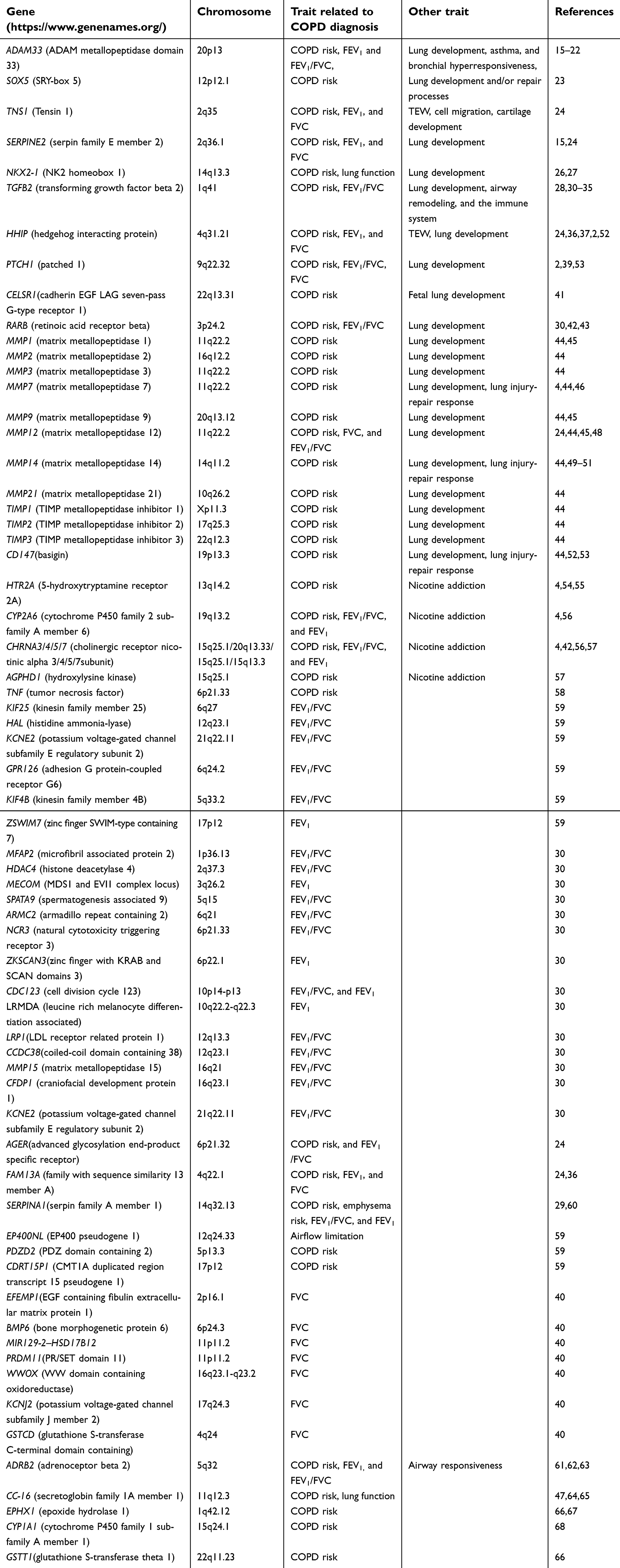 | Table 2 Genes involved in COPD |
The early abnormal lung development including airway and alveolar development might underlie the susceptibility to COPD and impaired lung function.3 The normal expression of NKX2-1, the first symbol of lung development, plays a critical role in morphogenesis of the anterior foregut and the lung and in differentiation of lung epithelial cells.26,27 The mice lung with its heterozygous mutation failed to undergo normal branching embryogenesis and was unable to sustain normal gas exchange function, and subsequently causes immediate postnatal lethality.26 The hedgehog (hh) pathway transmits signals to embryonic cells and is one of the key pathways of animal development.69 The functional loss in HHIP may lead to impaired branching morphogenesis and lung hypoplasia in mice.31,38 Retinoic acid receptor-beta (RARB) plays a key role in septation mature and alveoli formation of mice lung. RARB knockout mice showed premature septation, and they formed alveoli two times faster than that of wild-type mice during the period of septation.43 The transcription factor SOX5, a susceptibility gene for COPD, is critical for proper in utero lung morphogenesis. SOX5 deficiency mice exhibited delayed lung development before the saccular stage.23 Another COPD gene TGFB2 involves cellular growth, differentiation, and apoptosis, as well as other cellular functions from development to tissue homeostasis, also plays an important role in normal lung development, airway remodeling, and the immune system.28,30–35 The roles of matrix metalloproteinase (MMP) family and tissue inhibitors of MMPs (TIMPs), and CD147 in the lung development, lung repair responses to injury, and occurrence of multiple lung diseases including COPD and emphysema were well reviewed by Hendrix and Kheradmand.44During lung development, MMP1/2/3/7/9/12/14/21, CD147, and TIMP1/2/3 play important roles. These genes have different expression levels in different cell types of the lung during different developmental stages.70,71 The lack of MMP14 in mice may decrease alveolar enlarged airspaces and surface area, as well as delay angiogenesis.49,50 In addition, CD147, MMP7, and MMP14 might be involved in lung injury-repair response.46,51–53 ADAM33 also plays different roles in different developmental stages, including antenatal airway lung morphogenesis and airway wall modeling,15,16 and contributes to asthma and bronchial hyperresponsiveness in early life and in adults.16,21 The interaction of ADAM33 with prenatal smoking exposure could lead to reduced lung function and development of asthma at the age of 8.22 Moreover, polymorphisms of this gene, especially in its functionally relevant 5′ end, were related to the preschool children with increased airway resistance and impaired lung function and COPD susceptibility.20 Taken together, many GWAS-identified COPD genes play key roles in lung development and lung injury-repair response, and their abnormal expression modulated by variation or disturbance of maternal environmental exposure before the birth of individuals may pave the way for COPD development and poor lung function.
In addition, many COPD genes contribute to COPD development and poor lung function in childhood and adulthood of individuals through cross talking with environmental exposure factors. As shown in Table 2, many other COPD genes were showed to be associated with FEV1/FVC, FVC, and FEV1 of the population in different life stages. The polymorphisms at positions Arg16Gly/Gln27Glu within ADRB2 were found to be associated with airway responsiveness at the age of 6, with higher spirometry at the age of 6 and 11, as well as with the presence of COPD, asthma, and other respiratory symptoms in middle-aged and older adults,61 whereas associated with worse lung function and less likelihood of the asthma diagnosis at the age of 11.62 CC-16 was showed to accelerate decline in lung function in childhood and adulthood, as well as promotethe progress of moderate airflow limitation in adults.65 A follow-up study on fetalbirths indicated that several detoxification genes including EPHX1, CYP1A1, and GSTT1, which are implicated in the development of emphysema and COPD, may modify the impact of cigarette smoke exposure and ambient air pollutants such as PM2.5 and polycyclic aromatic hydrocarbons (PAHs) on acute bronchitis in their later life.72–75 In some cases, nicotine addiction increases cigarette smoking, thus increases the COPD risk and impair lung function. Some genes associated with nicotine addiction were reviewed, including CHRNA3/4/5/7, DRD4, SLC6A3, SLC6A4, NRXN1, HTR2A, CHRNA7, CYP2A6,47 of which, HTR2A, CYP2A6, and CHRNA3/4/5/7 are involved in COPD pathogenesis depending partly on cigarette exposure due to a gene-by-environment interaction.54,55–57 HTR2A may increase the risk for the early onset of cigarette smoking and the risk for relapsing after smoking cessation.55 Furthermore, the gene cluster of CHRNA3/CHRNA5/CHRNB4 plays an important role in the cigarette smoke-causing injury process. Smoking behavior may mediate the relationship between COPD and the rs1051730 mapped to CHRNA3/5.76 However, the mechanism of CHRNA3/5 increasing the respiratory diseases risk is controversial: either CHRNA3/5 has an independent effect or the regulation of nicotine addiction on COPD development.77,78
Epigenetic origins
During the lung development, some epigenetic alterations including: DNA methylation, histone modifications, and noncoding RNAs are key regulators of the process. An individual’s epigenetic alterations of genes that may originate from his parents or grandparents could subsequently persist well into childhood.79–81 Developmental programming, occurring primarily via epigenetic alterations, can be induced by the intrauterine conditions such as cigarette smoking, nutrition and stress, and result in inter- and transgenerational epigenetic effects on genetic origins in mice and their offspring.82,83 Reprogramming of the epigenome, genetic imprinting, retained nucleosomes may be the potential mechanism of inter- and transgenerational epigenetic effects.82 Epidemiological and experimental evidence indicated that exposure to environmental factors during prenatal and early postnatal period upon the epigenome is critical in embryonic development and tissue differentiation may lead to permanent epigenetic modifications and contribute to the possibility of developing adult-onset disorders such as metabolic, cardiovascular, lung cancer, lung function, and COPD.79,82,84,85 Epigenetic regulation is important in chronic remodeling of respiratory tracts.86 DNA methylation is an established mechanism for COPD development,87 which may be regulated by genetic polymorphisms.82,88 As the key regulators of lung development, histones are usually modified by methylation, phosphorylation, acetylation, and ubiquitination of specific amino acids. Especially, histone acetylation is crucial in regulation of lung development and function, and is implicated with asthma and COPD.89–91 Histone acetyltransferases (HATs) mediates histone acetylation that increases gene expression, whereas histone deacetylases (HDACs) induces hypocetylation that promotes gene silencing.89 Thus, the imbalance between HATs and HDACs activity caused by any adverse factors may lead to disorders of embryonic lung development, including the block in proximal airway development,92 alveolar hyperplasia,93 and disrupted alveolarization.94 In addition, the methyltransferases Suv39H1 and Suv39H2 that result in transcriptional silencing through histone H3 lysine 9 methylationare are involved in all lung development processes.95 Thus, the disturbance of necessary epigenetic alterations resulting from genetic variation or adverse risk factors during the prenatal and early postnatal period may influence the lung development of the fetus.
Postnatal environmental factors including cigarette smoking, aging and diet, as well as genetic risk factors such as genetic variation, can modulate the methylation modification of promoter CpG via DNA methyltransferases and methyl CpG binding protein 2, which affects the transcription and expression/activation of some key genes involved in pathogenesis of COPD and impaired lung function.96–98 Different risk factors can induce distinctive DNA methylation profiling on genome of individuals including patients with COPD and healthy people.98–100 Methylation at cg08257009 in the SERPINA gene cluster was found to be associated with FEV1/FVC in adults.101 Furthermore, one epigenome-wide association analysis (EWAS) in whole blood found that methylation at 15 CpG-sites was significantly associated with cigarette smoking and lung function, of which, 5 methylated CpG-sites (cg05575921, cg21161138, cg05951221, cg21566642, and cg06126421) showed significant associations between DNA methylation and gene expression in lung tissues.102 Another EWAS of four SNP (rs8034191:T>C-HYKK, rs12914385:C>T-CHRNA3, rs13180: C>T-IREB2 and rs8042238:C>T-IREB2), previously related to COPD, showed a significant association with blood DNA methylation of those genes, of which, PSMA4 and IREB2 were also differentially methylated in COPD cases and controls. In addition, all four variants also showed a significant correlation with differential expression of the IREB2 3ʹUTR in lung tissues.103
Taken together, genetic and environmental factors, and their cross talking may influence the early lung development, and result in COPD and poor lung function later via epigenetic modifications that modulate thiss activation and transcription of COPD genes.
MicroRNAs origins
The increasing human/animal models and cell studies demonstrated that microRNAs (miRNAs) play a central regulatory role in various biological processes, including cellular proliferation, differentiation and apoptosis. MiRNAs play key roles in the lung development, and pulmonary diseases such as COPD, whereas the degree of translation into pulmonary diseases is still unclear. Recently, we reviewed the roles of miRNAs in COPD development induced by different environmental exposure as well as genetic predisposition encounter. Environmental exposure including air pollutants and cigarette smoking can induce dysregulated miRNA expression profiles, which cause adverse biological response such as oxidative stress, inflammation, and the imbalance between apoptosis and replenishment of structural cells in the lung by disturbing their regulation on COPD genes, and contribute to COPD development and poor lung function in susceptible individuals. In addition, functional SNP variant with miRNA genes can affect the mature form of corresponding miRNAs and disturb the regulation of them on COPD genes, thus leading to COPD susceptibility. Some key miRNAs, such as miR-34 a/b/c, miR-146a, miR-203, miR-218 and let-7 family, may serve as potential fluid biopsy-based markers for risk indicators of environmental exposure and COPD.104
The environmental origins of COPDand poor lung function
Although without absolute consistence with the pulmonary development phase division, it is traditionally divided into five histological stages from the embryonic stage to the alveolar stage (Table 3).105,106 Two follow-up studies conducted on participants aged from 13 to 71 years showed that the plateau of FEV1 is 20–23 years old for males, and 15 for females, whereas the decline in FEV1 occurs at about 25 years old for both sexes, suggesting a longer plateau phase for FEV1 in females than in males.107,108 Moreover, the FEV1/FVC ratio increased until 17 years old in males and then declines approximately linearly, whereas this ratio indicated a uniform decline in the age range in females.107,108 Interestingly, although cigarette smoking can increase the rate of lung function decline in both sexes, it can only reduce the achieved peak of FEV1 value in males, but not in females.108 Those observations suggest a congenital difference of lung function among sexes. The difference may originate from hereditary difference from the gender-biased environmental exposure ways between males and females.
 | Table 3 The different risk factors during varied life periods of the lung |
Another follow-up study showed that the antenatal adverse factors and early childhood disadvantage factors lead to permanent lung function impairment, with a slightly greater decline in lung function but no catching up with age.109 COPD risk increases with increasing early-life adverse factors, of which, the impacts resulting from childhood asthma, maternal and paternal asthma, maternal smoking, and respiratory infections are the same as strongly implicated in an accelerated decline rate of lung function as that of severe smoking. The tendency of decline in lung function increased with the accumulative degree of smoking exposure (healthy never smokers, quitting smoking before the age of 30, quitting smoking between 30 and 40 years, quitting smoking after the age of 40, continuous smokers),107 suggesting that the different degrees of exposure to environmental factors might lead to different degrees of lung function impairment.
Maternal amniotic fluid
Maternal amniotic fluid has important impacts on fetal lung development and respiratory disease occurrence in offspring. Oligohydramnios can lead to fetal lung hypoplasia, while the extent depends on the degree and duration of little amniotic fluid, and the fetal lung development stages.110,111 Fetal lung development may be regulated by amniotic fluid components such as pro-inflammatory mediators.112 Plasminogen activator inhibitor 1 (PAI-1) is a main inhibitor of the fibrinolytic system and plays an important role in tissue remodeling,113 and its reduction is associated with cough at 1 year of age and wheeze at 2 years of age.114
Preterm birth and birth weight gain
Preterm birth is the main risk for bronchopulmonary dysplasia (BPD) that accounts for the prevalence of the vast majority of chronic pulmonary diseases115,116 and is a risk factor of permanent lung function decline.117–120 Preterm birth is also associated with school-age and adult asthma,112,118 wheeze and breath shortness,121 COPD,122 as well as bronchial hyperresponsiveness and decreased FEV1.120
In addition, low birth weight may lead to persistent decline in lung function and different degree of airway obstruction, and increase risk of respiratory symptoms.123–125 Interestingly, a meta-analysis report of 147,000 European children observed an independent relationship between higher infant weight gain and the higher risk of school-age asthma and preschool wheezing.112 The mechanism between birth weight and COPD needs further study.
Maternal cigarette smoking
Antenatal adverse exposure may lead to the lung’s response, making it more predisposed to subsequent injury.116 Fetal exposure to maternal smoking during pregnancy is one of the most serious events for abnormal lung development,126 it can increase the risk of poor lung function, COPD, asthma, and childhood wheeze.122,127–130 The mechanisms might partly result from epigenetic alterations because of the global DNA methylation in umbilical-cord blood was observed to be associated with prenatal exposure to PAH,131 which is the main harmful component of incomplete combustion of cigarettes.
Maternal air pollution exposure
Preconceptional and prenatal exposure to industrial and traffic air pollutants increases risk of childhood asthma, allergic rhinitis, and eczema.132 Particulate matter smaller than 2.5 µm (PM2.5), composed of ammonium, nitrate and bromine, mainly results from traffic and biomass combustion.133 Maternal sulfur dioxide (SO2) and PM2.5 exposure were found to be associated with preterm birth and low birth weight, and childhood asthma.133–135 Residential PM2.5 exposure was showed to influence the expression of placental imprinted genes, suggesting a plausible line of investigation of how air pollution affects fetal growth and development.136 Maternal PM10 exposure was reported to increase the risk of congenital anomaly, notably fetal growth and development, and is related to placental DNA methylation, such as the LINE1 and HSD11B2 genes.137 In the pilot study of 44 mother-infant pairs, Kingsley et al138 observed an association of prenatal perfluorooctanoic acid exposure with cord blood leukocyte DNA methylation in two CpG sites of RASA3 that plays a key role in cell growth and differentiation.139
Delivery patterns
Delivery mode shapes individual microbiota’s acquisition and establishment, which may influence children’s health.140 Maternal vaginal microbiota provides a natural first-class microbial exposure resembling the mother’s vaginal microbiota’s habitat on infant’s body via natural labor. Whereas cesarean section that lacks a vaginal exposure leads to the first microbial community resembling the maternal skin microbiota.140 Furthermore, cesarean section could increase the risk of allergic rhinitis, asthma, and hospitalization for asthma.141 These suggest that delivery patterns may lead to difference in normal physiology or contribute to respiratory diseases due to variations in the microbiota development. Previous studies commonly focused on the specific bacterial taxa of the gut, however, the role of respiratory tract flora in pulmonary disease occurrence is little known.
Maternal obesity
The role of maternal obesity in their children has been reviewed by Duijts et al.142 Pre-pregnancy obesity, and higher gestational weight gain and maternal overweight or obesity during pregnancy is associated with the higher risk of respiratory diseases, such as wheezing and asthma in their offspring.143,144,145 Thus, maternal obesity may be a risk of COPD, but further investigation is still needed.
Maternal diet and drug use
Maternal diet or drug use during pregnancy may regulate the risk of respiratory diseases in offspring, which may be caused by the interactions between maternal nutrition intake and genetic alterations, as well as by immune regulation, epigenetic modifications, and microbial changes. The overfull folic acid and free sugars intake in pregnant women were shown to increase the risk of asthma in offspring,146,147 which may be due to the role of the nutrition in airway inflammation and hyperreactivity in late generations.148 Hypercaloric diet (HFD) of pregnant dams could lead to metabolic abnormalities that may persist throughout development,148 and inflammatory response in the pups’ lungs.149 The intake of HFD + antioxidant N-acetylcysteine (NAC) in pregnant dams was showed to delay the alveolarization of pups, although their branching morphogenesis is normal.150 While maternal intake of some vitamins, microelements, and folic acid was found to have protective effects on some respiratory diseases in offspring151 and may modulate epigenetic modifications on gene expression and airway epithelial cell signaling in fetal lung, which may affect intrauterine programming of growth and development.152,153 Polymorphisms within some genes involve the regulation of maternal antioxidant intake on offspring respiratory disease.154 Furthermore, intrauterine antibiotic exposure plays important roles in the health of offspring through interfering with normal metabolic and immune maturation,155 affecting the fetal organogenesis and development by methylation alterations and placental microbiome changes.156,157 Prenatal cocaine exposure in the placenta might affect neurochemical effects, vasoconstrictive, and fetal programming. Maternal diet and drug use during pregnancy are an increasing focused topic, because they are modifiable causes of disease in offspring. However, these complex links and mechanisms between maternal intake and COPD are necessary to reveal.
Childhood air pollution exposure
Early life air pollution exposure including traffic-derived CO, NO, NO2, PM2.5, PM10, SO2, and black carbon appears to influence the development of airway diseases and increase risk of respiratory diseases, including COPD and asthma in later life.158–161 A prospective birth cohort study during the first 6 years of life indicated that early childhood air pollution exposure to PM2.5 increased the risk of early respiratory diseases,162 which was similar to another prospective study observation in children of Sweden.163 PM2.5 can induce both chemical and physical damage by penetrating the alveoli into the systemic circulation, whereas PM10 usually causes physical damage to the lungs, such as the alveoli and larynx.164 As we known, at least three mechanisms are thought to be involved in the causal processes: occurrence of oxidative stress, inflammation, and epigenetic alterations. Firstly, PM-induced excessive ROScauses oxidative stress that leads to cell function impairment and cell death.165 Secondly, oxidative stress alters the expression of proteins related to inflammatory response in the airways.166 Additionally, PM may induce epigenetic changes including aberrant DNA methylation and histone modifications of key genes like LINE-1, IL-8 and COX-2, and influence the inflammatory response.167–169
Childhood asthma
Childhood asthma is an established risk factor for low lung function and predisposition to COPD in adult.109,170–176 Although the clear mechanism between COPD development stemming from childhood asthma history is poorly understood, the overlapped genetic variations between COPD and asthma were identified by previous GWAS.172,177 Interestingly, Bui et al performed a cohort study in Tasmanian children (N=8,583) aged 7–45 years and found that the lowest quartile of FEV1 at 7 years old in a selected subsample (N=1,389) was related to asthma-COPD overlap syndrome (ACOS) but not asthma or COPD alone, and observed the association of the lowest quartile of FEV1/FVC ratio at 7 years with COPD (OR: 5.76; 95%CI: 1.9–17.4) and ACOS (OR: 16.3; 95%CI: 4.7–55.9), but not with asthma alone,172 suggesting that screening for lung function in children may provide help in identifying the high-risk group of COPD. Interestingly, the assessment of airway hyperresponsiveness that correlates with airway inflammation, asthma, and remodelling may contribute to estimate of asthma control and future exacerbation risk, although this procedure is still a study tool for asthma.178,179
Childhood respiratory infection
Normal respiratory tract microbiome is important in immunological development and allergic inflammatory response, which modulates the COPD risk.180,181 Childhood respiratory infection was demonstrated to be associated with lower lung function and increased COPD risk in later life.122,182,183 Early respiratory infection including virus and bacterial flora is predominantly related to a series of respiratory diseases. Viral respiratory tract infections especially respiratory syncytial virus (RSV) and human rhinovirus (HRV) in infancy and early childhood may promote the risk of asthma and wheezing later.184,185 Children with a history of HRV infection could contribute to the occurrence of asthma in preschool age.186 Children who suffer from RSV-bronchiolitis could increase the risk of lower lung function, asthma, wheezing, hospitalization and respiratory morbidity in later life.186–189 Even though the mechanisms between respiratory diseases and infection are poorly known, it may at least partly result from genetic factors. Some variants of the 17q21 locus were observed to be implicated in childhood asthma, and also associated with early-life infection and HRV-induced wheezing.183,190 Protecting children from being “at risk” during infancy or early childhood is a way to prevent serious respiratory infection, meaning an effectively preventive strategy for respiratory diseases.
Childhood cigarette smoking exposure
Early childhood smoking exposure majorly comes from parental secondary smoking with less active smoking. Early family cigarette smoking exposure can easily impair lung function and increase the later risk of respiratory diseases in children.191 Previous prospective cohort studies indicated that childhood cigarette smoking exposure from families leaded to reduced lung function, active smoking predisposition, airway obstruction susceptibility and early onset COPD, as well as prevalence of bronchodilator responsiveness, asthma and wheeze in later life.192–194 Parental smoking cessation and public-place banning cigarette smoke may be an effective measure for prevention of children’s respiratory diseases and COPD occurrence in later life.
Childhood obesity/nutritional factors
Obesity is not only prevalent among adults but also occurs in children.195 Childhood obesity is an increased risk of chronic respiratory diseases. Asthma is consistently one of the most common diseases among children. Presently, the relationship between childhood obesity and COPD are still largely unknown, but some evidence about the effects of childhood obesity on early asthma and airflow obstruction was found.112,196–200 The leukotriene pathway and some overlapping genes between obesity and asthma including β2-adrenergic receptor (ADRB2), TNF-a, lymphotoxin-a (LTA), vitamin D receptor (VDR),201 and protein kinase C alpha (PAKCA)202 were demonstrated to play important roles in the obesity-asthma phenotype.197 Additionally, age is a significant effect modifier of obesity and asthma. As asthma increases, the impact of obesity on asthma may decrease.197 The etiologies for COPD and asthma caused by obesity partly root in obesity-induced circulating inflammation in the lung, and airway smooth muscle dysfunction.197,203–207
Nutritional factors may play an important role in the development, progression and administration of pulmonary diseases such as COPD and asthma.208 High-fat diet pattern was shown to be associated with increased risk of childhood asthma and COPD,209,210 likely by augmenting neutrophil airway inflammation and suppressing bronchodilator’s recovery.211 Furthermore, eating fast food is correlated with the prevalence of asthma, airway hyperresponsiveness, and wheezing in childhood.212,213 Some antioxidants in lungs including uric acid, vitamins C and E, glutathione and beta-carotene are the first line of defending against the oxidants to increase risk of COPD, idiopathic pulmonary fibrosis and asthma.214 Abnormal concentration of these antioxidants may increase risk of lower lung function, current wheezing, and asthma.215,216 This may be explained by several potential biological mechanisms, including impaired pathogen elimination of respiratory airways,217 abnormal regulation of Th17 cells,218 as well as reduced maturation of airway smooth muscle cells and suppressor T cells.197 Modifying dietary fat intake and reducing obesity may be helpful to control and manage asthma and COPD.
Adulthood cigarette smoking exposure
Cigarette smoking and secondhand smoke exposure in adulthood contributes to the development of COPD and the increasing mortality of COPD, although persist smoking cessation.191,210,219 The excess risk of developing COPD in high cigarette smoke exposure categories was estimated 60–400%.220 Whilethere is controversy between smoking predisposition and gender,. one study showed that females have a higher susceptibility to cigarette smoking, another reported the same level of predisposition for both sexes.219,221 The burden of COPD would increase in women as cigarette smoking prevalence increased, and as young women started smoking at an earlier agecigarette smoking.161 The main causative processes at least involve oxidants-antioxidants, proteases-antiproteases, improper repair, and chronic inflammation of airways.219 These processes result in alveolar wall destruction and mucus hypersecretion, functional disorder and death of biomolecules, destruction of extracellular matrix, and fibrosis of lung with submucosal, adventitial and smooth muscle thickening.219 Therefore, inhibiting the pathogenesis of COPD should be a good strategy for the treatment and symptom improvement of the disease. Quitting smoking early is of great benefit in COPD development and the decline in lung function, especially before the age of 30 when the rate of lung function decline in those who had quitsmoking is indistinguishable from healthy nonsmokers.107
Adulthood air pollution exposure
Approximately 50% of all households and 90% of rural households use biomass fuel for heating and cooking, which accounts for over three billion people exposed to biomass smoke.160,222 Even in modern homes in some developed countries, biomass fuel is unable to be replaced by the ever-increasing cost of clean fuels.223 Women seem to suffer from more biomass smoke exposure because they could inhale over 25 million liters of highly polluted air during their lifetime when they spend an average of 60,000 hours cooking near a biomass stove.224 Biomass fuel including fossil coal, animal dung, wood and crop residues has low efficiency due to less heat production and incomplete burning, and releases more than 200 established chemical compounds, including gaseous and particulate pollutants and strong oxidant properties. Over 90% of those chemical compounds could penetrate deep into the lungs and result in chronic inflammation and destructive changes in airways and alveoli.160,224
Compared with no exposure to biomass smoke, exposure to biomass fuel smoke was observed to be associated with 2.44-fold and 2.4-fold increased odds of COPD in both sexes and women, respectively.225,226 Exposure to biomass smoke may be a greater risk factor for COPD compared with cigarette smoking exposure from a global perspective because of the number of people exposed to biomass smoke is three times more than smokers.224,227 COPD patients exposed to biomass smoke share part similar profile of cell and airway inflammation with smokers.228 Compared with control women cooking with clean fuels, women cooking with biomass have more severe airway inflammation and oxidative stress when evaluated with the induced sputum.229 The ventilation improvement has been demonstrated to be effective in reducing indoor biomass smoke,230 which might decrease the burden of COPD.
Outdoor air pollution is mainly caused bymotor vehicle and industrial emissions and is related to various respiratory impairments, particularly in children aged 10–18 years231 and women.232 The heavier traffic density was shown to be associated with the greater declines in lung function.232 Long-term exposure to ambient PM2.5 is associated with the decline in lung functionand increases risk of COPD.233,234 In addition, the COPD risk increases with the increase of PM10 levels,235 whereas the prevalence of COPD and respiratory symptoms reduces with the decline in levels of PM10.236 A population-based cohort study in Metropolitan Vancouver, Canada, reported that black carbon is responsible for the increase of COPD hospitalization and mortality, while wood smoke exposures increases the risk of COPD hospitalization.237 The oxidative stress, hyperresponsiveness, inflammation, impaired cilia activity and amplification of viral infections in airways may explain the adverse effects of ambient air pollutants.238
Occupational exposure
Occupational exposure, such as gases/fumes, biomass smoke, dust exposure, animal and crop planting, chemical exposure, is strongly associated with COPD.98,233,239,240 The recent National Health and Nutrition Examination Survey for the non-institutionalized civilian US indicated that prevalence of airflow obstruction varies by occupation and industry, and that mining, construction, manufacturing, prepress, bookbinders, installers, and repairers may influence airflow obstruction.241 In addition, occupational exposure to gas, vapor, dust, or fumes was shown to be associated with COPD, airflow limitation, and emphysema.242,243 Compared to developed countries, occupational exposure is more serious in developing countries due to lack of adequate protection and lack of strict regulations in the workplace.160 Therefore there would be a larger burden of COPD attributed to occupational exposure in developing countries compared to developed countries.
Conclusions
COPD is a heterogeneous and multifactorial disease. As shown in Table 1, COPD induced by active smoking, ambient particulate matter pollution, occupational particulate matter/gases/fumes, ambient ozone pollution, household air pollution from solid fuels, secondhand smoke, and lead exposure was responsible for about 3.46 million of global all-age deaths and 79.78 million of disability-adjusted life-years (DALYs) in 2017. Active smoking and ambient particulate matter pollution were the main causes of deaths and DALYs for COPD (Figure 1). The status of death and DALY for COPD is getting worse withpopulation growth and aging. Therefore, COPD emerges as an enormous challenge to global health.
Individuals may suffer special exposure factors during different life stages (Table 3). In turn, these special factors could exhibit their own effects at different life stages. As summarized in Figure 2 and Table 2, host family history of respiratory diseases such as COPD, asthma, and emphysema, which may share some overlapping predisposing genes, is an established risk factor for COPD development and poor lung function. Some COPD genes such as ADAM33, SOX5, TNS1, SERPINE2, NKX2-1, TGFB2, HHIP, PTCH1, CELSR1, RARB, CD147, MMP1/2/3/7/9/12/14, and TIMP1/2/3 are critical for lung development (organogenesis, alveolarization, branching morphogenesis, and angiogenesis) or/and lung repair responses to injury (airway inflammation, oxidative stress, impaired cilia activity, and amplification). Whereas, some COPD genes, including HTR2A, CYP2A6, CHRNA3/4/5/7, and AGPHD1, are involved in nicotine addiction and toxicant metabolism. Environmental exposure such as cigarette smoking, biomass smoke, and indoor/outdoor air pollutant during all life stages of an individual has well-documented adverse effects on the lung development, lung function, and COPD susceptibility through cross talking with COPD genes by which environmental exposure pollutants might induce abnormal epigenetic modifications on genome and dysregulated miRNA expression profiles, disturbing the expression and function of COPD genes. Based on the previous findings, we may get an inference that adverse exposure during the different life stages might cause permanent impact on the lung, such as failure to reach the normal spirometric plateau, and the accumulative impairment in the lung that paves the way for COPD development. Lung function apparently reduces with more risk factors (Figure 3).
 | Figure 2 The origins for chronic pulmonary disease resulting from genetic and environmental factors. |
 | Figure 3 The plausible trajectories to lung function by varied risk factors in different life stages of the lung. |
COPD is common and preventable. Undoubtedly, avoidance of exposure to any adverse environmental factor would be advisable for individuals with/without COPD susceptibility. Preschool age is likely to be the key period for prevention of lung function and respiratory diseases, and measures starting in adulthood may be too late. Early childhood lung function screening, banning cigarette smoking in public places, respiratory infection prevention, searching biomarkers for evaluation of environmental exposure could be the effective protective measures against lung function impairment and COPD development. Interestingly, two oxidative damage-related produce such as 8-Oxo-2′-deoxyguanosine (8-OHdG) and LINE-1 may become epigenetic biomarkers induced by ROS generation resulting from environmental exposure.167,244,245
In the future, investigation of genomics, epigenomics and transcriptomics for COPD development will remain urgent. Extensive studies on the diversity of structure and function for miRNAs associated with COPD development will give better insights into the selection of appropriate miRNAs serving as prognostic or therapeutic biomarkers for COPD. Notably, a technique that may safely remove DNA methylation, resulting in the direct re-installation of unmodified deoxycytidine (dC) from 5-formyl-deoxycytidine (fdC) undergoing C-C bond cleavage, has a potential to treat and prevent COPD caused by DNA methylation.246 Furthermore, gene editing in bronchioalveolar stem cells (BASCs) and basal stem cells (BSCs) that might regenerate both trachea cilia and secrete epithelium and generate alveolar epithelium after extreme injury may contribute to the recovery from both alveolar and bronchiolar injury.247–251
Acknowledgments
This work was funded by the National Natural Science Foundation of China (81460007) and the Fund for Applied Basic Research in Yunnan Province, China (2013FZ182).
Authors contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD executive summary. Am J Respir Crit Care Med. 2017;195(5):557–582. doi:10.1164/rccm.201701-0218PP
2. Mirza S, Clay RD, Koslow MA, Scanlon PD. COPD guidelines: a review of the 2018 GOLD report. Mayo Clinic Proc. 2018;93(10):1488–1502.
3. Postma DS, Bush A, van den Berge M. Risk factors and early origins of chronic obstructive pulmonary disease. Lancet. 2015;385(9971):899–909.
4. Perez-Rubio G, Cordoba-Lanus E, Cupertino P, Cartujano-Barrera F, Campos MA, Falfan-Valencia R. Role of genetic susceptibility in nicotine addiction and chronic obstructive pulmonary disease. Rev Invest Clin. 2019;71(1):36–54.
5. Salvi SS, Barnes PJ. Chronic obstructive pulmonary disease in non-smokers. Lancet. 2009;374(9691):733–743.
6. Stanaway JD, Afshin A, Gakidou E, et al. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392(10159):1923–1994.
7. Diaz-Guzman E, Mannino DM. Epidemiology and prevalence of chronic obstructive pulmonary disease. Clin Chest Med. 2014;35(1):7–16.
8. McCloskey SC, Patel BD, Hinchliffe SJ, Reid ED, Wareham NJ, Lomas DA. Siblings of patients with severe chronic obstructive pulmonary disease have a significant risk of airflow obstruction. Am J Respir Crit Care Med. 2001;164(8 Pt 1):1419–1424.
9. Silverman EK, Chapman HA, Drazen JM, et al. Genetic epidemiology of severe, early-onset chronic obstructive pulmonary disease. Risk to relatives for airflow obstruction and chronic bronchitis. Am J Respir Crit Care Med. 1998;157(6 Pt 1):1770–1778.
10. Patel BD, Coxson HO, Pillai SG, et al. Airway wall thickening and emphysema show independent familial aggregation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;178(5):500–505. doi:10.1164/rccm.200801-059OC
11. Hersh CP, Hokanson JE, Lynch DA, et al. Family history is a risk factor for COPD. Chest. 2011;140(2):343–350. doi:10.1378/chest.10-2761
12. McGeachie MJ, Yates KP, Zhou X, et al. Patterns of growth and decline in lung function in persistent childhood asthma. N Engl J Med. 2016;374(19):1842–1852. doi:10.1056/NEJMoa1513737
13. Stern DA, Morgan WJ, Wright AL, Guerra S, Martinez FD. Poor airway function in early infancy and lung function by age 22 years: a non-selective longitudinal cohort study. Lancet. 2007;370(9589):758–764. doi:10.1016/S0140-6736(07)61379-8
14. Klimentidis YC, Vazquez AI, de Los Campos G, Allison DB, Dransfield MT, Thannickal VJ. Heritability of pulmonary function estimated from pedigree and whole-genome markers. Front Genet. 2013;4:174. doi:10.3389/fgene.2013.00174
15. Haitchi HM, Bassett DJ, Bucchieri F, et al. Induction of a disintegrin and metalloprotease 33 during embryonic lung development and the influence of IL-13 or maternal allergy. J Allergy Clin Immunol. 2009;124(3):
16. Haitchi HM, Powell RM, Shaw TJ, et al. ADAM33 expression in asthmatic airways and human embryonic lungs. Am J Respir Crit Care Med. 2005;171(9):958–965. doi:10.1164/rccm.200409-1251OC
17. Klaassen EM, Penders J, Jobsis Q, et al. An ADAM33 polymorphism associates with progression of preschool wheeze into childhood asthma: a prospective case-control study with replication in a birth cohort study. PLoS One. 2015;10(3):e0119349. doi:10.1371/journal.pone.0119349
18. Wang X, Li W, Huang K, et al. Genetic variants in ADAM33 are associated with airway inflammation and lung function in COPD. BMC Pulm Med. 2014;14:173. doi:10.1186/1471-2466-14-173
19. Zhou DC, Zhou CF, Toloo S, Shen T, Tong SL, Zhu QX. Association of a disintegrin and metalloprotease 33 (ADAM33) gene polymorphisms with the risk of COPD: an updated meta-analysis of 2,644 cases and 4,804 controls. Mol Biol Rep. 2015;42(2):409–422. doi:10.1007/s11033-014-3782-5
20. van Diemen CC, Postma DS, Vonk JM, Bruinenberg M, Schouten JP, Boezen HM. A disintegrin and metalloprotease 33 polymorphisms and lung function decline in the general population. Am J Respir Crit Care Med. 2005;172(3):329–333.
21. Lee JY, Park SW, Chang HK, et al. A disintegrin and metalloproteinase 33 protein in patients with asthma: relevance to airflow limitation. Am J Respir Crit Care Med. 2006;173(7):729–735.
22. Reijmerink NE, Kerkhof M, Koppelman GH, et al. Smoke exposure interacts with ADAM33 polymorphisms in the development of lung function and hyperresponsiveness. Allergy. 2009;64(6):898–904.
23. Hersh CP, Silverman EK, Gascon J, et al. SOX5 is a candidate gene for chronic obstructive pulmonary disease susceptibility and is necessary for lung development. Am J Respir Crit Care Med. 2011;183(11):1482–1489.
24. Kerkhof M, Boezen HM, Granell R, et al. Transient early wheeze and lung function in early childhood associated with chronic obstructive pulmonary disease genes. J Allergy Clin Immunol. 2014;133(1):
25. Solleti SK, Srisuma S, Bhattacharya S, et al. Serpine2 deficiency results in lung lymphocyte accumulation and bronchus-associated lymphoid tissue formation. FASEB J. 2016;30(7):2615–2626.
26. Minoo P, Su G, Drum H, Bringas P, Kimura S. Defects in tracheoesophageal and lung morphogenesis in Nkx2.1(-/-) mouse embryos. Dev Biol. 1999;209(1):60–71.
27. Herriges M, Morrisey EE. Lung development: orchestrating the generation and regeneration of a complex organ. Development. 2014;141(3):502–513.
28. Cho MH, McDonald ML, Zhou X, et al. Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir Med. 2014;2(3):214–225.
29. Borel F, Sun H, Zieger M, et al. Editing out five Serpina1 paralogs to create a mouse model of genetic emphysema. Proc Natl Acad Sci U S A. 2018;115(11):2788–2793.
30. Soler Artigas M, Loth DW, Wain LV, et al. Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat Genet. 2011;43(11):1082–1090.
31. Hobbs BD, de Jong K, Lamontagne M, et al. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat Genet. 2017;49(3):426–432.
32. Moore B, Murphy RF, Agrawal DK. Interaction of tgf-beta with immune cells in airway disease. Curr Mol Med. 2008;8(5):427–436.
33. Letterio JJ, Geiser AG, Kulkarni AB, Roche NS, Sporn MB, Roberts AB. Maternal rescue of transforming growth factor-beta 1 null mice. Science (New York, NY). 1994;264(5167):1936–1938.
34. Sanford LP, Ormsby I, Gittenberger-de Groot AC, et al. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124(13):2659–2670.
35. Kaartinen V, Voncken JW, Shuler C, et al. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11(4):415–421.
36. van der Plaat DA, de Jong K, Lahousse L, et al. Genome-wide association study on the FEV1/FVC ratio in never-smokers identifies HHIP and FAM13A. J Allergy Clin Immunol. 2017;139(2):533–540.
37. Zhao J, Li M, Chen J, et al. Smoking status and gene susceptibility play important roles in the development of chronic obstructive pulmonary disease and lung function decline: a population-based prospective study. Medicine. 2017;96(25):e7283.
38. Chuang PT, Kawcak T, McMahon AP. Feedback control of mammalian hedgehog signaling by the hedgehog-binding protein, Hip1, modulates Fgf signaling during branching morphogenesis of the lung. Genes Dev. 2003;17(3):342–347.
39. Tam A, Hughes M, McNagny KM, et al. Hedgehog signaling in the airway epithelium of patients with chronic obstructive pulmonary disease. Sci Rep. 2019;9(1):3353.
40. Loth DW, Soler Artigas M, Gharib SA, et al. Genome-wide association analysis identifies six new loci associated with forced vital capacity. Nat Genet. 2014;46(7):669–677.
41. Hardin M, Cho MH, Sharma S, et al. Sex-based genetic association study identifies CELSR1 as a possible chronic obstructive pulmonary disease risk locus among women. Am J Respir Cell Mol Biol. 2017;56(3):332–341.
42. Wilk JB, Shrine NR, Loehr LR, et al. Genome-wide association studies identify CHRNA5/3 and HTR4 in the development of airflow obstruction. Am J Respir Crit Care Med. 2012;186(7):622–632.
43. Massaro GD, Massaro D, Chan WY, et al. Retinoic acid receptor-beta: an endogenous inhibitor of the perinatal formation of pulmonary alveoli. Physiol Genomics. 2000;4(1):51–57.
44. Hendrix AY, Kheradmand F. The role of matrix metalloproteinases in development, repair, and destruction of the lungs. Prog Mol Biol Transl Sci. 2017;148:1–29.
45. Belvisi MG, Bottomley KM. The role of matrix metalloproteinases (MMPs) in the pathophysiology of chronic obstructive pulmonary disease (COPD): a therapeutic role for inhibitors of MMPs? Inflamm Res. 2003;52(3):95–100.
46. Gharib SA, Altemeier WA, Van Winkle LS, et al. Matrix metalloproteinase-7 coordinates airway epithelial injury response and differentiation of ciliated cells. Am J Respir Cell Mol Biol. 2013;48(3):390–396.
47. Pang M, Liu HY, Li T, et al. Recombinant club cell protein 16 (CC16) ameliorates cigarette smokeinduced lung inflammation in a murine disease model of COPD. Mol Med Rep. 2018;18(2):2198–2206.
48. Molet S, Belleguic C, Lena H, et al. Increase in macrophage elastase (MMP-12) in lungs from patients with chronic obstructive pulmonary disease. Inflamm Res. 2005;54(1):31–36.
49. Atkinson JM, Pennington CJ, Martin SW, et al. Membrane type matrix metalloproteinases (MMPs) show differential expression in non-small cell lung cancer (NSCLC) compared to normal lung: correlation of MMP-14 mRNA expression and proteolytic activity. Eur J Cancer. 2007;43(11):1764–1771.
50. Vu TH, Shipley JM, Bergers G, et al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93(3):411–422.
51. Oblander SA, Zhou Z, Galvez BG, et al. Distinctive functions of membrane type 1 matrix-metalloprotease (MT1-MMP or MMP-14) in lung and submandibular gland development are independent of its role in pro-MMP-2 activation. Dev Biol. 2005;277(1):255–269.
52. Betsuyaku T, Kadomatsu K, Griffin GL, Muramatsu T, Senior RM. Increased basigin in bleomycin-induced lung injury. Am J Respir Cell Mol Biol. 2003;28(5):600–606.
53. Foda HD, Rollo EE, Drews M, et al. Ventilator-induced lung injury upregulates and activates gelatinases and EMMPRIN: attenuation by the synthetic matrix metalloproteinase inhibitor, Prinomastat (AG3340). Am J Respir Cell Mol Biol. 2001;25(6):717–724.
54. Verde Z, Santiago C, Chicharro LM, et al. Association of HTR2A-1438G/A genetic polymorphism with smoking and chronic obstructive pulmonary disease. Arch Bronconeumol. 2019;55(3):128–133.
55. Perez-Rubio G, Lopez-Flores LA, Garcia-Carmona S, et al. Genetic variants as risk factors for cigarette smoking at an early age and relapse to smoking cessation treatment: a pilot study. Gene. 2019;694:93–96.
56. Lutz SM, Cho MH, Young K, et al. A genome-wide association study identifies risk loci for spirometric measures among smokers of European and African ancestry. BMC Genet. 2015;16:138.
57. Yang L, Lu X, Qiu F, et al. Duplicated copy of CHRNA7 increases risk and worsens prognosis of COPD and lung cancer. Eur J Hum Genet. 2015;23(8):1019–1024.
58. Resendiz-Hernandez JM, Ambrocio-Ortiz E, Perez-Rubio G, et al. TNF promoter polymorphisms are associated with genetic susceptibility in COPD secondary to tobacco smoking and biomass burning. Int J Chron Obstruct Pulmon Dis. 2018;13:627–637.
59. Burkart KM, Sofer T, London SJ, et al. A genome-wide association study in hispanics/latinos identifies novel signals for lung function. The hispanic community health study/study of latinos. Am J Respir Crit Care Med. 2018;198(2):208–219.
60. Li X, Ortega VE, Ampleford EJ, et al. Genome-wide association study of lung function and clinical implication in heavy smokers. BMC Med Genet. 2018;19(1):134.
61. Zhao H, Wu X, Dong CL, Wang BY, Zhao J, Cao XE. Association between ADRB2 genetic polymorphisms and the risk of chronic obstructive pulmonary disease: a case-control study in a Chinese population. Genet Test Mol Biomarkers. 2017;21(8):491–496.
62. Hussein MH, Sobhy KE, Sabry IM, El Serafi AT, Toraih EA. Beta2-adrenergic receptor gene haplotypes and bronchodilator response in Egyptian patients with chronic obstructive pulmonary disease. Adv Med Sci. 2017;62(1):193–201.
63. Li JX, Fu WP, Zhang J, et al. A functional SNP upstream of the ADRB2 gene is associated with COPD. Int J Chron Obstruct Pulmon Dis. 2018;13:917–925.
64. Park HY, Churg A, Wright JL, et al. Club cell protein 16 and disease progression in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(12):1413–1419.
65. Guerra S, Halonen M, Vasquez MM, et al. Relation between circulating CC16 concentrations, lung function, and development of chronic obstructive pulmonary disease across the lifespan: a prospective study. Lancet Respir Med. 2015;3(8):613–620.
66. An L, Lin Y, Yang T, Hua L. Exploring the interaction among EPHX1, GSTP1, SERPINE2, and TGFB1 contributing to the quantitative traits of chronic obstructive pulmonary disease in Chinese Han population. Hum Genomics. 2016;10(1):13.
67. Akparova A, Abdrakhmanova B, Banerjee N, Bersimbaev R. EPHX1 Y113H polymorphism is associated with increased risk of chronic obstructive pulmonary disease in Kazakhstan population. Mutat Res Genet Toxicol Environ Mutagen. 2017;816-817:1–6.
68. Wang CD, Chen N, Huang L, et al. Impact of CYP1A1 polymorphisms on susceptibility to chronic obstructive pulmonary disease: a meta-analysis. Biomed Res Int. 2015;2015:942958.
69. Ingham PW, Nakano Y, Seger C. Mechanisms and functions of hedgehog signalling across the metazoa. Nat Rev Genet. 2011;12(6):393–406.
70. Greenlee KJ, Werb Z, Kheradmand F. Matrix metalloproteinases in lung: multiple, multifarious, and multifaceted. Physiol Rev. 2007;87(1):69–98.
71. Masumoto K, de Rooij JD, Suita S, Rottier R, Tibboel D, de Krijger RR. Expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases during normal human pulmonary development. Histopathology. 2005;47(4):410–419.
72. Lodovici M, Luceri C, Guglielmi F, et al. Benzo(a)pyrene diolepoxide (BPDE)-DNA adduct levels in leukocytes of smokers in relation to polymorphism of CYP1A1, GSTM1, GSTP1, GSTT1, and mEH. Cancer Epidemiol Biomarkers Prev. 2004;13(8):1342–1348.
73. Lakhdar R, Denden S, Knani J, et al. Combined analysis of EPHX1, GSTP1, GSTM1 and GSTT1 gene polymorphisms in relation to chronic obstructive pulmonary disease risk and lung function impairment. Dis Markers. 2011;30(5):253–263.
74. Vibhuti A, Arif E, Mishra A, et al. CYP1A1, CYP1A2 and CYBA gene polymorphisms associated with oxidative stress in COPD. Clin Chim Acta. 2010;411(7–8):474–480.
75. Ghosh R, Topinka J, Joad JP, Dostal M, Sram RJ, Hertz-Picciotto I. Air pollutants, genes and early childhood acute bronchitis. Mutat Res. 2013;749(1–2):80–86.
76. Wang J, Spitz MR, Amos CI, Wilkinson AV, Wu X, Shete S. Mediating effects of smoking and chronic obstructive pulmonary disease on the relation between the CHRNA5-A3 genetic locus and lung cancer risk. Cancer. 2010;116(14):3458–3462.
77. Pillai SG, Ge D, Zhu G, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009;5(3):e1000421.
78. Hardin M, Zielinski J, Wan ES, et al. CHRNA3/5, IREB2, and ADCY2 are associated with severe chronic obstructive pulmonary disease in Poland. Am J Respir Cell Mol Biol. 2012;47(2):203–208.
79. de Planell-Saguer M, Lovinsky-Desir S, Miller RL. Epigenetic regulation: the interface between prenatal and early-life exposure and asthma susceptibility. Environ Mol Mutagen. 2014;55(3):231–243.
80. Breton CV, Siegmund KD, Joubert BR, et al. Prenatal tobacco smoke exposure is associated with childhood DNA CpG methylation. PLoS One. 2014;9(6):e99716.
81. Rzehak P, Saffery R, Reischl E, et al. Maternal smoking during pregnancy and DNA-methylation in children at age 5.5 years: epigenome-wide-analysis in the European Childhood Obesity Project (CHOP)-Study. PLoS One. 2016;11(5):e0155554.
82. Krauss-Etschmann S, Meyer KF, Dehmel S, Hylkema MN. Inter- and transgenerational epigenetic inheritance: evidence in asthma and COPD? Clin Epigenetics. 2015;7:53.
83. Rehan VK, Liu J, Sakurai R, Torday JS. Perinatal nicotine-induced transgenerational asthma. Am J Physiol Lung Cell Mol Physiol. 2013;305(7):L501–7.
84. Meek PM, Sood A, Petersen H, Belinsky SA, Tesfaigzi Y. Epigenetic change (GATA-4 gene methylation) is associated with health status in chronic obstructive pulmonary disease. Biol Res Nurs. 2015;17(2):191–198.
85. Huang X, Wu C, Fu Y, Guo L, Kong X, Cai H. Methylation analysis for multiple gene promoters in non-small cell lung cancers in high indoor air pollution region in China. Bull Cancer. 2018;105(9):746–754.
86. Hagood JS. Beyond the genome: epigenetic mechanisms in lung remodeling. Physiology (Bethesda, Md). 2014;29(3):177–185.
87. Vucic EA, Chari R, Thu KL, et al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. Am J Respir Cell Mol Biol. 2014;50(5):912–922.
88. Morales E, Bustamante M, Vilahur N, et al. DNA hypomethylation at ALOX12 is associated with persistent wheezing in childhood. Am J Respir Crit Care Med. 2012;185(9):937–943.
89. Ito K, Caramori G, Lim S, et al. Expression and activity of histone deacetylases in human asthmatic airways. Am J Respir Crit Care Med. 2002;166(3):392–396.
90. Ito K, Yamamura S, Essilfie-Quaye S, et al. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J Exp Med. 2006;203(1):7–13.
91. Kim RY, Horvat JC, Pinkerton JW, et al. MicroRNA-21 drives severe, steroid-insensitive experimental asthma by amplifying phosphoinositide 3-kinase-mediated suppression of histone deacetylase 2. J Allergy Clin Immunol. 2017;139(2):519–532.
92. Wang Y, Tian Y, Morley MP, et al. Development and regeneration of Sox2+ endoderm progenitors are regulated by a Hdac1/2-Bmp4/Rb1 regulatory pathway. Dev Cell. 2013;24(4):345–358.
93. Londhe VA, Sundar IK, Lopez B, et al. Hyperoxia impairs alveolar formation and induces senescence through decreased histone deacetylase activity and up-regulation of p21 in neonatal mouse lung. Pediatr Res. 2011;69(5 Pt 1):371–377.
94. Zhu L, Li H, Tang J, Zhu J, Zhang Y. Hyperoxia arrests alveolar development through suppression of histone deacetylases in neonatal rats. Pediatr Pulmonol. 2012;47(3):264–274.
95. Benlhabib H, Mendelson CR. Epigenetic regulation of surfactant protein A gene (SP-A) expression in fetal lung reveals a critical role for Suv39h methyltransferases during development and hypoxia. Mol Cell Biol. 2011;31(10):1949–1958.
96. Sundar IK, Yin Q, Baier BS, et al. DNA methylation profiling in peripheral lung tissues of smokers and patients with COPD. Clin Epigenetics. 2017;9(1):38
97. Yao H, Rahman I. Current concepts on oxidative/carbonyl stress, inflammation and epigenetics in pathogenesis of chronic obstructive pulmonary disease. Toxicol Appl Pharmacol. 2011;254(2):72–85.
98. Guo C, Zhang Z, Lau AKH, et al. Effect of long-term exposure to fine particulate matter on lung function decline and risk of chronic obstructive pulmonary disease in Taiwan: a longitudinal, cohort study. Lancet Planet Health. 2018;2(3):e114–e25.
99. Lee MK, Xu CJ, Carnes MU, et al. Genome-wide DNA methylation and long-term ambient air pollution exposure in Korean adults. Clin Epigenetics. 2019;11(1):37.
100. Joehanes R, Just AC, Marioni RE, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9(5):436–447. doi:10.1161/CIRCGENETICS.116.001506
101. Beckmeyer-Borowko A, Imboden M, Rezwan FI, et al. SERPINA1 methylation and lung function in tobacco-smoke exposed European children and adults: a meta-analysis of ALEC population-based cohorts. Respir Res. 2018;19(1):156. doi:10.1186/s12931-018-0850-8
102. de Vries M, van der Plaat DA, Nedeljkovic I, et al. From blood to lung tissue: effect of cigarette smoke on DNA methylation and lung function. Respir Res. 2018;19(1):212. doi:10.1186/s12931-018-0904-y
103. Nedeljkovic I, Carnero-Montoro E, Lahousse L, et al. Understanding the role of the chromosome 15q25.1 in COPD through epigenetics and transcriptomics. Eur J Hum Genet. 2018;26(5):709–722. doi:10.1038/s41431-017-0089-8
104. Huang X, Zhu Z, Guo X, Kong X. The roles of microRNAs in the pathogenesis of chronic obstructive pulmonary disease. Int Immunopharmacol. 2019;67:335–347. doi:10.1016/j.intimp.2018.12.013
105. Mullassery D, Smith NP. Lung development. Semin Pediatr Surg. 2015;24(4):152–155. doi:10.1053/j.sempedsurg.2015.01.011
106. Narayanan M, Owers-Bradley J, Beardsmore CS, et al. Alveolarization continues during childhood and adolescence: new evidence from helium-3 magnetic resonance. Am J Respir Crit Care Med. 2012;185(2):186–191. doi:10.1164/rccm.201107-1348OC
107. Kohansal R, Martinez-Camblor P, Agusti A, Buist AS, Mannino DM, Soriano JB. The natural history of chronic airflow obstruction revisited: an analysis of the Framingham offspring cohort. Am J Respir Crit Care Med. 2009;180(1):3–10.
108. Wang X, Mensinga TT, Schouten JP, Rijcken B, Weiss ST. Determinants of maximally attained level of pulmonary function. Am J Respir Crit Care Med. 2004;169(8):941–949. doi:10.1164/rccm.2201011
109. Martinez FD. Early-life origins of chronic obstructive pulmonary disease. N Engl J Med. 2016;375(9):871–878. doi:10.1056/NEJMra1603287
110. Perlman M, Williams J, Hirsch M. Neonatal pulmonary hypoplasia after prolonged leakage of amniotic fluid. Arch Dis Child. 1976;51(5):349–353.
111. Nimrod C, Varela-Gittings F, Machin G, Campbell D, Wesenberg R. The effect of very prolonged membrane rupture on fetal development. Am J Obstet Gynecol. 1984;148(5):540–543.
112. Sonnenschein-van der Voort AM, Arends LR, de Jongste JC, et al. Preterm birth, infant weight gain, and childhood asthma risk: a meta-analysis of 147,000 European children. J Allergy Clin Immunol. 2014;133(5):1317–1329. doi:10.1016/j.jaci.2013.12.1082
113. Ma Z, Paek D, Oh CK. Plasminogen activator inhibitor-1 and asthma: role in the pathogenesis and molecular regulation. Clin Exp Allergy. 2009;39(8):1136–1144. doi:10.1111/j.1365-2222.2009.03272.x
114. Turner SW, Carter J, Danielian P, et al. Protease concentration in amniotic fluid at term and early childhood respiratory symptoms. J Matern Fetal Neonatal Med. 2014;27(4):416–420. doi:10.3109/14767058.2013.818647
115. Baraldi E, Filippone M. Chronic lung disease after premature birth. N Engl J Med. 2007;357(19):1946–1955. doi:10.1056/NEJMra067279
116. Walsh MC, Szefler S, Davis J, et al. Summary proceedings from the bronchopulmonary dysplasia group. Pediatrics. 2006;117(3 Pt 2):S52–6. doi:10.1542/peds.2005-0620I
117. Lamarche-Vadel A, Blondel B, Truffer P, et al. Re-hospitalization in infants younger than 29 weeks‘ gestation in the EPIPAGE cohort. Acta Paediatr. 2004;93(10):1340–1345.
118. Halvorsen T, Skadberg BT, Eide GE, Roksund OD, Carlsen KH, Bakke P. Pulmonary outcome in adolescents of extreme preterm birth: a regional cohort study. Acta Paediatr. 2004;93(10):1294–1300.
119. Shepherd EG, Clouse BJ, Hasenstab KA, et al. Infant pulmonary function testing and phenotypes in severe bronchopulmonary dysplasia. Pediatrics. 2018;141(5). doi:10.1542/peds.2017-3350.
120. Simpson SJ, Turkovic L, Wilson AC, et al. Lung function trajectories throughout childhood in survivors of very preterm birth: a longitudinal cohort study. Lancet Child Adolesc Health. 2018;2(5):350–359. doi:10.1016/S2352-4642(18)30064-6
121. Vrijlandt EJ, Gerritsen J, Boezen HM, Duiverman EJ. Gender differences in respiratory symptoms in 19-year-old adults born preterm. Respir Res. 2005;6:117. doi:10.1186/1465-9921-6-117
122. Savran O, Ulrik CS. Early life insults as determinants of chronic obstructive pulmonary disease in adult life. Int J Chron Obstruct Pulmon Dis. 2018;13:683–693. doi:10.2147/COPD.S153555
123. Hacking DF, Gibson AM, Robertson C, Doyle LW. Respiratory function at age 8-9 after extremely low birthweight or preterm birth in Victoria in 1997. Pediatr Pulmonol. 2013;48(5):449–455. doi:10.1002/ppul.22619
124. Doyle LW, Faber B, Callanan C, Freezer N, Ford GW, Davis NM. Bronchopulmonary dysplasia in very low birth weight subjects and lung function in late adolescence. Pediatrics. 2006;118(1):108–113. doi:10.1542/peds.2005-2522
125. Schultz ES, Hallberg J, Andersson N, et al. Early life determinants of lung function change from childhood to adolescence. Respir Med. 2018;139:48–54. doi:10.1016/j.rmed.2018.04.009
126. Upton MN, Watt GC, Davey Smith G, McConnachie A, Hart CL. Permanent effects of maternal smoking on offsprings‘ lung function. Lancet. 1998;352(9126):453. doi:10.1016/S0140-6736(05)79187-X
127. Martinez FD. The origins of asthma and chronic obstructive pulmonary disease in early life. Proc Am Thorac Soc. 2009;6(3):272–277. doi:10.1513/pats.200808-092RM
128. Aanerud M, Carsin AE, Sunyer J, et al. Interaction between asthma and smoking increases the risk of adult airway obstruction. Eur Respir J. 2015;45(3):635–643. doi:10.1183/09031936.00055514
129. Neuman A, Hohmann C, Orsini N, et al. Maternal smoking in pregnancy and asthma in preschool children: a pooled analysis of eight birth cohorts. Am J Respir Crit Care Med. 2012;186(10):1037–1043. doi:10.1164/rccm.201203-0501OC
130. Hayatbakhsh MR, Sadasivam S, Mamun AA, Najman JM, Williams GM, O‘Callaghan MJ. Maternal smoking during and after pregnancy and lung function in early adulthood: a prospective study. Thorax. 2009;64(9):810–814. doi:10.1136/thx.2009.116301
131. Skinner MK. Environmental epigenomics and disease susceptibility. EMBO Rep. 2011;12(7):620–622. doi:10.1038/embor.2011.125
132. Deng Q, Lu C, Ou C, Chen L, Yuan H. Preconceptional, prenatal and postnatal exposure to outdoor and indoor environmental factors on allergic diseases/symptoms in preschool children. Chemosphere. 2016;152:459–467. doi:10.1016/j.chemosphere.2016.03.032
133. Basu R, Pearson D, Ebisu K, Malig B. Association between PM2.5 and PM2.5 constituents and preterm delivery in California, 2000–2006. Paediatr Perinat Epidemiol. 2017;31(5):424–434. doi:10.1111/ppe.12380
134. Blum JL, Chen LC, Zelikoff JT. Exposure to ambient particulate matter during specific gestational periods produces adverse obstetric consequences in mice. Environ Health Perspect. 2017;125(7):077020. doi:10.1289/EHP36
135. Lee A, Leon Hsu HH, Mathilda Chiu YH, et al. Prenatal fine particulate exposure and early childhood asthma: effect of maternal stress and fetal sex. J Allergy Clin Immunol. 2018;141(5):1880–1886. doi:10.1016/j.jaci.2017.07.017
136. Kingsley SL, Deyssenroth MA, Kelsey KT, et al. Maternal residential air pollution and placental imprinted gene expression. Environ Int. 2017;108:204–211. doi:10.1016/j.envint.2017.08.022
137. Cai J, Zhao Y, Liu P, et al. Exposure to particulate air pollution during early pregnancy is associated with placental DNA methylation. Sci Total Environ. 2017;607–608:1103–1108. doi:10.1016/j.scitotenv.2017.07.029
138. Kingsley SL, Kelsey KT, Butler R, et al. Maternal serum PFOA concentration and DNA methylation in cord blood: a pilot study. Environ Res. 2017;158:174–178. doi:10.1016/j.envres.2017.06.013
139. Walker SA, Kupzig S, Lockyer PJ, Bilu S, Zharhary D, Cullen PJ. Analyzing the role of the putative inositol 1,3,4,5-tetrakisphosphate receptor GAP1IP4BP in intracellular Ca2+ homeostasis. J Biol Chem. 2002;277(50):48779–48785. doi:10.1074/jbc.M204839200
140. Dominguez-Bello MG, Costello EK, Contreras M, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;107(26):11971–11975. doi:10.1073/pnas.1002601107
141. Bager P, Wohlfahrt J, Westergaard T. Caesarean delivery and risk of atopy and allergic disease: meta-analyses. Clin Exp Allergy. 2008;38(4):634–642. doi:10.1111/j.1365-2222.2008.02939.x
142. Duijts L, Reiss IK, Brusselle G, de Jongste JC. Early origins of chronic obstructive lung diseases across the life course. Eur J Epidemiol. 2014;29(12):871–885. doi:10.1007/s10654-014-9981-5
143. Polinski KJ, Liu J, Boghossian NS, McLain AC. Maternal obesity, gestational weight gain, and asthma in offspring. Prev Chronic Dis. 2017;14:E109. doi:10.5888/pcd14.170196
144. Harpsoe MC, Basit S, Bager P, et al. Maternal obesity, gestational weight gain, and risk of asthma and atopic disease in offspring: a study within the Danish National Birth Cohort. J Allergy Clin Immunol. 2013;131(4):1033–1040. doi:10.1016/j.jaci.2012.09.008
145. Leermakers ET, Sonnenschein-van der Voort AM, Gaillard R, et al. Maternal weight, gestational weight gain and preschool wheezing: the generation R study. Eur Respir J. 2013;42(5):1234–1243.
146. Parr CL, Magnus MC, Karlstad O, et al. Maternal folate intake during pregnancy and childhood asthma in a population-based cohort. Am J Respir Crit Care Med. 2017;195(2):221–228. doi:10.1164/rccm.201604-0788OC
147. Bedard A, Northstone K, Henderson AJ, Shaheen SO. Maternal intake of sugar during pregnancy and childhood respiratory and atopic outcomes. Eur Respir J. 2017;50(1). doi:10.1183/13993003.00711-2017
148. Griffiths PS, Walton C, Samsell L, Perez MK, Piedimonte G. Maternal high-fat hypercaloric diet during pregnancy results in persistent metabolic and respiratory abnormalities in offspring. Pediatr Res. 2016;79(2):278–286. doi:10.1038/pr.2015.226
149. Song Y, Yu Y, Wang D, et al. Maternal high-fat diet feeding during pregnancy and lactation augments lung inflammation and remodeling in the offspring. Respir Physiol Neurobiol. 2015;207:1–6. doi:10.1016/j.resp.2014.12.003
150. Williams L, Charron MJ, Sellers RS. High post-natal mortality associated with defects in lung maturation and reduced adiposity in mice with gestational exposure to high fat and N-acetylcysteine. Res Vet Sci. 2017;114:262–265. doi:10.1016/j.rvsc.2017.05.020
151. Beckhaus AA, Garcia-Marcos L, Forno E, Pacheco-Gonzalez RM, Celedon JC, Castro-Rodriguez JA. Maternal nutrition during pregnancy and risk of asthma, wheeze, and atopic diseases during childhood: a systematic review and meta-analysis. Allergy. 2015;70(12):1588–1604. doi:10.1111/all.12729
152. Steegers-Theunissen RP, Obermann-Borst SA, Kremer D, et al. Periconceptional maternal folic acid use of 400 microg per day is related to increased methylation of the IGF2 gene in the very young child. PLoS One. 2009;4(11):e7845. doi:10.1371/journal.pone.0007845
153. Turner SW, Campbell D, Smith N, et al. Associations between fetal size, maternal {alpha}-tocopherol and childhood asthma. Thorax. 2010;65(5):391–397. doi:10.1136/thx.2008.111385
154. Hong SA, Lee E, Kwon SO, et al. Effect of prenatal antioxidant intake on infants‘ respiratory infection is modified by a CD14 polymorphism. World J Pediatr. 2017;13(2):173–182. doi:10.1007/s12519-016-0054-6
155. Blaser MJ, Bello MG. Maternal antibiotic use and risk of asthma in offspring. Lancet Respir Med. 2014;2(10):e16. doi:10.1016/S2213-2600(14)70219-X
156. Vidal AC, Murphy SK, Murtha AP, et al. Associations between antibiotic exposure during pregnancy, birth weight and aberrant methylation at imprinted genes among offspring. Int J Obes (Lond). 2013;37(7):907–913. doi:10.1038/ijo.2013.47
157. Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta harbors a unique microbiome. Sci Transl Med. 2014;6(237):237ra65. doi:10.1126/scitranslmed.3008599
158. Kravitz-Wirtz N, Teixeira S, Hajat A, Woo B, Crowder K, Takeuchi D. Early-life air pollution exposure, neighborhood poverty, and childhood asthma in the United States, 1990(-)2014. Int J Environ Res Public Health. 2018;15:6. doi:10.3390/ijerph15061188
159. Clark NA, Demers PA, Karr CJ, et al. Effect of early life exposure to air pollution on development of childhood asthma. Environ Health Perspect. 2010;118(2):284–290. doi:10.1289/ehp.0900916
160. Salvi S. Tobacco smoking and environmental risk factors for chronic obstructive pulmonary disease. Clin Chest Med. 2014;35(1):17–27. doi:10.1016/j.ccm.2013.09.011
161. Feenstra TL, van Genugten ML, Hoogenveen RT, Wouters EF, Rutten-van Molken MP. The impact of aging and smoking on the future burden of chronic obstructive pulmonary disease: a model analysis in the Netherlands. Am J Respir Crit Care Med. 2001;164(4):590–596. doi:10.1164/ajrccm.164.4.2003167
162. Morgenstern V, Zutavern A, Cyrys J, et al. Atopic diseases, allergic sensitization, and exposure to traffic-related air pollution in children. Am J Respir Crit Care Med. 2008;177(12):1331–1337. doi:10.1164/rccm.200701-036OC
163. Nordling E, Berglind N, Melen E, et al. Traffic-related air pollution and childhood respiratory symptoms, function and allergies. Epidemiology. 2008;19(3):401–408. doi:10.1097/EDE.0b013e31816a1ce3
164. Atkinson RW, Fuller GW, Anderson HR, Harrison RM, Armstrong B. Urban ambient particle metrics and health: a time-series analysis. Epidemiology. 2010;21(4):501–511. doi:10.1097/EDE.0b013e3181debc88
165. Kim HJ, Choi MG, Park MK, Seo YR. Predictive and prognostic biomarkers of respiratory diseases due to particulate matter exposure. J Cancer Prev. 2017;22(1):6–15. doi:10.15430/JCP.2017.22.1.6
166. Ma J, Xu H, Wu J, Qu C, Sun F, Xu S. Linalool inhibits cigarette smoke-induced lung inflammation by inhibiting NF-kappaB activation. Int Immunopharmacol. 2015;29(2):708–713. doi:10.1016/j.intimp.2015.09.005
167. Baccarelli A, Wright RO, Bollati V, et al. Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med. 2009;179(7):572–578. doi:10.1164/rccm.200807-1097OC
168. Gilmour PS, Rahman I, Donaldson K, MacNee W. Histone acetylation regulates epithelial IL-8 release mediated by oxidative stress from environmental particles. Am J Physiol Lung Cell Mol Physiol. 2003;284(3):L533–40. doi:10.1152/ajplung.00277.2002
169. Cao D, Bromberg PA, Samet JM. COX-2 expression induced by diesel particles involves chromatin modification and degradation of HDAC1. Am J Respir Cell Mol Biol. 2007;37(2):232–239. doi:10.1165/rcmb.2006-0449OC
170. McGeachie MJ. Childhood asthma is a risk factor for the development of chronic obstructive pulmonary disease. Curr Opin Allergy Clin Immunol. 2017;17(2):104–109. doi:10.1097/ACI.0000000000000348
171. Just J, Bourgoin-Heck M, Amat F. Clinical phenotypes in asthma during childhood. Clin Exp Allergy. 2017;47(7):848–855. doi:10.1111/cea.12939
172. Hayden LP, Cho MH, Raby BA, et al. Childhood asthma is associated with COPD and known asthma variants in COPDGene: a genome-wide association study. Respir Res. 2018;19(1):209. doi:10.1186/s12931-018-0890-0
173. Apostol GG, Jacobs DR
174. Guerra S, Sherrill DL, Kurzius-Spencer M, et al. The course of persistent airflow limitation in subjects with and without asthma. Respir Med. 2008;102(10):1473–1482. doi:10.1016/j.rmed.2008.04.011
175. Omori K, Iwamoto H, Yamane T, et al. Clinically remitted childhood asthma is associated with airflow obstruction in middle-aged adults. Respirology (Carlton, Vic). 2017;22(1):86–92. doi:10.1111/resp.12860
176. Shirtcliffe P, Weatherall M, Marsh S, et al. COPD prevalence in a random population survey: a matter of definition. Eur Respir J. 2007;30(2):232–239. doi:10.1183/09031936.00157906
177. Hardin M, Cho M, McDonald ML, et al. The clinical and genetic features of COPD-asthma overlap syndrome. Eur Respir J. 2014;44(2):341–350. doi:10.1183/09031936.00216013
178. Laprise C, Laviolette M, Boutet M, Boulet LP. Asymptomatic airway hyperresponsiveness: relationships with airway inflammation and remodelling. Eur Respir J. 1999;14(1):63–73.
179. Covar RA, Spahn JD, Martin RJ, et al. Safety and application of induced sputum analysis in childhood asthma. J Allergy Clin Immunol. 2004;114(3):575–582. doi:10.1016/j.jaci.2004.06.036
180. Hilty M, Burke C, Pedro H, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5(1):e8578. doi:10.1371/journal.pone.0008578
181. Nembrini C, Sichelstiel A, Kisielow J, Kurrer M, Kopf M, Marsland BJ. Bacterial-induced protection against allergic inflammation through a multicomponent immunoregulatory mechanism. Thorax. 2011;66(9):755–763. doi:10.1136/thx.2010.152512
182. Bacharier LB, Cohen R, Schweiger T, et al. Determinants of asthma after severe respiratory syncytial virus bronchiolitis. J Allergy Clin Immunol. 2012;130(1):91–100.e3. doi:10.1016/j.jaci.2012.02.010
183. Smit LA, Bouzigon E, Pin I, et al. 17q21 variants modify the association between early respiratory infections and asthma. Eur Respir J. 2010;36(1):57–64. doi:10.1183/09031936.00154509
184. Stein RT, Sherrill D, Morgan WJ, et al. Respiratory syncytial virus in early life and risk of wheeze and allergy by age 13 years. Lancet. 1999;354(9178):541–545. doi:10.1016/S0140-6736(98)10321-5
185. Kusel MM, de Klerk NH, Kebadze T, et al. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J Allergy Clin Immunol. 2007;119(5):1105–1110. doi:10.1016/j.jaci.2006.12.669
186. Jackson DJ, Gangnon RE, Evans MD, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Respir Crit Care Med. 2008;178(7):667–672. doi:10.1164/rccm.200802-309OC
187. Cassimos DC, Tsalkidis A, Tripsianis GA, et al. Asthma, lung function and sensitization in school children with a history of bronchiolitis. Pediatr Int. 2008;50(1):51–56. doi:10.1111/j.1442-200X.2007.02509.x
188. Koponen P, Helminen M, Paassilta M, Luukkaala T, Korppi M. Preschool asthma after bronchiolitis in infancy. Eur Respir J. 2012;39(1):76–80. doi:10.1183/09031936.00040211
189. Regnier SA, Huels J. Association between respiratory syncytial virus hospitalizations in infants and respiratory sequelae: systematic review and meta-analysis. Pediatr Infect Dis J. 2013;32(8):820–826. doi:10.1097/INF.0b013e31829061e8
190. Caliskan M, Bochkov YA, Kreiner-Moller E, et al. Rhinovirus wheezing illness and genetic risk of childhood-onset asthma. N Engl J Med. 2013;368(15):1398–1407. doi:10.1056/NEJMoa1211592
191. Diver WR, Jacobs EJ, Gapstur SM. Secondhand smoke exposure in childhood and adulthood in relation to adult mortality among never smokers. Am J Prev Med. 2018;55(3):345–352. doi:10.1016/j.amepre.2018.05.005
192. Cohen RT, Strunk RC, Field JJ, et al. Environmental tobacco smoke and airway obstruction in children with sickle cell anemia. Chest. 2013;144(4):1323–1329.
193. Foreman MG, Zhang L, Murphy J, et al. Early-onset chronic obstructive pulmonary disease is associated with female sex, maternal factors, and African American race in the COPDGene Study. Am J Respir Crit Care Med. 2011;184(4):414–420.
194. Hehua Z, Qing C, Shanyan G, Qijun W, Yuhong Z. The impact of prenatal exposure to air pollution on childhood wheezing and asthma: a systematic review. Environ Res. 2017;159:519–530.
195. Williams EP, Mesidor M, Winters K, Dubbert PM, Wyatt SB. Overweight and obesity: prevalence, consequences, and causes of a growing public health problem. Curr Obes Rep. 2015;4(3):363–370.
196. Ali Z, Ulrik CS. Obesity and asthma: a coincidence or a causal relationship? A systematic review. Respir Med. 2013;107(9):1287–1300.
197. Lang JE. Obesity, nutrition, and asthma in children. Pediatr Allergy Immunol Pulmonol. 2012;25(2):64–75.
198. Tantisira KG, Litonjua AA, Weiss ST, Fuhlbrigge AL. Association of body mass with pulmonary function in the Childhood Asthma Management Program (CAMP). Thorax. 2003;58(12):1036–1041.
199. Lang JE, Hossain J, Dixon AE, et al. Does age impact the obese asthma phenotype? Longitudinal asthma control, airway function, and airflow perception among mild persistent asthmatics. Chest. 2011;140(6):1524–1533.
200. Chu YT, Chen WY, Wang TN, Tseng HI, Wu JR, Ko YC. Extreme BMI predicts higher asthma prevalence and is associated with lung function impairment in school-aged children. Pediatr Pulmonol. 2009;44(5):472–479.
201. Litonjua AA, Gold DR. Asthma and obesity: common early-life influences in the inception of disease. J Allergy Clin Immunol. 2008;121(5):
202. Murphy A, Tantisira KG, Soto-Quiros ME, et al. PRKCA: a positional candidate gene for body mass index and asthma. Am J Hum Genet. 2009;85(1):87–96.
203. Lang JE, Williams ES, Mizgerd JP, Shore SA. Effect of obesity on pulmonary inflammation induced by acute ozone exposure: role of interleukin-6. Am J Physiol Lung Cell Mol Physiol. 2008;294(5):L1013–20.
204. Shore SA, Terry RD, Flynt L, Xu A, Hug C. Adiponectin attenuates allergen-induced airway inflammation and hyperresponsiveness in mice. J Allergy Clin Immunol. 2006;118(2):389–395.
205. Shore SA, Schwartzman IN, Mellema MS, Flynt L, Imrich A, Johnston RA. Effect of leptin on allergic airway responses in mice. J Allergy Clin Immunol. 2005;115(1):103–109.
206. Shore SA. Obesity, airway hyperresponsiveness, and inflammation. J Appl Physiol (1985). 2010;108(3):735–743.
207. Michelson PH, Williams LW, Benjamin DK, Barnato AE. Obesity, inflammation, and asthma severity in childhood: data from the National Health and Nutrition Examination Survey 2001-2004. Ann Allergy Asthma Immunol. 2009;103(5):381–385.
208. Berthon BS, Wood LG. Nutrition and respiratory health–feature review. Nutrients. 2015;7(3):1618–1643.
209. Carey OJ, Cookson JB, Britton J, Tattersfield AE. The effect of lifestyle on wheeze, atopy, and bronchial hyperreactivity in Asian and white children. Am J Respir Crit Care Med. 1996;154(2 Pt 1):537–540.
210. Varraso R, Fung TT, Barr RG, Hu FB, Willett W, Camargo CA
211. Wood LG, Garg ML, Gibson PG. A high-fat challenge increases airway inflammation and impairs bronchodilator recovery in asthma. J Allergy Clin Immunol. 2011;127(5):1133–1140.
212. Wickens K, Barry D, Friezema A, et al. Fast foods - are they a risk factor for asthma? Allergy. 2005;60(12):1537–1541.
213. Hijazi N, Abalkhail B, Seaton A. Diet and childhood asthma in a society in transition: a study in urban and rural Saudi Arabia. Thorax. 2000;55(9):775–779.
214. Rahman I, Biswas SK, Kode A. Oxidant and antioxidant balance in the airways and airway diseases. Eur J Pharmacol. 2006;533(1–3):222–239.
215. Gilliland FD, Berhane KT, Li YF, Gauderman WJ, McConnell R, Peters J. Children‘s lung function and antioxidant vitamin, fruit, juice, and vegetable intake. Am J Epidemiol. 2003;158(6):576–584.
216. Niruban SJ, Alagiakrishnan K, Beach J, Senthilselvan A. Association of vitamin D with respiratory outcomes in Canadian children. Eur J Clin Nutr. 2014;68(12):1334–1340.
217. Vitamin HM. D and the immune system: new perspectives on an old theme. Rheum Dis Clin North Am. 2012;38(1):125–139.
218. Brehm JM, Celedon JC, Soto-Quiros ME, et al. Serum vitamin D levels and markers of severity of childhood asthma in Costa Rica. Am J Respir Crit Care Med. 2009;179(9):765–771.
219. Copd BM. and tobacco smoke. Monaldi Arch Chest Dis. 2005;63(4):213–225.
220. Jaakkola MS. Environmental tobacco smoke and health in the elderly. Eur Respir J. 2002;19(1):172–181.
221. Anthonisen NR, Connett JE, Kiley JP, et al. Effects of smoking intervention and the use of an inhaled anticholinergic bronchodilator on the rate of decline of FEV1. The Lung Health Study. Jama. 1994;272(19):1497–1505.
222. Naghavi M, Abajobir AA, Abbafati C, et al. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390(10100):1151–1210.
223. Sood A, Petersen H, Blanchette CM, et al. Wood smoke exposure and gene promoter methylation are associated with increased risk for COPD in smokers. Am J Respir Crit Care Med. 2010;182(9):1098–1104.
224. Salvi S, Barnes PJ. Is exposure to biomass smoke the biggest risk factor for COPD globally? Chest. 2010;138(1):3–6.
225. Hu G, Zhou Y, Tian J, et al. Risk of COPD from exposure to biomass smoke: a metaanalysis. Chest. 2010;138(1):20–31.
226. Po JY, FitzGerald JM, Carlsten C. Respiratory disease associated with solid biomass fuel exposure in rural women and children: systematic review and meta-analysis. Thorax. 2011;66(3):232–239.
227. Fingerhut M, Nelson DI, Driscoll T, et al. The contribution of occupational risks to the global burden of disease: summary and next steps. Med Lav. 2006;97(2):313–321.
228. Rivera RM, Cosio MG, Ghezzo H, Salazar M, Perez-Padilla R. Comparison of lung morphology in COPD secondary to cigarette and biomass smoke. Int J Tuberc Lung Dis. 2008;12(8):972–977.
229. Dutta A, Roychoudhury S, Chowdhury S, Ray MR. Changes in sputum cytology, airway inflammation and oxidative stress due to chronic inhalation of biomass smoke during cooking in premenopausal rural Indian women. Int J Hyg Environ Health. 2013;216(3):301–308.
230. Chapman RS, He X, Blair AE, Lan Q. Improvement in household stoves and risk of chronic obstructive pulmonary disease in Xuanwei, China: retrospective cohort study. BMJ. 2005;331(7524):1050.
231. Gauderman WJ, Vora H, McConnell R, et al. Effect of exposure to traffic on lung development from 10 to 18 years of age: a cohort study. Lancet. 2007;369(9561):571–577.
232. Kan H, Heiss G, Rose KM, Whitsel E, Lurmann F, London SJ. Traffic exposure and lung function in adults: the atherosclerosis risk in communities study. Thorax. 2007;62(10):873–879.
233. Alif SM, Dharmage SC, Bowatte G, et al. Occupational exposure and risk of chronic obstructive pulmonary disease: a systematic review and meta-analysis. Expert Rev Respir Med. 2016;10(8):861–872.
234. Lin H, Qian ZM, Guo Y, et al. The attributable risk of chronic obstructive pulmonary disease due to ambient fine particulate pollution among older adults. Environ Int. 2018;113:143–148.
235. Schikowski T, Sugiri D, Ranft U, et al. Long-term air pollution exposure and living close to busy roads are associated with COPD in women. Respir Res. 2005;6:152.
236. Schikowski T, Ranft U, Sugiri D, et al. Decline in air pollution and change in prevalence in respiratory symptoms and chronic obstructive pulmonary disease in elderly women. Respir Res. 2010;11:113.
237. Gan WQ, FitzGerald JM, Carlsten C, Sadatsafavi M, Brauer M. Associations of ambient air pollution with chronic obstructive pulmonary disease hospitalization and mortality. Am J Respir Crit Care Med. 2013;187(7):721–727.
238. Ko FW, Hui DS. Air pollution and chronic obstructive pulmonary disease. Respirology (Carlton, Vic). 2012;17(3):395–401.
239. Sana A, Somda SMA, Meda N, Bouland C. Chronic obstructive pulmonary disease associated with biomass fuel use in women: a systematic review and meta-analysis. BMJ Open Respir Res. 2018;5(1):e000246.
240. Lytras T, Kogevinas M, Kromhout H, et al. Occupational exposures and 20-year incidence of COPD: the European Community Respiratory Health Survey. Thorax. 2018;73(11):1008–1015.
241. Kurth L, Doney B, Halldin C, Hale J, Frenk SM. Airflow obstruction among ever-employed U.S. adults aged 18-79 years by industry and occupation: NHANES 2007-2008 to 2011-2012. Am J Ind Med. 2019;62(1):30–42.
242. Toren K, Vikgren J, Olin AC, Rosengren A, Bergstrom G, Brandberg J. Occupational exposure to vapor, gas, dust, or fumes and chronic airflow limitation, COPD, and emphysema: the Swedish CArdioPulmonary BioImage Study (SCAPIS pilot). Int J Chron Obstruct Pulmon Dis. 2017;12:3407–3413.
243. Sadhra S, Kurmi OP, Sadhra SS, Lam KB, Ayres JG. Occupational COPD and job exposure matrices: a systematic review and meta-analysis. Int J Chron Obstruct Pulmon Dis. 2017;12:725–734.
244. Valavanidis A, Vlahoyianni T, Fiotakis K. Comparative study of the formation of oxidative damage marker 8-hydroxy-2‘-deoxyguanosine (8-OHdG) adduct from the nucleoside 2‘-deoxyguanosine by transition metals and suspensions of particulate matter in relation to metal content and redox reactivity. Free Radic Res. 2005;39(10):1071–1081.
245. Bellavia AUB, Speck M, Brook RD, et al. DNA hypomethylation, ambient particulate matter, and increased blood pressure: findings from controlled human exposure experiments. J Am Heart Assoc. 2015;4(10):e001981.
246. Iwan K, Rahimoff R, Kirchner A, et al. 5-Formylcytosine to cytosine conversion by C-C bond cleavage in vivo. Nat Chem Biol. 2018;14(1):72–78.
247. Rock JR, Onaitis MW, Rawlins EL, et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc Natl Acad Sci U S A. 2009;106(31):12771–12775.
248. Rock JR, Randell SH, Hogan BL. Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis Model Mech. 2010;3(9–10):545–556.
249. Kumar PA, Hu Y, Yamamoto Y, et al. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell. 2011;147(3):525–538.
250. Rock JR, Barkauskas CE, Cronce MJ, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108(52):E1475–83.
251. Kim CF, Jackson EL, Woolfenden AE, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121(6):823–835.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.