")
Back to Archived Journals » Oncolytic Virotherapy » Volume 4
The emerging therapeutic potential of the oncolytic immunotherapeutic Pexa-Vec (JX-594)
Authors Breitbach C, Bell JC, Hwang T, Kirn D, Burke J
Received 30 April 2014
Accepted for publication 15 July 2014
Published 28 January 2015 Volume 2015:4 Pages 25—31
DOI https://doi.org/10.2147/OV.S59640
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Caroline J Breitbach,1 John C Bell,2,3 Tae-Ho Hwang,1 David H Kirn,1 James Burke1
1SillaJen Inc, Seoul, South Korea; 2Department of Biochemistry, Microbiology and Immunology, University of Ottawa, Ottawa, ON, Canada; 3Centre for Cancer Therapeutics, Ottawa Hospital Research Institute, Ottawa, ON, Canada
Abstract: Oncolytic immunotherapeutics (OIs) are viruses designed to preferentially replicate in and lyse cancer cells, thereby triggering antitumor immunity. Numerous oncolytic platforms are currently in clinical development. Here we review preclinical and clinical experience with Pexa-Vec (pexastimogene devacirepvec, JX-594). Pexa-Vec is derived from a vaccinia vaccine strain that has been engineered to target cancer cells and express the therapeutic transgene granulocyte macrophage colony-stimulating factor (GM-CSF) in order to stimulate antitumor immunity. Key to its ability to target metastatic disease is the evolution of unique vaccinia virus characteristics that allow for effective systemic dissemination. Multiple mechanisms of action (MOA) for Pexa-Vec have been demonstrated in preclinical models and patients: 1) tumor cell infection and lysis, 2) antitumor immune response induction, and 3) tumor vascular disruption. This review will summarize data on the Pexa-Vec MOA as well as provide an overview of the Pexa-Vec clinical development program from multiple Phase I studies, Phase II studies in renal cell cancer and colorectal cancer, through Phase IIb clinical testing in patients with advanced hepatocellular carcinoma (primary liver cancer).
Keywords: Pexa-Vec, JX-594, oncolytic immunotherapy, hepatocellular carcinoma
Vaccinia biology and selective cancer targeting
Vaccinia viruses are enveloped double-stranded DNA viruses belonging to the family of Poxviridae and were utilized as vaccines for the eradication of smallpox.1 Pexa-Vec (pexastimogene devacirepvec; JX-594) is a targeted and armed oncolytic and immunotherapeutic vaccinia virus with disruption of the viral thymidine kinase gene and expression of the human granulocyte macrophage colony-stimulating factor (hGM-CSF) and β-galactosidase transgenes under control of the synthetic early/late and p7.5 promoters, respectively.2–4 Selective targeting of tumor cells by Pexa-Vec is attributed to both engineered mechanisms as well as to inherent vaccinia selectivity for cancers.5 Thymidine kinase gene inactivation renders viral replication dependent on the high cellular thymidine kinase activity that is a hallmark of cancer cells.6 Vaccinia vaccine strains have been shown to be inherently tumor targeting.7,8 This may be attributable to the fact that many of the hallmarks of cancer9 (eg, blocks in apoptotic pathways, dysregulation of cell cycle control and immune evasion) are also optimal cellular conditions for successful vaccinia virus replication. Furthermore, vaccinia replication and spread is dependent on epidermal growth factor receptor (EGFR) signaling,10 a pathway that is activated in most cancers.9 Blocking EGFR signaling inhibits vaccinia replication.11 Indeed, the majority of solid tumor cell lines tested were susceptible to vaccinia infection. In contrast, leukemia and lymphoma cell lines were the only cancer cell types uniformly refractory to infection.5
Systemic tumor targeting and transgene expression
Vaccinia viruses, unlike many other viruses, are stable in blood following intravenous (IV) or intratumoral (IT) introduction. Vaccinia has evolved to produce an extracellular enveloped virus form, which provides an additional membrane coat that results in resistance to complement and antibody neutralization following exposure to the systemic vasculature.12–14 This inherent property results in enhanced effective tumor delivery following the systemic application of oncolytic vaccinia for the treatment of cancer. This results in highly efficient systemic delivery to and spread between tumor cells after treatment.8,15,16 Based on these preclinical studies, clinical evaluation of oncolytic vaccinia was initiated. IV Pexa-Vec infusion was well-tolerated up to a dose of 3×107 plaque forming units per kg (pfu/kg).17 Systemic delivery of Pexa-Vec to tumors was tested by collecting tumor biopsies in all patients between 7 and 10 days postinfusion. On this trial, reproducible Pexa-Vec delivery to tumors was established at a dose threshold of 1×109 pfu, with tumor biopsies in seven of eight patients containing detectable Pexa-Vec.17 The induction of humoral immunity to the Pexa-Vec transgene β-galactosidase was also used as a surrogate marker of Pexa-Vec replication in patients, as high concentrations of the transgene are only detected (and, thereby, provoke the induction of antibodies) after productive Pexa-Vec infection. A dose-dependent increase in anti-β-galactosidase antibodies was observed in patients.17 Detectable concentrations of granulocyte macrophage colony-stimulating factor (GM-CSF) in plasma were measured acutely (4–15 days after treatment) in a subset of patients receiving IV or IT Pexa-Vec.17,18 Together, these data demonstrate that Pexa-Vec leads to successful infection and gene expression in metastatic tumors following systemic administration.
Pexa-Vec mechanisms of action
Pexa-Vec mechanisms of actions (MOA) have been evaluated in both preclinical and clinical studies (Figure 1 and Table 1). Firstly, Pexa-Vec has been shown to selectively infect and amplify within tumors (termed oncolysis).2,17,18 Infection of tumors by viruses results in the release of damage-associated molecular patterns (DAMPs)19,20 as well as pathogen-associated molecular patterns (PAMPs)21 originating from components of the virus particle, resulting in an acute inflammatory response. The production and release of tumor antigens within this highly stimulatory context is capable of inducing an adaptive immune response against the tumor itself. Protective antitumor immunity has been demonstrated following vaccinia infection of murine tumors in vivo.22 Inflammation within tumors was detected following IT Pexa-Vec administration in patients with melanoma.23,24 Furthermore, functional anticancer immunity in the context of Pexa-Vec treatment was demonstrated in preclinical models and patients by measuring induction of antibody-mediated complement-dependent cytotoxicity (CDC), utilizing a panel of tumor cell lines of different histologies.25 Low concentrations of sera incubated with tumor cell lines ex vivo resulted in a dramatic reduction in tumor cell viability; normal cells did not exhibit decreased viability. This activity was shown to be dependent on both active complement as well as IgG antibody. Although the testing was performed in a Phase I study of patients with diverse cancer types and disease burdens at baseline, it was notable (hypothesis-generating) that the patients who survived the longest had the highest CDC activity. Reproducible CDC activity was also observed in a Phase II study in patients with advanced hepatocellular carcinoma (HCC).26 Furthermore, T cell responses to β-galactosidase peptides were detected in HCC patients treated with Pexa-Vec, as shown by Enzyme-Linked ImmunoSpot (ELISPOT) analysis; this provides proof-of-concept that T cell responses can be induced to transgenes encoded by oncolytic vaccinia viruses.
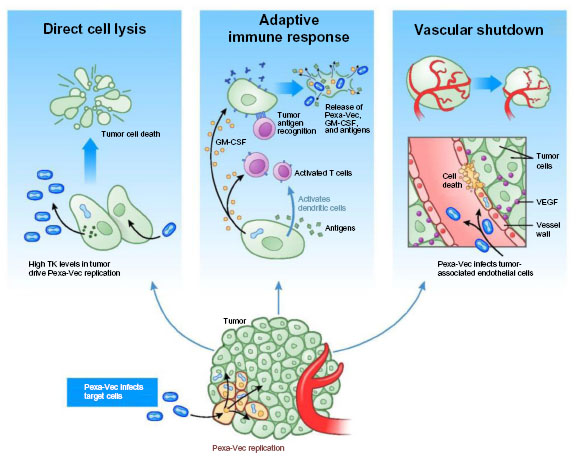 | Figure 1 Pexa-Vec mechanisms of action. |
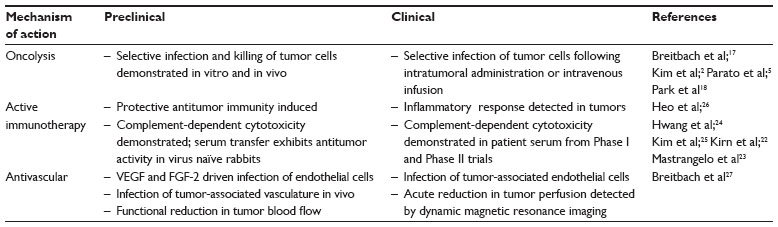 | Table 1 Overview of oncolytic vaccinia mechanisms of action |
Finally, Pexa-Vec has demonstrated acute antivascular effects. In vitro, Pexa-Vec was shown to be capable of infecting human endothelial cells (human umbilical vein endothelial cells [HUVECs] and human dermal microvascular endothelial cells [HDMECs]); this process was both vascular endothelial growth factor and fibroblast growth factor 2 driven.27 Infection of tumor-associated vasculature was demonstrated in vivo in murine tumor models.27 A functional reduction in tumor perfusion was assessed by ultrasound imaging and ex vivo tissue analysis.27,28 These results were confirmed in Pexa-Vec–treated patients. Biopsy analysis of tumors revealed reproducible infection of tumor-associated endothelial cells.27 Furthermore, a subset of HCC patients treated with Pexa-Vec by IT injection on a Phase II study exhibited an acute reduction in tumor perfusion as measured by dynamic magnetic resonance imaging (MRI) 5 days after the initial Pexa-Vec injection. Adjacent normal liver tissue did not demonstrate reduced perfusion.27
In summary, Pexa-Vec has been shown to target tumors by multiple, complementary MOA in preclinical and clinical investigations. Acute debulking of tumors secondary to oncolysis and vascular targeting are followed by an adaptive antitumor immune response that may impact both existing and new tumor development.
Clinical development
Pexa-Vec has been evaluated in 12 completed and ongoing clinical trials to date. Over 300 patients have been treated by IV infusion and/or IT injection; with >600 IV infusions and >500 IT injections. Overall, Pexa-Vec treatment was well-tolerated with acute, transient flu-like symptoms being the most commonly reported adverse events. Patients at higher risk of complication from vaccinia vaccination (eg, immunocompromised individuals and those with inflammatory skin conditions) were excluded from enrollment on trials. With >1,000 Pexa-Vec treatments, there was no evidence of complications stemming from systemic vaccinia infection.
Initial studies investigated the safety of Pexa-Vec when administered by IT injection into skin (melanoma)23 and liver tumors (primary HCC or liver metastases)18 (Table 2). Antitumor activity was demonstrated in both injected and noninjected tumor sites.18,23 After successful IT dose escalation, and demonstration of subsequent systemic dissemination following local injection, an IV dose-escalation trial in advanced solid tumors was initiated to evaluate the tolerability of a single dose of Pexa-Vec delivered via a 60-minute IV infusion.17 Notably, IV Pexa-Vec was associated with dose-dependent delivery to multiple solid tumor types (including colorectal cancer, lung cancer, pancreatic cancer, and mesothelioma) and resulted in antitumor activity at high doses. Anticancer activity was suggested by the observation that patients were less likely to present with new tumors if treated at the Pexa-Vec maximum feasible dose and demonstration of a partial response after a single IV infusion in a patient with metastatic mesothelioma.17 Pexa-Vec was well-tolerated up to a dose of 3×107 pfu/kg per patient. The most common adverse events reported were acute, transient flu-like symptoms, including fever and chills. Transient, fluid responsive hypotension was also noted in a subset of patients within 12 hours of treatment. Pexa-Vec–related skin pustules were observed in a minority of patients receiving IV infusion. The pustules were self-limited and resolved without sequelae within 2–3 weeks of treatment, consistent with the timeline for pustule resolution utilizing wild-type vaccinia vaccine.
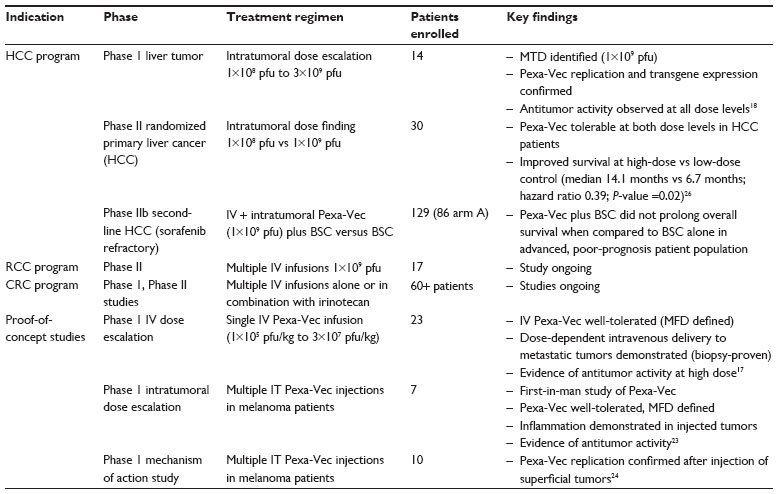 | Table 2 Overview of Pexa-Vec clinical trials |
A randomized Phase II dose-ranging study was initiated to evaluate the safety and antitumor efficacy of Pexa-Vec administered at high dose (maximum tolerable dose; 1×109 pfu) versus low dose (1×108 pfu) in patients with advanced HCC.26 IT injection was well-tolerated at both dose levels in this population of patients with HCC and concurrent underlying liver disease. Four patients responded to treatment based on modified response evaluation criteria in solid tumors (RECIST) criteria (one complete response; three partial responses). Responses were observed in injected and noninjected tumors (Figure 2). Further, overall survival (OS) was significantly longer in the high-dose arm compared with the low-dose arm (median 14.1 months versus 6.7 months; hazard ratio: 0.39; P-value =0.020; Gehan–Breslow–Wilcoxon test; one-sided test for superiority of high-dose).26 The median OS was 9.0 months for the entire population.
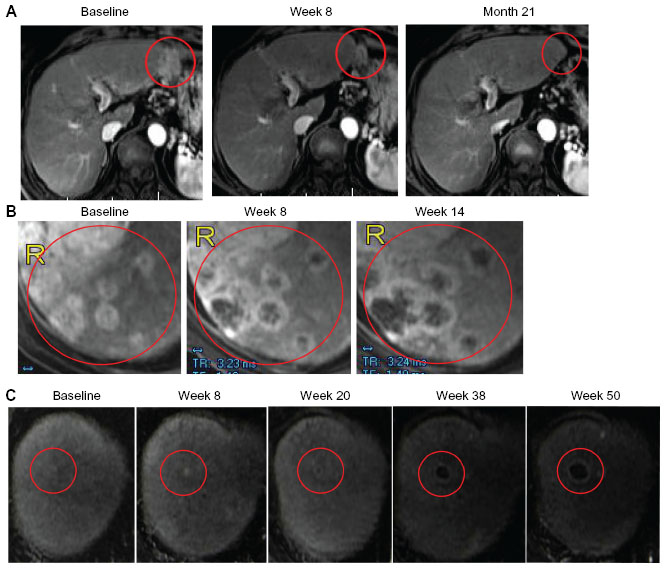 | Figure 2 Responses in injected and noninjected tumors. |
In contrast, a Phase IIb clinical trial in HCC patients who failed sorafenib therapy (n=120) was recently completed and did not achieve the primary endpoint of prolonging OS in Pexa-Vec–treated patients when compared to patients treated with best supportive care in this last-line, poor prognosis patient population.
Additional Phase II studies evaluating single-agent Pexa-Vec treatment administered by multiple IV infusions are currently underway in treatment-refractory patients with renal cell carcinoma (RCC), and colorectal cancer.
Combination therapy strategies
Based on the early observations of a tolerable safety profile (eg, transient flu-like symptoms generally resolving between 24 and 48 hours posttreatment) and antitumor activity, Pexa-Vec appears to be a relevant partner for combination with other anticancer therapeutics. Sequential Pexa-Vec dosing with sorafenib has been evaluated in patients with HCC29 and an ongoing study (NCT01171651). Since sorafenib has effects on both tumor cells and their associated vasculature, it is possible that Pexa-Vec sensitizes either or both of these tumor components to sorafenib. Notably, since sorafenib inhibits virus replication, one limitation of this approach is the requirement for sequential (ie, not concurrent) administration.
Unlike sorafenib, irinotecan (a topoisomerase inhibitor) does not interfere with Pexa-Vec replication. Though vaccinia encodes a viral topoisomerase, it is not inhibited by irinotecan (unlike other chemotherapeutics that bind DNA directly). Combination Pexa-Vec treatment with irinotecan was shown to have additive effects in preclinical models (unpublished data) and resensitization to irinotecan by Pexa-Vec is currently being evaluated in patients (NCT01394939).
Finally, given the ability of Pexa-Vec to cause acute debulking and release of tumor antigens in the context of a proinflammatory environment, Pexa-Vec may lend itself to combination therapy with other immunomodulating agents, including checkpoint inhibitors. Data thus far with immune-modulating agents (IMA) demonstrate a high correlation of tumor-infiltrating T cells and drug targets (eg, PD-L1), with subsequent response to these agents.30 Therefore, acute infection, inflammation, and antitumor immune response induction triggered by Pexa-Vec have potential to sensitize to subsequent treatment with immune checkpoint inhibitors. For example, treatment with Pexa-Vec leads to tumor cell death in the context of viral infection (immunogenic cell death). Furthermore, vaccinia has been shown to ligate toll-like receptors (TLRs) 2 and 8,31,32 initiating an acute inflammatory response and promoting immune cell infiltration into tumors. Finally, GM-CSF produced by Pexa-Vec infected cells boosts white blood cell counts (as shown in patients)17,18 as well as activates mature dendritic cells, thereby promoting priming and activation of T cells. In an effort to enhance response to the IMA, a priming treatment with Pexa-Vec may lead to increased T cell infiltration and IMA target upregulation in patient tumors. Indeed, combination therapy of an oncolytic vaccinia with antibody-promoting costimulation (CD137/41BB agonistic antibody) has been shown to have more enhanced antitumor activity than treatment with either agent alone in a preclinical model.33
Future directions
Single agent Pexa-Vec has demonstrated a tolerable safety profile and activity across multiple tumor types after both IV and IT administration. Completion of single agent Phase II studies in renal and colon cancers are awaited. Lessons learned from the Phase IIb study in HCC patients having failed sorafenib therapy point to treatment of more fit, less heavily pretreated patients with Pexa-Vec, consistent with the ongoing view in general that healthier, more immunocompetent patients may be the best target population for this class of agents. Further investigation of the synergistic or complementary activity of Pexa-Vec in combination with other agents is highly anticipated, including combination with tyrosine-kinase inhibitors (eg, sunitinib plus Pexa-Vec in RCC), and IMA (eg, anti-PD1 plus Pexa-Vec). Further, exploration of front-line HCC with single agent Pexa-Vec, based on earlier results from the randomized Phase II dose-ranging study, is also warranted. Finally, oncolytic vaccinia viruses exhibit a large transgene-encoding capacity,34 which allows product engineering for complementary MOA through insertion of therapeutic transgenes. Transgene expression is highly tumor-selective, and transgenes are amplified within the tumor during replication. Therefore, next generation vaccinia viruses can be engineered to express antibodies targeting checkpoint inhibitors, cytokines/chemokines, as well as tumor antigens to further activate antitumor immunity.
Disclosure
CJB, THH, DHK, and JB are employees of SillaJen Biotherapeutics, which own the rights to Pexa-Vec. The authors have no other conflicts of interest in this work.
References
Fenner F, Henderson DA, Arita I, Ježek Z, Ladnyi ID. Smallpox and its Eradication. Geneva, Switzerland: World Health Organization; 1988. | |
Kim JH, Oh JY, Park BH, et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol Ther. 2006;14(3):361–370. | |
Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9(1):64–71. | |
Breitbach CJ, Thorne SH, Bell JC, Kirn DH. Targeted and armed oncolytic poxviruses for cancer: the lead example of JX-594. Curr Pharm Biotechnol. 2012;13(9):1768–1772. | |
Parato KA, Breitbach CJ, Le Boeuf F, et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther. 2012;20(4):749–758. | |
Hengstschläger M, Pfeilstöcker M, Wawra E. Thymidine kinase expression. A marker for malignant cells. Adv Exp Med Biol. 1998;431:455–460. | |
Yu YA, Shabahang S, Timiryasova TM, et al. Visualization of tumors and metastases in live animals with bacteria and vaccinia virus encoding light-emitting proteins. Nat Biotechnol. 2004;22(3):313–320. | |
Thorne SH, Hwang TH, O’Gorman WE, et al. Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963. J Clin Invest. 2007;117(11):3350–3358. | |
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. | |
Katsafanas GC, Moss B. Vaccinia virus intermediate stage transcription is complemented by Ras-GTPase-activating protein SH3 domain-binding protein (G3BP) and cytoplasmic activation/proliferation-associated protein (p137) individually or as a heterodimer. J Biol Chem. 2004;279(50):52210–52217. | |
Yang H, Kim SK, Kim M, et al. Antiviral chemotherapy facilitates control of poxvirus infections through inhibition of cellular signal transduction. J Clin Invest. 2005;115(2):379–387. | |
Vanderplasschen A, Mathew E, Hollinshead M, Sim RB, Smith GL. Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope. Proc Natl Acad Sci U S A. 1998;95(13):7544–7549. | |
Bell E, Shamim M, Whitbeck JC, Sfyroera G, Lambris JD, Isaacs SN. Antibodies against the extracellular enveloped virus B5R protein are mainly responsible for the EEV neutralizing capacity of vaccinia immune globulin. Virology. 2004;325(2):425–431. | |
Putz MM, Midgley CM, Law M, Smith GL. Quantification of antibody responses against multiple antigens of the two infectious forms of Vaccinia virus provides a benchmark for smallpox vaccination. Nat Med. 2006;12(11):1310–1315. | |
McCart JA, Ward JM, Lee J, et al. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001;61(24):8751–8757. | |
Kirn DH, Wang Y, Liang W, Contag CH, Thorne SH. Enhancing poxvirus oncolytic effects through increased spread and immune evasion. Cancer Res. 2008;68(7):2071–2075. | |
Breitbach CJ, Burke J, Jonker D, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477(7362):99–102. | |
Park BH, Hwang T, Liu TC, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9(6):533–542. | |
Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1):nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5(4):331–342. | |
Rubartelli A, Lotze MT. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007;28(10):429–436. | |
Zhu J, Martinez J, Huang X, Yang Y. Innate immunity against vaccinia virus is mediated by TLR2 and requires TLR-independent production of IFN-beta. Blood. 2007;109(2):619–625. | |
Kirn DH, Wang Y, Le Boeuf F, Bell J, Thorne SH. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007;4(12):e353. | |
Mastrangelo MJ, Maguire HC Jr, Eisenlohr LC, et al. Intratumoral recombinant GM-CSF-encoding virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 1999;6(5):409–422. | |
Hwang TH, Moon A, Burke J, et al. A mechanistic proof-of-concept clinical trial with JX-594, a targeted multi-mechanistic oncolytic poxvirus, in patients with metastatic melanoma. Mol Ther. 2011;19(10):1913–1922. | |
Kim MK, Breitbach CJ, Moon A, et al. Oncolytic and immunotherapeutic vaccinia induces antibody-mediated complement-dependent cancer cell lysis in humans. Sci Transl Med. 2013;5(185):185ra63. | |
Heo J, Reid T, Ruo L, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19(3):329–336. | |
Breitbach CJ, Arulanandam R, De Silva N, et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 2013;73(4):1265–1275. | |
Breitbach CJ, Paterson JM, Lemay CG, et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol Ther. 2007;15(9):1686–1693. | |
Heo J, Breitbach CJ, Moon A, et al. Sequential therapy with JX-594, a targeted oncolytic poxvirus, followed by sorafenib in hepatocellular carcinoma: preclinical and clinical demonstration of combination efficacy. Mol Ther. 2011;19(6):1170–1179. | |
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. | |
Quigley M, Martinez J, Huang X, Yang Y. A critical role for direct TLR2-MyD88 signaling in CD8 T-cell clonal expansion and memory formation following vaccinia viral infection. Blood. 2009;113(10):2256–2264. | |
Martinez J, Huang X, Yang Y. Toll-like receptor 8-mediated activation of murine plasmacytoid dendritic cells by vaccinia viral DNA. Proc Natl Acad Sci U S A. 2010;107(14):6442–6447. | |
John LB, Howland LJ, Flynn JK, et al. Oncolytic virus and anti-4-1BB combination therapy elicits strong antitumor immunity against established cancer. Cancer Res. 2012;72(7):1651–1660. | |
Smith GL, Moss B. Infectious poxvirus vectors have capacity for at least 25 000 base pairs of foreign DNA. Gene. 1983;25(1):21–28. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.