Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 9
The effects of huntingtin-lowering: what do we know so far?
Authors Kaemmerer WF ![]() , Grondin RC
, Grondin RC
Received 17 September 2018
Accepted for publication 22 January 2019
Published 8 March 2019 Volume 2019:9 Pages 3—17
DOI https://doi.org/10.2147/DNND.S163808
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Müller
William F Kaemmerer,1 Richard C Grondin2
1CGTA Research Group, Eagan, MN, USA; 2Department of Neuroscience, University of Kentucky Medical Center, Lexington, KY, USA
Abstract: Therapies targeting mutant huntingtin DNA, mRNA, and protein have a chance at becoming the first disease-modifying treatments for Huntington’s disease, a fatal inherited neurodegenerative disorder for which only symptom management treatments are available today. This review focuses on evidence addressing several key questions pertinent to huntingtin-lowering, ranging from the functions of wild-type huntingtin (wtHTT) that may be disrupted by huntingtin-lowering treatments through the various ways huntingtin can be lowered, the tolerability of wtHTT-lowering in mice and primates, what has been found in the Ionis Pharmaceutical safety trial of a huntingtin-lowering therapy, and to the question of how much mutant huntingtin may need to be lowered for a therapy to be clinically effective. We conclude that adverse consequences of lowering wtHTT in animals appear to be brain region-specific, and/or dependent upon the animal’s stage of development and the amount by which huntingtin is lowered. Therefore, safe approaches to huntingtin-lowering in patients may be to lower huntingtin only moderately, or lower huntingtin only in the most affected brain regions, or lower huntingtin allele-selectively, or all of the above. Many additional questions about huntingtin-lowering remain open, and will only be answered by upcoming clinical trials, such as whether the delivery approaches currently planned will be adequate to get the treatment to the necessary brain regions, and whether non-allele-selective huntingtin-lowering will be safe in the long run. Meantime, there is a role for preclinical research to address key knowledge gaps, including the effects of non-allele-selective huntingtin-lowering on protein trafficking and viability at the cellular level, the tolerability of wtHTT-lowering in the corticostriatal connections of the primate brain, and the effects of this lowering on the functioning of neurotransmitter systems and the transport of neurotrophic factors to the striatum.
Keywords: Huntington’s disease, huntingtin-lowering, gene therapy, cortex, striatum, CAG repeat disorder
Introduction
Huntington’s disease (HD) is an inherited, fatal disorder in which the primary pathology is neurodegeneration, particularly cortical thinning and atrophy of the caudate nucleus and putamen. The disease is caused by the expansion of a CAG repeat region in exon 1 of the HTT gene, resulting in the expression of an expanded, mutant huntingtin protein (mHTT). Individuals with just one HTT allele containing more than 40 CAG repeats are invariably affected by the disease, while individuals with fewer than 36 CAG repeats on both alleles do not manifest the disease.1,2 The age of symptom onset is inversely related to the number of CAG repeats and influenced by genetic modifier loci on chromosomes 8 and 15.3,4 No disease-modifying treatment yet exists.
Studies in cells and animal models of HD have found mHTT to have a myriad of disruptive effects, including transcriptional dysregulation, impairment of protein degradation systems, mitochondrial dysfunction, and altered synaptic plasticity.5 Some treatments targeting downstream effects of mHTT have shown efficacy in animal models of HD, but none have shown efficacy in the human disease today. Consequently, the best approach to treatment appears to be the targeting of huntingtin itself, seeking to eliminate or at least lower the expression of mHTT. In principle, this can be accomplished by various technologies, including intrabodies targeting the huntingtin protein, antisense oligonucleotides (ASOs) or RNAi agents targeting the mRNA transcripts, zinc-finger repressors to prevent mRNA transcription, or gene editing of HTT DNA itself. With the exception of the latter approach, these techniques will reduce but not completely eliminate the production of mHTT. Therefore, the term “huntingtin-lowering” is the preferred term for this class of potential therapies. As discussed later in this review, the first trial of a huntingtin-lowering agent in patients has recently been completed by Ionis Pharmaceuticals, showing via an assay for mHTT in the cerebrospinal fluid (CSF) that lowering of HTT was achieved and there were no adverse events attributable to the agent.
Although many questions will only be answered by clinical trials, it is of interest to review what has been learned about the effects of huntingtin-lowering since the discovery of the HTT gene in 1993. This review focuses on key questions regarding the rationale, safety, possible efficacy, and possible side-effects of huntingtin-lowering as a treatment for HD, surveying the empirical evidence that bears on these questions.
Is lowering the expression of mHTT protein the right objective?
HD is due to toxic properties of mHTT rather than just loss of wild-type huntingtin (wtHTT) function. While wtHTT is necessary in early life (knockout of the mouse homologue of huntingtin is embryonically lethal6–8), individuals lacking wtHTT because both their alleles contain an expanded CAG repeat region can be healthy for decades.
Could provision of “extra” wtHTT to a patient be a therapeutic option? In neuronal and non-neuronal cells transfected with mHTT, co-transfection of wtHTT significantly reduces cell death.9 In mice with a yeast artificial chromosome (YAC) for mutant human huntingtin, a pro-apoptotic effect of mHTT on cells in the testes is completely inhibited in mice that are hemizygous or homozygous for wild-type murine huntingtin (Htt).10 YAC18 mice overexpressing a normal repeat length transgene (18 CAGs) are protected against neurodegeneration induced by striatal injection of quinolinic acid.11 However, other evidence indicates that a treatment based on supplementing wtHTT expression would be insufficient. A genetic cross of YAC128 mice (128 CAGs) and YAC18 mice produces offspring that overexpress wtHTT in the context of mHTT. Though these offspring have mild improvement in striatal cross-sectional area compared to YAC128 mice, they show no significant improvement in motor coordination, striatal volume, striatal neuronal numbers, or striatal DARPP-32 expression.12
Is the mHTT protein the only pathogen in HD? Some loss of wtHTT function and possible effects of protein products produced by repeat-associated non-ATG (RAN) translation of the expanded mRNA have been hypothesized to also play a role in the disease. Products of RAN translation from mHTT mRNA have been found in HD human brains, and the toxicity of expanded HTT exon1 mRNA in human neuroblastoma cells can be blocked by anti-CAG small RNA.13 Intrastriatal injection of a locked nucleic acid consisting of CTG repeats (complementary to CAGs) induces rapid motor improvement in the R6/2 mouse model of HD without lowering either mHTT or wild-type protein levels in these mice.14 Therefore, while lowering of mHTT is essential, huntingtin-lowering therapies utilizing agents that target HTT at the mRNA level (ASOs and RNAi agents) might be more advantageous than those that act only on the protein product (eg, anti-huntingtin intrabodies). However, for these agents, consideration must be given to the potential for the agent to target other genes and transcripts besides HTT, producing “off-target” effects. Although recommended by the Oligonucleotide Safety Working Group,15 in silico approaches to predicting the off-target effects and toxicity of ASOs based on their sequence16 are not perfect. Therefore, ASO developers utilize a combination of in silico screening, in vivo screens for liver toxicity, and transcriptome-wide evaluations of gene suppression to identify lead candidates.17 Developers of RNAi agents expressed from DNA delivered by a viral vector rely heavily on in vitro and in vivo screening of candidate sequences, because chemical modification is not available as a way to improve any inadequate specificity of a chosen candidate.
Different types of huntingtin-lowering therapies also differ in their advantages and disadvantages with regards to route of delivery, longevity of effect, reversibility, control of dosing, etc. See Wild and Tabrizi18 for a review.
Can HTT-lowering be accomplished allele specifically if necessary?
Ever since it was determined that huntingtin protein is required for embryogenesis6–8 and early development,19 consideration has been given to the possibility of lowering expression of the mutant huntingtin allele while preserving expression of wtHTT in heterozygous individuals. Many approaches have proven feasible in cell and animal models of HD. Because a single nucleotide mismatch between an siRNA and a complementary mRNA can substantially reduce its RNAi activity, an siRNA targeting a single-nucleotide polymorphism (SNP) site in HTT in a heterozygous cell can reduce expression of the allele with the matching nucleotide of the SNP while leaving expression of the other allele much less affected. van Bilsen et al20 designed siRNA targeting SNPs in the HD gene and identified siRNA that are allele-selective in reducing HTT in fibroblasts from heterozygous HD patients. They also developed a simple method for identifying which SNP variant is on the same allele as the expanded CAG repeat allele using SNP variant-specific primers for the reverse transcription step of an RT-PCR assay. Subsequently, Zhang et al21 demonstrated allele specificity using an siRNA targeting a polymorphic deletion (Δ2642) linked to the CAG repeat allele in HD fibroblasts. Carroll et al22 identified numerous ASOs that are potent and selective for their respective SNP target in vitro and in vivo when injected into the brains of transgenic mouse models of HD.
A disadvantage of leveraging SNPs to achieve allele specificity in huntingtin-lowering is that there is currently no known single SNP that can be used as the basis for an allele-specific therapy in all HD patients. Genotyping followed by combinatorial analysis of the frequency and co-occurrence of 26 SNP sites on the alleles of 327 unrelated European Caucasian HD patients showed that a repertoire of seven allele-specific siRNAs would be needed for 85.6% of those patients to be treatable by at least one of the siRNA.23 A similar analysis sequencing 22 SNP sites in 225 HD samples led to the estimate that 5 siRNAs targeting 3 SNPs could provide therapy for 75% of the US and European HD populations.24 Although encouraging, these findings are still daunting with regards to the cost of developing and achieving regulatory approval for multiple agents.
Could an allele-specific agent be found that would apply to all patients? Initial attempts to use ASOs to target the expanded CAG repeat itself achieved only modest allele selectivity.25 However, advances in chemically modified oligonucleotides and the use of multiple mismatches within an agent’s sequence has led to the identification of ss-RNAs that can inhibit the translation of mHTT with high potency and selectivity.26 The ss-RNAs can distinguish between 44 CAG repeats and 15 CAG repeats on the alleles of patient-derived cells (GM04719); however, many patients have a difference between the repeat lengths of their alleles that is smaller than this (44 CAGs is the median number found in patient samples).2 Also, these agents might have off-target effects, lowering expression of other genes that contain CAG repeats.
Another option for targeting the CAG repeat region is the use of zinc-finger proteins (ZFPs).27 Using a repressor element targeted to the CAG repeat region of the HD gene with ZFPs that bind CAGs, Zhang et al28 achieved about 90% repression of mHTT expression with minimal effect on normal CAG length alleles in fibroblasts derived from HD patients. In another approach, Garriga-Canut et al29 screened long ZFP chains designed to bind both the CAG and complementary GTC strand of long HD repeat regions more strongly than shorter repeat regions. In a cell line from a knockin model of HD (STHdhQ7/Q111), these ZFPs fused with a Kox-1 repressor element reduced mutant huntingtin mRNA by almost 80% and mutant protein expression by 95% while reducing neither protein nor RNA produced from the wild-type allele (Q7). Both investigators found their ZFPs function in vivo to improve the phenotype of the R6/2 mouse model of HD.28,29 However, further development of these entities toward clinical trials has not yet been reported.
Finally, another way to lower huntingtin allele specifically may be to use CRISPR/Cas9 editing to delete DNA from the mutant allele. Allele-specific gene editing can occur if the single-stranded guide RNA used with Cas9 targets a SNP site whereby one of the SNP variants either eliminates a protospacer-adjacent motif (PAM) sequence (on the allele to be preserved) or creates a PAM sequence (on the allele to be edited). Monteys et al30 found that appropriately designed sgRNA applied in pairs can result in allele-specific deletion of genomic DNA spanning the 5′UTR and exon-1 of HTT in heterozygous patient-derived fibroblasts in vitro and reduce human mHTT expression in a BACHD mouse model of HD to 40% in vivo. Similarly, Shin et al31 used sgRNA to selectively excise 44 kilobases of DNA spanning the promoter region, transcription start site, and first three exons of HTT. However, both approaches have the disadvantage that the allele specificity depends upon SNPs in the HD gene, so no single therapeutic agent would be applicable to all patients. Of greater concern is the recent finding that gene editing based on the dsDNA breaks produced by Streptococcus pyrogenes Cas9 (SpCas9) can result in a wide variety of unintended editing events, including large deletions that are non-contiguous with the targeted cut site.32 This safety issue might be avoided using variants of Cas9 (nickases) that produce single-stranded cuts in the DNA rather than dsDNA breaks. Using such nickases, Dabrowska et al33 demonstrated that the CAG repeat region can be precisely excised from the HTT gene by targeting two sites, one at the 5′ end of the CAG repeat and the other about 40 bases downstream of the 3′ end of the repeat. However, the excision is independent on CAG repeat length, and thus is not allele specific. Substantial additional research will be needed to develop a CRISPR/Cas9 agent that is allele specific and safe for in vivo human use.
Of note, one allele-specific approach to lowering huntingtin using oligonucleotides targeting SNP sites has advanced to clinical trials. Patients are being recruited for two trials (NCT03225833 and NCT03225846, Wave Life Sciences, Ltd.) each using an oligonucleotide targeting one of the more prevalent SNPs in the HD gene. However, other companies investing in HD therapy development have opted for non-allele-specific approaches.
What functions of wtHTT may be disrupted by non-allele-specific huntingtin-lowering?
Concerns that lowering wtHTT in patients may have undesirable consequences have arisen from studies identifying many functions of the normal protein. These have roles not only in development but also in regulation of intracellular trafficking processes, formation of cortical and striatal excitatory synapses, and in transcriptional regulation (for a review, see Jimenez-Sanchez et al5). In vitro, wtHTT protects striatal-derived cells from mitochondrial toxins.34 Both in vivo and in vitro, wtHTT is phosphorylated in response to DNA damage, suggesting a role in the DNA damage response signal.35 Proteomic analyses have identified hundreds of proteins that interact with wtHTT,36 supporting a view of HTT as a scaffolding protein involved in numerous protein–protein interactions.
wtHTT is also involved in many types of trafficking in cells, including bidirectional transport of vesicles in neurons,37 trafficking between Golgi and extracellular space,38 transport via endocytic and secretory pathways,39 and secretory vesicle fusion at the plasma membrane40 where loss of wtHTT results in ~50% decrease in the number of vesicle fusion events per unit time. Investigations of wtHTT interaction with dynein and dydactin41 have led to the proposal of models whereby wtHTT plays a global role in vesicle and organelle transport throughout the cytoskeletal network.42
The wide variety and apparent centrality of the functions of wtHTT (see Liu and Zeitlin43 for a detailed review) have led to the question of whether HTT could be indispensable in the adult brain. If so, excessive non-allele-specific lowering of huntingtin may not be safe. What evidence exists to inform this issue?
What evidence is there concerning the safety of lowering wtHTT?
wtHTT-lowering may not be safe in the developing brain
In a study of mice with one null copy of Hdh and one copy altered to express Htt at a greatly reduced level (~15% of endogenous levels), the mice were small from birth and had movement abnormalities and variable increases in cerebroventricular volume.44 Studies using Cre-mediated conditional knockout of Hdh in mice have shown that knockout of Hdh at various stages of embryonic and post-natal development results in deficiencies in cortical neuron migration,45 abnormalities in cortical and striatal synaptic development,46 and behavioral and neurological abnormalities that are not reversed by later restoration of ~50% Htt expression (at P21).47
In vitro evidence related to the safety of wtHTT-lowering
Much evidence giving rise to safety concerns about huntingtin-lowering comes from in vitro studies, particularly those identifying a role wtHTT plays in the production and transport of brain-derived neurotrophic factor (BDNF) and BDNF mRNA.48 Wild type but not mHTT promotes cortical BDNF gene transcription by inhibiting the BDNF silencing activity of the neuron restrictive silencer element.49 Huntingtin interacts indirectly with the REST/NRSF-interacting LIM domain protein through dynactin;50 in the case of mHTT, this interaction is weaker. This is consistent with the finding that BDNF mRNA and protein levels are reduced in HD.51 Further reduction due to non-allele-specific huntingtin-lowering could jeopardize the survival of medium spiny neurons of the striatum, which are dependent upon cortically derived BDNF52and particularly vulnerable in HD. In neuroblastoma cells, reduction of HTT by siRNAs reduces the velocity of vesicle transport and increases the time vesicles spend without moving.53 Her et al54 studied primary cortical neurons from Hdh floxed mice and found that complete knockout of Htt from these cortical neurons did not affect their viability, but reduced both anterograde and retrograde transport of particles in their axons. Interestingly, they also found that axonal transport of BDNF was not affected in primary cortical neurons transfected with mHTT.54
Caviston et al41 have found that reduction of HTT protein by 83% in HeLa cells results in the Golgi becoming stretched out or vesicular in these cells, disrupting protein transport particularly of proteins destined for the extracellular compartment. This consequence may not arise in huntingtin-lowering treatments using agents that do not achieve this extent of HTT reduction, but could arise in treatments involving gene editing. The clinical relevance of this effect may depend upon whether, in the long-term, there are compensatory adaptations in cells that would mitigate this disruption of the Golgi.
In vivo evidence regarding the safety of wtHTT-lowering in adults: mouse studies
At least three studies have investigated the consequences of complete knockout of Htt in adult mice at various ages, with conflicting conclusions. Wang et al55 crossed conditional Hdh knockout mice56 with mice that express a tamoxifen-inducible Cre, producing inducible Htt knockout mice. Depletion of Htt in mice by tamoxifen injection at 2 months of age resulted in 95% mortality due to acute pancreatitis. In contrast, 95% of mice in which Htt was depleted at 8 months of age survived long term (studied for 10–11 more months) with no significant differences in rotarod performance, body weight, or the expression of various proteins (LC3I/II, P62, caspase-3, NfκB, FAT10) in the brain and peripheral tissues. They also crossed floxed Hdh mice with transgenic mice expressing Nestin-CreER, producing mice in which tamoxifen injection eliminated Htt expression in neurons and not peripheral tissues. These mice, injected with tamoxifen at 2, 4, or 8 months of age, showed no differences in brain volume or histology, and no differences over 7–8 months time in post-injection body weight, rotarod performance, or gripping ability compared to heterozygous Hdh knockout controls. These results suggest that lowering and even elimination of wtHTT may be safe if depletion of wtHTT occurs only in the brain or only in older individuals.
However, a study by Dietrich et al57 produced distinctly different results. In their study, CAG-CreER mice were first crossed with heterozygous knockout mice (Hdh+/-) and offspring were subsequently crossed with Hdhflox/flox mice, producing CreER;Hdhflox/- mice, termed cKO mice. These cKO mice express ~50% of the level of endogenous Htt compared to wild-type mice from conception. Administration of tamoxifen to cKO mice results in nearly complete elimination of Htt expression (evaluated by Western blot), reduction in Htt mRNA in total brain extracts to <15% of wild-type levels (evaluated by RT-PCR), and reduction of Htt expression in the cortex and striatum to <20% of wild-type levels (evaluated by electrochemiluminescence assay). Regardless of when tamoxifen was administered to these mice (at age 3, 6, or 9 months), the mice developed progressive gait abnormalities, resting tremors, and substantially reduced rotarod performance within 1–3 months after huntingtin-lowering. They also had a shortened life span compared to cKO mice not administered tamoxifen. No pancreatitis was reported in this study in mice administered tamoxifen at 3 months of age, although other adverse effects of huntingtin-lowering were observed in peripheral tissues, including thickening of the cornea observed in about one-third of tamoxifen-treated cKO mice and testicular atrophy seen in tamoxifen-treated males. Histologically, the most pronounced findings were early development of reactive gliosis in the cerebellum and thalamus within 3 months of huntingtin-lowering and later development of calcification in the thalamus (seen as early as 13 months of age and confined to the thalamus). Ferric iron levels were significantly reduced throughout the brains of tamoxifen-treated cKO mice, including in the striatum and cortical neuronal cells. Conversely, although reactive gliosis was seen in the striatum 9 months after huntingtin-lowering, no overt loss of cells was observed in the cortex even 14 months after huntingtin-lowering, and no change in DARPP-32 expression or overt cell loss was seen in the striatum 12 months after huntingtin-lowering. Overall, these observations suggest that huntingtin-lowering may be tolerated in the striatum and cortex more than in other regions of the brain.
A third study of huntingtin-lowering in the adult mouse was performed by Pla et al58 These investigators crossed CaMKCreER mice (in which tamoxifen activation of Cre recombinase occurs only in mature cortical and hippocampal neurons) with Hdhflox/flox mice to obtain offspring in which Hdh was inactivated in these neurons by administering tamoxifen to the mice at 2 months of age. Six months later, investigation of hippocampal neurogenesis in the dentate gyrus of the mice showed impaired maturation and differentiation of newborn neurons (assessed by dendritic arborization) and reduced survival of newborn neurons. These mice showed no obvious motor deficits in rotarod tests, but spent significantly less time than controls in the center of an open field and less time in open arms of an elevated plus maze, indicative of an anxious phenotype. Western blotting of protein extracts from the hippocampi of the mice indicated that although BDNF levels were no different from controls, Erk and Akt (kinases involved in intracellular signaling upon BDNF binding to cell surface receptors) were less extensively phosphorylated in the tamoxifen-treated mice than in controls. This suggests that BDNF signaling was impaired due to a reduction in BDNF transport or release. The investigators suggest that some of the anxiety disorders prevalent among HD patients may be caused by loss of normal HTT function in the hippocampus and cortex, particularly with regards to BDNF transport, due to a dominant negative effect of mHTT (although Her and Goldstein54 did not find a dominant negative effect of mHTT in BDNF transport in primary cortical neurons).
It is difficult to reconcile the results of these three studies. Wang et al did not report testing their mice for behavior indicative of an anxious phenotype; Dietrich et al did not find pancreatitis or dramatically early mortality in their conditional cKO mice treated with tamoxifen at 3 months of age in contrast to the findings of Wang et al in mice treated at 2 months of age; Wang et al did not observe the progressive behavioral deficits, thalamic calcifications, or other adverse effects of huntingtin elimination or lowering reported by Dietrich et al. Barring procedural, environmental, or genetic background differences, the best explanation might be that, as noted by Dietrich et al, the effects of Htt elimination may be more deleterious in a mouse that has had only 50% of normal endogenous levels of Htt in its life prior to Htt elimination (in their study) than in a mouse with normal levels of Htt prior to tamoxifen administration (in the Wang et al study).
In vivo evidence that wtHTT-lowering in adulthood is tolerated: non-human primate studies
We and others have studied the effects of lowering wild-type Htt in non-human primates whose neurophysiology, basal ganglia anatomy, and behavioral repertoire more closely resembles that of humans. McBride et al59 stereotactically injected 10–12 µL of 1E12 vg/mL AAV2/1 encoding a huntingtin-lowering miRNA (miHDS1) into each of three sites in the putamen in one hemisphere of adult male rhesus macaques (n=4). Animals were assessed pre- and post-operatively on general behavior and motor skill assays and euthanized 6 weeks post-AAV administration for molecular and histological analyses. AAV-miHDS1-injected recipients showed a significant 45% reduction in wild-type Htt mRNA expression in the putamen compared to animals that received a control miRNA. Compared to controls, AAV-miHDS1-injected animals showed no motor skill deficits, no significant decrease in the mRNA for DARPP-32 (a marker for medium spiny neurons), and no loss of NeuN-positive neurons in the putamen.
We conducted a much longer study in rhesus macaques60 using bilateral injections of a larger quantity and higher titer of AAV. Adult females were injected bilaterally with 2E12 vg/mL of either AAV2-HD5 (a huntingtin-lowering shRNA, n=4) or AAV2-CTRL5 (a scrambled shRNA, n=4) into three sites per hemisphere, one in the caudate (30 µL) and two in the putamen (60 µL each). Six months later, shRNA transcripts were detected in the targeted brain regions, and HD5 vector recipients showed a 30% reduction in wtHtt mRNA and an average wtHtt protein reduction of 45% (range 32%–67% reduction) relative to controls. Home-cage activity levels and motor performance on an automated hand-retrieval task were not adversely affected by either AAV2-HD5 or CTRL5 delivery. Motor memory was also preserved as all animals recalled and performed the retrieval task without intervening practice between the monthly test sessions. There was no significant effect on immunostaining for DARPP-32, and no discernible neuronal loss or abnormal astrocytosis noted in any of the animals upon blinded microscopic evaluation by a board-certified pathologist. Our study therefore showed that 32%–67% reduction of wtHtt is safe and well-tolerated in the primate caudate and putamen for at least 6 months.
In addition to using viral vectors, our group has also evaluated the use of programmable pumps and intraparenchymal catheters to chronically deliver huntingtin-lowering siRNA directly into the rhesus striatum.61,62 Intraparenchymal catheters were implanted unilaterally into the right putamen of adult, female rhesus monkeys and connected to an abdominally implanted pump from which siRNA (or PBS, in controls) was delivered. Continuous infusion of siRNA for 28 days was well-tolerated and resulted in up to 75% lowering of Htt mRNA in individual brain tissue samples, and ~45% suppression of Htt mRNA on average throughout most of the rhesus striatum. Decreased neuronal levels of Htt protein were indicated by a pronounced attenuation of immunostaining intensity in the putamen of siRNA recipients. No behavioral changes were observed. Microscopic evaluations conducted on Fluoro-Jade B-stained brain sections did not reveal any continuing neuronal necrosis in any of the control or siRNA-treated animals. Normal patterns of Nissl-stained neurons were retained in corresponding brain regions.
Together, these results support the feasibility and safety of wtHtt reduction in the primate brain. In addition, Ambrose et al63 have noted a case in humans of a phenotypically normal female in whom the HD locus was bisected by a balanced translocation, indicating that heterozygous disruption of HTT from conception onward was fully tolerated in this individual. It should be noted, however, that it is possible that the toler- ability of wtHTT reduction may be different in patients in the context of mHTT expression, and that the time required for a reduction in wtHtt to have ill effects in primates may be longer than the primate studies reported so far.
What has been learned about huntingtin-lowering in large animal models of HD?
Because of the inherent limitations of rodent models of HD, including but not limited to basal ganglia anatomy, brain size, and lifespan, there has been interest in developing transgenic models of HD in sheep, minipigs, and non-human primates. These larger animals have a longer pubertal age and gestation time, so breeding can be a slow and expensive process, especially in primates. Data from only five male and three female transgenic HD monkeys have been reported to date.64 The three females had a severe motor phenotype necessitating euthanasia within a month of birth.65 A longitudinal study of four males showed deterioration of motor and cognitive skills with age and enlargement of lateral ventricles as seen in the human disease.66 However, the small numbers of these animals has precluded extensive studies of huntingtin-lowering in these transgenic primates.
Conversely, a limitation for the use of transgenic sheep and minipigs so far is that they have been slow in developing a clear disease-related phenotype.67–69 For example, only sparse cortical mHTT aggregates were found in 18-month-old transgenic sheep along with nuclear inclusions in these sheep at 36 months of age.70 At the age of 24 months, transgenic minipigs have modest neuropathological changes, including moderate microglial activation in the caudate and mHTT fragments (but not aggregates) in the cytoplasm of striatal and cortical cells, but remain asymptomatic.71 A recently developed CRISPR/Cas9 knockin pig model may more closely recapitulate HD features including gait abnormalities, enlarged lateral ventricles, and degeneration of striatal medium spiny neurons.72
To date, few studies of huntingtin-lowering in these larger transgenic models of HD have been reported. Pfister et al73 stereotactically injected the striatum of 8- to 14-month-old transgenic sheep expressing full-length human HTT with AAV9 encoding an artificial miRNA targeting the transgene. The treatment reduced human mHTT mRNA and protein by 50%–80% in the striatum at 1 and 6 months post-injection, but not endogenous levels of sheep Htt protein. There was no significant loss of striatal neurons at 6 months after treatment, and Iba1-positive microglia levels were comparable to controls suggesting that safe and sustained silencing of human mHTT protein can be achieved in a large animal’s brain by direct delivery of an AAV carrying an artificial miRNA. Similarly, bilateral administration of AAV5-miHTT in 20- to 39-month-old transgenic minipigs significantly reduced human mHTT mRNA and protein in the putamen (by 47.5% and 53.0%, respectively), caudate nucleus (by 44.2% and 50.5%, respectively), and thalamus (by 72.8% and 53.5%, respectively) 3 months following a single injection into both the putamen and thalamus.74 The AAV5-miHTT treatment did not significantly increase microglial expression or alter DARPP-32 staining of medium spiny neurons. These studies support the translation of AAV-delivered huntingtin-lowering therapies to the clinic, but neither one was informative with regards to the effect of the treatment on any disease phenotype of the animal. Also, since endogenous huntingtin was not targeted by these therapeutic agents, additional studies will be needed to address the tolerability of total huntingtin-lowering in these larger transgenic models of HD.
A summary of animal studies regarding the tolerability of the lowering of wtHTT protein by genetic means vs administration of a huntingtin-targeting agent, at different ages, and by various amounts, is provided in Table 1.
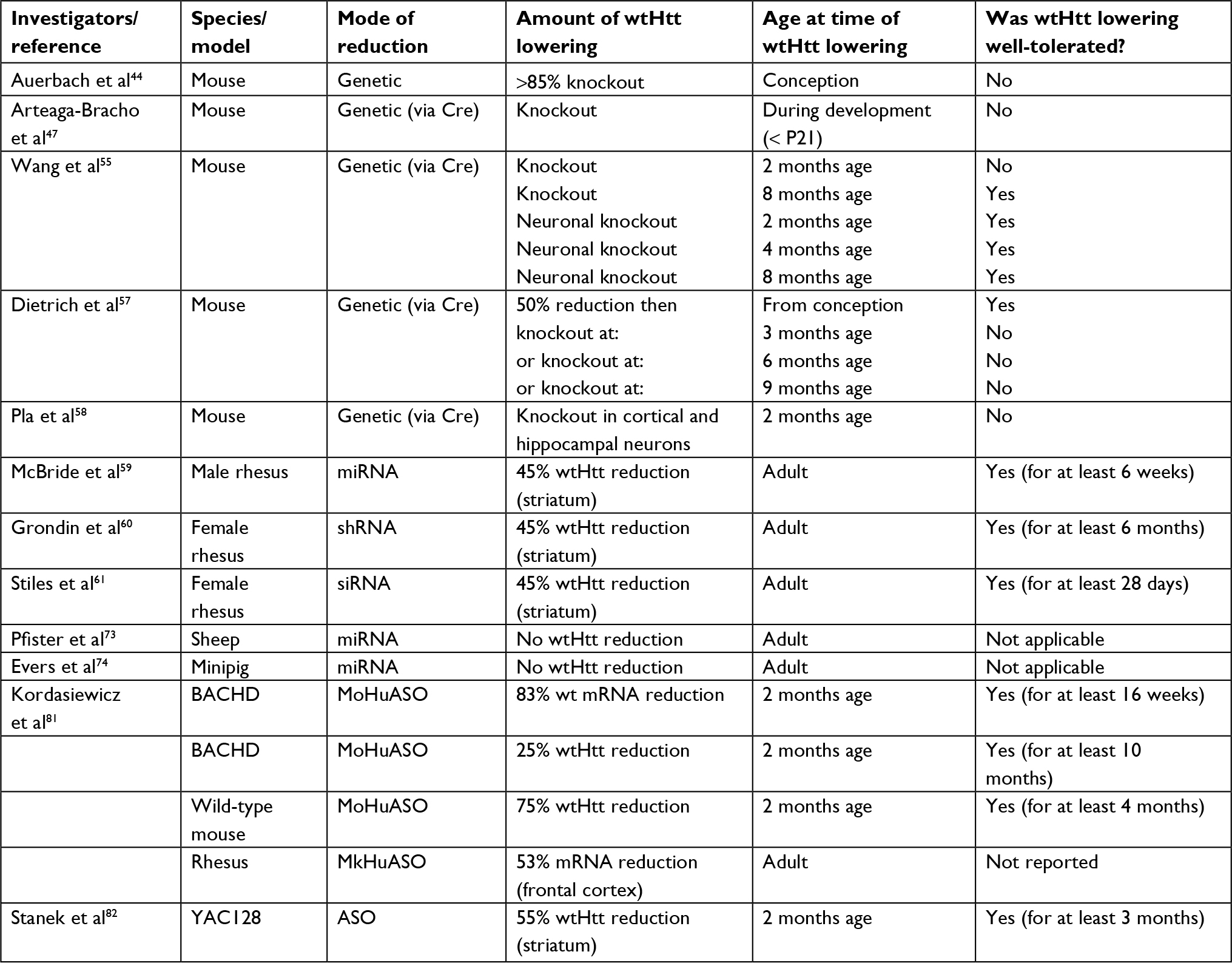 | Table 1 Tolerability studies of huntingtin-lowering in animals Abbreviations: wt, wild type; ASO, antisense oligonucleotide. |
The apparent tolerability of wtHTT-lowering in adult animals, particularly primates, has led many organizations to develop therapies for clinical use to focus on total huntingtin-lowering with non-allele-specific agents. Compared to allele-specific approaches requiring multiple agents and associated diagnostic kits for selecting the appropriate agent for a given patient, a non-allele-specific approach may cost less to develop and be easier to deploy.
Non-allele-specific therapy development efforts include those by Ionis Pharmaceuticals in collaboration with Hoffman-La Roche to develop a huntingtin-lowering therapy using ASOs, as well as efforts by uniQure, Inc., and by Voyager Therapeutics in collaboration with Genzyme to develop AAV-delivered RNAi agents. The Ionis Pharmaceuticals’ first clinical trial of a non-allele-specific huntingtin-lowering agent has been completed. Despite the potential safety concerns that could be raised based on preclinical studies of the function of wtHTT and the effects of non-allele-specific lowering in animals, this trial was successful. What was learned?
What has the first trial of a non-allele-specific HTT-lowering treatment shown in patients?
The Phase I/IIa trial of IONIS-HTTRX (NCT02519036), completed in November 2017, was a randomized, placebo-controlled, dose-escalation trial of the safety and tolerability of IONIS-HTTRX delivered by intrathecal injections via lumbar puncture to patients with early HD.75 Five patient cohorts were enrolled, with treated patients receiving four monthly doses of 10, 30, 60, 90, or 120 mg of ASO per injection, respectively. Patients were followed for 4 months after their last injection. The treatment was found to be safe and well-tolerated at all doses tested, with no serious adverse events in patients receiving the ASOs. Importantly, a dose-dependent reduction in mHTT in the patient’s CSF was observed in CSF samples taken just prior to the drug administration at each month, and again in a follow-up lumbar puncture. The extent of wtHTT-lowering was not determined, because so far there is no assay available for measuring total HTT protein in human CSF. In the highest dose cohort, mHTT was lowered by an average of just under 40% relative to the patient’s baseline level. In a majority of the patients receiving IONIS-HTTRX, the amount of mHTT in the CSF was continuing to decline at the time of the last treatment, suggesting that further monthly treatments could lead to further reduction in mHTT.
However, there are limitations to the conclusions that can be drawn from the trial. As a trial primarily concerned with safety, it involved a small number of patients and a short duration (4 months) of therapy administration. Strictly speaking, the main conclusions afforded by the trial are that the treatment was safe for the duration of the study and follow-up, and the treatment produces a reduction of mHTT in the CSF. However, as a double-blind, placebo-controlled trial in patients, the data from the trial merit considerable weight among the body of evidence about huntingtin-lowering available so far. Potential challenges for this therapy going forward are the long-term tolerability of the monthly intrathecal deliveries (over half of the patients experienced pain and/or post-lumbar puncture syndrome due to the procedure), and patient-to-patient variability in the effect of the treatment delivery with regards to mHTT-lowering. Although the mean lowering of mHTT in the CSF of the patients in the highest dose cohort was ~40% reduction from baseline, the range was extensive, including a paradoxical ~20% increase in mHTT over baseline in one patient. Nevertheless, an average reduction of 40% is encouraging, and Roche is currently planning for a larger and longer pivotal trial to determine the efficacy and continued safety of IONIS-HTTRX (RG6042) in HD patients. All participants who took part in the Phase I/IIa study are continuing to receive RG6042 as part of an “open-label extension” study run by Ionis. This study assesses the safety and tolerability of longer term dosing of RG6042. A randomized, double-blind, Phase III trial to evaluate the efficacy and safety of RG6042 treatment given once per month or once every 2 months (bi-monthly) over a period of 25 months (~2 years) is expected to start enrolling patients in 2019.76
Ionis Pharmaceutical investigators and their partner, Roche, are hypothesizing that a 40% reduction of protein in the CSF is indicative of sufficient huntingtin-lowering in the brain for clinical efficacy, as indicated by their proceeding to invest in further trials aimed at efficacy. What is known from animal models regarding the amount of mutant huntingtin-lowering needed for efficacy?
By how much must mutant huntingtin be lowered for a treatment to be efficacious?
Numerous studies in rodent models of HD support the efficacy of reducing mutant huntingtin mRNA and protein levels in the brain for improving the disease phenotype, whether using ASOs, siRNAs, shRNAs, miRNAs, or other approaches. Partial reversal of disease progression and delayed motor dysfunction was achieved in the severely affected R6/1 transgenic mice by intrastriatal AAV5-mediated delivery of anti-Htt shRNA which lowered levels of mHTT mRNA in the striatum by 78% and protein levels by 28%.77 Also, using AAV to deliver RNAi constructs to the striatum, Boudreau et al78 showed that a 75% reduction of human mHTT and endogenous wild-type mouse Htt mRNA levels was well-tolerated and prevented motor and neuropathological deficits in HDN171-82Q mice. Harper et al79 found that a more modest 51%–55% reduction in mHTT mRNA in these HDN171-82Q mice was also sufficient for phenotypic benefit, resulting in significant improvements in gait deficits and rotarod performance. Along the same lines, lentiviral delivery of inhibitory RNAs in a rat model of HD that conferred just a 35% knockdown of huntingtin gene expression of both mutant and wild-type alleles was shown to be safe and provided both neuroanatomical and behavioral benefits up to to 9 months after injection.80 Using ASOs, Kordasiewicz et al81 report that reduction of the expression of human mutant exon1 mRNA in the R6/2 mouse brain by 43% (±5%) was sufficient to prevent further brain weight loss and significantly extend the lifespan of these severely affected mice. They also reported that reduction in mHTT mRNA and protein levels in 6-month-old YAC128 mice by 58% and 56% (ie, to 42% and 44% of controls, respectively) restored their motor deficits to the performance level of nontransgenic controls.81 Similarly, Stanek et al82 found that reduction of both wild-type and mutant huntingtin by ~40% in YAC128 mice resulted in significant improvements in behavioral deficits, with no notable overt neurotoxicity.
Unfortunately, because the various rodent models differ in the nature of their mutant allele (number of CAGs, full length or exon1 only, transgenic or knockin, etc), the number of wtHTT alleles in their genotype, the level of expression of mHTT, and the severity of their phenotype, any numerical extrapolation from these findings to the human case is fraught with uncertainty even ignoring the species difference between rodents and humans. For example, the YAC128 mouse expresses its full-length 128 CAG-repeat mHTT transgene at about 75% of endogenous mouse Htt levels and manifests a disease phenotype,83 suggesting that a 25% reduction in mHTT mRNA might not be sufficient as a therapy. However, a lowering of mHTT by an intervention may have different effects than the lowering due to a genetic difference. On the other hand, even in a mouse model with a severe phenotype (R6/2), reduction of mRNA expression by 43% extended the animal’s lifespan. Therefore, there is reason to be hopeful that the level of huntingtin-lowering attainable by the ASOs, RNAi agents, and other approaches progressing toward or in clinical trials will be sufficient for therapeutic benefit, if the delivery of the agent to key regions of the brain is adequate.
Timing of huntingtin-lowering: how early to start treatment?
Because HD is a progressive disease and individuals needing treatment can be positively identified, it would seem desirable to start treatment as early in life as can be safely done. However, some animal data suggest a need for caution with regards to huntingtin-lowering treatments. For example, a knockout of wtHTT in mice at 2 months of age (early adulthood) produced acute pancreatitis.55 This may not pose an issue for treatments in which wtHTT is lowered but not eliminated, particularly for treatments delivered directly to the central nervous system (CNS) with minimal “spill over” and persistence systemically. Other studies in mice have found that wtHtt is important for brain development,19,84 and if wtHtt levels have been low during a developmental period, restoration of wtHtt levels at a later age does not compensate for the ill effects of an earlier deficiency.47 These findings suggest that except in the case of juvenile HD (in which the benefit may outweigh the risks), perhaps treatment should not be initiated in people before they are in their early twenties. In most cases, this precaution would still allow treatment to be initiated many years before predicted clinical symptom onset.
Timing of huntingtin-lowering: how long is long enough?
Because HD is a chronic and progressive neurodegenerative disease, it is unrealistic to expect that a short course of huntingtin-lowering treatment will be curative – a successful huntingtin-lowering treatment will be a life-long endeavor. However, preclinical studies of huntingtin-lowering using siRNA or ASOs support the conclusion that the administration of the treatment need not be continuous. The lowering of HTT mRNA in primate brain has been found to persist for weeks beyond the cessation of delivery of “naked” siRNA into the tissue, even though the siRNA itself was not specially synthesized or encapsulated to prevent its degradation.62 More importantly, Kordasiewicz et al81 showed that transient administration of ASOs in HD transgenic mice was able to degrade HTT mRNA and elicit phenotypic improvements that persisted beyond the period of not only ASO administration but also mRNA lowering. This has led to the interpretation that cells can benefit from a “huntingtin holiday”, whereby transient lowering of huntingtin expression enables compensatory mechanisms (yet to be fully delineated) to “catch up” so cells can better handle the re-expressed huntingtin.85 This observation has supported the use of periodic rather than continuous administration of ASOs in the trials of IONIS-HTTRX. In the case of huntingtin-lowering agents that can be delivered using viral vectors (shRNA, miRNA, intrabodies), continuous and perhaps lifelong delivery of the treatment will be a function of the persistence of expression of the delivered transgene. Finally, despite the other hurdles faced by DNA editing approaches (eg, CRISPR-based gene editing), the treatment is expected to be permanent, at least in the cells transduced by the agent.
When both wtHTT and mHTT are lowered, will the net result be beneficial?
Development of therapies involving non-allele-specific huntingtin-lowering is based on the hypothesis that the net result of lowering both wtHTT and mHTT in a patient will be beneficial. If either an allele-specific or a non-allele-specific agent results in lowering of mHTT protein to a greater extent than it lowers wtHTT protein, there is reason to believe this would be beneficial. Becanovic et al86 have studied samples of patients with very early or very late age of disease onset relative to the age of onset expected from their CAG repeat numbers, and identified a SNP in the promoter region of HTT that results in the lowering of expression of the huntingtin protein on the cis-allele. If that allele is the mutant allele, mHTT protein is lowered relative to wtHTT, and this lowering is associated with a delayed age of onset. Conversely, if the SNP variant is on the non-expanded allele, wtHTT protein is lowered relative to mHTT protein, and the lowering is associated with an earlier age of onset.
In a study identifying allele-selective ASOs in a humanized mouse model of HD, Southwell et al87 reported that a non-allele-selective ASO resulted in reduced levels of HTT with maintenance of the basal wild type to mutant protein ratios in all areas of the CNS examined. Over the long term, what if mHTT might accumulate disproportionately to wtHTT due to differences in the efficiency of proteosome degradation of the expanded repeat protein? A study of adult-onset HD brain samples88 found that even though there was a small but significantly lower expression of mutant huntingtin mRNA compared to wild-type mRNA, wild type and mutant protein levels did not differ significantly. In contrast, Liu et al89 reported finding that mutant huntingtin mRNA exceeds wild-type mRNA in post-mortem HD brains. An assay for total HTT in CSF, if available for use in the next human trial of IONIS-HTTRX, will answer the question about proportionate huntingtin-lowering.
The observations that wtHTT stimulates BDNF transcription49 and modulates anxiety and depression-like behaviors in mice,90 among other observations, suggest that loss of wtHTT function could be part of the pathogenesis of the disease. If both mHTT and wtHTT are lowered by an amount sufficient for efficacy, will the net result be beneficial? Studies showing the tolerability of huntingtin-lowering by ~45% in wild-type animals do not address this question, because this result is not in the context of a co-expressed mutant protein. Studies showing benefit of non-allele-specific huntingtin-lowering in mouse models of HD (eg, Boudreau et al78) also do not address this question to the extent that in these models the mutant transgene is overexpressed, or expressed in the presence of two copies of the wild-type murine Hdh. The humanized Hu97/18 mouse expresses full-length human and mutant huntingtin at similar endogenous levels,91 but to our knowledge the net effect of long-term non-allele-specific huntingtin-lowering on the phenotype of this mouse has not been reported.) Induced pluripotent cell lines (iPS cells) derived from HD patients and stably transduced with shRNA non-allele specifically lowering HTT have been generated and are viable, but the benefit of huntingtin-lowering in these cells is unclear.92 Lowering HTT by 50%–60% in these cells has no beneficial effect on the level of p53 protein nor the functioning of the MAP kinase signaling pathway, two aspects of the abnormal phenotype of these cells. Conversely, Trager et al93 found that lowering HTT by ~50% restores normal cytokine function in patient-derived monocytes. However, it remains unknown as to whether non-allele-specific huntingtin-lowering by ~50% will restore cells of the CNS to normalcy.
Summary and conclusion
This review supports several conclusions about huntingtin-lowering. It is clear that it is possible to lower huntingtin in vivo in rodents, sheep, pigs, and non-human primates using agents that could be administered to humans, such as shRNA, miRNA, ASOs, zinc fingers, and CRISPR/Cas9 gene editing agents. With some agents, it is possible to lower mutant huntingtin in an allele-selective manner. The lowering of mutant huntingtin by various means is efficacious at improving the abnormal phenotype in many rodent models of HD. In rodents, genetic reduction of wtHTT by 50% or more adversely affects the developing brain, and near elimination of wild-type huntingtin is detrimental even if not initiated until adulthood. Lowering wild-type huntingtin by ~45% in the adult non-human primate striatum is well-tolerated for at least 6 months. Four months of periodic administration of a non-allele-specific ASO that lowers the level of mutant huntingtin detected in the CSF is safe and well-tolerated in patients, and longer term studies of the tolerability of the same ASO in primates are reportedly underway. In mice, some adverse effects of wtHTT-lowering (eg, thalamic calcification) take 9 months to a year to emerge, indicating that long-term preclinical studies of the safety of huntingtin-lowering are advisable, even as human trials are proceeding. It is notable that the adverse effects in mice are the result of huntingtin-lowering to the point of near elimination, rather than the more moderate lowering typically achieved with clinically relevant agents. Collectively, the evidence indicates that the potential adverse effects of wild-type huntingtin-lowering are age, amount, and perhaps brain region-specific (to the extent that near elimination of Htt in wild-type mice eventually resulted in calcification, specifically in the thalamus).57 For now, what can be concluded is that for safety, huntingtin-lowering in the human disease should be either limited in the amount by which huntingtin is lowered (eg, preserving about 50% of wild-type expression), limited to the most affected regions of the brain (ie, the striatum and cortex, and probably avoiding in particular the thalamus), or allele-specific, or all of the above. This statement is dissatisfying relative to the desire for parsimony in the interpretation of evidence, but it is not unrealistic given that the disease itself is age, region, and dose (CAG repeat number) specific.
Other important questions about huntingtin-lowering remain open. Non-allele-specific lowering of huntingtin in patients has been found to be safe, but whether it will be safe over the long term remains unknown. Also, it remains to be seen whether IONIS-HTTRX given via a lumbar route of administration penetrates not only the cortex but also the caudate nucleus and putamen of patients sufficiently for clinical efficacy. It is possible that huntingtin-lowering only in the cortex could have some benefit – in rodents, selective expression of mHTT only in striatal neurons produces cell-autonomous deficits in striatal electrophysiology, but spares the animal locomotor deficits and striatal degeneration.94 However, HD is a multi-system disease.95 Huntingtin-lowering in at least both the cortex and the striatum may be necessary in the human disease. It remains to be seen whether miRNA against huntingtin delivered by AAV5 to the striatum (as planned by uniQure) will be transported retrogradely to the cortex sufficiently to lower huntingtin in layer V cortical neurons projecting to the striatum, and whether other serotypes of AAV can deliver miRNA against huntingtin across the blood–brain barrier sufficiently for a systemic route of delivery to be feasible for clinical use in patients. Answers to most of these questions will only be provided by clinical trials over the next several years.
What gaps in our knowledge about huntingtin-lowering should be pursued by preclinical research in the meantime? A fundamental question is whether lowering wild-type huntingtin in layer V cortical neurons in particular (not just generally in cortical tissue) and thus the corticostriatal connections to the striatum will be tolerated in the primate brain. Studies in BACHD mice96,97 have indicated that huntingtin-lowering in both cortical neurons and the striatum is likely to be necessary for optimal treatment efficacy. However, other studies have shown that wtHTT functions to promote the production of BDNF,49 and transport of BDNF to the striatum is slowed53 when wtHTT is lowered. So far, studies in non-human primates showing that lowering of wtHTT is well-tolerated have not measured and demonstrated huntingtin-lowering specifically in layer V cortical neurons whose efferents form the corticostriatal tract. Studies using treatment delivery methods that achieve simultaneous lowering of huntingtin in both the striatum and the cortical neurons projecting to it are needed to establish whether reducing wtHTT by about 40%–50% in the corticostriatal pathway has adverse effects on BDNF levels or otherwise adversely effects the functioning of striatal neurons. If so, this would indicate that an allele-specific approach will be necessary.
To our knowledge, the use of patient-derived embryonic stem cells or iPS cells to address the question of whether lowering of both wild-type and mutant huntingtin results in a net benefit at the cellular level has not been fully exploited, particularly in cells differentiated to a phenotype resembling cortical neurons or medium spiny neurons of the striatum. Also, so far, huntingtin-lowering studies in non-human primates have not been long enough or inclusive of the thalamus enough to cover another knowledge gap – whether lowering of wtHTT by a clinically relevant amount in the thalamus of the primate brain will eventually result in thalamic calcification, as seen in mice.57
Although many open questions and hurdles remain, much progress is being made toward a huntingtin-lowering therapy for HD. Scientists, clinicians, patients, and their families can look forward with interest to further developments in this field.
Acknowledgments
The authors wish to thank Douglas Macdonald, PhD, Director, Research Operations and Scientific Alliances, CHDI Foundation, for his helpful reviews of the manuscript. However, any misinterpretations or errors remain the responsibility of the authors.
Disclosure
Neither author has any financial, advisory, investment or other relationship with any of the companies mentioned in this article, nor with any other company developing a therapy for HD. WFK is a member of the Scientific Advisory Board of Alcyone Lifesciences, Inc., which makes catheters for the delivery of therapies to the brain via neurosurgery and to the CSF via lumbar puncture, and a co-founder of the CGTA Research Group. The latter organization is a 501(c)3 not-for-profit public foundation dedicated to facilitating the development of treatments for rare diseases affecting the CNS. The authors report no other conflicts of interest in this work.
References
Brinkman RR, Mezei MM, Theilmann J, Almqvist E, Hayden MR. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am J Hum Genet. 1997;60(5):1202–1210. | ||
Kremer B, Goldberg P, Andrew SE, et al. A worldwide study of the Huntington’s disease mutation. The sensitivity and specificity of measuring CAG repeats. N Engl J Med. 1994;330(20):1401–1406. | ||
Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell. 2015;162(3):516–526. | ||
Long JD, Lee JM, Aylward EH, et al. Genetic modification of Huntington disease acts early in the prediagnosis phase. Am J Hum Genet. 2018;103(3):349–357. | ||
Jimenez-Sanchez M, Licitra F, Underwood BR, Rubinsztein DC. Huntington’s disease: mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb Perspect Med. 2017;7(7):a024240. | ||
Duyao MP, Auerbach AB, Ryan A, et al. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science. 1995;269(5222):407–410. | ||
Nasir J, Floresco SB, O’Kusky JR, et al. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81(5):811–823. | ||
Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat Genet. 1995;11(2):155–163. | ||
Ho LW, Brown R, Maxwell M, Wyttenbach A, Rubinsztein DC. Wild type huntingtin reduces the cellular toxicity of mutant huntingtin in mammalian cell models of Huntington’s disease. J Med Genet. 2001;38(7):450–452. | ||
Leavitt BR, Guttman JA, Hodgson JG, et al. Wild-type huntingtin reduces the cellular toxicity of mutant huntingtin in vivo. Am J Hum Genet. 2001;68(2):313–324. | ||
Leavitt BR, van Raamsdonk JM, Shehadeh J, et al. Wild-type huntingtin protects neurons from excitotoxicity. J Neurochem. 2006;96(4):1121–1129. | ||
van Raamsdonk JM, Pearson J, Murphy Z, Hayden MR, Leavitt BR. Wild-type huntingtin ameliorates striatal neuronal atrophy but does not prevent other abnormalities in the YAC128 mouse model of Huntington disease. BMC Neurosci. 2006;7(1):80. | ||
Bañez-Coronel M, Ayhan F, Tarabochia AD, et al. RAN translation in Huntington disease. Neuron. 2015;88(4):667–677. | ||
Rué L, Bañez-Coronel M, Creus-Muncunill J, et al. Targeting CAG repeat RNAs reduces Huntington’s disease phenotype independently of huntingtin levels. J Clin Invest. 2016;126(11):4319–4330. | ||
Lindow M, Vornlocher HP, Riley D, et al. Assessing unintended hybridization-induced biological effects of oligonucleotides. Nat Biotechnol. 2012;30(10):920–923. | ||
Hagedorn PH, Yakimov V, Ottosen S, et al. Hepatotoxic potential of therapeutic oligonucleotides can be predicted from their sequence and modification pattern. Nucleic Acid Ther. 2013;23(5):302–310. | ||
Kamola PJ, Maratou K, Wilson PA, et al. Strategies for in vivo screening and mitigation of hepatotoxicity associated with antisense drugs. Mol Ther Nucleic Acids. 2017;8:383–394. | ||
Wild EJ, Tabrizi SJ. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017;16(10):837–847. | ||
Reiner A, Dragatsis I, Zeitlin S, Goldowitz D. Wild-type huntingtin plays a role in brain development and neuronal survival. Mol Neurobiol. 2003;28(3):259–276. | ||
van Bilsen PH, Jaspers L, Lombardi MS, Odekerken JC, Burright EN, Kaemmerer WF. Identification and allele-specific silencing of the mutant huntingtin allele in Huntington’s disease patient-derived fibroblasts. Hum Gene Ther. 2008;19(7):710–718. | ||
Zhang Y, Engelman J, Friedlander RM. Allele-specific silencing of mutant Huntington’s disease gene. J Neurochem. 2009;108(1):82–90. | ||
Carroll JB, Warby SC, Southwell AL, et al. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/allele-specific silencing of mutant huntingtin. Mol Ther. 2011;19(12):2178–2185. | ||
Lombardi MS, Jaspers L, Spronkmans C, et al. A majority of Huntington’s disease patients may be treatable by individualized allele-specific RNA interference. Exp Neurol. 2009;217(2):312–319. | ||
Pfister EL, Kennington L, Straubhaar J, et al. Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington’s disease patients. Curr Biol. 2009;19(9):774–778. | ||
Gagnon KT, Pendergraff HM, Deleavey GF, et al. Allele-selective inhibition of mutant Huntingtin expression with antisense oligonucleotides targeting the expanded CAG repeat. Biochemistry. 2010;49(47):10166–10178. | ||
Yu D, Pendergraff H, Liu J, et al. Single-stranded RNAs use RNAi to potently and allele-selectively inhibit mutant huntingtin expression. Cell. 2012;150(5):895–908. | ||
Klug A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu Rev Biochem. 2010;79(1):213–231. | ||
Zhang HS, Zeitler B, Froelich S. Engineered zinc finger transcriptional repressors selectively inhibit mutant huntingtin expression and reverse disease phenotypes in Huntington’s disease patient-derived neurons and in rodent models. In: Society for Neuroscience Annual Conference; 2014; Washington, DC. November 15-19, 2014. | ||
Garriga-Canut M, Agustin-Pavon C, Herrmann F, et al. Synthetic zinc finger repressors reduce mutant huntingtin expression in the brain of R6/2 mice. Proc Natl Acad Sci U S A. 2012;109(45):E3136–E3145. | ||
Monteys AM, Ebanks SA, Keiser MS, Davidson BL. CRISPR/Cas9 editing of the mutant huntingtin allele in vitro and in vivo. Mol Ther. 2017;25(1):12–23. | ||
Shin JW, Kim KH, Chao MJ, et al. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum Mol Genet. 2016;25(20):4566–4576. | ||
Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol. 2018;36(8):765–771. | ||
Dabrowska M, Juzwa W, Krzyzosiak WJ, Olejniczak M. Precise excision of the CAG tract from the huntingtin gene by Cas9 Nickases. Front Neurosci. 2018;12:75. | ||
Rigamonti D, Bauer JH, de-Fraja C, et al. Wild-type huntingtin protects from apoptosis upstream of caspase-3. J Neurosci. 2000;20(10):3705–3713. | ||
Anne SL, Saudou F, Humbert S. Phosphorylation of huntingtin by cyclin-dependent kinase 5 is induced by DNA damage and regulates wild-type and mutant huntingtin toxicity in neurons. J Neurosci. 2007;27(27):7318–7328. | ||
Culver BP, Savas JN, Park SK, et al. Proteomic analysis of wild-type and mutant huntingtin-associated proteins in mouse brains identifies unique interactions and involvement in protein synthesis. J Biol Chem. 2012;287(26):21599–21614. | ||
Colin E, Zala D, Liot G, et al. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008;27(15):2124–2134. | ||
Strehlow AN, Li JZ, Myers RM. Wild-type huntingtin participates in protein trafficking between the Golgi and the extracellular space. Hum Mol Genet. 2007;16(4):391–409. | ||
Velier J, Kim M, Schwarz C, et al. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp Neurol. 1998;152(1):34–40. | ||
Brandstaetter H, Kruppa AJ, Buss F. Huntingtin is required for ER-to-Golgi transport and for secretory vesicle fusion at the plasma membrane. Dis Model Mech. 2014;7(12):1335–1340. | ||
Caviston JP, Ross JL, Antony SM, Tokito M, Holzbaur ELF. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc Natl Acad Sci U S A. 2007;104(24):10045–10050. | ||
Caviston JP, Holzbaur EL. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 2009;19(4):147–155. | ||
Liu JP, Zeitlin SO. Is huntingtin dispensable in the adult brain? J Huntingtons Dis. 2017;6(1):1–17. | ||
Auerbach W, Hurlbert MS, Hilditch-Maguire P, et al. The HD mutation causes progressive lethal neurological disease in mice expressing reduced levels of huntingtin. Hum Mol Genet. 2001;10(22):2515–2523. | ||
Barnat M, Le Friec J, Benstaali C, Humbert S. Huntingtin-mediated multipolar-bipolar transition of newborn cortical neurons is critical for their postnatal neuronal morphology. Neuron. 2017;93(1):99–114. | ||
McKinstry SU, Karadeniz YB, Worthington AK, et al. Huntingtin is required for normal excitatory synapse development in cortical and striatal circuits. J Neurosci. 2014;34(28):9455–9472. | ||
Arteaga-Bracho EE, Gulinello M, Winchester ML, et al. Postnatal and adult consequences of loss of huntingtin during development: implications for Huntington’s disease. Neurobiol Dis. 2016;96:144–155. | ||
Ma B, Culver BP, Baj G, Tongiorgi E, Chao MV, Tanese N. Localization of BDNF mRNA with the Huntington’s disease protein in rat brain. Mol Neurodegener. 2010;5(1):22. | ||
Zuccato C, Tartari M, Crotti A, et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35(1):76–83. | ||
Shimojo M. Huntingtin regulates RE1-silencing transcription factor/neuron-restrictive silencer factor (REST/NRSF) nuclear trafficking indirectly through a complex with REST/NRSF-interacting LIM domain protein (RILP) and dynactin p150 Glued. J Biol Chem. 2008;283(50):34880–34886. | ||
Zuccato C, Ciammola A, Rigamonti D, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293(5529):493–498. | ||
Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci. 2004;24(17):4250–4258. | ||
Gauthier LR, Charrin BC, Borrell-Pagès M, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118(1):127–138. | ||
Her LS, Goldstein LS. Enhanced sensitivity of striatal neurons to axonal transport defects induced by mutant huntingtin. J Neurosci. 2008;28(50):13662–13672. | ||
Wang G, Liu X, Gaertig MA, Li S, Li XJ. Ablation of huntingtin in adult neurons is nondeleterious but its depletion in young mice causes acute pancreatitis. Proc Natl Acad Sci U S A. 2016;113(12):3359–3364. | ||
Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26(3):300–306. | ||
Dietrich P, Johnson IM, Alli S, Dragatsis I. Elimination of huntingtin in the adult mouse leads to progressive behavioral deficits, bilateral thalamic calcification, and altered brain iron homeostasis. PLoS Genet. 2017;13(7):e1006846. | ||
Pla P, Orvoen S, Benstaali C, et al. Huntingtin acts non cell-autonomously on hippocampal neurogenesis and controls anxiety-related behaviors in adult mouse. PLoS One. 2013;8(9):e73902. | ||
McBride JL, Pitzer MR, Boudreau RL, et al. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol Ther. 2011;19(12):2152–2162. | ||
Grondin R, Kaytor MD, Ai Y, et al. Six-month partial suppression of huntingtin is well tolerated in the adult rhesus striatum. Brain. 2012;135(Pt 4):1197–1209. | ||
Stiles DK, Zhang Z, Ge P, et al. Widespread suppression of huntingtin with convection-enhanced delivery of siRNA. Exp Neurol. 2012;233(1):463–471. | ||
Grondin R, Ge P, Chen Q, et al. Onset time and durability of huntingtin suppression in rhesus putamen after direct infusion of antihuntingtin siRNA. Mol Ther Nucleic Acids. 2015;4:e245. | ||
Ambrose CM, Duyao MP, Barnes G, et al. Structure and expression of the Huntington’s disease gene: evidence against simple inactivation due to an expanded CAG repeat. Somat Cell Mol Genet. 1994;20(1):27–38. | ||
Snyder BR, Chan AWS. Progress in developing transgenic monkey model for Huntington’s disease. J Neural Transm (Vienna). 2018;125(3):401–417. | ||
Yang SH, Cheng PH, Banta H, et al. Towards a transgenic model of Huntington’s disease in a non-human primate. Nature. 2008;453(7197):921–924. | ||
Chan AW, Jiang J, Chen Y, et al. Progressive cognitive deficit, motor impairment and striatal pathology in a transgenic Huntington disease monkey model from infancy to adulthood. PLoS One. 2015;10(5):e0122335. | ||
Jacobsen JC, Bawden CS, Rudiger SR, et al. An ovine transgenic Huntington’s disease model. Hum Mol Genet. 2010;19(10):1873–1882. | ||
Baxa M, Hruska-Plochan M, Juhas S, et al. A transgenic minipig model of Huntington’s disease. J Huntingtons Dis. 2013;2(1):47–68. | ||
Schuldenzucker V, Schubert R, Muratori LM, et al. Behavioral testing of minipigs transgenic for the Huntington gene – a three-year observational study. PLoS One. 2017;12(10):e0185970. | ||
Huntington’s Disease Sheep Collaborative Research Group, Reid SJ, Patassini S, et al. Further molecular characterisation of the OVT73 transgenic sheep model of Huntington’s disease identifies cortical aggregates. J Huntingtons Dis. 2013;2(3):279–295. | ||
Vidinská D, Vochozková P, Šmatlíková P, et al. Gradual phenotype development in Huntington disease transgenic minipig model at 24 months of age. Neurodegener Dis. 2018;18(2–3):107–119. | ||
Yan S, Tu Z, Liu Z, et al. A huntingtin knockin pig model recapitulates features of selective neurodegeneration in Huntington’s disease. Cell. 2018;173(4):989–1002. | ||
Pfister EL, DiNardo N, Mondo E, et al. Artificial miRNAs reduce human mutant huntingtin throughout the striatum in a transgenic sheep model of Huntington’s disease. Hum Gene Ther. 2018;29(6):663–673. | ||
Evers MM, Miniarikova J, Juhas S, et al. AAV5-miHTT gene therapy demonstrates broad distribution and strong human mutant huntingtin lowering in a Huntington’s disease minipig model. Mol Ther. 2018;26(9):2163–2177. | ||
Smith A, Tabrizi S. Development of IONIS-HTTrx from first principles to the first successful huntingtin-lowering drug trial. Paper presented at: CHDI 13th Annual HD Therapeutics Conference; March 1, 2018; Palm Springs, CA. This presentation may be viewed in its entireity at https://chdifoundation.org/2018-conference/. | ||
Roche HD Team. Update on RG6042 Huntington’s disease global development programme. Paper presented at: European Huntington’s Disease Network Plenary Meeting; 2018; Vienna, Austria. September 14-16, 2018. | ||
Rodriguez-Lebron E, Denovan-Wright EM, Nash K, Lewin AS, Mandel RJ. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington’s disease transgenic mice. Mol Ther. 2005;12(4):618–633. | ||
Boudreau RL, McBride JL, Martins I, et al. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol Ther. 2009;17(6):1053–1063. | ||
Harper SQ, Staber PD, He X, et al. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc Natl Acad Sci U S A. 2005;102(16):5820–5825. | ||
Drouet V, Perrin V, Hassig R, et al. Sustained effects of nonallele-specific Huntingtin silencing. Ann Neurol. 2009;65(3):276–285. | ||
Kordasiewicz HB, Stanek LM, Wancewicz EV, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. 2012;74(6):1031–1044. | ||
Stanek LM, Sardi SP, Mastis B, et al. Silencing mutant Huntingtin by adeno-associated virus-mediated RNA interference ameliorates disease manifestations in the YAC128 mouse model of Huntington’s disease. Hum Gene Ther. 2014;25(5):461–474. | ||
Slow EJ, van Raamsdonk J, Rogers D, et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum Mol Genet. 2003;12(13):1555–1567. | ||
Tong Y, Ha TJ, Liu L, Nishimoto A, Reiner A, Goldowitz D. Spatial and temporal requirements for huntingtin (Htt) in neuronal migration and survival during brain development. J Neurosci. 2011;31(41):14794–14799. | ||
Lu XH, Yang XW. “Huntingtin holiday”: progress toward an antisense therapy for Huntington’s disease. Neuron. 2012;74(6):964–966. | ||
Bečanović K, Nørremølle A, Neal SJ, et al. A SNP in the HTT promoter alters NF-κB binding and is a bidirectional genetic modifier of Huntington disease. Nat Neurosci. 2015;18(6):807–816. | ||
Southwell AL, Skotte NH, Kordasiewicz HB, et al. In vivo evaluation of candidate allele-specific mutant huntingtin gene silencing antisense oligonucleotides. Mol Ther. 2014;22(12):2093–2106. | ||
Evers MM, Schut MH, Pepers BA, et al. Making (anti-) sense out of huntingtin levels in Huntington disease. Mol Neurodegener. 2015;10(1):21. | ||
Liu W, Chaurette J, Pfister EL, et al. Increased steady-state mutant huntingtin mRNA in Huntington’s disease brain. J Huntingtons Dis. 2013;2(4):491–500. | ||
Ben M’Barek K, Pla P, Orvoen S, et al. Huntingtin mediates anxiety/depression-related behaviors and hippocampal neurogenesis. J Neurosci. 2013;33(20):8608–8620. | ||
Southwell AL, Warby SC, Carroll JB, et al. A fully humanized transgenic mouse model of Huntington disease. Hum Mol Genet. 2013;22(1):18–34. | ||
Szlachcic WJ, Wiatr K, Trzeciak M, Figlerowicz M, Figiel M. The generation of mouse and human huntington disease iPS cells suitable for in vitro studies on huntingtin function. Front Mol Neurosci. 2017; 10:253. | ||
Träger U, Andre R, Lahiri N, et al. HTT-lowering reverses Huntington’s disease immune dysfunction caused by NFκB pathway dysregulation. Brain. 2014;137(3):819–833. | ||
Gu X, André VM, Cepeda C, et al. Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington’s disease. Mol Neurodegener. 2007;2(1):8. | ||
Rüb U, Seidel K, Heinsen H, Vonsattel JP, den Dunnen WF, Korf HW. Huntington’s disease (HD): the neuropathology of a multisystem neurodegenerative disorder of the human brain. Brain Pathol. 2016;26(6):726–740. | ||
Wang N, Gray M, Lu XH, et al. Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington’s disease. Nat Med. 2014;20(5):536–541. | ||
Estrada-Sánchez AM, Burroughs CL, Cavaliere S, et al. Cortical efferents lacking mutant huntingtin improve striatal neuronal activity and behavior in a conditional mouse model of Huntington’s disease. J Neurosci. 2015;35(10):4440–4451. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.