Back to Journals » Lung Cancer: Targets and Therapy » Volume 11
The Current Understanding Of Asbestos-Induced Epigenetic Changes Associated With Lung Cancer
Authors Cheng YY, Rath EM ![]() , Linton A, Yuen ML, Takahashi K
, Linton A, Yuen ML, Takahashi K ![]() , Lee K
, Lee K
Received 7 August 2019
Accepted for publication 8 November 2019
Published 8 January 2020 Volume 2020:11 Pages 1—11
DOI https://doi.org/10.2147/LCTT.S186843
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sai-Hong Ou
Yuen Yee Cheng, 1, 2 Emma M Rath, 3 Anthony Linton, 1, 2, 4 Man Lee Yuen, 1 Ken Takahashi, 1 Kenneth Lee 1, 2, 4
1Asbestos Disease Research Institute, Sydney Medical School, University of Sydney, Sydney, New South Wales, Australia; 2Sydney Medical School, The University of Sydney, Sydney, New South Wales, Australia; 3Garvan Institute of Medical Research, Sydney, New South Wales, Australia; 4Concord Repatriation General Hospital, Sydney, New South Wales, Australia
Correspondence: Yuen Yee Cheng
Asbestos Disease Research Institute, PO Box 3628, Rhodes, New South Wales 2139, Australia
Tel +61 2 9767 9800
Fax +61 2 9767 9860
Email [email protected]
Abstract: Asbestos is a naturally occurring mineral consisting of extremely fine fibres that can become trapped in the lungs after inhalation. Occupational and environmental exposures to asbestos are linked to development of lung cancer and malignant mesothelioma, a cancer of the lining surrounding the lung. This review discusses the factors that are making asbestos-induced lung cancer a continuing problem, including the extensive historic use of asbestos and decades long latency between exposure and disease development. Genomic mutations of DNA nucleotides and gene rearrangements driving lung cancer are well-studied, with biomarkers and targeted therapies already in clinical use for some of these mutations. The genes involved in these mutation biomarkers and targeted therapies are also involved in epigenetic mechanisms and are discussed in this review as it is hoped that identification of epigenetic aberrations in these genes will enable the same gene biomarkers and targeted therapies to be used. Currently, understanding of how asbestos fibres trapped in the lungs leads to epigenetic changes and lung cancer is incomplete. It has been shown that oxidoreduction reactions on fibre surfaces generate reactive oxygen species (ROS) which in turn damage DNA, leading to genetic and epigenetic alterations that reduce the activity of tumour suppressor genes. Epigenetic DNA methylation changes associated with lung cancer are summarised in this review, and some of these changes will be due to asbestos exposure. So far, little research has been carried out to separate the asbestos driven epigenetic changes from those due to non-asbestos causes of lung cancer. Asbestos-associated lung cancers exhibit less methylation variability than lung cancers in general, and in a large proportion of samples variability has been found to be restricted to promoter regions. Epigenetic aberrations in cancer are proving to be promising biomarkers for diagnosing cancers. It is hoped that further understanding of epigenetic changes in lung cancer can result in useful asbestos-associated lung cancer biomarkers to guide treatment. Research is ongoing into the detection of lung cancer epigenetic alterations using non-invasive samples of blood and sputum. These efforts hold the promise of non-invasive cancer diagnosis in the future. Efforts to reverse epigenetic aberrations in lung cancer by epigenetic therapies are ongoing but have not yet yielded success.
Keywords: lung cancer, epigenetic biomarkers, microRNA, DNA methylation, immunohistochemistry, IHC, fluorescence in situ hybridization, FISH
Asbestos And Lung Cancer
Asbestos is a group of naturally occurring fibrous silicate minerals with multiple commercial applications. Asbestos material is resistant to heat and corrosion. Its natural fibrous nature enables it to be woven into cloth, incorporated into cement materials, ceiling tiles, break and clutch linings, flooring, resins, polymers, and filter papers. In the 19th and 20th centuries, asbestos was used in a large number of industries with minimal control of exposure. From 1950 to 1985, it was extensively used in construction and ship building for insulation and fire protection, and as material for anti-friction and filtering. It is estimated that one in every three houses in Australia built before 1990 contains asbestos,1 putting Australians at risk of exposure.
Lung cancers are aggressive respiratory tumours with poor survival.2 While tobacco smoking remains the principal cause for lung cancers, exposure to asbestos is the most important occupational risk factor for these cancers.3 Asbestos exposure causes 6 to 23% of all male lung cancers (estimates depend on the exposure and population),4,5 and >107,000 deaths annually in Europe from asbestos‐related diseases. In 2018, there were 12,741 reported lung cancer deaths in Australia, which is equivalent to 500 deaths for every one million people, placing lung cancer in the top five deadly cancers.6 All forms of asbestos are carcinogenic to humans.4 It is estimated that asbestos exposure causes six times more lung cancer than malignant mesothelioma,7 and mesothelioma deaths are estimated to be 38,400 per year worldwide.8 Research has linked asbestos exposures to development of lung cancer and malignant mesothelioma,9 and has shown the potential for asbestos to induce formation of reactive oxygen species (ROS) that cause damage of DNA and alterations that reduce the activity of tumour suppressor genes.10 Given that the latency of cancer development may be as long as 30–40 years after asbestos exposure and the fact that demolition of asbestos‐containing buildings is common, asbestos exposure will continue to inflict substantial disease morbidity and mortality in future years.4,11 Although the link between asbestos and lung cancer risk is well‐established, asbestos‐associated neoplasms have proven difficult to diagnose early and treat successfully. As with other cancers, most asbestos-related lung cancers are diagnosed at late stages of disease, underscoring the need for better understanding of the molecular mechanisms of these diseases, and for identification of critical gene targets for new diagnostic biomarkers and therapies.
When a person inhales airborne asbestos particles, the asbestos fibres can become lodged in their lung tissue.12,13 Exposure to asbestos puts people at risk of developing cancers in the lungs (lung cancer),12–15 pleural and peritoneal lining (pleural and peritoneal mesothelioma),12,14–16 larynx,17 and ovaries.18 All four major histological types of lung cancers (squamous, adeno, large-cell, and small cell) can be caused by asbestos exposure.14 Almost all mesotheliomas are caused by asbestos exposure.16 Over many years, asbestos fibres can cause genetic, epigenetic, and cellular damage, causing lung cells to become malignant. For these cancers, there is a long latency period between exposure and development of symptoms. Lung cancer caused by asbestos is different from pleural malignant mesothelioma, with the former developing inside the lung and the latter in the outer lining of the lung.
Biomarkers Used In Lung Cancers To Choose Treatment (Table 1)
The identification of genetic alterations in lung cancers allows clinicians to select optimal treatments tailored to an individual’s pathology. Replacing the generalised approach, the use of agents targeting specific driver gene mutations or overexpression has served to increase therapeutic effect and overall survival, often with a more tolerable side effect profile than traditional chemotherapy.
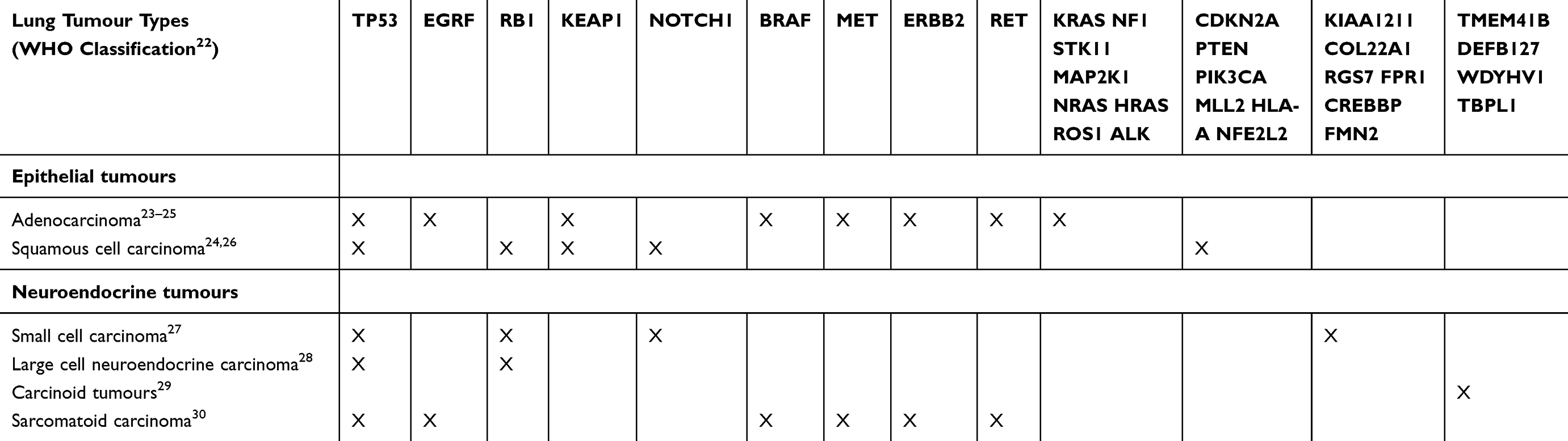 |
Table 1 Genes Frequently Possessing Alterations In Lung Cancer |
Genes frequently mutated in lung cancer and thus used as biomarkers are summarised in Table 1. Amongst these biomarkers, alterations in EGFR, ALK, ROS1 and BRAF genes are currently the most relevant in clinical practice with clinically effective specific targeted treatments available.19
EGFR mutations for example, present in approximately 15% of patients with adenocarcinoma in Western countries and over 50% in some Asian populations,20 have been managed with first generation EGFR tyrosine kinase inhibitors (TKIs) (e.g. gefinitib or erlotinib) with demonstrable superiority over systemic first line chemotherapy.21 These agents traditionally proved ineffective in tumours with T790M EGFR mutations, though third generation agents (e.g. osimertinib) have proven capable of not only overcoming the development of this resistance mutation, but offer improved progression free survival over first generation agents in treatment naïve EGFR mutation positive populations, with or without a T790M mutation.
Additional driver mutations are currently under investigation with preliminary evidence of treatment efficacy when targeted therapies are utilised, including HER2 mutations, MET14 mutation or amplification, and RET rearrangements, whilst RAS and PIK3CA mutations may confer worse prognosis and response to other therapies (e.g. EGFR TKIs in EGFR-mutant non-small cell lung cancer (NSCLC)).
Table 2 summaries other relevant treatment options indicated by biomarker alterations. Most of these biomarkers are nucleotide mutations and gene rearrangements rather than epigenetic alterations. It is hoped that future characterisation of epigenetic alterations will increase the number of targetable individual cancers for which a potentially beneficial treatment can be identified.
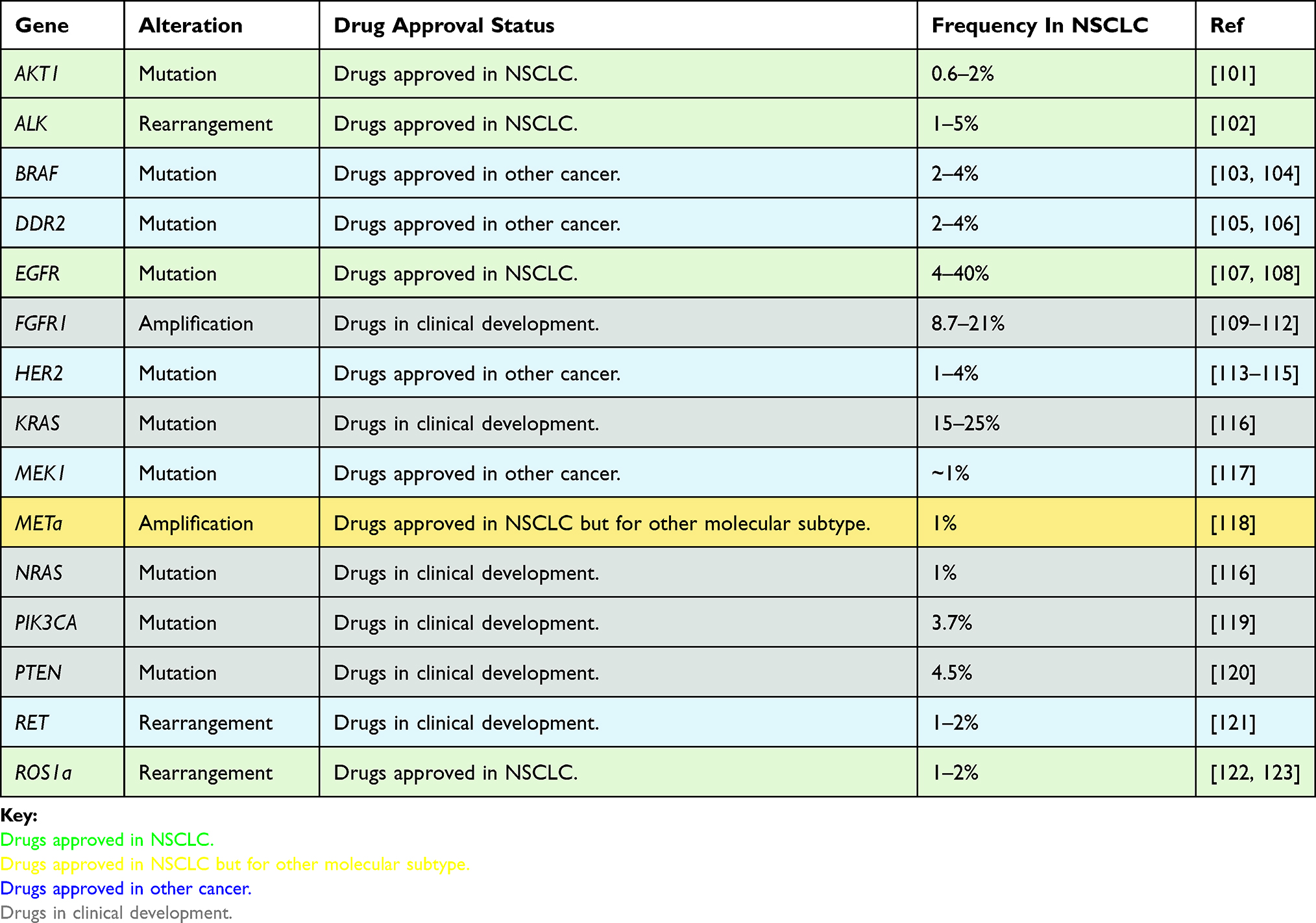 |
Table 2 Gene alteration and drug approval status in NSCLC |
Asbestos-Induced Epigenetic Changes (Figure 1)
How asbestos induces epigenetic changes is not fully understood. Several studies have shown both clastogenic and cytotoxic responses of cells to asbestos fibres.32,33 Phagocytosis of fibres by macrophages and oxidoreduction reactions on fibre surfaces are known to generate genotoxic ROS that result in DNA damage and oxidative stress, leading to genetic alterations in the cells.34–36 When asbestos interacts with human cells, asbestos silicates attract and bind cations, and in the lungs, asbestos fibres will both retain the ions on the fibre surface and leach them into the cellular milieu.37 These processes can generate reactive oxygen species and free radicals that initiate the processes of cellular and DNA damage and genotoxicity.38,39 The high iron content of some asbestos fibres, as well as the propensity for asbestos to adsorb iron in vivo, have led to the suggestion that iron-induced Fenton reactions also contribute to increased ROS, inflammation, and carcinogenesis.40 Chrysotile and crocidolite types of asbestos were shown to induce oxidative stress and induce local inflammatory mediators (cytokines and growth factors), leading to a reactive microenvironment of inflammation and proliferation of cells.41,42 Exposure of cells to asbestos induces extensive alterations in expression of genes involved in integrin-mediated signalling, DNA damage repair, and cell cycle regulation pathways.43,44 The chronic inflammation caused by exposure of serosal surfaces to asbestos fibres is likely to represent a central factor in the carcinogenesis and is likely to be mediated through epigenetic changes.45
Many studies have investigated microRNAs associated with asbestos induced epigenetic alteration in mesothelioma (a highly specific cancer induced by asbestos exposure).46–48 However, there has been only limited exploration in asbestos-related lung cancers, including adenocarcinoma, adenosquamous carcinoma, small cell lung cancer and large cell lung cancer.49 Although these demonstrated effects show the potential of asbestos to induce epigenetic alterations, it is nonetheless unclear how these factors contribute to cell toxicity and the transformation to a malignant state.
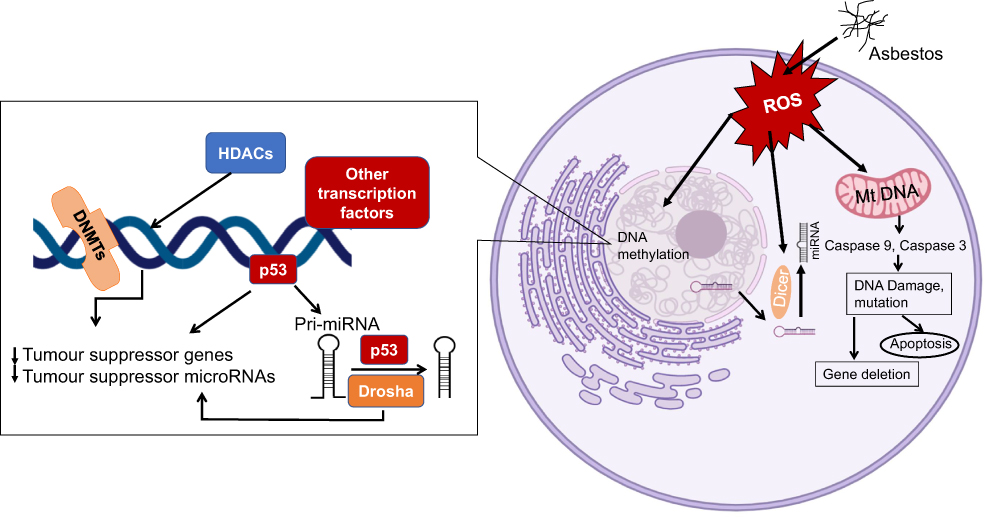 |
Figure 1 Potential molecular responses of affected cells to the presence of asbestos. When cells are exposed to asbestos, the generation of reactive oxygen species (ROS) will lead to alteration of DNA methylation and microRNA (miRNA) expression/processing, resulting in cell apoptosis or epigenetic alterations that allow cells to progress to diseased states. |
Epigenetic Biomarkers In Cancer
DNA methylation is a fundamental epigenetic mechanism for regulating gene expression. Dysregulation of epigenetic transcriptional control, particularly aberrant promoter DNA methylation and histone modifications, is a fundamental feature of human malignancies.10 Many cancers present with a global DNA hypomethylation of non‐coding regions, and site‐specific hypermethylation of CpG islands (CGI) in tumour suppressor regions.50 To what extent epigenetic changes cause carcinogenesis is currently being investigated.51 Complicated epigenetics mechanisms, including CGI shores’ and gene body methylation may contribute to the carcinogenesis process.51,52 Genomic regions having different DNA methylation status in cancer versus non-cancer are referred to as differentially methylated regions (DMRs). The most widely studied epigenetic alterations are DNA methylation at CpG dinucleotides. These are highly concentrated in CpG islands within promoter regions or near the first exon of genes. Their state of methylation controls gene expression.53 Differences in DNA methylation status lead to various levels of gene silencing in cancer. Promoter hypermethylation has been linked to the silencing of tumour suppressor genes and oncogenesis.54–56 DNA methylation heterogeneity and variability are observed at distinctive genomic regions in cancer tissue.57,58 These differentially methylated CpGs (DMCs) are consistently methylated in a non-cancer group, and variably methylated in cancer groups, with the highly variable CpGs hypothesised to contribute to tumour heterogeneity.59 Studies have identified differentially variable and differentially methylated CpGs (DVMCs) using algorithms that identify regions of differential variability and rank or filter them by the statistical significance of their differential methylation.59 There are a number of epigenetic biomarkers that show potential for the early detection of cancers due to their involvement in the initiation of carcinogenic pathways.60,61 Epigenetic biomarkers have high potential and wide scope to be implemented as early diagnostic biomarkers.
Amongst all epigenetic alterations in cancer, aberrant DNA hypermethylation is more studied than aberrant hypomethylation, and diagnostic tests being developed also tend to look for hypermethylated regions rather than hypomethylated ones.62 The reason for the focus on hypermethylation instead of hypomethylation is technical. Methylation assays produce a signal for methylated DNA, and a lack of signal signifies a lack of methylated DNA due to either hypomethylation of the DNA present or due to absence of the targeted DNA in the assay. Thus, molecular tests to identify methylated DNA are carried out more often than tests for hypomethylation. On the other hand, hypomethylation of genes leads to overexpression of their protein. Thus, immunohistochemistry (IHC) to measure protein expression is a common tool in the diagnostic setting.
DNA Methylation In Lung Cancers
There are a number of genes that have been reported as aberrantly hypermethylated in human lung cancers (Table 3). Studies have shown that epigenetic alterations (Figure 1), especially DNA methylation, can be used to identify patients at risk of developing lung cancer.44 The relationship between promoter DNA hypermethylation and inflammation has been documented in many forms of cancer, including asbestos-related lung cancer.76 It is hypothesised that asbestos exposure contributes to lung cancer formation through this relationship.45 Genomic investigations have shown DNA copy number alterations, changes in miRNA profiles, and deregulation of expression of certain genes in asbestos‐related lung cancer.49,51,77,78 However, little is known about how asbestos fibres directly or indirectly interact with cells at the molecular level.78
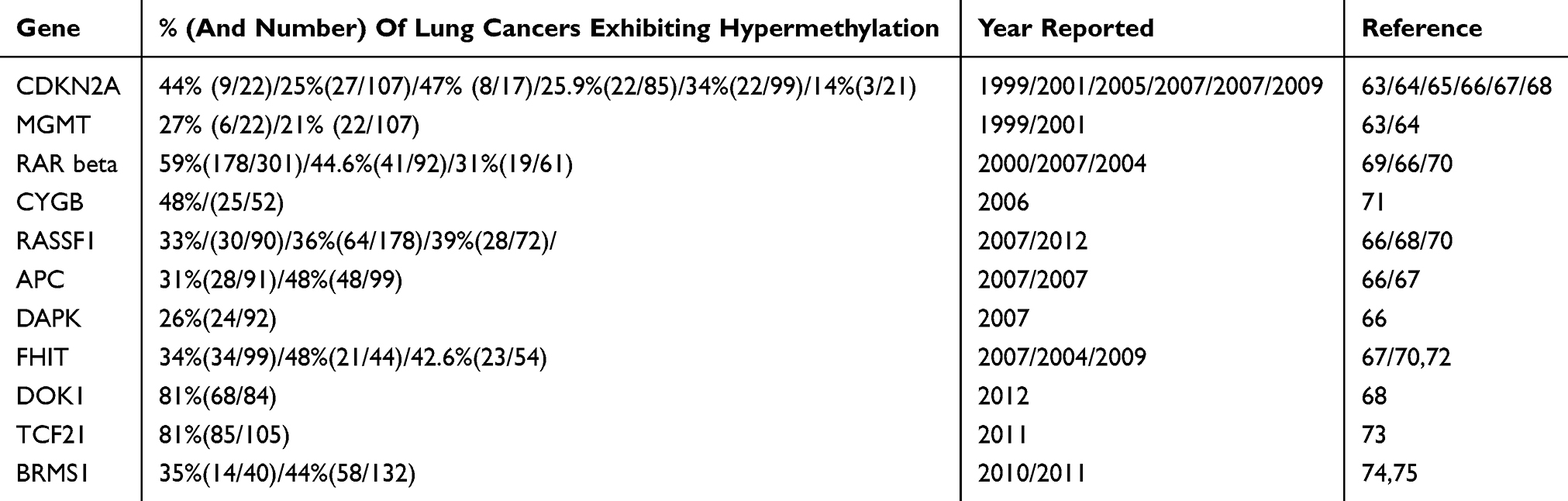 |
Table 3 Genes Found To Have Hypermethylated DNA In Human Lung Cancer |
Lung cancer cases with no documented occupational exposure to asbestos have been found to have larger methylation variability and a higher proportion of hypomethylated DVMCs than asbestos-exposed subjects.76 A large proportion of DVMCs in lung tumours of the asbestos‐exposed subjects have been found within 1 kilobasepair (kb) of promoter regions.76 The genes NPTN, NRG2, and TRPC3 exhibit significant asbestos-related DVMCs.76 NRG2 has a cell proliferation role and thus its DVMC in asbestos-related lung cancers may be playing a role in the cancer.76 Little is known about the role of NPTN (neuroplastin), and it may potentially be involved in modulating intracellular Ca2+ as a result of its interaction with FGFR.79 This may result in stimulating the Ca2+ sensing receptor that promotes the expression of TRPC3, a member of the canonical transient receptor potential channels, leading to perturbation in Ca2+ homeostasis.80 Hypermethylation of TRPC3 was observed in lung cancer cases that were not associated with asbestos exposure. In asbestos-associated tumours, TRPC3 methylation remained at the same level as in peripheral normal lung tissue.81
In asbestos-associated lung cancers, DMRs were identified in genes RARB, GPR135, MYT1L, TPO, and RPTOR.76 Hypomethylation of TPO was observed in asbestos-associated lung tumours.76 TPO is a thyroid peroxidase that is responsible for oxidative metabolic reactions and was mostly studied in thyroid cells.82 The carcinogenesis mechanism of asbestos is hypothesised to involve reactive oxygen species (ROS).39 An increase in expression of TPO has been observed in tumour and lung tissue of adenocarcinoma patients with high intakes of red meat and the increased expression was attributed to a gene product linked to heme‐iron toxicity and oxidative stress.83 Iron‐related toxicity mechanisms have also been proposed for asbestos.4 Importantly, TPO expression was not found to be associated with smoking in lung adenocarcinoma.83
Epigenetic Biomarkers Detectable In Lung Cancer Using Minimally Invasive Biopsy Samples
Of all the known types of epigenetic alterations, DNA methylation is the most widely studied in cancer due to the stability of DNA and it being readily detectable in blood circulation. To definitively diagnose lung cancer, a tissue biopsy needs to be obtained from the patient based upon the initial clinical and radiological findings. The lesion is often detected by an initial thoracic screening for respiratory lesions performed by computed tomography (CT). This method is considered highly sensitive for lung cancer. However, it has a high false positive rate as a proportion of the lesions are benign tumours.84,85 There are several tissue and circulating epigenetic biomarkers for lung cancers, including EGF-like and two follistatin domains (TMEFF2),86 that are detectable in the blood of lung cancer patients as tumours shed tumour DNA into the blood. TMEFF2 is inactivated through hypermethylation in many cancers including NSCLC and is common in non-EGFR mutated patients who have never smoked.86 RASFF1A hypermethylation was detected in 33.8% of NSCLC patients and not in healthy control benign pulmonary disease.54 Confirmation by tissue or other biopsy is currently still required for definitive diagnosis.87 In NSCLC, sputum represents a good source for biomarker detection, as cancer cells shed tumour DNA from the lung into the sputum.54,88 Various studies utilised sputum as a minimally invasive biospecimen and identified DNA methylation biomarkers including p16, DAPK, PAX5b, GATA5,88 RASSF1A, PRDM14, and 3OST236 for early detection of lung cancer in stage 1 of the disease. MGMT has been shown to be capable of identifying squamous cell lung carcinoma three years before clinical diagnosis.89 Table 4 summarises the most useful of the promising epigenetic biomarkers detected using minimally invasive lung cancer biospecimens.
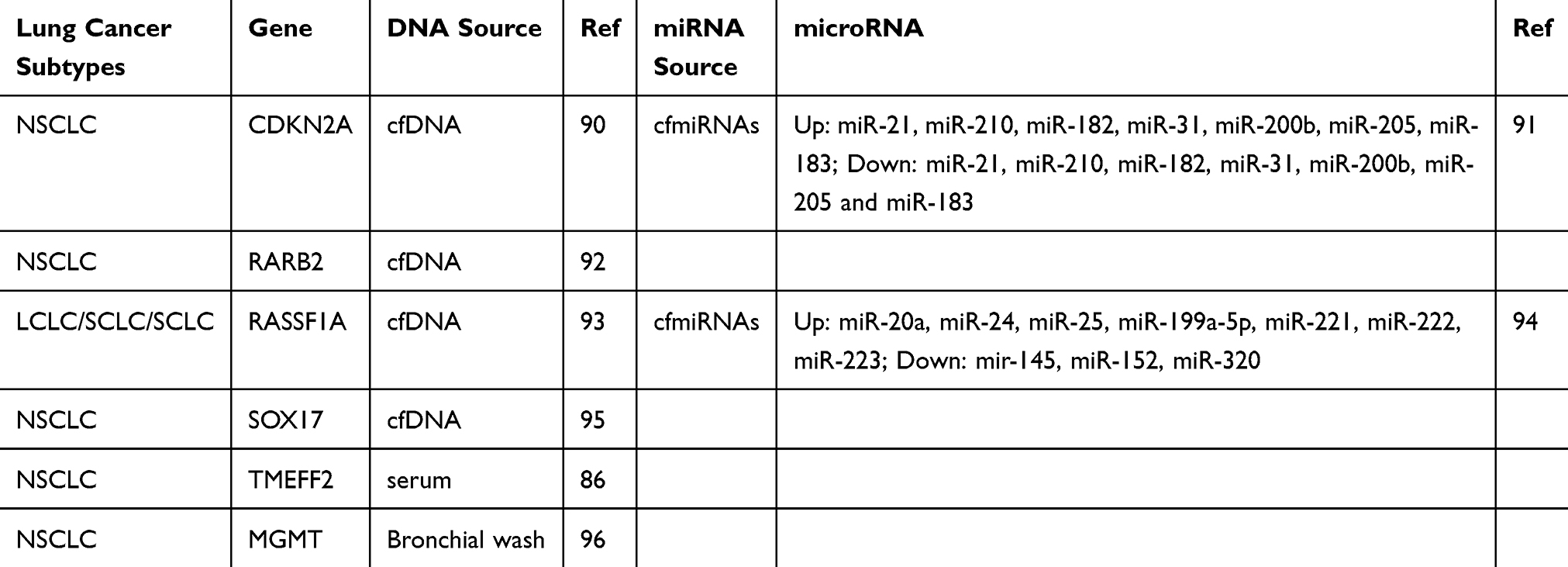 |
Table 4 Epigenetic Biomarkers In Lung Cancer Using Minimally Invasive Biospecimens |
Epigenetic Alterations As Treatment Options For Lung Cancer
Unlike gene mutations, epigenetic dysregulation can be reversed by selectively targeted treatment. Studies suggest that epigenetic dysregulation may contribute to drug-resistance in subpopulations of cells within the heterogeneous tumour population.97 Single-agent demethylation drugs such as azacytidine have been investigated for treating NSCLC solid tumours. A comparison of outcomes for 103 patients treated between 1972 and 1977 indicated that single epigenetic agents have limited efficacy in NSCLC, with an objective response rate of only 8%.98 A study of more than 200 NSCLC patients enrolled in a single epigenetic-agent trial produced disappointing initial findings.98 Given these initial findings for single epigenetic agents in treating solid tumours, researchers are investigating combination therapies on the basis that the ineffectiveness of single-agent epigenetic therapies may be due to complications associated with DNMT inhibitors at cytotoxic doses.99 The results of a phase I/II study of azacytidine and etinostat combination therapy in 45 heavily pre-treated advanced NSCLC patients indicate that combinations with low-dose epigenetic therapy may be beneficial for treating solid tumours.99 More combination clinical trials are needed to confirm the efficacy of epigenetic therapy, and such trials are in progress.100
Conclusion
In summary, DNA methylation and microRNA alterations are key epigenetic alteration features in asbestos-related lung cancers. There are a number of epigenetic biomarkers (CDKN2A, RARB2, RASSF1A, SOX17, TMEFF2 and MGMT) that are potentially useful for identifying asbestos-related lung cancers. However, further studies are needed to clarify the direct link between asbestos and these biomarkers. Development of new treatments is needed for better outcomes in asbestos-related lung cancers. Currently, despite their promise, no epigenetic therapies have been implemented clinically. Therefore, further characterisation and development in utilising epigenetic biomarkers for drug treatment discovery is warranted.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Asbestos Safety and Eradication Agency. About asbestos. 2019; Available from: https://www.asbestossafety.gov.au/about-asbestos/about-asbestos.
2. WHO. World Cancer Report 2014. Bernard CPW, Stewart W, Editor. International Agency for Research on Cancer Scientific Publication. 2014:1–630.
3. Human IWG. Personal habits and indoor combustion. IARC Monogr Eval Carcinog Risks Hum. 2012;100(Pt. E):1–538.
4. Humans IWG. Arsenic, metals, fibes, and dust. IARC Monogr Eval Carcinog Risks Hum. 2012;100C(p):2019–2309.
5. Kameda T, Takahashi K, Kim R, et al. Asbestos: use, bans and disease burden in Europe. Bull World Health Organ. 2014;92(11):790–7. doi:10.2471/BLT.13.132118
6. Australian Government. Cancer Australia. Lung cancer in Australia statistics. Available from: https://lung-cancer.canceraustralia.gov.au/statistics. Accessed July 4, 2019.
7. McCormack V, Peto J, Byrnes G, et al. Estimating the asbestos-related lung cancer burden from mesothelioma mortality. Br J Cancer. 2012;106(3):575–584. doi:10.1038/bjc.2011.563
8. Odgerel C-O, Takahashi K, Sorahan T, et al. Estimation of the global burden of mesothelioma deaths from incomplete national mortality data. Occup Environ Med. 2017;74(p):851–885. doi:10.1136/oemed-2017-104298
9. Norbet C, Joseph A, Rossi SS, et al. Asbestos-related lung disease: a pictorial review. Curr Probl Diagn Radiol. 2015;44(4):371–82. doi:10.1067/j.cpradiol.2014.10.002
10. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3(6):415–28. doi:10.1038/nrg816
11. Takahashi K, Landrigan PJ, Collegium R. The global health dimensions of asbestos and asbestos-related diseases. Ann Glob Health. 2016;82(1):209–13. doi:10.1016/j.aogh.2016.01.019
12. American Lung Association. Mesothelioma symptoms, causes and risk factors. 2019; Available from: https://www.lung.org/lung-health-and-diseases/lung-disease-lookup/mesothelioma/mesothelioma-symptoms-causes-risks.html.
13. The Mesothelioma Center. Asbestos-related lung cancer. 2019; Available from: https://www.asbestos.com/cancer/lung-cancer/.
14. Wolff H, Vehmas T, Oksa P, et al. Asbestos, asbestosis, and cancer, the Helsinki criteria for diagnosis and attribution 2014: recommendations. Scand J Work Environ Health. 2015;41(1):5–15. doi:10.5271/sjweh.3462
15. Collegium Ramazzini. Collegium Ramazzini response to ‘Asbestos, asbestosis, and cancer, the Helsinki criteria for diagnosis and attribution 2014: recommendations’. Scandinavian journal of work. Environ Health. 2016;42:1.
16. The Mesothelioma Center. Mesothelioma. 2019; Available from: https://www.asbestos.com/mesothelioma/.
17. Peng WJ, Mi J, Jiang YH. Asbestos exposure and laryngeal cancer mortality. Laryngoscope. 2016;126(5):1169–1174. doi:10.1002/lary.25693
18. Reid BM, Permuth JB, Sellers TA. Epidemiology of ovarian cancer: a review. Cancer Biol Med. 2017;14(1):9–32. doi:10.20892/j.issn.2095-3941.2016.0084
19. Calvayrac O, Pradines A, Pons E, et al. Molecular biomarkers for lung adenocarcinoma. Eur Respir J. 2017;49(4):1601734. doi:10.1183/13993003.01734-2016
20. Serizawa M, Koh Y, Kenmotsu H, et al. Assessment of mutational profile of Japanese lung adenocarcinoma patients by multitarget assays: a prospective, single-institute study. Cancer. 2014;120(10):1471–1481. doi:10.1002/cncr.28604
21. Grigoriu B, Berghmans T, Meert AP. Management of EGFR mutated nonsmall cell lung carcinoma patients. Eur Respir J. 2015;45(4):1132–1141. doi:10.1183/09031936.00156614
22. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10(9):1243–1260. doi:10.1097/JTO.0000000000000630
23. Kashima J, Kitadai R, Okuma Y. Molecular and morphological profiling of lung cancer: a foundation for “Next-Generation” pathologists and oncologists. Cancers (Basel). 2019;11(5):599. doi:10.3390/cancers11050599
24. Testa U, Castelli G, Pelosi E. Lung cancers: molecular characterization, clonal heterogeneity and evolution, and cancer stem cells. Cancers (Basel). 2018;10(8):248. doi:10.3390/cancers10080248
25. Yoshizawa A, Motoi N, Riely GJ, et al. Impact of proposed IASLC/ATS/ERS classification of lung adenocarcinoma: prognostic subgroups and implications for further revision of staging based on analysis of 514 stage I cases. Mod Pathol. 2011;24(5):653–664. doi:10.1038/modpathol.2010.232
26. TCGA. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(4717):519–525. doi:10.1038/nature11404
27. George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524(7563):47–53. doi:10.1038/nature14664
28. Miyoshi T, Umemura S, Matsumura Y, et al. Genomic profiling of large-cell neuroendocrine carcinoma of the lung. Clin Cancer Res. 2017;23(3):757–765. doi:10.1158/1078-0432.CCR-16-0355
29. Asiedu MK, Thomas CF, Dong J, et al. Pathways impacted by genomic alterations in pulmonary carcinoid tumors. Clin Cancer Res. 2018;24(7):1691–1704. doi:10.1158/1078-0432.CCR-17-0252
30. Schrock AB, Li SD, Frampton GM, et al. Pulmonary sarcomatoid carcinomas commonly harbor either potentially targetable genomic alterations or high tumor mutational burden as observed by comprehensive genomic profiling. J Thorac Oncol. 2017;12(6):932–942. doi:10.1016/j.jtho.2017.03.005
31. Vanderbilt-Ingram Cancer Center. My cancer genome. Genetically informed cancer medicine. 2019; Available from: https://www.mycancergenome.org/.
32. Msiska Z, Pacurari M, Mishra A, et al. DNA double-strand breaks by asbestos, silica, and titanium dioxide: possible biomarker of carcinogenic potential? Am J Respir Cell Mol Biol. 2010;43(2):210–219. doi:10.1165/rcmb.2009-0062OC
33. Vaslet CA, Messier NJ, Kane AB. Accelerated progression of asbestos-induced mesotheliomas in heterozygous p53± mice. Toxicol Sci. 2002;68(2):331–338.
34. Burmeister B, Schwerdtle T, Poser I, et al. Effects of asbestos on initiation of DNA damage, induction of DNA-strand breaks, P53-expression and apoptosis in primary, SV40-transformed and malignant human mesothelial cells. Mutat Res. 2004;558(1–2):81–92. doi:10.1016/j.mrgentox.2003.11.003
35. Pietruska JR, Johnston T, Zhitkovich A, et al. XRCC1 deficiency sensitizes human lung epithelial cells to genotoxicity by crocidolite asbestos and Libby amphibole. Environ Health Perspect. 2010;118(12):1707–1713. doi:10.1289/ehp.1002312
36. Su Y, Fang H, Jiang F. Integrating DNA methylation and microRNA biomarkers in sputum for lung cancer detection. Clin Epigenetics. 2016;8:109. doi:10.1186/s13148-016-0275-5
37. Baldys A, Aust AE. Role of iron in inactivation of epidermal growth factor receptor after asbestos treatment of human lung and pleural target cells. Am J Respir Cell Mol Biol. 2005;32(5):436–442. doi:10.1165/rcmb.2004-0133OC
38. Aljandali A, Pollack H, Yeldandi A, et al. Asbestos causes apoptosis in alveolar epithelial cells: role of iron-induced free radicals. J Lab Clin Med. 2001;137(5):330–339. doi:10.1067/mlc.2001.114826
39. Upadhyay D, Kamp DW. Asbestos-induced pulmonary toxicity: role of DNA damage and apoptosis. Exp Biol Med (Maywood). 2003;228(6):650–659. doi:10.1177/153537020322800602
40. Liu G, Cheresh P, Kamp DW. Molecular basis of asbestos-induced lung disease. Annu Rev Pathol. 2013;24(8):161–187. doi:10.1146/annurev-pathol-020712-163942
41. Nymark P, Lindholm PM, Korpela MV, et al. Gene expression profiles in asbestos-exposed epithelial and mesothelial lung cell lines. BMC Genomics. 2007;8:62. doi:10.1186/1471-2164-8-62
42. Trevisan E, Zabucchi G, Pascolo L, et al. Histopathological data of iron and calcium in the mouse lung after asbestos exposure. Data Brief. 2016;6:769–775. doi:10.1016/j.dib.2016.01.026
43. Acencio MM, Soares B, Marchi E, et al. Inflammatory cytokines contribute to asbestos-induced injury of mesothelial cells. Lung. 2015;193(5):831–837. doi:10.1007/s00408-015-9744-4
44. Wang H, Gillis A, Zhao C, et al. Crocidolite asbestos-induced signal pathway dysregulation in mesothelial cells. Mutat Res. 2011;723(2):171–176. doi:10.1016/j.mrgentox.2011.04.008
45. Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer. 2007;121(11):2373–2380. doi:10.1002/ijc.23173
46. Kirschner MB, Cheng YY, Armstrong NJ, et al. MiR-score: a novel 6-microRNA signature that predicts survival outcomes in patients with malignant pleural mesothelioma. Mol Oncol. 2015;9(3):715–726. doi:10.1016/j.molonc.2014.11.007
47. Kao SC, Cheng YY, Williams M, et al. Tumor suppressor microRNAs contribute to the regulation of PD-L1 expression in malignant pleural mesothelioma. J Thorac Oncol. 2017;12(9):1421–1433. doi:10.1016/j.jtho.2017.05.024
48. Johnson TG, Schelch K, Cheng YY, et al. Dysregulated expression of the MicroRNA miR-137 and its target YBX1 contribute to the invasive characteristics of malignant pleural mesothelioma. J Thorac Oncol. 2018;13(2):258–272. doi:10.1016/j.jtho.2017.10.016
49. Nymark P, Guled M, Borze I, et al. Integrative analysis of microRNA, mRNA and aCGH data reveals asbestos- and histology-related changes in lung cancer. Genes Chromosomes Cancer. 2011;50(8):585–597. doi:10.1002/gcc.v50.8
50. Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21:5400–5413. doi:10.1038/sj.onc.1205651
51. Mossman BT, Lippmann M, Hesterberg TW, et al. Pulmonary endpoints (lung carcinomas and asbestosis) following inhalation exposure to asbestos. J Toxicol Environ Health B Crit Rev. 2011;14(1–4):76–121. doi:10.1080/10937404.2011.556047
52. Irizarry RA, Ladd-Acosta C, Wen B, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41(2):178–186. doi:10.1038/ng.298
53. Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. PNAS. 2006;103(5):1412–1417. doi:10.1073/pnas.0510310103
54. Wang Y, Yu Z, Wang T, et al. Identification of epigenetic aberrant promoter methylation of RASSF1A in serum DNA and its clinicopathological significance in lung cancer. Lung Cancer. 2007;56(2):289–294. doi:10.1016/j.lungcan.2006.12.007
55. Cheng YY, Yu J, Wong YP, et al. Frequent epigenetic inactivation of secreted frizzled-related protein 2 (SFRP2) by promoter methylation in human gastric cancer. Br J Cancer. 2007;97(7):895–901. doi:10.1038/sj.bjc.6603968
56. Yu J, Tao Q, Cheng YY, et al. Promoter methylation of the Wnt/beta-catenin signaling antagonist Dkk-3 is associated with poor survival in gastric cancer. Cancer. 2009;115(1):49–60.
57. Yang X, Han H, De Carvalho D, et al. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26(4):577–590. doi:10.1016/j.ccr.2014.07.028
58. Fernandez AF, Assenov Y, Martin-Subero JI, et al. A DNA methylation fingerprint of 1628 human samples. Genome Res. 2012;22(2):407–419. doi:10.1101/gr.119867.110
59. Hansen KD, Timp W, Bravo HC, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43(8):768–775. doi:10.1038/ng.865
60. Chen HM, Fang JY. Epigenetic biomarkers for the early detection of gastrointestinal cancer. Gastrointest Tumors. 2014;1(4):201–208. doi:10.1159/000380784
61. Yang X, Dai W, Kwong DL, et al. Epigenetic markers for noninvasive early detection of nasopharyngeal carcinoma by methylation-sensitive high resolution melting. Int J Cancer. 2015;136(4):E127–E135. doi:10.1002/ijc.29192
62. Aref-Eshghi E, Bend EG, Colaiacovo S, et al. Diagnostic utility of genome-wide DNA methylation testing in genetically unsolved individuals with suspected hereditary conditions. Am J Hum Genet. 2019;104(4):685–700. doi:10.1016/j.ajhg.2019.03.008
63. Esteller M, Sanchez-Cespedes M, Rosell R, et al. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res. 1999;59(1):67–70.
64. Zochbauer-Muller S, Fong KM, Virmani AK, et al. Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res. 2001;61(1):249–255.
65. Dammann R, Strunnikova M, Schagdarsurengin U, et al. CpG island methylation and expression of tumour-associated genes in lung carcinoma. Eur J Cancer. 2005;41(8):1223–1236. doi:10.1016/j.ejca.2005.02.020
66. Fischer JR, Ohnmacht U, Rieger N, et al. Prognostic significance of RASSF1A promoter methylation on survival of non-small cell lung cancer patients treated with gemcitabine. Lung Cancer. 2007;56(1):115–123. doi:10.1016/j.lungcan.2006.11.016
67. Kim DS, Cha S-I, Lee J-H, et al. Aberrant DNA methylation profiles of non-small cell lung cancers in a Korean population. Lung Cancer. 2007;58(1):1–6. doi:10.1016/j.lungcan.2007.04.008
68. Saulnier A, Vaissière T, Yue J, et al. Inactivation of the putative suppressor gene DOK1 by promoter hypermethylation in primary human cancers. Int J Cancer. 2012;130(11):2484–2494. doi:10.1002/ijc.v130.11
69. Virmani AK, Rathi A, Zochbauer-Muller S, et al. Promoter methylation and silencing of the retinoic acid receptor-beta gene in lung carcinomas. J Natl Cancer Inst. 2000;92(16):1303–1307. doi:10.1093/jnci/92.16.1303
70. Tomizawa Y, et al. Clinicopathological significance of aberrant methylation of RARbeta2 at 3p24, RASSF1A at 3p21.3, and FHIT at 3p14.2 in patients with non-small cell lung cancer. Lung Cancer. 2004;46(3):305–312. doi:10.1016/j.lungcan.2004.05.003
71. Xinarianos G, McRonald FE, Risk JM, et al. Frequent genetic and epigenetic abnormalities contribute to the deregulation of cytoglobin in non-small cell lung cancer. Hum Mol Genet. 2006;15(13):2038–2044. doi:10.1093/hmg/ddl128
72. Verri C, Roz L, Conte D, et al. Fragile histidine triad gene inactivation in lung cancer: the European early lung cancer project. Am J Respir Crit Care Med. 2009;179(5):396–401. doi:10.1164/rccm.200807-1153OC
73. Richards KL, Zhang B, Sun M, et al. Methylation of the candidate biomarker TCF21 is very frequent across a spectrum of early-stage nonsmall cell lung cancers. Cancer. 2011;117(3):606–617. doi:10.1002/cncr.v117.3
74. Nagji AS, Liu Y, Stelow EB, et al. BRMS1 transcriptional repression correlates with CpG island methylation and advanced pathological stage in non-small cell lung cancer. J Pathol. 2010;221(2):229–237. doi:10.1002/path.2707
75. Yang J, Shen Y, Liu B, et al. Promoter methylation of BRMS1 correlates with smoking history and poor survival in non-small cell lung cancer patients. Lung Cancer. 2011;74(2):305–309. doi:10.1016/j.lungcan.2011.03.002
76. Kettunen E, Hernandez-Vargas H, Cros M-P, et al. Asbestos-associated genome-wide DNA methylation changes in lung cancer. Int J Cancer. 2017;141(10):2014–2029. doi:10.1002/ijc.v141.10
77. Kettunen E, Aavikko M, Nymark P, et al. DNA copy number loss and allelic imbalance at 2p16 in lung cancer associated with asbestos exposure. Br J Cancer. 2009;100(8):1336–1342. doi:10.1038/sj.bjc.6605012
78. Nymark P, Aavikko M, Mäkilä J, et al. Accumulation of genomic alterations in 2p16, 9q33.1 and 19p13 in lung tumours of asbestos-exposed patients. Mol Oncol. 2013;7(1):29–40. doi:10.1016/j.molonc.2012.07.006
79. Owczarek S, Richards KL, Zhang B, et al. Neuroplastin-55 binds to and signals through the fibroblast growth factor receptor. Faseb J. 2010;24(4):1139–1150. doi:10.1096/fj.09-140509
80. Shapovalov G, Lehen’kyi V, Skryma R, et al. TRP channels in cell survival and cell death in normal and transformed cells. Cell Calcium. 2011;50(3):295–302. doi:10.1016/j.ceca.2011.05.006
81. Kamp DW, Liu G, Cheresh P, et al. Asbestos-induced alveolar epithelial cell apoptosis. The role of endoplasmic reticulum stress response. Am J Respir Cell Mol Biol. 2013;49(6):892–901. doi:10.1165/rcmb.2013-0053OC
82. Cipollini M, Pastor S, Gemignani F, et al. TPO genetic variants and risk of differentiated thyroid carcinoma in two European populations. Int J Cancer. 2013;133(12):2843–2851. doi:10.1002/ijc.28317
83. Lam TK, Rotunno M, Ryan BM, et al. Heme-related gene expression signatures of meat intakes in lung cancer tissues. Mol Carcinog. 2014;53(7):548–556. doi:10.1002/mc.v53.7
84. Bach PB, Mirkin JN, Oliver TK, et al. Benefits and harms of CT screening for lung cancer: a systematic review. JAMA. 2012;307(22):2418–2429. doi:10.1001/jama.2012.5521
85. Cui JW, Li W, Han F-J, et al. Screening for lung cancer using low-dose computed tomography: concerns about the application in low-risk individuals. Transl Lung Cancer Res. 2015;4(3):275–286. doi:10.3978/j.issn.2218-6751.2015.02.05
86. Lee SM, Park JY, Kim DS. Methylation of TMEFF2 gene in tissue and serum DNA from patients with non-small cell lung cancer. Mol Cells. 2012;34(2):171–176. doi:10.1007/s10059-012-0083-5
87. Eggert JA, Palavanzadeh M, Blanton A. Screening and Early Detection of Lung Cancer. Semin Oncol Nurs. 2017;33(2):129–140. doi:10.1016/j.soncn.2017.03.001
88. Belinsky SA, Grimes MJ, Casas E, et al. Predicting gene promoter methylation in non-small-cell lung cancer by evaluating sputum and serum. Br J Cancer. 2007;96(8):1278–1283. doi:10.1038/sj.bjc.6603721
89. Palmisano WA, Divine KK, Saccomanno G, et al. Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res. 2000;60(21):5954–5958.
90. Nikolaidis G, Raji OY, Markopoulou S, et al. DNA methylation biomarkers offer improved diagnostic efficiency in lung cancer. Cancer Res. 2012;72(22):5692–5701. doi:10.1158/0008-5472.CAN-12-2309
91. Zhu W, Liu X, He J, et al. Overexpression of members of the microRNA-183 family is a risk factor for lung cancer: a case control study. BMC Cancer. 2011;11:393. doi:10.1186/1471-2407-11-393
92. Nadal E, Chen G, Gallegos M, et al. Epigenetic inactivation of microRNA-34b/c predicts poor disease-free survival in early-stage lung adenocarcinoma. Clin Cancer Res. 2013;19(24):6842–6852. doi:10.1158/1078-0432.CCR-13-0736
93. Saint-Pierre MD, Pease C, Mithoowani H, et al. Malignant pleural mesothelioma outcomes in the era of combined platinum and folate antimetabolite chemotherapy. Lung Cancer Int. 2015;2015:590148. doi:10.1155/2015/590148
94. Sozzi G, Boeri M, Rossi M, et al. Clinical utility of a plasma-based miRNA signature classifier within computed tomography lung cancer screening: a correlative MILD trial study. J Clin Oncol. 2014;32(8):768–773. doi:10.1200/JCO.2013.50.4357
95. Sandoval J, Mendez-Gonzalez J, Nadal E, et al. A prognostic DNA methylation signature for stage I non-small-cell lung cancer. J Clin Oncol. 2013;31(32):4140–4147. doi:10.1200/JCO.2012.48.5516
96. Miglio U, Mezzapelle R, Paganotti A, et al. Frequency of O 6 -methylguanine-DNA methyltransferase promoter methylation in cytological samples from small cell lung cancer. Diagn Cytopathol. 2015;43(11):947–952. doi:10.1002/dc.v43.11
97. Sharma SV, Lee DY, Li B, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141(1):69–80. doi:10.1016/j.cell.2010.02.027
98. Cowan LA, Talwar S, Yang AS. Will DNA methylation inhibitors work in solid tumors? A review of the clinical experience with azacitidine and decitabine in solid tumors. Epigenomics. 2010;2(1):71–86. doi:10.2217/epi.09.44
99. Juergens RA, Wrangle J, Vendetti FP, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011;1(7):598–607. doi:10.1158/2159-8290.CD-11-0214
100. Mohammad HP, Barbash O, Creasy CL. Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat Med. 2019;25(3):403–418. doi:10.1038/s41591-019-0376-8
101. De Marco C, Malanga D, Rinaldo N, et al. Mutant AKT1-E17K is oncogenic in lung epithelial cells. Oncotarget. 2015;6(37):p.39634–50. doi:10.18632/oncotarget.v6i37
102. Hofman P. ALK in Non-Small Cell Lung Cancer (NSCLC) pathobiology, epidemiology, detection from tumor tissue and algorithm diagnosis in a daily practice. Cancers (Basel). 2017;9:8. doi:10.3390/cancers9080107
103. Alvarez JGB, Otterson GA. Agents to treat BRAF-mutant lung cancer. Drugs Context. 2019;8(p):212566.
104. Baik CS, Myall NJ, Wakelee HA. Targeting BRAF-mutant non-small cell lung cancer: from molecular profiling to rationally designed therapy. Oncologist. 2017;22(7):786–796. doi:10.1634/theoncologist.2016-0458
105. La Fleur L, Falk-Sörqvist E, Smeds P, et al. Mutation patterns in a population-based non-small cell lung cancer cohort and prognostic impact of concomitant mutations in KRAS and TP53 or STK11. Lung Cancer. 2019;130:50–58. doi:10.1016/j.lungcan.2019.01.003
106. Fathi Z, Mousavi SAJ, Roudi R, et al. Distribution of KRAS, DDR2, and TP53 gene mutations in lung cancer: an analysis of Iranian patients. PLoS One. 2018;13(7):e0200633. doi:10.1371/journal.pone.0200633
107. Yatabe Y, Kerr KM, Utomo A, et al. EGFR mutation testing practices within the Asia Pacific region: results of a multicenter diagnostic survey. J Thorac Oncol. 2015;10(3):438–445. doi:10.1097/JTO.0000000000000422
108. Mok TS, Cheng Y, Zhou X, et al. Improvement in overall survival in a randomized study that compared dacomitinib with gefitinib in patients with advanced non-small-cell lung cancer and EGFR-activating mutations. J Clin Oncol. 2018;36(22):2244–2250. doi:10.1200/JCO.2018.78.7994
109. Weeden CE, Solomon B, Asselin-Labat ML. FGFR1 inhibition in lung squamous cell carcinoma: questions and controversies. Cell Death Discov. 2015;1:15049. doi:10.1038/cddiscovery.2015.49
110. Heist RS, Mino-Kenudson M, Sequist LV, et al. FGFR1 amplification in squamous cell carcinoma of the lung. J Thorac Oncol. 2012;7(12):1775–1780. doi:10.1097/JTO.0b013e31826aed28
111. Dutt A, et al. Inhibitor-sensitive FGFR1 amplification in human non-small cell lung cancer. PLoS One. 2011;6(6):e20351. doi:10.1371/journal.pone.0020351
112. Miao JL, Liu RJ, Zhou JH, Meng SH. Fibroblast growth factor receptor 1 gene amplification in nonsmall cell lung cancer. Chin Med J (Engl). 2016;129(23):2868–2872. doi:10.4103/0366-6999.194649
113. Auliac JB, Dô P, Bayle S,et al. Non-small cell lung cancer patients harboring HER2 mutations: clinical characteristics and management in a real-life setting. Cohort HER2 EXPLORE GFPC 02-14. Adv Ther. 2019;36(8):2161–2166.
114. Pillai RN, Behera M, Berry LD, et al. HER2 mutations in lung adenocarcinomas: a report from the lung cancer mutation consortium. Cancer. 2017;123(21):4099–4105. doi:10.1002/cncr.30869
115. Ogoshi Y, Shien K, Yoshioka T, et al. Anti-tumor effect of neratinib against lung cancer cells harboring HER2 oncogene alterations. Oncol Lett. 2019;17(3):2729–2736. doi:10.3892/ol.2019.9908
116. Ferrer I, Zugazagoitia J, Herbertz S, et al. KRAS-mutant non-small cell lung cancer: from biology to therapy. Lung Cancer. 2018;124:53–64. doi:10.1016/j.lungcan.2018.07.013
117. Arcila ME, Drilon A, Sylvester BE, et al. MAP2K1 (MEK1) mutations define a distinct subset of lung adenocarcinoma associated with smoking. Clin Cancer Res. 2015;21(8):1935–1943. doi:10.1158/1078-0432.CCR-14-2124
118. Rivalland G, Mitchell P, Murone C, et al. Mesenchyme to epithelial transition protein expression, gene copy number and clinical outcome in a large non-small cell lung cancer surgical cohort. Transl Lung Cancer Res. 2019;8(2):167–175. doi:10.21037/tlcr
119. Scheffler M, Bos M, Gardizi M, et al. PIK3CA mutations in non-small cell lung cancer (NSCLC): genetic heterogeneity, prognostic impact and incidence of prior malignancies. Oncotarget. 2015;6(2):1315–1326. doi:10.18632/oncotarget.v6i2
120. Jin G, Kim MJ, Jeon H-S, et al. PTEN mutations and relationship to EGFR, ERBB2, KRAS, and TP53 mutations in non-small cell lung cancers. Lung Cancer. 2010;69(3):279–283. doi:10.1016/j.lungcan.2009.11.012
121. Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. doi:10.1038/nature13385
122. Rossi G, Jocollé G, Conti A, et al. Detection of ROS1 rearrangement in non-small cell lung cancer: current and future perspectives. Lung Cancer (Auckl). 2017;8:45–55. doi:10.2147/LCTT.S120172
123. Joshi A, Pande N, Noronha V, et al. ROS1 mutation non-small cell lung cancer-access to optimal treatment and outcomes. Ecancermedicalscience. 2019;13:900. doi:10.3332/ecancer.2019.900
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.