")
Back to Journals » OncoTargets and Therapy » Volume 10
The critical role of EGF-β-catenin signaling in the epithelial-mesenchymal transition in human glioblastoma
Authors Wang X, Wang S, Li X, Jin S, Xiong F, Wang X
Received 5 April 2017
Accepted for publication 5 May 2017
Published 31 May 2017 Volume 2017:10 Pages 2781—2789
DOI https://doi.org/10.2147/OTT.S138908
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr William C. Cho
Xingqiang Wang, Shanshi Wang, Xiaolong Li, Shigang Jin, Feng Xiong, Xin Wang
Department of Neurosurgery, People’s Hospital of Rizhao, Jining Medical University, Rizhao, China
Abstract: To date, β-catenin has been reported to be implicated in mediating the epithelial-mesenchymal transition (EMT) in a variety of human cancers, which can be triggered by EGF. However, the mechanisms underlying EGF-β-catenin pathway-induced EMT of glioblastoma multiforme (GBM) have not been reported previously. In the present study, immunohistochemistry, reverse transcription polymerase chain reaction, and Western blot were applied to investigate the effect of EGF-β-catenin pathway on EMT of GBM. Here, we identified that β-catenin mRNA and protein levels were up-regulated in GBM tissues and four kinds of glioblastoma cell lines, including T98G, A172, U87, and U251 cells, compared with normal brain tissue and astrocytes. In U87 cell line, inhibition of β-catenin by siRNA suppressed EGF-induced proliferation, migration, invasiveness, and the expression of EMT activators (Snail and Slug). In addition, the expression of epithelial markers (E-cadherin) was up-regulated and the expression of mesenchymal markers (N-cadherin and MMP9) was down-regulated. Finally, inhibitor of PI3K/Akt signaling pathways inactivated the EGF-β-catenin-induced EMT. In conclusion, β-catenin-EMT pathway induced by EGF is important for GBM progression by the PI3K/Akt pathways. Inhibition of β-catenin leads to suppression of EGF pathway-induced EMT, which provides a new way to treat GBM patients.
Keywords: EGF, β-catenin, EMT, GBM
Introduction
Glioblastoma, also known as glioblastoma multiforme (GBM), is the most common aggressive brain tumor.1–3 GBM features high cell proliferation, angiogenesis and/or necrosis, and has no efficient and specific curative therapy. The aggressiveness of GBM is correlated to the tumor heterogeneity derived from a series of genetic and epigenetic alterations, and is also implicated in some abnormal signaling pathways induced by extracellular growth factors.4,5 Up to now, little is known about how the invasive phenotype is regulated in the progression of GBM.6 As reported, an invasive phenotype needs the change of cellular and molecular levels. Thus it is essential to seek key regulators which are involved in the progression of GBM.
Recently, increasing reports demonstrated that activation of Wnt/β-catenin signaling and increased β-catenin expression is involved in the development of gliomas,7 and some signaling pathways can regulate Wnt/β-catenin signaling to participate in cell apoptosis,7 human neural stem cell growth,8 glioma cell proliferation,9,10 and glioma cell cycle.10 Based on the molecular level, Wnt/β-catenin signaling can up-regulate many kinds of cell cycle-related regulators, involving cyclin D1 and cyclin E, and down-regulate cell cycle-related inhibitors, like p21 and p27.11 Furthermore, current studies suggest that Wnt/β-catenin signaling pathway forms cross-talk with the EGF pathway, indicating the potential signaling pathway clinically involved in glioma.12 On the other hand, EGFs and EGFRs play an important role in the development of tumors by regulating cell proliferation, migration, differentiation, morphogenesis, and angiogenesis.13–15 EGFR is highly expressed in some types of human tumors, such as colon cancer, liver cancer, breast cancer, and pancreatic cancer.16 However, the role of EGF-β-catenin in human cancers remains poorly understood.
In the present study, we used in vitro experiments to investigate the potential significance of EGF-β-catenin signaling pathway. Moreover, inhibition of β-catenin by siRNA suppressed EGF-induced proliferation, migration, and invasiveness, and the expression of epithelial-mesenchymal transition (EMT) effectors, including the up-regulated expression of epithelial markers and the down-regulated expression of mesenchymal markers. These results identified that EGF-β-catenin pathway could help reduce tumorigenesis.
Materials and methods
Patient samples
The protocol for this study was approved by the ethics committee of Rizhao People’s Hospital, Jining Medical University. Twenty patients from Rizhao People’s Hospital, Jining Medical University diagnosed with GBM between 2015 and 2016 were selected for the study. All patients involved in the study provided written informed consent. Patients with other medical conditions or those who received treatment prior to surgery were excluded. The pathological specimens were reviewed, and histological classifications were made based on the World Health Organization (WHO) classification of brain tumors. We identified 20 cases of GBM. Ten healthy normal brain (NB) tissue samples obtained from patients who underwent needle biopsy without malignancy served as controls. All the slides were re-examined by two pathologists to ensure correct diagnosis. Tissue samples were harvested for immunohistochemistry assay, reverse transcription polymerase chain reaction (RT-PCR), and Western blot analysis.
Cell culture, antibodies, and reagents
Primary normal human astrocytes (NHA) were purchased from Sciencell Research Laboratories (Carlsbad, CA, USA) and cultured under the conditions as instructed by the manufacturer. Cell lines T98G, A172, U87, and U251 were obtained from the KeyGEN Company (Shanghai, China) and cultured in DMEM supplemented with 10% fetal bovine serum (FBS) (Gemini Bio-Products, Woodland, CA, USA) and 1% penicillin/streptomycin solution (Thermo Fisher Scientific, Waltham, MA, USA). Cells were incubated at 37°C in a humidified incubator with 5% CO2. Antibodies against β-catenin, E-cadherin, N-cadherin, Slug, Snail, and MMP9 were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-β-actin antibody and EGF were purchased from Sigma-Aldrich Co. (St Louis, MO, USA).
siRNA
U87 cells growing in 6-well plates were incubated with human siRNAs (100 nmol/L) for β-catenin. Mock-transfection was performed using a negative control siRNA (Santa Cruz Biotechnology Inc., Dallas, TX, USA) as control. Cells were harvested at 48 h post-transfection, washed, and stored for future experiments. The knockdown efficiency was assessed using the Western blot assay.
Immunohistochemistry
Briefly, sections were deparaffinized in xylene, rehydrated in alcohol and water, antigen repaired, and blocked. Antibodies (1:200) were incubated overnight, followed by incubation with HRP-labeled polymer for 20 min. Sections were then stained with DAB (Dako Denmark A/S, Glostrup, Denmark) for 5 min. All sections were counterstained with hematoxylin, dehydrated, and mounted. As for scoring, Image-Pro software showed the percentage of expression area and the mean optical density (MOD).
RT-PCR and immunoblotting
Total RNAs were isolated from the cells by using Trizol reagent (TaKaRa, Kusatsu, Japan). The sequences of primers used in this study were as follows.
β-catenin:
forward: 5′-AAGTTCTTGGCTATTACGACA-3′,
reverse: 5′-ACAGCACCTTCAGCACT-CT-3′;
Snail:
forward: 5′-TTTACCTTCCAGCAGCCCTA-3′,
reverse: 5′-GACAGAGTCCCAGATGAGCA-3′;
Slug:
forward: 5′-ACAGCGAACTGGACACACAT-3′,
reverse: 5′-GGAATGGAGCAGCGGTAGT-3′;
E-cadherin:
forward: 5′-TGCCCAGAAAATGAAAAAGG-3′,
reverse: 5′-GTGTATGTGGCAATGCGTTC-3′;
N-cadherin:
forward: 5′-CACTGCTCAGGACCCAGAT-3′,
reverse: 5′-TAAGCCGAGTGATGGTCC-3′;
MMP9:
forward: 5′-CATTCAGGGAGACGCCCA-3′,
reverse: 5′-AACCACGACGCCCTTGC-3′;
GAPDH:
forward: 5′-CAATGACCCCTTCATTGACC-3′,
reverse: 5′-GACAAGCTTCCCGTTCTCAG-3′.
Total RNA (1 μg) was reverse-transcribed into cDNA using a Quantscript RT Kit (Tiangen, Beijing, China). PCR was performed using a PCR MasterMix Kit (BioTeke, Beijing, China) in a GeneAmp PCR system 9600 (ABI Int., Beijing, China). cDNA was amplified under thermocycling conditions as follows: 3 min initial denaturation at 94°C (one cycle), 30 s denaturation at 94°C (35 cycles), 30 s annealing at 57°C, and 45 s extension at 72°C. The last amplification was followed by a final 7 min incubation at 72°C. PCR products were separated by electrophoresis through 1% agarose gel, stained with ethidium bromide, and visualized by ultraviolet (UV) transillumination in a Tocan Gel Imaging System (Tocan Co., Shanghai, China). GAPDH was used as an internal control. The mRNA level was calculated by determining the integrated intensity of the bands of each treated group as a ratio of the control. Each sample was measured in triplicate.
Sample protein extraction and concentration determination of whole cells were performed as previously described.17 Cytoplasmic and nuclear proteins were obtained using the Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, Beijing, China) according to the manufacturer’s instructions. Briefly, equal amounts of protein were run on sodium dodecyl sulfate polyacrylamide gels and transferred to nitrocellulose membrane. The resulting blots were blocked with 5% non-fat dry milk and probed with antibodies. Protein bands were detected by incubating with horseradish peroxidase (HRP)-conjugated antibodies (Santa Cruz Biotechnology Inc.) and visualized with enhanced chemiluminescence reagent (EMD Millipore, Billerica, MA, USA).
Cell proliferation assays
Cell counting kit-8 (CCK-8) assay (Beyotime) was used to detect the cell proliferation after 24 h transfection, accompanied by the employment of the TECAN infinite M200 Multimode microplate reader (Tecan, Mechelen, Belgium) to measure the absorbance at 450 nm. All experiments were performed in triplicate independently.
Wound-healing assays
Wound-healing assays were performed by creating identical wound areas in the cell monolayer using Ibidi culture-inserts (cat no 80209; Ibidi GmbH, Munich, Germany). Cells were seeded in complete culture medium at a density of 3×104cells on each side of the Ibidi culture-insert, in a 24-well plate. After attachment, cells were treated for 24 h; then the culture-insert was detached in order to form a cell-free gap in the cell monolayer. Each well was rinsed once with PBS to remove cell debris and immediately refilled with fresh medium, with new addition of the inhibitors (samples with “maintained” treatment) or without new addition of the inhibitors (samples with “removed” treatment). Cells were allowed to migrate for a further 48 h. The wound images were captured using a Leica DMI4000 automated inverted microscope equipped with a Leica DFC300 FX camera.
Invasion assay
For the invasion assays, cells were serum-starved overnight and 1×105 cells were seeded in a Matrigel-coated chamber (BD Biosciences, San Jose, CA, USA) with 8.0 mm pores (Corning Incorporated, Corning, NY, USA) and cultured for 48 h. The invaded cells on the lower surface of the inserts were fixed in 4% paraformaldehyde and stained with hematoxylin before mounting on glass slides. More than five views were analyzed under a light microscope. All experiments were carried out in triplicate.
Statistical analysis
Statistical analysis was performed using the SPSS statistical software program (version 19.0; IBM Corporation, Armonk, NY, USA). Student’s two-tailed t-test was used to compare data between two groups. One-way ANOVA and Dunnett’s test were used to compare data between three or more groups. P<0.05 was considered statistically significant.
Results
Expression of β-catenin in GBM tissues
To figure out the expression and role of β-catenin in the development of GBM, we enrolled 20 patients diagnosed with GBM. Then, ten samples of healthy NB tissue obtained from patients without malignant tumors undergoing needle biopsy, acted as the control group. In the present study, the expression of β-catenin was detected using immunohistochemistry, RT-PCR, and Western blotting. The immunohistochemistry analysis revealed that positive immunoreactivity of β-catenin was observed in the soma of neurons of the GBM tissues. β-catenin expression was detected in most of the cancer cells of GBM tissues. Using Image-Pro software, we demonstrated that the MOD value of β-catenin was obviously higher in GBM tissues compared with that in the NB tissues (P<0.01) (Figure 1A).
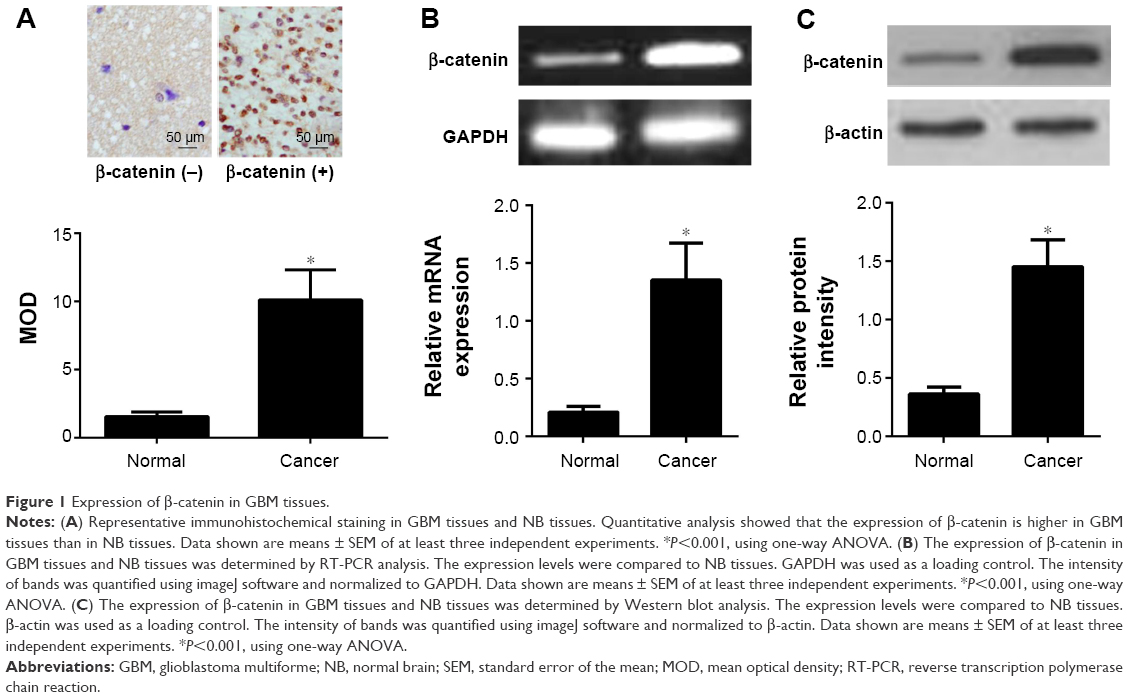 | Figure 1 Expression of β-catenin in GBM tissues. |
These findings were further confirmed using RT-PCR and Western blotting (Figure 1B and C). We found that GBM tissues had higher mRNA expression levels of β-catenin than the NB tissues (P<0.01). Consistent with mRNA expression, the expression level of β-catenin protein in GBM tissues was also obviously higher than in the NB tissues (P<0.01).
β-catenin regulates U87 cell proliferation
As mentioned previously, the expression levels of β-catenin in the NHA and GBM cell lines (T98G, A172, U87, and U251) were further tested using RT-PCR and Western blotting. As shown in Figure 2A, our results identified that all GBM cell lines had a significantly higher expression level of β-catenin mRNA compared with NHA. Consistently, all GBM cell lines had a significantly higher expression level of β-catenin protein compared with NHA (Figure 2B). In order to investigate the mechanisms of β-catenin in U87 cells, a specific siRNA for β-catenin was used. Lysates of cells with β-catenin siRNA were subjected to Western blot analysis. Firstly, we found that the mean expression level of β-catenin protein in the si-β-catenin group was 0.43, while the mean expression level of β-catenin protein in the si-control group was 1.21. Thus, the knockdown efficiency of β-catenin was nearly 65% in U87 cells. Subsequently, we employed the CCK-8 assay to assess the proliferation potential of U87 cells. We observed that down-regulation of β-catenin expression decreased cell proliferation in a time-dependent manner (Figure 2C; P<0.01).
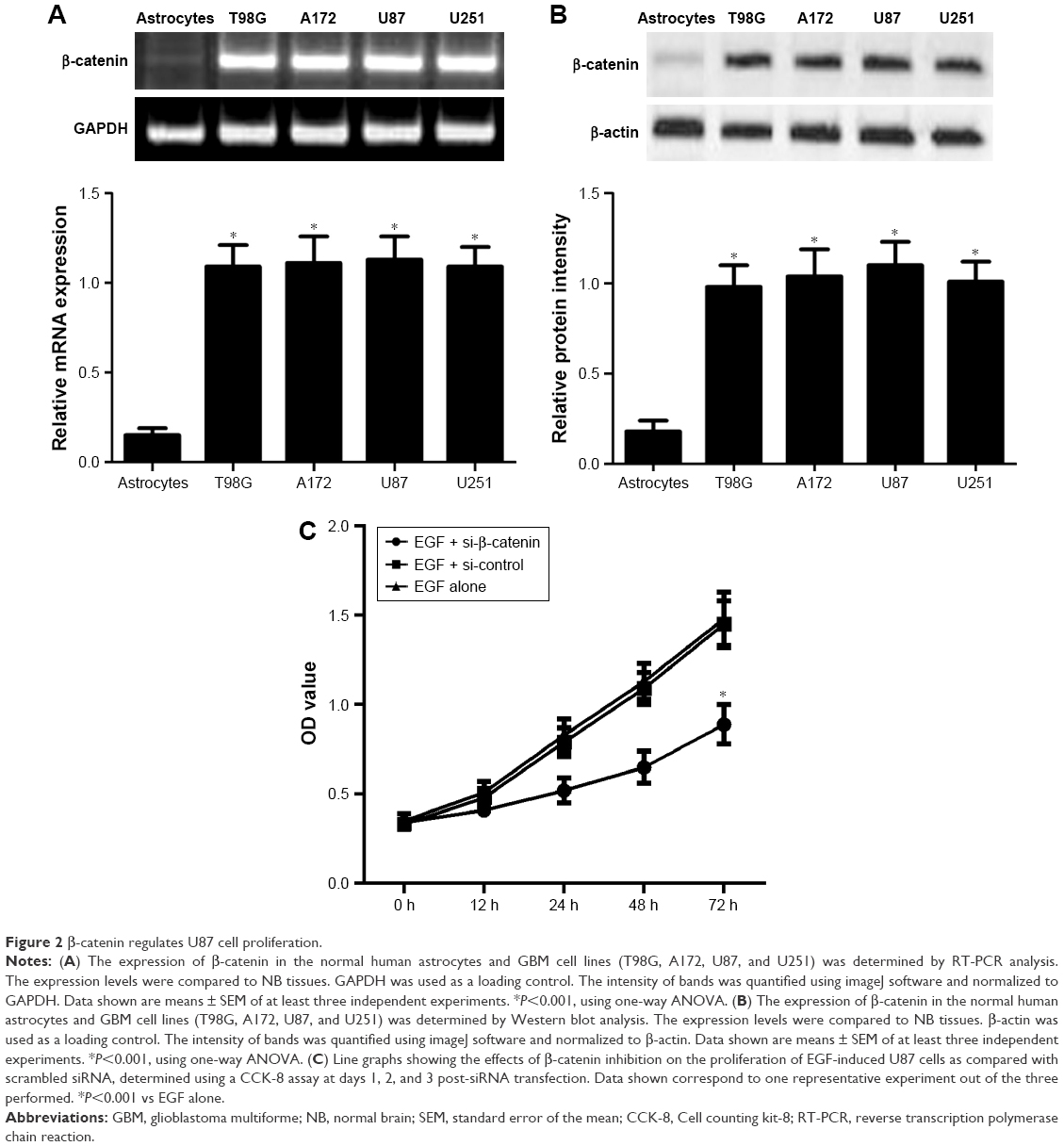 | Figure 2 β-catenin regulates U87 cell proliferation. |
Inhibition of β-catenin expression suppresses the migration and invasion in U87 cells
We used the wound-healing assay and the transwell invasion assays to identify whether β-catenin affected U87 cell migration and invasion (Figure 3A). In the present study, upon down-regulation of β-catenin expression by siRNA, the migration of U87 cells toward artificially created wounds in confluent cell monolayers was obviously repressed as shown by the increased wound width (P<0.01). Following the transwell invasion assay, the cells invading the lower layer of the inserts were fixed in 4% paraformaldehyde and stained with hematoxylin. We observed that down-regulation of β-catenin expression also resulted in fewer cell colonies in the transwell assay compared with the control (P<0.01, Figure 3B).
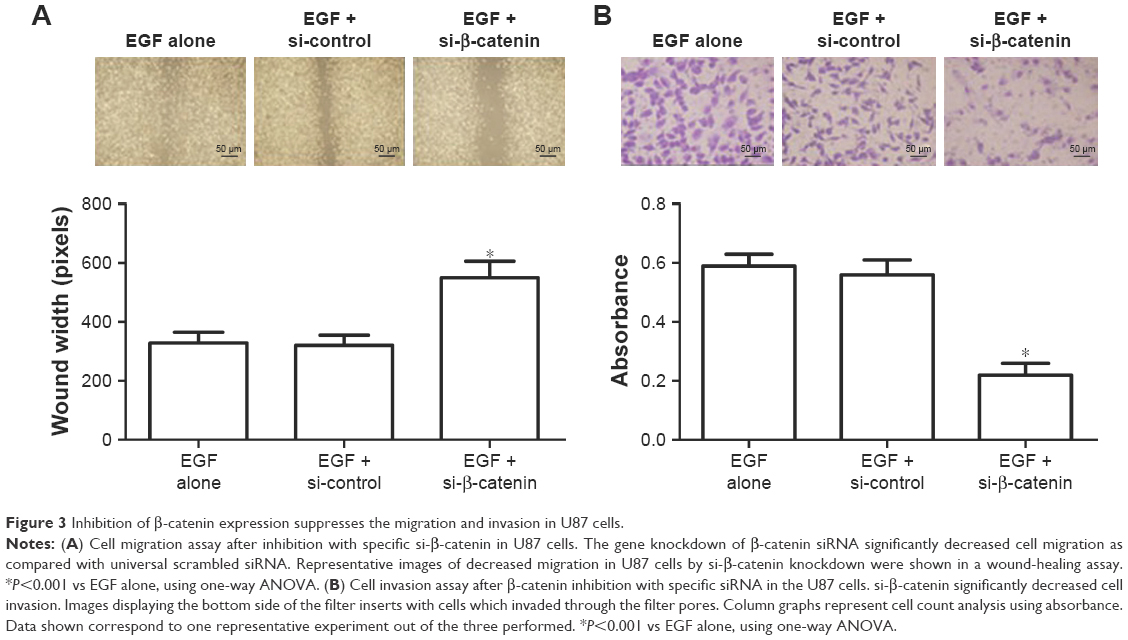 | Figure 3 Inhibition of β-catenin expression suppresses the migration and invasion in U87 cells. |
Inhibition of β-catenin expression suppresses EMT in U87 cells
To figure out the role of β-catenin expression in EMT of U87 cells, we detected the mRNA and protein expression of EMT activators and effectors in U87 cells depleted of β-catenin. Firstly, we determined the role of EGF in inducing the phosphorylation of β-catenin in U87 cells. Because EGFs are reported to correlate with Wnt/β-catenin during carcinogenesis, we tried to determine whether EGF affects the phosphorylation of β-catenin in U87 cells. Treatment of cells with EGF (0–100 ng/mL) showed a significant increase in the phosphorylation of β-catenin as early as 15 min (Figure 4A). However, no associations between the level of β-catenin phosphorylation and its activity were found (Figure 4B; r2=0.169, P=0.184). Next, we hypothesized that β-catenin plays an important role in EGF-mediated EMT in U87 cells. To this end, we knocked down β-catenin expression using specific siRNA for β-catenin in U87 cells and treated U87 cells with EGF. In the present study, si-catenin effectively knocked down the expression of β-catenin (Figure 4B). We found that EMT activators Slug and Snail were significantly down-regulated compared with the control group (P<0.01, Figure 4C and D). Besides, down-regulation of β-catenin also led to an up-regulation of the epithelial marker E-cadherin and the down-regulation of the mesenchymal markers N-cadherin and MMP9 (P<0.01, Figure 4C and D).
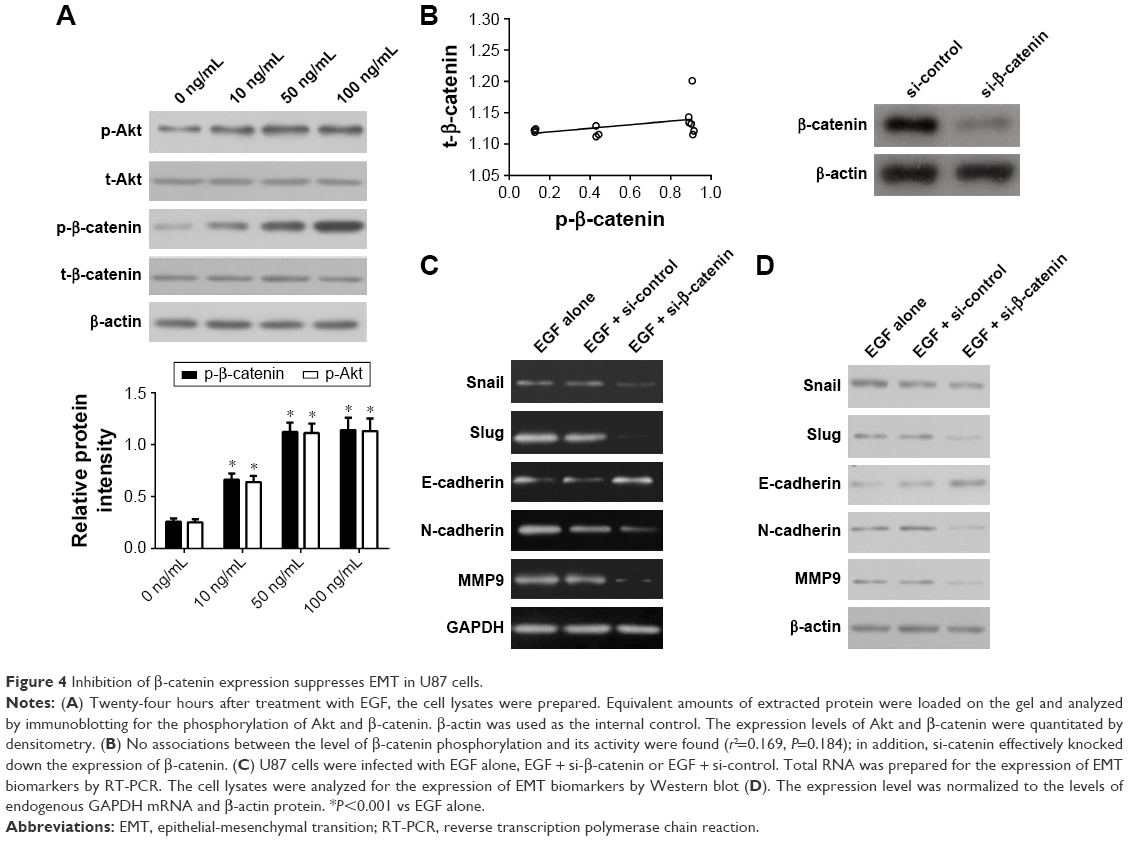 | Figure 4 Inhibition of β-catenin expression suppresses EMT in U87 cells. |
β-catenin activates EMT through the PI3K/Akt signaling pathways
It has been reported that the PI3K/Akt signaling pathways play an important role in modulating cell invasion and the progression of EMT. In our study, we investigated whether β-catenin-mediated EMT was induced by the PI3K/Akt signaling pathways. Besides, in order to explore the effect of the primary signaling mediators on up-regulation of EMT, PI3K/Akt antagonist LY294002 was used to individually block PI3K/Akt signaling pathway in U87 cells, and then treated with 50 ng/mL of EGF. Subsequently, the expression level of EMT markers was detected. We found that LY294002 down-regulated the Akt phosphorylation but not their total protein levels (P<0.01, Figure 5A), resulting in the down-regulation of β-catenin phosphorylation expression. Likewise, dimethyl sulfoxide (DMSO)-treated control cells with the EGF obviously up-regulated the Akt phosphorylation (P<0.01, Figure 5A), and the total Akt protein levels remained unaffected. In addition, we found that mRNAs, proteins of Snail, Slug, N-cadherin, and MMP9 were down-regulated, and E-cadherin expression was up-regulated in the LY294002 group, in comparison with DMSO-treated control (P<0.01, Figure 5B and C).
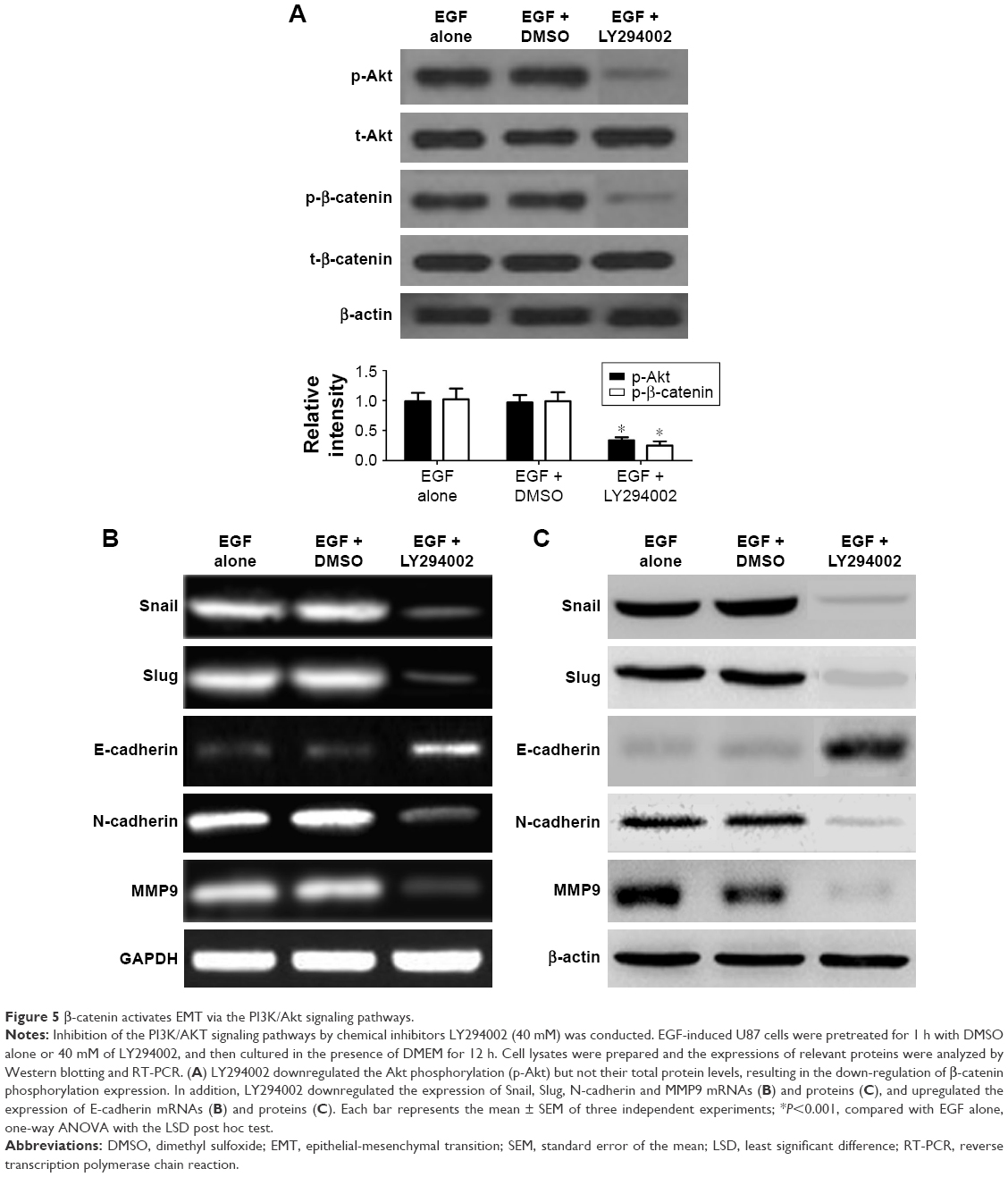 | Figure 5 β-catenin activates EMT via the PI3K/Akt signaling pathways. |
Discussion
To date, accumulating evidence indicates that EMT plays an important role in the initiation and development of cancer cells, and some extracellular components surrounding tumor microenvironment exert active effects on EMT.17–19 EGF, a kind of extracellular factor, can be derived from many types of cells within the tumor microenvironment. It has been reported as a kind of autocrine and paracrine factor to promote the development of tumor cells and stromal cells. Mechanistically, some studies reported that EGF is required for activation of Wnt/β-catenin and controls metastasis of cancer cells in vivo. With the help of immunohistochemistry, RT-PCR, and Western blot, our findings identified that a high expression of β-catenin is found in GBM tissues, and a lower β-catenin level was found in the glia cells of NB tissues and NHA, indicating that β-catenin is involved in the development of GBM.
Afterwards, we examined the expression of β-catenin in GBM cell lines, including T98G, A172, U87, and U251, and our results exhibited a relatively high expression of β-catenin mRNA and protein compared with human astrocytes. This altered expression of β-catenin in GBM cells might present a neuronal differentiation property in human GBM development. Therefore, we assumed that β-catenin expression was implicated in the progression of EMT, and then facilitated the development of GBM. The EMT is characterized by the invasion phenotype, which showed a decrease in the expression of the epithelial marker E-cadherin and an increase in the expression of the mesenchymal markers vimentin, N-cadherin, and β-catenin. Thus, inhibiting EMT would inhibit cancer cell invasion and ameliorate prognosis of GBM patients.20,21
Wnt/β-catenin signaling, a key regulator of tumorigenesis, plays an important role in the progression of human GBM. Increasing reports indicate that a complex cross-talk exists between the β-catenin pathway and other signaling molecules, and EMT-corresponding effectors.22–24 In the present study, we identified the in vitro effect of β-catenin on different malignant phenotypes in U87 glioblastoma cells. To validate the role of β-catenin in biological progression of tumors, we applied a specific siRNA for β-catenin to down-regulate endogenous β-catenin expression. We found that EMT activators Slug and Snail were obviously decreased compared with the control group. In addition, depletion of β-catenin expression increased expression of the epithelial marker E-cadherin but decreased expression of the mesenchymal markers N-cadherin and MMP9.
Besides, a previous study also validated that Akt signaling can directly stimulate the activity of β-catenin. Gantner et al demonstrated that resveratrol inhibited activation of Akt to affect activation of β-catenin.25 Namely, Akt phosphorylation is regulated by a cascade of signals, resulting in the activated PI3K/Akt pathway and the phosphorylation of GSK-3β, and eventually facilitates the nuclear translocation of β-catenin. In the present study, we utilized the PI3K/Akt antagonist LY294002 to block its signaling pathway in U87 cells, and then examined the change of EMT biomarkers. We observed that LY294002 decreased the phosphorylation of Akt but not total Akt proteins, and finally decreased β-catenin expression.
Furthermore, our cell function analysis revealed that down-regulation of β-catenin reduced cell proliferation in GBM cells in a time-dependent manner. Then, we found that down-regulation of β-catenin expression by siRNA suppressed the migration of U87 cells toward artificially created wounds in confluent cell monolayers. Meanwhile, down-regulation of β-catenin expression also led to fewer cell colonies in the transwell assay compared with the control. These findings are consistent with previous studies.26
In conclusion, our studies demonstrated that the expression of β-catenin contributes to tumorigenesis of GBM as well as the progression of EMT. High expression of β-catenin has been identified in GBM tissues. Additionally, the expression of β-catenin was involved in the proliferation and invasion of GBM cells. Thus, β-catenin acts as a crucial regulator in cancer progression and might be a useful and novel target for the treatment of GBM.
Acknowledgments
We gratefully thank other members of our lab for valuable suggestions and writing.
Disclosure
The authors report no conflicts of interest in this work.
References
Verhoeff JJ, van Tellingen O, Claes A, et al. Concerns about anti-angiogenic treatment in patients with glioblastoma multiforme. BMC Cancer. 2009;9:444. | ||
Dwarakanath BS, Singh D, Banerji AK, et al. Clinical studies for improving radiotherapy with 2-deoxy-D-glucose: present status and future prospects. J Cancer Res Ther. 2009;5 Suppl 1:S21–S26. | ||
Maucort-Boulch D, Baron MH, Pommier P, et al. Rationale for carbon ion therapy in high-grade glioma based on a review and a meta-analysis of neutron beam trials. Cancer Radiother. 2010;14(1):34–41. | ||
Martin SE, Bonnin JM, Hall DC, Hattab EM. Glioblastoma with signet-ring morphology: a case report and review of the literature. Hum Pathol. 2010;41(3):443–446. | ||
Romanelli P, Conti A, Pontoriero A, et al. Role of stereotactic radiosurgery and fractionated stereotactic radiotherapy for the treatment of recurrent glioblastoma multiforme. Neurosurg Focus. 2009;27(6):E8. | ||
Lakhan SE, Harle L. Difficult diagnosis of brainstem glioblastoma multiforme in a woman: a case report and review of the literature. J Med Case Rep. 2009;3:87. | ||
Jiang Y, Sheng H, Meng L, et al. RBM5 inhibits tumorigenesis of gliomas through inhibition of Wnt/β-catenin signaling and induction of apoptosis. World J Surg Oncol. 2017;15(1):9. | ||
Yang JW, Ma W, Luo T, et al. BDNF promotes human neural stem cell growth via GSK-3β-mediated crosstalk with the wnt/β-catenin signaling pathway. Growth Factors. 2016;34(1–2):19–32. | ||
Zhu Y, Qiu Z, Zhang X, et al. Jab1 promotes glioma cell proliferation by regulating Siah1/β-catenin pathway. J Neurooncol. 2017;131(1):31–39. | ||
Peng Z, Wu T, Li Y, et al. MicroRNA-370-3p inhibits human glioma cell proliferation and induces cell cycle arrest by directly targeting β-catenin. Brain Res. 2016;1644:53–61. | ||
Denysenko T, Annovazzi L, Cassoni P, Melcarne A, Mellai M, Schiffer D. WNT/β-catenin signaling pathway and downstream modulators in low- and high-grade glioma. Cancer Genomics Proteomics. 2016;13(1):31–45. | ||
Yang JW, Ru J, Ma W, et al. BDNF promotes the growth of human neurons through crosstalk with the Wnt/β-catenin signaling pathway via GSK-3β. Neuropeptides. 2015;54:35–46. | ||
Nie YC, Wu H, Li PB, et al. Naringin attenuates EGF-induced MUC5AC secretion in A549 cells by suppressing the cooperative activities of MAPKs-AP-1 and IKKs-IκB-NF-κB signaling pathways. Eur J Pharmacol. 2012;690(1–3):207–213. | ||
Nakata A, Gotoh N. Recent understanding of the molecular mechanisms for the efficacy and resistance of EGF receptor-specific tyrosine kinase inhibitors in non-small cell lung cancer. Expert Opin Ther Targets. 2012;16(8):771–781. | ||
Deharvengt S, Marmarelis M, Korc M. Concomitant targeting of EGF receptor, TGF-beta and SRC points to a novel therapeutic approach in pancreatic cancer. PLoS One. 2012;7(6):e39684. | ||
Glogowska A, Stetefeld J, Weber E, Ghavami S, Hoang-Vu C, Klonisch T. Epidermal growth factor cytoplasmic domain affects ErbB protein degradation by the lysosomal and ubiquitin-proteasome pathway in human cancer cells. Neoplasia. 2012;14(5):396–409. | ||
Shiota M, Itsumi M, Takeuchi A, et al. Crosstalk between epithelial-mesenchymal transition and castration resistance mediated by Twist1/AR signaling in prostate cancer. Endocr Relat Cancer. 2015;22(6):889–900. | ||
Zhang Z, Yang XF, Huang KQ, et al. The upregulated α-catulin expression was involved in head-neck squamous cell carcinogenesis by promoting proliferation, migration, invasion, and epithelial to mesenchymal transition. Tumour Biol. 2016;37(2):1671–1681. | ||
Buhrmann C, Shayan P, Kraehe P, Popper B, Goel A, Shakibaei M. Resveratrol induces chemosensitization to 5-fluorouracil through up-regulation of intercellular junctions, Epithelial-to-mesenchymal transition and apoptosis in colorectal cancer. Biochem Pharmacol. 2015;98(1):51–68. | ||
Wang TH, Lin YS, Chen Y, et al. Long non-coding RNA AOC4P suppresses hepatocellular carcinoma metastasis by enhancing vimentin degradation and inhibiting epithelial-mesenchymal transition. Oncotarget. 2015;6(27):23342–23357. | ||
Bousquet E, Calvayrac O, Mazières J, et al. RhoB loss induces Rac1-dependent mesenchymal cell invasion in lung cells through PP2A inhibition. Oncogene. 2016;35(14):1760–1769. | ||
Eiró N, Fernandez-Garcia B, Vázquez J, Del Casar JM, González LO, Vizoso FJ. A phenotype from tumor stroma based on the expression of metalloproteases and their inhibitors, associated with prognosis in breast cancer. Oncoimmunology. 2015;4(7):e992222. | ||
Ji W, Li Y, He Y, et al. AIP1 expression in tumor niche suppresses tumor progression and metastasis. Cancer Res. 2015;75(17):3492–3504. | ||
Jin L, Zhu C, Wang X, et al. Urocortin attenuates TGFβ1-induced Snail1 and slug expressions: inhibitory role of Smad7 in Smad2/3 signaling in breast cancer cells. J Cell Biochem. 2015;116(11):2494–2503. | ||
Gantner BN, Jin H, Qian F, Hay N, He B, Ye RD. The Akt1 isoform is required for optimal IFN-β transcription through direct phosphorylation of β-catenin. J Immunol. 2012;189(6):3104–3111. | ||
Jansen SR, Poppinga WJ, de Jager W, et al. Epac1 links prostaglandin E2 to β-catenin-dependent transcription during epithelial-to-mesenchymal transition. Oncotarget. 2016;7(29):46354–46370. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.