Back to Journals » Drug Design, Development and Therapy » Volume 8
The CB1 cannabinoid receptor agonist reduces L-DOPA-induced motor fluctuation and ERK1/2 phosphorylation in 6-OHDA-lesioned rats
Authors Song L, Yang X, Ma Y, Wu N, Liu Z
Received 17 January 2014
Accepted for publication 13 June 2014
Published 4 November 2014 Volume 2014:8 Pages 2173—2180
DOI https://doi.org/10.2147/DDDT.S60944
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Lu Song,* Xinxin Yang,* Yaping Ma, Na Wu, Zhenguo Liu
Department of Neurology, Xinhua Hospital, Shanghai Jiao Tong University School of Medicine, People’s Republic of China
*These authors contributed equally to this work
Abstract: The dopamine precursor L-3,4-dihydroxyphenylalanine (L-DOPA) has been used as an effective drug for treating dopamine depletion-induced Parkinson’s disease (PD). However, long-term administration of L-DOPA produces motor complications. L-DOPA has also been found to modify the two key signaling cascades, protein kinase A/dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32) and extracellular signal-regulated kinases 1 and 2 (ERK1/2), in striatal neurons, which are thought to play a pivotal role in forming motor complications. In the present study, we tested the possible effect of a CB1 cannabinoid receptor agonist on L-DOPA-stimulated abnormal behavioral and signaling responses in vivo. Intermittent L-DOPA administration for 3 weeks induced motor fluctuation in a rat model of PD induced by intrastriatal infusion of dopamine-depleting neurotoxin 6-hydroxydopamine (6-OHDA). A single injection of a CB1 cannabinoid receptor agonist WIN-55,212-2 had no effect on L-DOPA-induced motor fluctuation. However, chronic injections of WIN-55,212-2 significantly attenuated abnormal behavioral responses to L-DOPA in 6-OHDA-lesioned rats. Similarly, chronic injections of WIN-55,212-2 influence the L-DOPA-induced alteration of DARPP-32 and ERK1/2 phosphorylation status in striatal neurons. These data provide evidence for the active involvement of CB1 cannabinoid receptors in the regulation of L-DOPA action during PD therapy.
Keywords: striatum, the CB1 receptor agonist WIN-55,212-2, 6-hydroxydopamine, Parkinson’s disease, motor fluctuation, L-DOPA
Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder which is characterized by progressive loss of dopaminergic neurons in the substantia nigra.1 As one of the most common neurological diseases, PD affects approximately 1% of the human population.2 To relieve dopamine depletion, L-3,4-dihydroxyphenylalanine (L-DOPA), a dopamine precursor, has been used to treat PD and it to date remains the most effective drug for the treatment of PD.3 However, L-DOPA therapy causes a noticeable side effect. With the progression of the disease, L-DOPA reduces the parkinsonism at the cost of evoking motor complications.4,5 In fact, L-DOPA-induced motor complications can become so severely disabling as to negate any clinical benefit from dopaminergic therapy and significant efforts must be made to maintain control of symptoms in the advanced PD patient. Thus, we are forced to find an alternative drug which can reduce L-DOPA-induced motor complications without affecting L-DOPA-induced movements. At this point in time, the pathogenesis of L-DOPA-induced motor complications is not fully understood. Available data from recent studies seem to link distinct plastic changes in striatal neurons to L-DOPA-induced motor complications.6,7 In animals showing L-DOPA-induced motor complications after repeated L-DOPA administration, phosphorylation levels of dopamine- and cAMP-regulated phosphoprotein of 32 kDa (DARPP-32) and extracellular signal-regulated kinases 1 and 2 (ERK1/2) were increased in striatal neurons.8–10 These increases are believed to play a role, and are considered to be characteristic neurochemical changes in the development of L-DOPA-induced motor complications.
Previous studies have reported that the cannabinoid receptor agonist seems to be effective in attenuating L-DOPA-induced dyskinesia in experimental animals and patients,11–14 even though precise mechanisms underlying the anti-dyskinetic properties of the cannabinoid receptor agonist are unclear. It has been proposed that cannabinoids may affect psychomotor activity via regulating a signaling cascade in striatal projection neurons involving protein kinase A(PKA)-dependent phosphorylation of DARPP-32.15 However, the direct effect of the cannabinoid receptor agonist on the L-DOPA-stimulated phosphorylation of ERK1/2 has not been explored at present.
In this study, we attempted to investigate whether pharmacological activation of cannabinoid receptors with a selective agonist alters motor fluctuation induced by repeated injections of L-DOPA during the treatment of PD. A rat model of unilateral 6-hydroxydopamine (6-OHDA)-induced PD was used. In addition to neurobehavioral studies, we tested whether the cannabinoid receptor agonist affects the L-DOPA-associated phosphorylation of the molecular marker (DARPP-32, ERK1/2) in striatal neurons.
Materials and methods
Animals
Adult Sprague Dawley female rats (180–220 g) were used in this study. Animals were individually housed in clear plastic cages in a controlled environment at a constant temperature of 23°C and humidity of 50%±10% with food and water available ad libitum. The animal room was on a 12/12 hours light/dark cycle with lights on at 7:00 am. The rats were allowed 6–7 days of habituation to the animal colony before any treatment began. All protocols involving animals were approved by the Institutional Review Board of Xinhua Hospital and were performed according to the guidelines of the National Institutes of Health for the care and use of laboratory animals (NIH publication No 80-23).
Dopaminergic lesions
For creation of the 6-OHDA lesion, the rat was anesthetized with an intraperitoneal (IP) injection of pentobarbital (50 mg/kg). The animal’s head was fixed in a stereotactic apparatus (Narishige, Tokyo, Japan). All animals received injections of a total of 8 μg of 6-OHDA (dissolved in 4 μl of 0.9% physiological saline containing 0.02% ascorbic acid [Sigma-Aldrich Co., St Louis, MO, USA]). The coordinates were calculated with reference to bregma for the anterioposterior and the mediolateral coordinates using the rat brain atlas16 as follows: 1) anterioposterior – 3.7, mediolateral – 1.7; 2) anterioposterior – 4.4, mediolateral – 1.2. The dorsoventral position of all injections was −7.8 mm below the dura and the tooth bar set to −2.4 mm. Three weeks after injections, the rats that exhibited a stable apomorphine-induced rotational asymmetry of at least seven full turns per minute away from the lesioned side were selected for the next experiment. It has been previously demonstrated that rats meeting this criterion have a greater than 90% depletion of striatal dopamine.17
Treatment
Two experiments were conducted in this work. In the first experiment, the effect of acute IP administration of the CB1 receptor agonist WIN-55,212-2 (Cayman Chemical Company, MI, USA) on L-DOPA-treated rats was evaluated. Fourteen 6-OHDA-lesioned rats were treated with L-DOPA (50 mg/kg plus benserazide 12.5 mg/kg, IP), twice daily for 21 consecutive days. At day 22, fourteen rats were randomly divided into two groups and received co-administration of WIN-55,212-2 (1 mg/kg) or vehicle with L-DOPA (50 mg/kg) and benserazide (12.5 mg/kg). The dose of WIN-55,212-2 (1 mg/kg) was chosen because the agonist at this dose effectively activated the CB1 receptor in vivo.18 In the second experiment, the effect of chronic administration of WIN-55,212-2 on rats treated with L-DOPA was investigated. Fourteen 6-OHDA-lesioned rats were randomly divided into two groups. Animals in the first group were treated with co-administration (IP) of WIN-55,212-2 (1 mg/kg) with L-DOPA (50 mg/kg) and benserazide (12.5 mg/kg) for 21 days. Animals in the second group were treated with co-administration of vehicle with L-DOPA (50 mg/kg) and benserazide (12.5 mg/kg) for 21 days. Seven 6-OHDA-lesioned rats with injections of vehicle for 21 days served as the control.
Behavioral assessment
Rotational behavior was measured immediately after L-DOPA administration at the date indicated (1, 7, 14, 21, and 22 days). The duration of the rotational response was measured as the time between the first 5-minute interval when turning exceeded 20% of the peak rate and the first 5-minute interval when turning fell below 20% of the peak rate. The peak intensity of rotation was measured as the peak number of contralateral turns in any 5-minute interval. L-DOPA response failure was defined as the rats showing no obvious rotations during 5 minutes after L-DOPA administrations. Behavioral assessment was measured manually by a researcher who was blinded to this study.
Western blot
Animals were deeply anesthetized with pentobarbital (50 mg/kg, IP) and decapitated. Brains were quickly removed and the striatum was dissected. Striatal tissues were homogenized (1:10, weight-to-volume) in a homogenization buffer containing 20 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1% Triton-100, 1 mM NaF, 100 μM phenylmethylsulfonyl fluoride, and freshly-added protease inhibitor cocktail (Calbiochem, La Jolla, CA, USA). Cytosol fractions were prepared by centrifugation at 12,000× g for 10 minutes at 4°C. Total protein samples (40 μg) were separated on a 10% sodium dodecyl sulphate–polyacrylamide gel and electrophoretically transferred to polyvinylidene difluoride membranes in a Tris–glycine transfer buffer. The membranes were blocked in 5% (weight-to-volume) instant non-fat dried milk for 2 hours at room temperature, and incubated with primary antibodies against DARPP-32 (1:1000), ERK1/2 (1:1000), phospho-DARPP-32 at Thr75 (pDARPP-32; 1:500), phospho-ERK1/2 at Thr202/Tyr204 (pERK1/2, 1:500), β-actin (1:1000), α-tubulin (1:1000) at 4°C overnight. β-actin and α-tubulin were used as a loading control. All antibodies were purchased from Cell Signaling Technology, Inc., (Danvers, MA, USA). The membranes were subsequently washed with tris-buffered saline (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, and 0.05% Tween 20) and incubated with secondary horseradish peroxidase-conjugated IgG (1:1000) for 1 hour at room temperature. Immunoreactive proteins were visualized by LumiGLO (Cell Signaling Technology, Beverly, MA, USA) chemiluminescent reagent and peroxide. The light-emitting bands were detected with X-ray films. Quantitative analysis of Western blots was performed by calculating the relative density of the immunoreactive bands after acquisition of the blot image with a Nikon charge-coupled device video camera module and analysis with NIH Image (1.34). Values of each band of sample, after background subtraction, are compared with that of β-actin or α-tubulin and expressed as the mean percent of unlesioned striatum for each immunoblot.
Statistics
Data are expressed as mean ± standard error of mean. Statistical analysis was conducted by one-way analysis of variance, followed by Dunnett’s t-test. A P-value of less than 0.05 was considered statistically significant.
Results
Effects of acute administration of WIN-55,212-2 on L-DOPA-induced motor fluctuation
We first tested whether acute administration of the CB1 cannabinoid receptor agonist WIN-55,212-2 alters L-DOPA-induced motor fluctuation. At day 1, 7, 14, 21, and 22, L-DOPA-induced motor responses were measured immediately after L-DOPA injection (day 22) or after the second L-DOPA injection (day 1, 7, 14, and 21). We found that a daily injection of L-DOPA (days 1–21) shortened the duration of rotational responses (Figure 1A). Similarly, L-DOPA increased peak turning responses (Figure 1C). Since the shortened response duration resembled human motor fluctuation, daily L-DOPA in our experiments produced reliable wearing-off response. We then tested whether acute WIN-55,212-2 injection has any impact on L-DOPA-induced motor fluctuation responses. At day 22, pretreatment with WIN-55,212-2 did not modify the duration of rotational responses to L-DOPA (Figure 1B). No significant effect of WIN-55,212-2 was found on the peak turning (Figure 1D). Thus, a single dose of WIN-55,212-2 seems to be ineffective in affecting L-DOPA-induced motor fluctuation behavior.
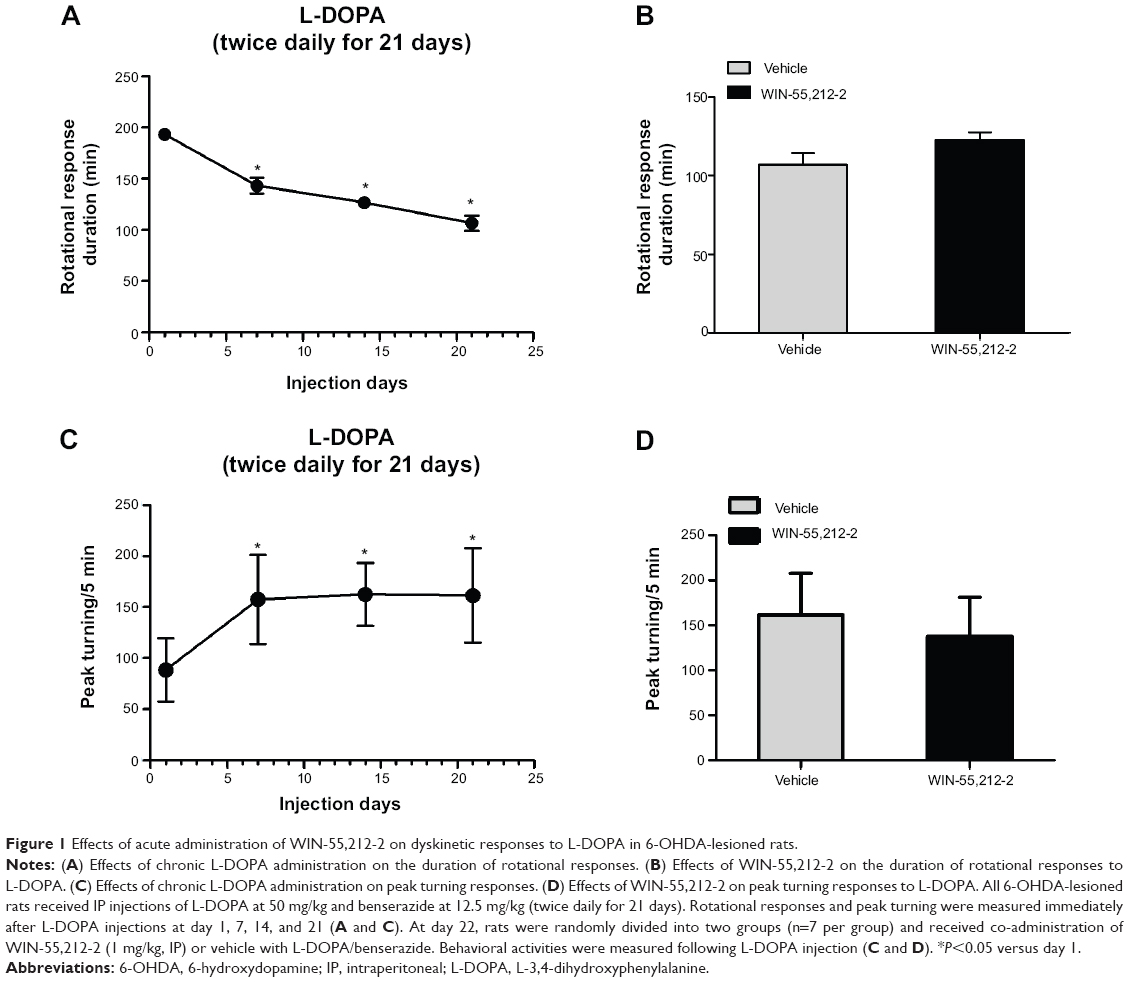 | Figure 1 Effects of acute administration of WIN-55,212-2 on dyskinetic responses to L-DOPA in 6-OHDA-lesioned rats. |
Effects of chronic administration of WIN-55,212-2 on L-DOPA-induced motor fluctuation
We then examined the effect of chronic administration of WIN-55,212-2. To this end, WIN-55,212-2 was administered 30 minutes prior to each injection of L-DOPA for 21 days. We found that WIN-55,212-2 partially and significantly blocked the L-DOPA-induced motor fluctuation in terms of the duration of rotational responses. As shown in Figure 2A, at day 14 and day 21, the duration of rotational responses in WIN-55,212-2-treated rats was 153.33±5.77 minutes and 153.33±5.77 minutes, which was significantly longer than the vehicle plus L-DOPA group (126.67±5.77 and 106.67±7.64 minutes). Although not statistically significant, WIN-55,212-2 showed the potential to reduce the amplitude of increased peak turning induced by L-DOPA at day 7, 14 and 21 (Figure 2B). These results demonstrate that the CB1 cannabinoid receptor agonist possesses the ability to alleviate L-DOPA-induced motor fluctuation.
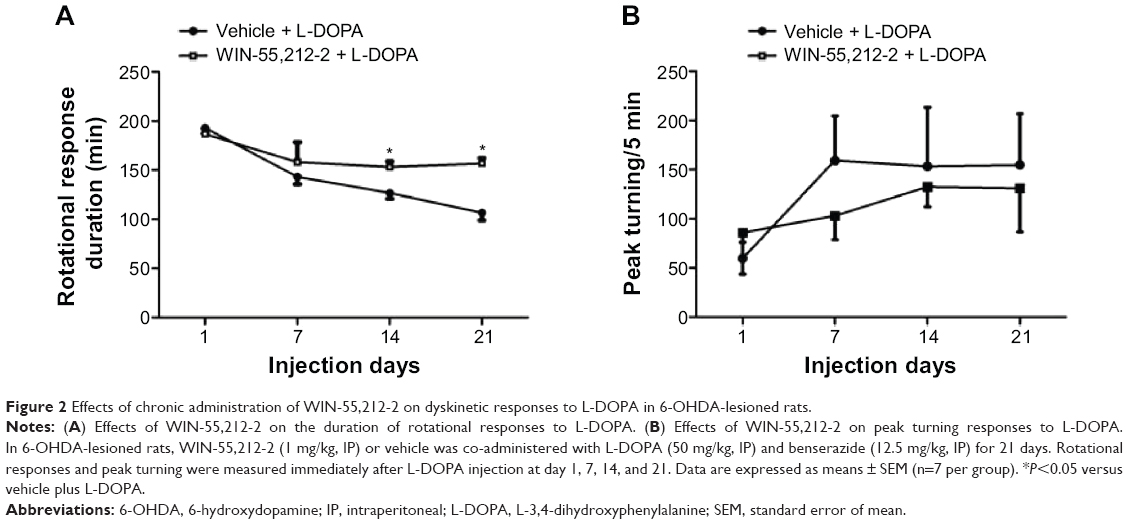 | Figure 2 Effects of chronic administration of WIN-55,212-2 on dyskinetic responses to L-DOPA in 6-OHDA-lesioned rats. |
Effects of WIN-55,212-2 on L-DOPA response failures
Prolonged L-DOPA administration causes characteristic L-DOPA response failures. We then evaluated whether chronic WIN-55,212-2 administration alters the response failure rate. During the first week of twice daily vehicle plus L-DOPA administration, approximately 3.6% of injections failed to elicit a motor response. Animals did not show any other signs of L-DOPA-induced response. By the second and third week of L-DOPA treatment, the frequency of ineffective injections (response failures) had increased to 23.8% and 28.6% separately, and this was observed in 100% of the animals treated with L-DOPA plus vehicle. However, only one injection failed to elicit a motor response in one rat during the chronic WIN-55,212-2 plus L-DOPA treatment. Thus, chronic administration of WIN-55,212-2 is able to reduce the occurrence of L-DOPA response failures.
Effects of WIN-55,212-2 on L-DOPA-stimulated DARPP-32 phosphorylation
The phosphorylation site and level of DARPP-32 is a biochemical marker of L-DOPA-induced dyskinesia. We thus tested whether WIN-55,212-2 affects L-DOPA-induced DARPP-32 phosphorylation in striatal neurons. As shown in Figure 3, daily injections of L-DOPA (50 mg/kg, IP, benserazide 12.5 mg/kg, IP) produced a significant decrease in DARPP-32 phosphorylation at Thr75 site in the striatum (48.97%±1.57%, F=814.77). Noticeably, co-administration of WIN-55,212-2 (1 mg/kg, IP) with L-DOPA induced an advanced decrease in phosphorylation DARPP-32 level (35.23%±3.67%, F=814.77). These data indicate that WIN-55,212-2 alters the response of DARPP-32 to L-DOPA.
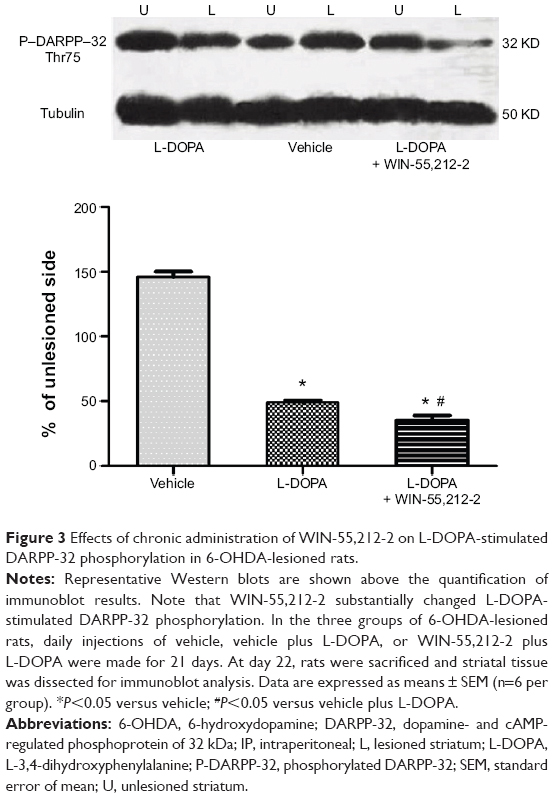 | Figure 3 Effects of chronic administration of WIN-55,212-2 on L-DOPA-stimulated DARPP-32 phosphorylation in 6-OHDA-lesioned rats. |
Effects of WIN-55,212-2 on L-DOPA-stimulated ERK1/2 phosphorylation
To determine the role of CB1 receptors in regulating ERK1/2 responses to L-DOPA, we tested the effect of WIN-55,212-2 on L-DOPA-stimulated ERK1/2 phosphorylation. As shown in Figure 4, L-DOPA (50 mg/kg, IP, benserazide 12.5 mg/kg, IP, twice daily for 21 days) induced a significant increase in phosphorylated ERK1/2 protein levels in the striatum (124.81%±3.74%, F=1858.39). WIN-55,212-2 co-administered with L-DOPA blocked this increase (117.07±1.46, F=1858.39), but did not normalize to the vehicle group level. But these results still indicate that ERK1/2 phosphorylation induced by L-DOPA was a signaling event sensitive to the downregulation by CB1 receptors.
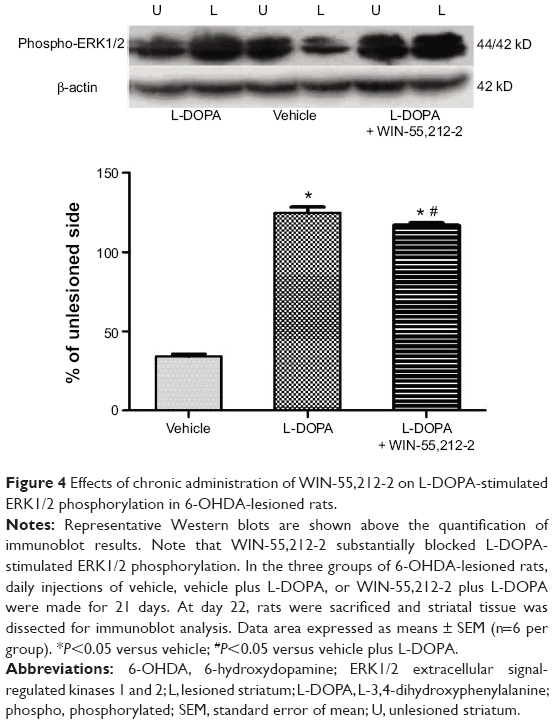 | Figure 4 Effects of chronic administration of WIN-55,212-2 on L-DOPA-stimulated ERK1/2 phosphorylation in 6-OHDA-lesioned rats. |
Discussion
This study investigated the effect of the CB1 cannabinoid receptor agonist on L-DOPA-induced motor fluctuation behavior and neurochemical changes in rat striatal neurons in vivo. We found that acute injection of the CB1 receptor agonist WIN-55,212-2 did not affect rotational responses to L-DOPA. Of note, chronic WIN-55,212-2 administration substantially blocked motor fluctuation induced by L-DOPA. Additionally, chronic administration of WIN-55,212-2 blocked L-DOPA-induced ERK1/2 phosphorylation alteration in the striatum. These data suggest that the CB1 receptor is a sensitive site for influencing the development of motor fluctuation. Stimulation of these receptors exerts an inhibitory modulation of abnormal responses to L-DOPA.
In the 6-OHDA-lesioned rat model of PD, chronic L-DOPA administration readily induced motor fluctuation behavior in this study and others.19–21 These changes in the motor response are considered as a valid model of the “wearing off” motor fluctuation.17 L-DOPA-induced dyskinesia is common, but not necessarily present in patients who have developed a “wearing off” response, and, indeed, both motor complications are not believed to originate by the same mechanisms.22 It has been reported that administration of the cannabinoid agonist can attenuate L-DOPA-induced dyskinesia by assessing axial, limb, and oral abnormal involuntary movements.11–13 Our result demonstrated co-administration of the CB1 cannabinoid receptor agonist WIN-55,212-2 ameliorated the motor fluctuation and response failures induced by L-DOPA. In contrast to chronic treatment, acute administration of WIN-55,212-2 had no significant effect on the L-DOPA-induced motor fluctuation. Thus, repeated stimulation of CB1 receptors seems to be needed to render the receptor efficacy in suppressing motor fluctuation.
How CB1 receptors inhibit L-DOPA-induced motor fluctuation is unclear. CB1 receptors are known to be enriched in brain areas important for the regulation of motor activity, including the basal ganglia.23 The receptor and its endogenous ligand endocannabinoid play a critical role in the modulation of basal ganglia activity.24,25 CB1 receptors are also known to co-express with dopamine D1, D2 receptors in the striatum.23 This co-expression has a functional implication as stimulation of CB1 receptors modified D1, D2-mediated cellular and behavioral responses.26 Since non-physiological stimulation of dopamine receptors contributes enormously to the development of L-DOPA-induced motor complications,27–29 CB1 receptors are believed to counteract with dopamine receptors to antagonize the side effect of L-DOPA. In addition, CB1 receptors are shown to modify striatal glutamatergic and GABAergic activity.30 Through modifying these systems, CB1 receptors may exert their action.
At the transcriptional level, we found that CB1 stimulation downregulates the phosphorylation of DARPP-32 at Thr75 site, and suppresses the L-DOPA-induced upregulation of ERK phosphorylation. DARPP-32, which is abundantly expressed in the medium spiny neurons of the striatum is one of important downstream effectors of the cAMP/PKA pathway.31 Its phosphorylation at Thr34 by PKA converts it into an effective inhibitor of protein phosphatase-1, a phosphatase highly expressed in striatal medium spiny neurons.32,33 By inhibiting protein phosphatase-1, DARPP-32 increases phosphorylation of distinct signaling proteins important for the regulation of gene expression. In addition to Thr-34, phosphorylation of DARPP-32 at Thr-75 site by cyclin–dependent kinase 5 blocks PKA-mediated phosphorylation of Thr-34 of DARPP-32, thereby oppositely modulating the efficacy of the PKA signaling cascade.34 The striatonigral and striatopallidal pathways exert opposing influence on the basal ganglia’s control of locomotor function.35
Therefore, the stimulation of the PKA pathway in striatonigral projecting neurons enhances the locomotor activity, and in striatopallidal projecting neurons acts oppositely.36 It was reported that the motor depression produced by CP55,940, a CB1 receptor agonist, was reduced by genetic inactivation either of dopamine D2 receptors or of adenosine A2A receptors.15 This indicates that a considerable proportion of the psychomotor effect of cannabinoids can be accounted for by a signaling cascade in striatopallidal projecting neurons involving PKA-dependent phosphorylation of DARPP-32, achieved via modulation of dopamine D2 and adenosine A2A transmission. Therefore, the motor suppression effect induced by co-administration of WIN-55,212-2 may occur through stimulating the PKA pathway in striatopallidal projecting neurons.
Active ERK1/2 have been well documented to be translocated into the nucleus to activate discrete transcription factors such as cAMP-responsive element-binding protein and Elk-1.37 Active cAMP-responsive element-binding protein and Elk-1 then regulate a program of gene expression to contribute to L-DOPA-induced motor complications. In this study, the CB1 receptor agonist showed the ability to suppress the L-DOPA-stimulated ERK1/2 phosphorylation, but did not normalize the ERK phosphorylation to PD group level. Thus, CB1 receptors may prevent motor fluctuation through a mechanism involving the modulation of the striatonigral and striatopallidal pathway.
Acknowledgments
The study was supported by the Key Science Development Program of Shanghai Municipal Education Commission China (No. 10ZZ72), the National Science Foundation of China (No. 81171203 and 81200871), and NIH grants (R01DA010355 and R01MH061469).
Disclosure
The authors report no conflicts of interest in this work.
References
Sherer TB, Betarbet R, Greenamyre JT. Pathogenesis of Parkinson’s disease. Curr Opin Investig Drugs. 2001;2(5):657–662. | ||
Simuni T, Sethi K. Nonmotor manifestations of Parkinson’s disease. Ann Neurol. 2008;64 Suppl 2:S65–S80. | ||
Eskow KL, Dupre KB, Barnum CJ, Dickinson SO, Park JY, Bishop C. The role of the dorsal raphe nucleus in the development, expression, and treatment of L-dopa-induced dyskinesia in hemiparkinsonian rats. Synapse. 2009;63(7):610–620. | ||
Carta M, Carlsson T, Kirik D, Björklund A. Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain. 2007;130(Pt 7):1819–1833. | ||
Jaunarajs KL, Dupre KB, Steiniger A, et al. Serotonin 1B receptor stimulation reduces D1 receptor agonist-induced dyskinesia. Neuroreport. 2009;20(14):1265–1269. | ||
Santini E, Valjent E, Fisone G. Parkinson’s disease: levodopa-induced dyskinesia and signal transduction. FEBS J. 2008;275(7):1392–1399. | ||
Fox SH, Chuang R, Brotchie JM. Parkinson’s disease – opportunities for novel therapeutics to reduce the problems of levodopa therapy. Prog Brain Res. 2008;172:479–494. | ||
Pavón N, Martín AB, Mendialdua A, Moratalla R. ERK phosphorylation and FosB expression are associated with L-DOPA-induced dyskinesia in hemiparkinsonian mice. Biol Psychiatry. 2006;59(1):64–74. | ||
Santini E, Valjent E, Usiello A, et al. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J Neurosci. 2007;27(26):6995–7005. | ||
Westin JE, Vercammen L, Strome EM, Konradi C, Cenci MA. Spatiotemporal pattern of striatal ERK1/2 phosphorylation in a rat model of L-DOPA-induced dyskinesia and the role of dopamine D1 receptors. Biol Psychiatry. 2007;62(7):800–810. | ||
Fox SH, Henry B, Hill M, Crossman A, Brotchie J. Stimulation of cannabinoid receptors reduces levodopa-induced dyskinesia in the MPTP-lesioned nonhuman primate model of Parkinson’s disease. Mov Disord. 2002;17(6):1180–1187. | ||
Morgese MG, Cassano T, Cuomo V, Giuffrida A. Anti-dyskinetic effects of cannabinoids in a rat model of Parkinson’s disease: role of CB(1) and TRPV1 receptors. Exp Neurol. 2007;208(1):110–119. | ||
Segovia G, Mora F, Crossman AR, Brotchie JM. Effects of CB1 cannabinoid receptor modulating compounds on the hyperkinesia induced by high-dose levodopa in the reserpine-treated rat model of Parkinson’s disease. Mov Disord. 2003;18(2):138–149. | ||
Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol. 2000;23(2):82–85. | ||
Andersson M, Usiello A, Borgkvist A, et al. Cannabinoid action depends on phosphorylation of dopamine- and cAMP-regulated phosphoprotein of 32 kDa at the protein kinase A site in striatal projection neurons. J Neurosci. 2005;25(37):8432–8438. | ||
Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. San Diego: Academic Press; 2007. | ||
Papa SM, Engber TM, Kask AM, Chase TN. Motor fluctuations in levodopa treated parkinsonian rats: relation to lesion extent and treatment duration. Brain Res. 1994;662(1–2):69–74. | ||
Ferrer B, Asbrock N, Kathuria S, Piomelli D, Giuffrida A. Effects of levodopa on endocannabinoid levels in rat basal ganglia: implications for the treatment of levodopa-induced dyskinesias. Eur J Neurosci. 2003;18(6):1607–1614. | ||
Engber TM, Papa SM, Boldry RC, Chase TN. NMDA receptor blockade reverses motor response alterations induced by levodopa. Neuroreport. 1994;5(18):2586–2588. | ||
Marin C, Jiménez A, Bonastre M, et al. LY293558, an AMPA glutamate receptor antagonist, prevents and reverses levodopa-induced motor fluctuations in Parkinsonian rats. Synapse. 2001;42(1):40–47. | ||
Marin C, Aguilar E, Bonastre M, Tolosa E, Obeso JA. 2005. Early administration of entacapone prevents levodopa-induced motor fluctuations in hemiparkinsonian rats. Exp Neurol. 2005;192(1):184–193. | ||
Jenner P. The MPTP-treated primate as a model of motor complications in PD: primate model of motor complications. Neurology. 2003;61(6 Suppl 3):S4–S11. | ||
García-Arencibia M, García C, Fernández-Ruiz J. Cannabinoids and Parkinson’s disease. CNS Neurol Disord Drug Targets. 2009;8(6):432–439. | ||
Corbillé AG, Valjent E, Marsicano G, et al. Role of cannabinoid type 1 receptors in locomotor activity and striatal signaling in response to psychostimulants. J Neurosci. 2007;27(26):6937–6947. | ||
Lerner TN, Horne EA, Stella N, Kreitzer AC. Endocannabinoid signaling mediates psychomotor activation by adenosine A2A antagonists. J Neurosci. 2010;30(6):2160–2164. | ||
Pérez-Rial S, García-Gutiérrez MS, Molina JA, et al. Increased vulnerability to 6-hydroxydopamine lesion and reduced development of dyskinesias in mice lacking CB1 cannabinoid receptors. Neurobiol Aging. 2011;32(4):631–645. | ||
Morissette M, Dridi M, Calon F, et al. Prevention of dyskinesia by an NMDA receptor antagonist in MPTP monkeys: effect on adenosine A2A receptors. Synapse. 2006;60(3):239–250. | ||
Johnston TH, van der Meij A, Brotchie JM, Fox SH. Effect of histamine H2 receptor antagonism on levodopa-induced dyskinesia in the MPTP-macaque model of Parkinson’s disease. Mov Disord. 2010;25(10):1379–1390. | ||
Ouattara B, Hoyer D, Grégoire L, et al. Changes of AMPA receptors in MPTP monkeys with levodopa-induced dyskinesias. Neuroscience. 2010;167(4):1160–1167. | ||
Cao X, Liang L, Hadcock JR, et al. Blockade of cannabinoid type 1 receptors augments the antiparkinsonian action of levodopa without affecting dyskinesias in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated rhesus monkeys. J Pharmacol Exp Ther. 2007;323(1):318–326. | ||
Greengard P. The neurobiology of slow synaptic transmission. Science. 2001;294(5544):1024–1030. | ||
da Cruz e Silva EF, Fox CA, Ouimet CC, Gustafson E, Watson SJ, Greengard P. Differential expression of protein phosphatase 1 isoforms in mammalian brain. J Neurosci. 1995;15(5 Pt 1):3375–3389. | ||
Hemmings HC Jr, Greengard P, Tung HY, Cohen P. DARPP-32, a dopamine-regulated neuronal phosphoprotein, is a potent inhibitor of protein phosphatase-1. Nature. 1984;310(5977):503–505. | ||
Bibb JA, Snyder GL, Nishi A, et al. Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature. 1999;402(6762):669–671. | ||
Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12(10):366–375. | ||
Bateup HS, Svenningsson P, Kuroiwa M, et al. Cell type-specific regulation of DARPP-32 phosphorylation by psychostimulant and antipsychotic drugs. Nat Neurosci. 2008;11(8):932–939. | ||
Xing J, Ginty DD, Greenberg ME. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996;273(5277):959–963. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.