Back to Journals » Journal of Inflammation Research » Volume 14
The Binary Classification of Protein Kinases
Authors Elkoshi Z
Received 25 January 2021
Accepted for publication 24 February 2021
Published 18 March 2021 Volume 2021:14 Pages 929—947
DOI https://doi.org/10.2147/JIR.S303750
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Zeev Elkoshi
Research and Development Department, Taro Pharmaceutical Industries Ltd, Haifa, Israel
Correspondence: Zeev Elkoshi Email [email protected]
Abstract: In an earlier publication a binary model for chronic diseases classification has been proposed. According to the model, chronic diseases were classified as “high Treg” or “low Treg” diseases, depending on whether the immune response is anti- or pro-inflammatory and assuming that regulatory T cells are major determinants of the response. It turned out that most cancers are “high Treg” diseases, while autoimmune diseases are “low Treg”. This paper proposes a molecular cause for this binary response. The mechanism proposed depends on the effect of protein kinases on the immune system. Thus, protein kinases are classified as anti- or pro-inflammatory kinases depending on whether they drive “high Treg” or “low Treg” diseases. Observations reported in the earlier publication can be described in terms of anti-inflammatory kinase (AIK) or pro-inflammatory kinase (PIK) activity. Analysis of literature data reveals that the two classes of kinases display distinctive properties relating to their interactions with pathogens and environmental factors. Pathogens that promote Treg activity (“high Treg” pathogens) activate AIKs, while pathogens that suppress Treg activity (“low Treg” pathogens) activate PIKs. Diseases driven by AIKs are associated with “high Treg” pathogens while those diseases driven by PIKs are associated with “low Treg” pathogens. By promoting the activity of AIKs, alcohol consumption increases the risk of “high Treg” cancers but decreases the risk of some “low Treg” autoimmune diseases. JAK1 gain-of-function mutations are observed at high frequencies in autoimmune diseases while JAK1 loss-of-function mutations are observed at high frequencies in cancers with high tumor-infiltrating Tregs. It should also be noted that the corresponding two classes of protein kinase inhibitors are mutually exclusive in terms of their approved therapeutic indications. There is no protein kinase inhibitor that is approved for the treatment of both autoimmune diseases and “high Treg” cancers. Although there are exceptions to the conclusions presented above, these conclusions are supported by the great bulk of published data. It therefore seems that the binary division of protein kinases is a useful tool for elucidating (at the molecular level) many distinctive properties of cancers and autoimmune diseases.
Keywords: protein kinases, regulatory T cells, autoimmunity, cancer, inflammation
Introduction
The protein kinase family is a large family of kinases (at least 518 members in man)1 that catalyze the phosphorylation of proteins, resulting in the production of phosphoproteins. Only three amino acids are modified in this way: serine, threonine, and tyrosine. These amino acids are characterized by a hydroxyl group attached to the hydrocarbon backbone. Adenosine triphosphate is used in this process as the phosphate donor. Dephosphorylation of phosphoproteins, the reverse-reaction, is catalyzed by protein phosphatases, using adenosine diphosphate as the phosphate acceptor. Since phosphorylation has a profound effect on protein activity, and since at least two-thirds of proteins encoded by human genome are subject to phosphorylation,2 the role of protein kinases in the control of different cellular activities (such as metabolism, proliferation, and apoptosis) is crucial. In fact, protein phosphorylation is one of the most important post-translational modifications of proteins.3
Deregulation of protein kinase activity is associated with many cancers (colorectal cancer, non-small cell lung cancer (NSCLC), renal cell carcinoma (RCC), thyroid cancer, breast cancer, glioblastoma, pancreatic cancer, ovarian cancer, chronic myelogenous leukemia (CML), chronic lymphoid leukemia (CLL), myelofibrosis, and acute myeloid leukemia (AML)) with autoimmune diseases (rheumatoid arthritis (RA) and psoriasis (Ps)), with age-related macular degeneration (AMD),4 with cutaneous disorders (atopic dermatitis, pruritus associated with allergic dermatitis, vitiligo, and alopecia),5 and with atherosclerosis.6,7
In an earlier paper, a binary classification of chronic diseases was proposed.8 Chronic diseases were classified according to the extent of regulatory T cell (Treg) activity, estimated in peripheral blood or within tissues implicated in the disease. Diseases with high Treg activity as a driver of pathogenicity were classified as “high Treg” diseases (most solid cancers, for example). Diseases with low Treg activity as a driver of pathogenicity were classified as “low Treg” diseases (autoimmune diseases, for example). This classification explains the association of particular pathogens with cancer and the association of others with autoimmune diseases. It also explains why certain specific pathogens are involved in coinfections. The effectiveness or ineffectiveness of certain immune-modulating drugs in the treatment of autoimmunity and cancer is also elucidated by this binary model.8 In addition, it explains why “high Treg” inflammation promotes many solid cancers while “low Treg” inflammation promotes lymphomas.9
This paper focuses on protein kinases as the cause of this binary response. A binary classification of protein kinases as anti-inflammatory kinases (AIKs) or pro-inflammatory kinases (PIKs) is therefore proposed. Frequently, but not always, the effect of protein kinases on the immune system is mediated by Tregs, where AIKs promote Treg activity, while PIKs suppress it. One way to practically classify protein kinases as PIKs or AIKs is by the immunological effect observed following their inhibition. Based on the analysis of literature data, this paper shows that, in general, aberrant activity of AIKs promotes “high Treg” cancers while deregulated activity of PIKs promotes autoimmune diseases. It seems that in addition to the direct effect of AIKs on tumor proliferation,2 a simultaneous or consecutive induction of immune tolerance (“high Treg” response) by these AIKs drives cancer development as well. Similarly, in autoimmune diseases, in addition to the direct effect of PIKs on the target tissue, an induction of a pro-inflammatory immune response (“low Treg” response) by these kinases increases the collateral damage to the target tissue and advances the disease.
Based on the analysis of literature data, it is also shown that the pathogens that promote Treg activity (“high Treg” pathogens) activate AIKs while pathogens that suppress Treg activity (“low Treg” pathogens) activate PIKs. This explains the association of most solid cancers with “high Treg” pathogens and of autoimmune diseases with “low Treg” pathogens, which was reported in earlier publications.8,9 Moreover, pathogens that activate both AIKs and PIKs may induce both cancers and autoimmune diseases.
The activation of AIKs by ethanol can explain the increased risk of “high Treg” cancers10 and the decreased risk of some autoimmune diseases11 observed in heavy and occasional drinkers of alcohol.
It is also observed that the gain-of-function or loss-of-function mutations of the same protein kinase may induce “low Treg” or “high Treg” diseases. JAK1 gain-of-function is frequent in diseases with low Treg frequency while JAK1 loss-of-function is frequent in diseases with high Treg frequency.
Lastly, it is noted that the corresponding two classes of protein kinase inhibitors are mutually exclusive with respect to their approved therapeutic indications: to date, there is no approved protein kinase inhibitor that is indicated for the treatment of both autoimmune diseases and “high Treg” cancers. These distinctive properties of the two classes of kinases are described in detail in the next sections.
AIKs are Involved in the Pathology of Solid Cancers, CLL, ALL, Mantle Cell Lymphoma (MCL), and CML
Many protein kinase inhibitors that are effective in the treatment of solid cancers, CLL, CML, ALL, and MCL induce a decrease in Treg frequency or an impairment in Treg function. This implies that the protein kinases involved are AIKs.
Table 1 presents protein kinase inhibitors effective in these diseases, their target kinases, their effect on Treg activity, references for the effect on Tregs, and Tregs marker used.
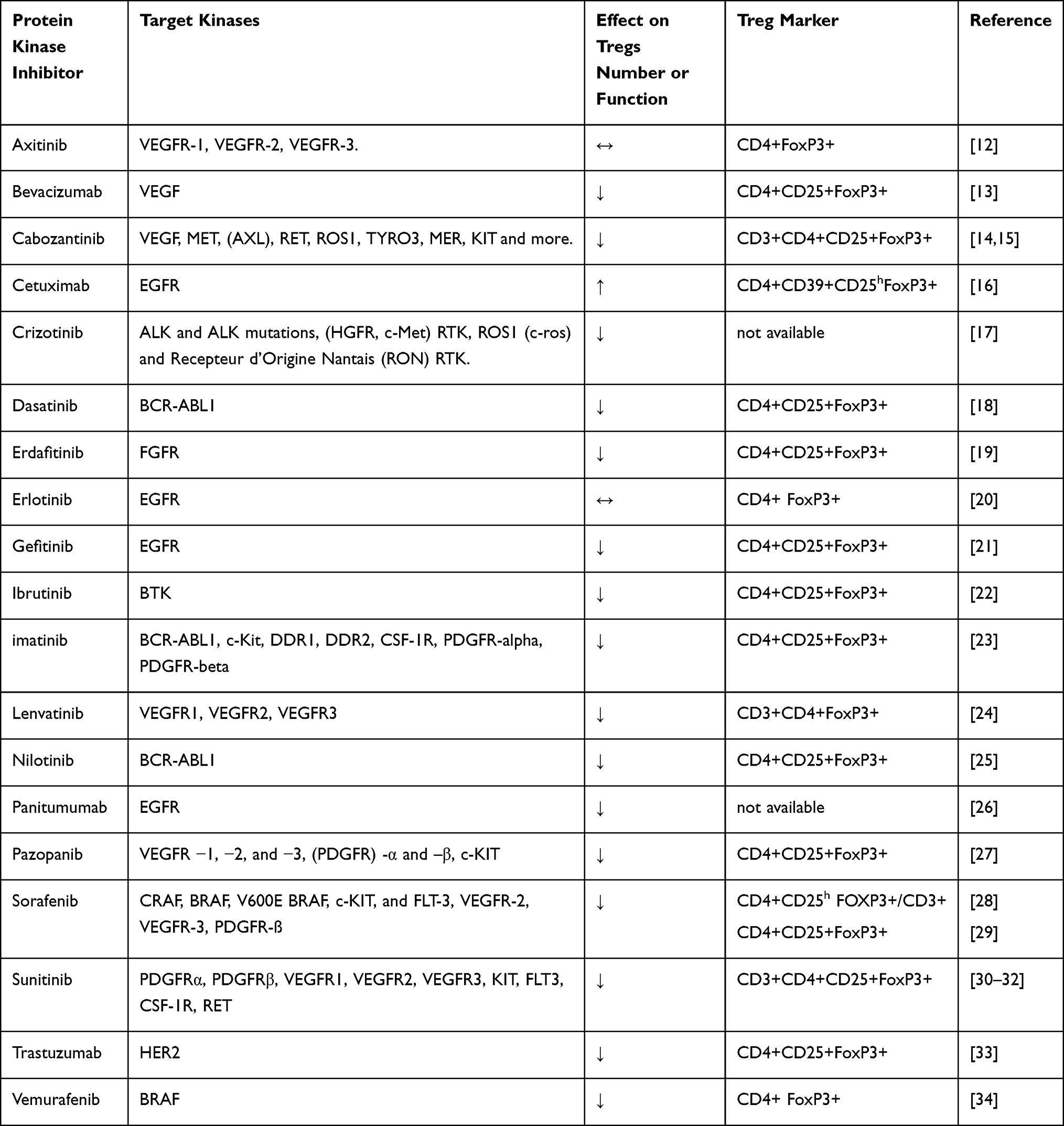 |
Table 1 Protein Kinase Inhibitors Effective in “High Treg” Diseases, Their Effects on Tregs Activity (↑ = an Increase, ↓ = a Decrease, ↔ = No Effect) and Treg Markers Used |
It should be realized that Tregs identification methods used in order to sort Tregs from conventional T cells expressing similar markers have been evolved over the years. Different markers affect the specificity in sorting out Tregs. For this reason, literature data presented in this paper in relation to Tregs frequency evaluation, include also the particular markers used.
In Table 1, it can be seen that 16 out of the 19 kinase inhibitors suppress Treg number or function; two show no effect on Tregs and one promotes Treg number. It is of note that the two studies that show no effect used a low specificity Treg marker (CD4+FoxP3+). It can be concluded that most of these kinases show a pro-inflammatory effect.
PIKs are Involved in Autoimmune Diseases and Myeloproliferative Neoplasms
JAKs are Pro-Inflammatory Tyrosine Kinases
The four members of the Janus kinase family, JAK1, JAK2, JAK3, and TYK2, form one subgroup of the non-receptor protein tyrosine kinases. Whereas JAK1, JAK2, and TYK2 are expressed ubiquitously in mammals, JAK3 is primarily expressed in hematopoietic cells.35
JAK1: Conflicting data regarding the pro- or anti-inflammatory effect of JAK1 activation, under different conditions, have been published over the years. Nevertheless, a JAK1 inhibitor, upadacitinib (Rinvoqu®), was recently approved in the USA and EU for the treatment of RA, indicating a pro-inflammatory effect of JAK1 within the setting of autoimmune diseases.
JAK2: Stimulation of the JAK2/STAT3 pathway induces a pro-inflammatory reaction with reduced Treg activity.36 BCR-ABL1 negative myeloproliferative neoplasms (hereafter, MPNs) are a group of rare hematological cancers where JAK2 mutation (JAK2V617F) is often observed. Barbui et al have shown that C reactive protein, a very common inflammation marker, positively correlates with JAK2V617F allele burden in MPN patients.37 Much clinical and pathological evidence indicates that deregulated JAK2 activity induces a pro-inflammatory reaction in MPNs.38 As far as autoimmune diseases are concerned, JAK2 is involved in the pathogenesis of PsA and spondyloarthropathy (SpAs).39 It seems that JAK2 is a PIK.
JAK3: Despite conflicting in vitro data, in vivo data indicate a pro-inflammatory effect of JAK3 activation. A highly selective JAK3 inhibitor (RB1) exerted significantly improved joints pathology in a collagen-induced arthritis mouse model.40 In addition, decernotinib, an experimental selective JAK3 inhibitor, was efficacious in improving RA clinical symptoms in a Phase I study with 204 RA patients.41
TYK2: IL-12-dependent signals, in particular those involved with IFN-γ production by Th1 cells, are TYK2 dependent.42 The involvement of TYK2 in IL-23-dependent inflammatory conditions, such as psoriasis and colitis, has been demonstrated in mice models.43 A single nucleotide polymorphism in TYK2 gene has been reported in Crohn’s disease and in systemic lupus erythematosus (SLE)44 and references therein. It is seen therefore that TYK2 aberrant signaling results in a pro-inflammatory reaction.
It seems that all four members of the JAK family play mainly a pro-inflammatory role within the context of autoimmune diseases and MPNs.
Spleen Tyrosine Kinase is a PIK
Spleen tyrosine kinase (SYK) is a member of the SYK family of tyrosine kinases. It is overexpressed in T cells of patients with SLE. It has been shown that induced overexpression of SYK in T cells from healthy individuals resulted in a pro-inflammatory effect that could be reverted by SYK inhibition. SYK deregulation may hamper IL-2 production which results in reduced Treg differentiation.45 It seems therefore that SYK tyrosine kinase is a PIK.
SYK signaling is prominent in the process of platelet destruction in adults with immune thrombocytopenia (ITP), an autoimmune disease.46 Most patients with ITP show increased Th1 and decreased Th2 and Treg frequencies in blood.47 An SYK inhibitor, fostamatinib, was approved by FDA in 2018 for the treatment of ITP.
Most JAK Inhibitors Induce an Anti-Inflammatory Effect That Might or Might Not Be Mediated by Tregs
As seen in Table 2, the effect of JAK inhibitors on Treg frequency is variable. It can depend on the inhibitor and can also vary between studies with the same inhibitor. Out of 11 studies performed with different JAK inhibitors, five indicate an increase in Tregs, four indicate a decrease in Tregs (3 with ruxolitinib and 1 with fedratinib). In two studies no change in Treg frequency was observed following JAK inhibition. However, in all four studies with a decreased Treg frequency, a parallel decrease in pro-inflammatory cytokines secretion was reported. It is not clear from these studies whether this down-regulation of pro-inflammatory cytokine is a direct effect of the inhibitor or whether this is an indirect effect of an over-suppressive Treg function. Keohane et al reported that “Tregs appear functional in vivo following JAK inhibition” in MPN patients.48 However, the authors did not compare suppressive Treg function following JAK inhibition, with suppressive Treg function in untreated MPNs patients. On the other hand, Sewgobind et al report of 56% (mean) increase in suppressive Treg effects on the proliferation of alloactivated Teff cells, following tofacitinib treatment of kidney transplant patients.49 Similarly, Meyer et al report of a significant suppression of Th17 cell percentage following tofacitinib treatment in RA patients.50 It seems that although some JAK inhibitors do not induce an increase in Treg frequency, they still exert a suppressive anti-inflammatory effect.
 |
Table 2 Protein Kinase Inhibitors Effective in “Low Treg” Diseases, Their Target Kinases,48,52,55,59 Their Effect on Treg and Th17 Cells Activities, Reference(s) for This Effect (↑ = an Increase, ↓ = a Decrease, ↔ = No Effect) and Treg Markers Used |
SYK Inhibitor Induces an Anti-Inflammatory Effect
An SYK inhibitor, R406, has been shown to attenuate psoriatic inflammation, upregulate Treg cells and downregulate Th17 cells in a mouse model of psoriasis.51 As an inhibitor of PIK, it is presented in Table 2, next to JAK inhibitors.
AIKs Drive Both a Direct Pathogenic Effect and a “High Treg” Immune Response in Most Solid Cancers
Deregulated activity of protein kinases drives many types of cancers by inducing cancer cell proliferation.4 On the other hand, the tumor microenvironment (TME) of most solid cancers (and of some hematological cancers) is enriched with regulatory T cells, and their accumulation in the TME is predictive of poor prognosis in most types of solid cancers (probably due to the immune-suppressive effect exerted by Tregs). These cancers are “high Treg” diseases.8 Accordingly, protein kinases that drive these malignancies are AIKs. It seems reasonable to assume that the same AIKs affect both cancer growth and Treg proliferation. As shown below, there are two ways of driving this double effect: (a) some AIKs are expressed by both tumor cells and Treg cells. Hyper-activation of these kinases may therefore affect simultaneously tumor epithelial cell propagation and Treg activity; (b) certain tumorigenic kinases have been reported to induce the activity of dendritic cells (DCs) which in turn promote Treg propagation, function, or recruitment. The examples below illustrate these two modes of operation:
EGFR- Epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase that stimulates cell growth and differentiation when binding to its ligands. In many epithelial cancers, especially in lung cancer, breast cancer, and glioblastoma, EGFR is a driver of carcinogenicity mainly due to gene mutations or amplification.60 EGFR inhibition by targeted drug therapy is currently used for the treatment of many types of epithelial cancers such as head and neck squamous cell carcinoma, non-small-cell lung cancer (NSCLC), pancreatic cancer, and colorectal cancer. Zaiss et al have demonstrated that Tregs express EGFR under inflammatory conditions while amphiregulin, a protein that is a member of the EGF tyrosine kinase family, supports suppressive Treg function in vitro and in vivo.61 Another research demonstrated that this support is mediated via the EGFR/GSK-3β/Foxp3 axis.21 It seems that EGFR upregulation induces both cancer and Treg proliferation.
VEGFR – Vascular endothelial growth factor receptor (VEGFR) is a receptor protein kinase that binds to its ligand, vascular endothelial growth factor (VEGF), to induce vasculogenesis and angiogenesis. In particular, cancer cells promote the formation of new blood vessels that are essential for tumor growth by VEFG excretion. VEGF and VEGFR inhibitors are FDA approved for the treatment of seven different types of solid cancers.62 VEGF-A is one of the three members of the VEGF family of kinases. One of the receptors of VEGF-A, VEGFR2, is detected on the membrane surface of Treg cells. In a mouse model of colon cancer, stimulation of VEGFR2 by VEGF induced Tregs gathering in the tumor surroundings63 and references therein. Therefore, VEGFR hyperactivity drives both angiogenesis and Treg accumulation in the TME. Each of these two processes promotes cancer growth.
PDGFR - Platelet-derived growth factor receptor (PDGFR) is a protein kinase receptor family of two members (α and β) that regulate cell growth and division upon binding to their ligands. The PDGF family (PDGFR ligands) exists as homodimer and/or heterodimer formed by dimerization of A-polypeptide, B-polypeptide, C-polypeptide, and D-polypeptide chains. Defects in one of the PDGF A/B/C/D and PDGFR α/β genes are reported in up to 30% of cancer patients, depending on the type of cancer.64 It is also reported that PDGF upregulates (in vitro) the expression of C-type lectin-like receptor member 2(CLEC-2) on DCs which in turn induce the polarization of T cells towards FoxP3 regulatory T cells.65 Together, PDGF gain-of-function mutations promote both cancer growth and Treg proliferation.
BRAF – The BRAF gene which is located on the long arm of chromosome 7 encodes for a cytoplasmatic serine/threonine protein kinase (B-Raf). The wild type of this gene is commonly involved with normal processes of cell differentiation, growth, and apoptosis, downstream within the ERK/MAPK signaling pathway. On the other hand, BRAF gain-of-function mutations are oncogenic.66 BRAF somatic missense mutations are reported in 66% of the malignant melanomas and at lower frequencies in other malignancies. The V600E mutation (where valine (V) is substituted by glutamic acid (E) at amino acid 600 of the BRAF gene) accounts for 80% of BRAF mutations observed in cancers.67 It is also reported that BRAFV600E controls Treg recruitment to melanoma skin sites.68 Hence, BRAFV600E mutation in melanocytes has a double effect: inducing melanoma and recruiting Tregs to melanoma cutaneous sites.
BCR-ABL1 – The fusion gene BCR-ABL1 is generated by reciprocal translocation of genetic material between chromosome 9 and chromosome 22 where the ABL1 gene on chromosome 9 and the BCR gene on chromosome 22 code for a hybrid protein that is continuously active, inducing uncontrolled division of cells. The mutation is found in all CML patients and in 11%-29% of acute lymphoblastic leukemia (ALL) adult patients.69
Using a mice model, it was demonstrated that BCR-ABL1+ leukemia induces the conversion of anti-BCR-ABL1 specific T cells into Treg cells, a process that inhibited an anti-leukemic immune response. The conversion was mediated via MHC-II antigen presentation by leukemia cells.70 Hence, the BCR-ABL1 hybrid protein drives BCR-ABL +leukemia while this type of leukemia promotes Treg proliferation.
PIKs Drive Both a Direct Pathogenic Effect and a “Low Treg” Immune Response in Autoimmune Diseases and MPNs
Deregulated activity of protein kinases is implicated in the pathogenesis of many autoimmune diseases, mainly via the JAK/STAT signaling pathway.44 As mentioned above, all four members of the JAK family present a pro-inflammatory profile.
Autoimmune diseases are typical “low Treg” diseases, since Treg function is impaired in many autoimmune diseases.71 As presented below, JAKs deregulation may inflict direct tissue damage in autoimmune diseases, and directly promotes cancer cell proliferation in “low Treg” malignancies:
Psoriasis: Histologically, psoriasis is characterized by keratinocyte hyperproliferation and deregulated differentiation, hyperplastic dilated blood vessels, and inflammatory leukocytes infiltration, mainly into the dermis. Even though psoriasis is considered to be a T cell-driven autoimmune disease where T cells, in particular Th17 cells, play a dominant pathogenic role in the initiation and sustainment of the disease,72 JAK1 and JAK2 as well as STAT1 and STAT3 are expressed by keratinocytes73 and may directly affect pathogenicity. The JAK1/JAK2/STAT1 and JAK1/TYK2/STAT3 pathways triggered by IFN-γ and IL-22, respectively, are aberrantly activated in psoriasis. A pretreatment with Tofacitinib, a JAK1/JAK3 inhibitor that impedes JAK phosphorylation, has been shown to restore normal proliferative and differentiation in psoriatic keratinocyte cultures stimulated with either IFN-γ or IL-22.74 Since no immune cells were present in the culture, this study demonstrated a direct effect of JAKs located within keratinocytes on psoriasis pathology.
Rheumatoid arthritis: Even though RA is initiated and propagated by a deregulated immune response, ie, by an autoimmune reaction, activated synovial fibroblasts contribute to the pathogenicity by the secretion of inflammatory cytokines that damage the cartilage and by the recruitment of immune cells that augment the inflammation within joints and cause pain and disability. An experimental inhibitor of TGFβ-activated kinase 1 (TAK1) was able to block fibroblasts activation in ex vivo cultures of synovial fibroblasts from RA patients.75 This experiment indicates that a serine/threonine kinase, TAK1, mediates synovial fibroblast activation directly in RA without an involvement of the immune system.
BCR-ABL1 negative myeloproliferative neoplasms (MPNs): As mentioned above, a JAK2 mutation (JAK2V617F) that promotes the proliferation of myeloid tissue cells is the main driver of these “low Treg” cancers.76
Taken together, PIK hyperactivation induces both a direct pathogenic effect on affected tissues (skin lesions in psoriasis, joints in RA, tumor tissue in MPNs) and a pro-inflammatory immune reaction.
Diseases Driven by AIKs are Associated with “High Treg” Pathogens
It is expected that diseases triggered by AIK hyperactivity (most solid cancers) will be associated with “high Treg” pathogens, since both proliferate under anti-inflammatory conditions. Here are examples of two AIK-driven solid cancers, and their associated pathogens:
Renal Cell Carcinoma (RCC)
Treg frequency in the TME of RCC is higher than Treg frequency in the peripheral blood, and a higher Treg frequency in RCC correlates with a poorer prognosis.77 This indicates a “high Treg” disease driven by AIKs.
Vascular endothelial growth factor (VEGF) is a growth factor that promotes RCC by inducing vasculature growth that is vital for cancer cell proliferation. VEGF or VEGFR inhibitors (eg, axitinib or bevacizumab) are efficacious in the treatment of RCC. There was a statistically significant advantage of axitinib over sorafenib (another VEGFR inhibitor) in prolonging the progression-free-survival period in RCC.78 A combination treatment of {IFNα-2a + bevacizumab} was superior to {IFNα-2a + placebo} in prolonging the progression-free-survival period and increasing the tumor response rate in advanced and/or metastatic RCC.79 Axitinib and bevacizumab suppress Treg frequency in the circulation (Table 1), indicating that VEGFR is an anti-inflammatory kinase.
RCC is associated with hepatitis C virus (HCV), Epstein-Barr virus (EBV), and probably with human papillomavirus (HPV). A meta-analysis of seven observational studies found a pooled relative risk (RR) of 1.86 for RCC among HCV positive participants.80 EBV was detected in 31% of tissue samples from 71 patients with histologically proven RCC compared with 4.2% of samples from peritumoral tissue.81 Shimakage et al found that the expression of EBV may be involved in the pathogenesis of RCC.82 There is some research pointing to an association between HPV and RCC but the results are controversial.83 In line with this, HCV, EBV (in the context of gastric cancer, and probably other solid cancers, see below) are “high Treg” pathogens. HPV is also a “high Treg” pathogen.8
Non-Small-Cell Lung Carcinoma (NSCLC)
Peripheral Treg frequency increases in NSCLC patients and it is positively correlated with a worse prognosis.84 In addition, tumor-infiltrating Tregs are associated with worse recurrence-free survival in NSCLC.85 It follows that NSCLC is a “high Treg” disease. EGFR mutations drive 10–35% of NSCLC. Fibroblast growth factor receptor 1 (FGFR1) gene amplification affects 20% of patients. Up to 7% of NSCLC patients have EML4-ALK translocations or mutations in the ROS1 gene.86 EGFR, ALK, ROS1, and FGFR1 are anti-inflammatory kinases since their inhibitors (erlotinib, crizotinib, and erdafitinib) reduce Treg frequency or function (Table 1).
The relative abundance of only three bacteria, Bifidobacterium, Streptococcus, and Prevotellan, out of 32 evaluated, demonstrated a statistically significant increase in lung cancer, compared with emphysema.87 All three demonstrate profiles characteristic of “high Treg” bacteria:
- Tregs are increased in the mucosa and spleen of mice following the consumption of Bifidobacterium infantis which was proceeded by the injection with Salmonella typhimurium or with lipopolysaccharides. In vivo imaging revealed a profound inhibition of infection.88
- Streptococcus pneumoniae (pneumococcus) induces CD8+CD28+ suppressive Tregs which produce IL-10 and TGF-β.89
- Prevotella inflammatory effects are site specific: the effect is pro-inflammatory when the inflammation site is in the guts (in inflammatory bowel disease, metabolic syndrome, and AIDS), in the oral cavity (in periodontitis), in the joints (in RA), or in the vagina (in vaginosis). However, when the lungs are involved (in the settings of asthma and COPD) Prevotella has an anti-inflammatory (“high Treg”) role.90 The effect of Prevotlla is therefore expected to be anti-inflammatory in the lungs of NSCLC patients. In addition, a Prevotella histicola challenge increased Tregs in human leukocyte antigen (HLA) transgenic mice and suppressed experimental autoimmune encephalomyelitis (EAE) in this model of multiple sclerosis.91
Viral DNA analysis of NSCLC tissue samples indicated the association of six viruses with NSCLC: Human papilloma virus (HPV), Hepatitis B virus (HBV), Human T-cell lymphotropic virus 2 (HTLV-2), Bovine leukemia virus (BLV), Y53 sarcoma virus, and Simian T-cell lymphotropic viruses (STLV-1, 2, or 6).92 In addition, EBV probably plays a pathological role in NSCLC.93
HPV, HBV, and EBV (in the context of cancer) are “high Treg” viruses.8 No data related to HTLV-2, BLV, STLV, or Sarcoma virus effect on Tregs could be found. However, the first three viruses are closely related to HTLV-1, which is a “high Treg” virus.8
Diseases Driven by PIKs are (Mostly) Associated with “Low Treg” Pathogens
Similar to diseases triggered by AIKs, it is expected that diseases triggered by PIK hyperactivity (autoimmune diseases and MPNs) will be associated with “low Treg” pathogens, since both are developed under pro-inflammatory conditions. Indeed, this expectation holds true for most pathogens associated with AIKs driven diseases. The association between “low Treg” pathogens and autoimmune diseases was discussed in earlier publications.8,9 Here we discuss the association between “low Treg” pathogens and MPNs.
In a recent paper, Landtblom et al performed a large population-based matched cohort study in Sweden including 8363 MPN patients and 32,405 controls to assess the risk of infections in MPN patients. The following pathogens were found in MPNs population in a descending order of related hazard ratios (HR):
Pneumocystis jirovecii, Hepatitis B, Staphylococci, Streptococci, Haemophilus influenzae, Varicella zoster virus, Influenza, Escherichia coli, Mycobacterium tuberculosis (Mbt).94
As shown below, out of these nine pathogens associated with MPNs, six are “low Treg”, two (viruses) are “high Treg”, and one (bacterium) evolves from a “low Treg” to a “high Treg” pathogen as the disease progresses.
Pneumocystis jirovecii – As part of the host defense against this fungal pathogen, inflammatory cells are recruited into the lung tissue in order to prevent the development of pneumonia.95 Moreover, pulmonary markers of inflammation correlate with the clinical severity of Pneumocystis jirovecii pneumonia.96 It seems that Pneumocystis jirovecii is a “low Treg” fungus that induces a strong inflammatory reaction, at least in the lungs.
Hepatitis B virus (HBV) - An expansion of regulatory T cells and impaired TCR signaling in newborns with HBV infection represent the immune tolerant state of the adaptive immune system.97 Hence, HBV is a “high Treg” virus.
Staphylococci – T cells exposed to Staphylococcus aureus (SA) release predominantly (but not solely) Th1 and Th17 cytokines.98 Toll-like receptor (TLR)-2 on dendritic cells mounts this inflammatory response. However, TLR-2 on macrophages can modulate immunity against SA by inducing IL-10.99 It was demonstrated that IL-10 shifts the Th1/Th17 balance towards Th17 response.100 This is important since high serum levels of IL-10 in SA patients correlate with mortality.101 At any rate, whether Th1 or Th17 prevails, the immune reaction is pro-inflammatory (“low Treg”).
Streptococci –S. pyogenes induce mainly Th17 reaction,8 ie, it is a “low Treg” bacterium.
Haemophilus influenzae – These bacteria drive Th17 immune responses that promote the development of neutrophilic inflammation and suppress eosinophilic inflammation during allergic airways disease.102,103 Guan et al have shown in a mice model of chronic obstructive pulmonary disorder (COPD) that non-typeable H. influenzae impairs Treg function, which facilitates the development of inflammatory acute exacerbation of the disease.104 H. influenza is a “low Treg” bacterium, at least in pulmonary disorders.
Varicella zoster virus – “The proportion of circulating Th17 cells and the Th17/Treg cell ratio were significantly higher in patients with herpes zoster than controls”.105 This indicates a “low Treg” response.
Influenza A virus – The ability of influenza virus-induced regulatory T cells to suppress antigen-specific CD4+ and CD8+ T cell proliferation and cytokine production correlates closely to their ability to respond to influenza virus antigens, suggesting that virus-induced Tregs are capable of attenuating effector responses in an antigen-dependent manner.106
A mice model demonstrated the inhibition of Th17 activity against bacterial pneumonia by Influenza A virus.107 It therefore seems that Influenza A virus is a “high Treg” pathogen.
Escherichia coli – When administered parenterally, Escherichia coli heat-labile enterotoxin (LT) promotes Ag-specific IL-17, as well as IFN-γ, IL-4, and IL-10 production in response to coadministered Ags. When added as an adjuvant to pertussis vaccine, LT induces the development of Ag-specific Th17 cells that mount protection against a challenge with Bordetella pertussis bacteria.108 This indicates a “low Treg” effect of E. coli when administered parenterally.
Mycobacterium tuberculosis (Mtb) – Mtb induces an increase in Tregs, Th1, and Th17 cell frequencies. However, the Th17/Treg ratio increases in active tuberculosis (TB) patients relative to latent TB or healthy controls.109 Th1 cells in blood and lungs of TB patients predominate over Th17 and {Th17Th1} cells.110 In fact, IFNγ release assays (in response to exposure of blood samples to TB antigens) are used for the diagnosis of TB.111 In a meta-analysis of 9 studies, Li et al demonstrate that BCG anti-TB vaccination induced dramatically high level of IL-17 and IFNγ. The levels of these cytokines were lower during active disease than in healthy controls or during latent disease. IL-17 was lower during latent disease compared to healthy controls.112 It seems therefore that TB starts as a “low Treg” disease and continuously evolves to a “high Treg” disease. In line with this, TGFβ (but not IFNγ or TNFα or IL-4) was highly expressed in tuberculous granulomas which are a late-stage symptom of TB.113 High TGFβ activity is a hallmark of active pulmonary TB.114 This evolvement from a “low Treg” to a “high Treg” disease can be compared to cirrhosis-associated immune dysfunction phenotypes switching from predominantly “pro-inflammatory” to predominantly “immunodeficient” in patients with stable ascitic cirrhosis and in patients with acute-on-chronic liver failure.115
As mentioned above, “low Treg” pathogens are expected to be associated with PIK-driven diseases. Six out of nine infectious agents associated with MPNs according to the population-based study are “low Treg” pathogens. Two viruses associated with MPNs are “high Treg”. This can be explained by the intracellular replication of viruses which shelters them from the inflammatory environment, at least for part of their life cycle. This way “high Treg” viruses like HBV and Influenza A virus can survive the hostile inflammatory environment of MPNs. It should be added that the data presented by Landtblom et al could be affected by the anti-MPN drugs used during the study. However, reviewing the data, it seems that treatment with hydroxyurea, interferon alpha, or anagrelide does not affect the risk of infection, compared to the risk in untreated patients (Ref. 94, Tab. 4). The evaluated hazard ratios for infection in this work are good estimates of the values in untreated patients (versus matching controls without MPN) since about 49% of the patients were not treated with any drug, and about 37% of the patients used one of these three agents, with HR=1 versus untreated patients.
Out of the nine drugs used by the patients participating in this study, ruxolitinib (which was used only in primary myelofibrosis patients) demonstrated the largest hazard ratio for the development of infection. This can be related to the anti-inflammatory effect of ruxolitinib, mediated by the increasing number of Tregs.
Pathogens That Activate Both AIKs and PIKs Can Induce Both “High Treg” Cancers and Autoimmune Diseases
Helicobacter pylori – Src homology region 2 domain-containing phosphatase-2 (SHP-2) is a protein tyrosine-phosphatase. Activating SHP-2 mutations have been observed in many “high Treg” cancers such as neuroblastoma, melanoma, acute myeloid leukemia, breast cancer, lung cancer, and colorectal cancer.116 Glycoprotein 130 (gp130) is a transmembrane protein that constitutes a subunit of the type I cytokine receptor within the IL-6 receptor family. Glycoprotein 130 modulates the balance between the SHP-2/ERK and JAK/STAT pathways.117 The Helicobacter pylori protein CagA can undergo tyrosine phosphorylation following its entry into human gastric epithelial cells and switch this balance towards the SHP-2/ERK anti-inflammatory (“high Treg”) pathway, promoting gastric cancer. However, in its unphosphorylated state, CagA skews the balance towards the pro-inflammatory (“low Treg”) path.117 In line with this, H. pylori infection is associated with autoimmune atrophic gastritis118 and Grave’s disease,119 two autoimmune diseases. Clinical improvements following H. pylori eradication was reported in two other autoimmune diseases: immune thrombocytopenic purpura and psoriasis.120
Epstein-Barr virus – Epstein–Barr virus latent membrane protein 1 (LMP1) is an EBV oncogenic protein that activates several cellular pathways in cervical carcinoma and B cell lymphoma. In particular, LMP1 induces EGFR expression121 and activates the RAF/MEK/ERK/MAPK pathway122 in cervical carcinoma cell lines. The latter pathway has shown to regulate epithelial cell motility and invasion.122 In addition, LMP1 induces the PIK3/AKT pathway in cervical carcinoma cell line.123 All these pathways promote an anti-inflammatory reaction by the immune system (a “high Treg” response). At the same time, LMP1 has been shown to activate the JAK/STAT pathway in EBV-associated post-transplant B-cell lymphoma.124 As discussed above, this is a pro-inflammatory (“low Treg”) pathway. Moreover, a JAK1/3 inhibitor, tofacitinib, inhibited tumor growth in EBV-associated lymphoma T cells and natural killer cells.125 Indeed, there is evidence implicating the involvement of EBV in 6 autoimmune diseases: MS, SLE, RA, Sjӧgren syndrome, autoimmune liver disease, and autoimmune thyroiditis.126 These are all “low Treg” diseases. EBV is also involved with several types of lymphomas: Burkitt’s lymphoma, Hodgkin lymphoma, and lymphoproliferative disease in immunocompromised hosts.126 It has been shown that lymphoma starts as a “low Treg” disease, and can dwell in this state for several years, before aggressive disease develops.9 In addition, EBV is involved in the etiology of nasopharyngeal carcinoma and gastric carcinoma.126 Tumor-infiltrating Tregs correlate with a poor prognosis in gastric cancer but no statistically significant correlation was observed for oropharyngeal cancer.127 Hence, gastric cancer can be classified as a “high Treg” solid cancer, while more data are needed for the classification of oropharyngeal cancer.
“High Treg” Viruses Activate Anti-Inflammatory Protein Kinases and Inhibit the Pro-Inflammatory JAK/STAT Pathway
EGFR – EGFR is activated by Influenza virus, Rhinovirus (RV), Cytomegalovirus (CMV), EBV, and HCV.128
VEGFR – The following viruses: EBV, HBV, HCV, HPV, Herpes Simplex Virus 1 (HSV-1), Kaposi’s sarcoma herpesvirus (KSHV), and Dengue virus upregulate VEGF (VEGFR ligand).129
PDGFR – Human CMV glycoprotein B interacts directly with PDGFR-alpha, resulting in receptor tyrosine phosphorylation.130
MAPK – MAPK/ERK signaling is stimulated by BK polyomavirus (BKPyV), human adenovirus, EBV, HBV, HPV, herpes simplex virus 1 (HSV-1), herpes simplex virus 2 (HSV-2), JC polyomavirus (JCPyV), KSHV, and Vaccinia virus (VACV).131
RAF kinase – The HBx protein of HBV is a small transcriptional trans-activator that is essential for HBV-mediated liver carcinogenicity. HBx upstream activation of the Ras-Raf-MAP kinase signaling pathway was found to be essential for downstream activation of AP-1 and NF-kB.132
Src family kinases – Activation of Src by HBV-related HBx protein is essential for activation of the Ras-Raf-MAPK pathway.133
EBV (in the context of cancer), HCV, HBV, HPV, HSV-1, KSHV, BKPyV are all “high Treg” viruses,8 Influenza A virus is also “high Treg” virus,106 and so is human adenovirus.134 RV induced a “high Treg” reaction in PBMC of blood samples taken from healthy children, following in vitro stimulation by peptides containing species-specific VP1 epitopes of RV, but induced a “low Treg” reaction in PBMC samples of asthmatic children.135 CMV induces CD4+CD27−CD28− T cells that have regulatory (Treg) function.136 Treg frequency was expanded in patients with Dengue virus infection relative to healthy controls but no relationship was observed between Treg frequency and clinical disease severity or the degree of viraemia indicating Tregs with poor suppressive function.137 Vaccinia virus is a “low Treg” virus.138
Many of these “high Treg” viruses that activate AIKs, as presented above (HBV, HCV, HPV, human adenovirus, HSV-1, KSHV, Vaccinia virus, CMV, Influenza A virus) also inhibit the JAK/STAT signaling pathway.139
“Low Treg” Pathogens Activates JAK1 and JAK2
Some “low Treg” pathogens induce host cells IFNγ production, as part of host defense against the invader:
Both Chlamydia and Mycobacterial infections induce host CD4+T cells to secrete IFNγ.140
Recognition of Varicella Zoster virus by host cells drives IFNγ production by immune cells like DCs and CD4+T cells.141
IFNγ generated by T cell lymphocytes is regarded as a key cytokine in the combat against Staphylococcus Aureus infections.142
All four pathogens are “low Treg” pathogens (see Diseases Driven by PIKs are (Mostly) Associated with “Low Treg” Pathogens of this work and ref.81).
At the same time, INFγ receptor activates JAK1 and JAK2 when binding to its ligand.35
By Activating Anti-Inflammatory Kinases, Alcohol Consumption Lowers the Risk of Some “Low Treg” Diseases but Increases the Risk of “High Treg” Diseases
Acute alcohol drinking activates the Src family of kinases that in turn activates STAT3 to promote IL-10 production in human monocytes.143 This pathway is an anti-inflammatory pathway. As such, alcohol drinking is expected to confer protection against “low Treg” conditions. Indeed, alcohol consumption reduces the risk of autoimmune hypothyroidism,11 of non-Hodgkin’s lymphoma,144 and of type-2 diabetes in men.145 Non-Hodgkin’s lymphoma is a “low Treg” disease during the long indolent stage of the disease.9 Type-2 diabetes is a “low Treg” disease.146
On the other hand, alcohol drinking increases the risk of 16 out of 21 types of solid cancers investigated. The increased risk correlates positively with daily alcohol dose.10
JAK1 Gain-of-Function Mutations are Frequent in “Low Treg” Diseases While Loss-of-Function Mutations are Frequent in Diseases with Increased Tumor-Infiltrating Tregs
Gain-of-function JAK1 mutations probably drive diverse “low Treg” diseases, since the non-selective JAK1 inhibitors tofacitinib, ruxolitinib, baricitinib and the selective JAK1 inhibitor, upadacitinib, are FDA approved for the treatment of “low Treg” diseases like: RA, PsA, ulcerative colitis, myelofibrosis and polycythaemia vera. JAK1 gain-of-function was reported in psoriasis.147 JAK1 gain-of-function mutations resulted in complex autoinflammatory syndrome,148 in hypereosinophilic syndrome,149 in myeloproliferative neoplasm.150
Loss-of-function JAK1 mutations occurred at high frequency in endometrial, colorectal, gastric, and prostate carcinomas.151,152 All of these four solid cancers demonstrate increased infiltration of Tregs into the TME and increased Treg frequency in peripheral blood.153–156 The infiltration of Tregs into the TME correlates with poor prognosis in gastric and prostate carcinomas,127 classifying them as “high Treg” diseases. However, the prognostic value of Treg infiltration into the TME is controversial in the case of endometrial cancer152 while Treg infiltration correlates with favorable prognosis in the case of colorectal cancer.157 For this reason, these two cancers cannot be classified as “high Treg” diseases.
Discussion
It seems that the pathogenic effect of protein kinase deregulated activity is double:
A direct pathogenic effect which involves the affected tissue, and an indirect effect mediated by the immune system reaction (a pro- or anti-inflammatory response). It is possible that this double effect is needed to promote pathogenicity.
This paper classifies protein kinases as anti- or pro-inflammatory kinases depending on whether they drive “high Treg” or “low Treg” diseases. This does not mean that a kinase classified as anti-inflammatory, since it drives cancer, would not promote a pro-inflammatory response in the setting of autoimmune diseases. Similarly, a pro-inflammatory kinase, defined as such in the setting of autoimmune disease may promote an anti-inflammatory response in cancer.
Indeed, there are data indicating the involvement of AIKs in the pathogenicity of autoimmune diseases in addition to their frequently reported activity in “high Treg” diseases. For example, imatinib, an AIK inhibitor, showed efficacy in a RA murine model.158 Imatinib was also found effective in the treatment of RA and spondyloarthritis in small-scale clinical trials.159,160 Similarly, topical sunitinib (VEGFR and PDGFR inhibitor) alleviated psoriasis-like inflammation in mouse model,161 and intra-gastric administration of lapatinib (EGFR and HER2 inhibitor) ameliorated arthritis in a rat model.162 However, no large-scale clinical trials that study the effect of AIK inhibitors on “low Treg” diseases have been published.
Likewise, there are data indicating the involvement of PIKs in “high Treg” cancers, in addition to their frequently reported activity in “low Treg” diseases. For example, a JAK1/2 inhibitor, ruxolitinib, inhibited tumor angiogenesis and prolonged survival in genetically engineered murine models of pancreatic cancer.163 In addition, ruxolitinib blocked tumor growth in another murine pancreatic model.164 The combination of momelotinib, a JAK1/2 and TBK1 inhibitor, with a MEK inhibitor, induced regression of an aggressive murine lung adenocarcinoma driven by KRAS mutation and p53 loss.165
Despite these preclinical studies indicating the efficacy of JAK inhibitors in controlling solid cancers, JAK inhibitors failed to improve efficacy in clinical trials when added to an approved treatment:
- The combination of the JAK1/2 inhibitor momelotinib with trametinib (MEK inhibitor) was not superior to trametinib monotherapy in KRAS-mutated NSCLC (on the basis of historic data).166
- The triple combination of the JAK1/2 inhibitor momelotinib, gemcitabine, and nab-paclitaxel in patients with previously untreated metastatic pancreatic ductal adenocarcinoma showed no superiority in prolonging patients survival over the combination of gemcitabine and nab-paclitaxel.167
- The combination of regorafenib and ruxolitinib did not prolong survival of colorectal cancer patients compared to the combination of regorafenib and placebo.168
Furthermore, ruxolitinib prolonged survival of metastatic pancreatic cancer patients when added to capecitabine in a Phase II study169 but failed to do so in a Phase III study.170
To date, there is no AIK inhibitor approved for the treatment of autoimmune diseases, and no PIK inhibitor approved for the treatment of “high Treg” cancers. In other words, no kinase inhibitor has been approved (so far) for the treatment of both, autoimmune diseases and “high Treg” cancers.
It might be thought that JAK inhibitors are not effective enough in treating “high Treg” cancers because of their anti-inflammatory effect. Similarly, it might be thought that AIK inhibitors are not effective enough in the treatment of autoimmune diseases because of their pro-inflammatory effect. However, imatinib (a PIK) reduced inflammation in murine collagen-induced arthritis by inhibiting mast cell production of pro-inflammatory cytokines156 and behaved as AIK. Therefore, it is possible that the same protein kinase inhibitor will have opposite effects on the immune reaction (ie, switch from PIK to AIK or vice versa), depending on the type of disease.
The distinctive properties of AIKs and PIKs are summarized in Table 3 and Figure 1.
 |
Table 3 The Distinctive Properties of AIKs and PIKs |
 |
Figure 1 A summary diagram of the protein kinase binary classification model and the related findings. Abbreviations: EGFR, endothelial growth factor receptors; VEGFR, vascular endothelial growth factor receptors; PDGFR, platelet-derived growth factor receptors; FGFR, fibroblast growth factor receptors; ALK, anaplastic lymphoma kinase; JAK, Janus kinases; SYK, spleen tyrosine kinase; CLL, chronic lymphoid leukemia; ALL, acute lymphoblastic leukemia; MCL, mantle cell lymphoma; CML, chronic myelogenous leukemia; AMD, age-related macular degeneration. |
Summary
A division of protein kinases into anti- and pro-inflammatory classes is proposed as a molecular model that can explain the unique immunological properties of “high Treg” diseases versus those of “low Treg” diseases. Promotion of Treg activity by anti-inflammatory kinases drives most cancers; suppression of Treg activity by pro-inflammatory kinases drives autoimmune diseases and MPNs. It has been demonstrated that protein kinases directly promote diseases b y activating pathogenic pathways within the affected tissue cells, and indirectly, through the mediation of the immune system. Diseases driven by each class of kinases are associated with a specific group of pathogens that activate these kinases. “High Treg” pathogens activate anti-inflammatory kinases while “low Treg” pathogens activate pro-inflammatory kinases. Pathogens that activate both anti- and pro-inflammatory kinases promote both cancers and autoimmune diseases. Alcohol activates anti-inflammatory kinases. This explains why alcohol consumption increases the risk of most cancers but confers protection against some autoimmune diseases. Gain-of-function or loss-of-function mutations of the same protein kinase may induce “low Treg” or “high Treg” diseases. No protein kinase inhibitor has been approved so far for the treatment of both “high Treg” cancers and autoimmune diseases, in spite of some promising pre-clinical and small-scale clinical studies. It is still unclear whether this regulatory situation is incidental and may be breached in the future or is it a consequence of a rule of general validity (ie, that the two classes of inhibitors are mutually exclusive with respect to the diseases they may treat).
Disclaimer
The views and opinions expressed, and/or conclusions drawn, in this article are those of the author and do not necessarily reflect those of Taro Pharmaceutical Industries Ltd., its affiliates, directors or employees.
Acknowledgments
Professor Shlomo Ben Sasson at the Hebrew University of Jerusalem is gratefully acknowledged for his support and advice.
Disclosure
Zeev Elkoshi is employed by Taro Pharmaceutical Industries Ltd, reports personal fees from Taro Pharmaceutics outside the submitted work, and reports no other potential conflicts of interest for this work.
References
1. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–1934. doi:10.1126/science.1075762
2. Ardito F, Giuliani M, Perrone D, Troiano G, Lo Muzio L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (review). Int J Mol Med. 2017;40(2):271–280. doi:10.3892/ijmm.2017.3036
3. Sacco F, Perfetto L, Castagnoli L, Cesareni G. The human phosphatase interactome: an intricate family portrait. FEBS Lett. 2012;586(17):2732–2739. doi:10.1016/j.febslet.2012.05.008
4. Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov. 2018;17(5):353–377. doi:10.1038/nrd.2018.21
5. Szilveszter KP, Németh T, Mócsai A. Tyrosine kinases in autoimmune and inflammatory skin diseases. Front Immunol. 2019;10:1862. doi:10.3389/fimmu.2019.01862
6. Siasos G, Tousoulis D, Siasou Z, Stefanadis C, Papavassiliou AG. Shear stress, protein kinases and atherosclerosis. Curr Med Chem. 2007;14(14):1567–1572. doi:10.2174/092986707780831087
7. Kinase-disease association/cell signaling technology. Available from: https://www.cellsignal.com/contents/resources-reference-tables/kinase-disease-associations/science-tables-kinase-disease. Accessed February 26, 2021.
8. Elkoshi Z. The binary classification of chronic diseases. J Inflamm Res. 2019;12:319–333. doi:10.2147/JIR.S227279
9. Elkoshi Z. “High treg” inflammations promote (most) non-hematologic cancers while “low treg” inflammations promote lymphoid cancers. J Inflamm Res. 2020;13:209–221. doi:10.2147/JIR.S249384
10. Bagnardi V, Rota M, Botteri E, et al. Alcohol consumption and site-specific cancer risk: a comprehensive dose-response meta-analysis. Br J Cancer. 2015;112(3):580–593. doi:10.1038/bjc.2014.579
11. Carlé A, Pedersen IB, Knudsen N, et al. Moderate alcohol consumption may protect against overt autoimmune hypothyroidism: a population-based case-control study. Eur J Endocrinol. 2012;167(4):483–490. doi:10.1530/EJE-12-0356
12. Zhang X, Fang X, Gao Z, et al. Axitinib, a selective inhibitor of vascular endothelial growth factor receptor, exerts an anticancer effect in melanoma through promoting antitumor immunity. Anticancer Drugs. 2014;25(2):204–211. doi:10.1097/CAD.0000000000000033
13. Napoletano C, Ruscito I, Bellati F, et al. Bevacizumab-based chemotherapy triggers immunological effects in responding multi-treated recurrent ovarian cancer patients by favoring the recruitment of effector T cell subsets. J Clin Med. 2019;8(3):380. doi:10.3390/jcm8030380
14. Bergerot P, Lamb P, Wang E, Pal SK. Cabozantinib in combination with immunotherapy for advanced renal cell carcinoma and urothelial carcinoma: rationale and clinical evidence. Mol Cancer Ther. 2019;18(12):2185–2193. doi:10.1158/1535-7163.MCT-18-1399
15. Kwilas AR, Ardiani A, Donahue RN, Aftab DT, Hodge JW. Dual effects of a targeted small-molecule inhibitor (cabozantinib) on immune-mediated killing of tumor cells and immune tumor microenvironment permissiveness when combined with a cancer vaccine. J Transl Med. 2014;12(1):294. doi:10.1186/s12967-014-0294-y
16. Jie HB, Schuler PJ, Lee SC, et al. CTLA-4+ regulatory T cells increased in cetuximab-treated head and neck cancer patients suppress NK cell cytotoxicity and correlate with poor prognosis. Cancer Res. 2015;75(11):2200–2210. doi:10.1158/0008-5472.CAN-14-2788
17. Liu P, Zhao L, Kepp O, Kroemer G. Crizotinib - a tyrosine kinase inhibitor that stimulates immunogenic cell death. Oncoimmunology. 2019;8(7):1596652. doi:10.1080/2162402X.2019.1596652
18. Fei F, Yu Y, Schmitt A, et al. Dasatinib inhibits the proliferation and function of CD4+CD25+ regulatory T cells. Br J Haematol. 2009;144(2):195–205. doi:10.1111/j.1365-2141.2008.07433.x
19. Palakurthi S, Kuraguchi M, Zacharek SJ, et al. The combined effect of FGFR inhibition and PD-1 blockade promotes tumor-intrinsic induction of antitumor immunity. Cancer Immunol Res. 2019;7(9):1457–1471. doi:10.1158/2326-6066.CIR-18-0595
20. Ayeni D, Miller B, Kuhlmann A, et al. Tumor regression mediated by oncogene withdrawal or erlotinib stimulates infiltration of inflammatory immune cells in EGFR mutant lung tumors. J Immunother Cancer. 2019;7(1):172. doi:10.1186/s40425-019-0643-8
21. Wang S, Zhang Y, Wang Y, et al. Amphiregulin confers regulatory T cell suppressive function and tumor invasion via the EGFR/GSK-3β/Foxp3 axis. J Biol Chem. 2016;291(40):21085–21095. doi:10.1074/jbc.M116.717892
22. Podhorecka M, Goracy A, Szymczyk A, et al. Changes in T-cell subpopulations and cytokine network during early period of ibrutinib therapy in chronic lymphocytic leukemia patients: the significant decrease in T regulatory cells number. Oncotarget. 2017;8(21):34661–34669. doi:10.18632/oncotarget.16148
23. Larmonier N, Janikashvili N, LaCasse CJ, et al. Imatinib mesylate inhibits CD4+ CD25+ regulatory T cell activity and enhances active immunotherapy against BCR-ABL- tumors. J Immunol. 2008;181(10):6955–6963. doi:10.4049/jimmunol.181.10.6955
24. Kimura T, Kato Y, Ozawa Y, et al. Immunomodulatory activity of lenvatinib contributes to antitumor activity in the Hepa1-6 hepatocellular carcinoma model. Cancer Sci. 2018;109(12):3993–4002. doi:10.1111/cas.13806
25. Lu Z, Xu N, Zhou X, et al. Therapeutic immune monitoring of CD4+CD25+ T cells in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors. Oncol Lett. 2017;14(2):1363–1372. doi:10.3892/ol.2017.6294
26. Wang X, Shan S, Semba T, et al. Abstract 4492: humanized anti-EGFR antibody panitumumab inhibits tumor growth of inflammatory breast cancer by inducing antitumor immunity. Cancer Res. 2019;79(13 Supplement):4492. doi:10.1158/1538-7445.AM2019-4492
27. Verzoni E, De Cecco L, Dugo M, et al. Broad immunomodulating effect of first-line pazopanib in metastatic renal cell carcinoma patients. Ann Oncol. 2017:28. doi:10.1093/annonc/mdx371.043.
28. Busse A, Asemissen AM, Nonnenmacher A, et al. Immunomodulatory effects of sorafenib on peripheral immune effector cells in metastatic renal cell carcinoma. Eur J Cancer. 2011;47(5):690–696. doi:10.1016/j.ejca.2010.11.021
29. Stehle F, Schulz K, Fahldieck C, et al. Reduced immunosuppressive properties of axitinib in comparison with other tyrosine kinase inhibitors. J Biol Chem. 2013;288(23):16334–16347. doi:10.1074/jbc.M112.437962
30. Finke JH, Rini B, Ireland J, et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin Cancer Res. 2008;14(20):6674–6682. doi:10.1158/1078-0432.CCR-07-5212
31. Salas RN, Finke JH, Rini B. The intersection of sunitinib with the immunosuppressive microenvironment of renal cell carcinoma: implications for future therapeutics. Target Oncol. 2007;2(4):225–234. doi:10.1007/s11523-007-0064-3
32. Ko JS, Zea AH, Rini BI, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15(6):2148–2157. doi:10.1158/1078-0432.CCR-08-1332
33. Horlock C, Stott B, Dyson PJ, et al. The effects of trastuzumab on the CD4+CD25+FoxP3+ and CD4+IL17A+ T-cell axis in patients with breast cancer. Br J Cancer. 2009;100(7):1061–1067. doi:10.1038/sj.bjc.6604963
34. Steinberg SM, Zhang P, Malik BT, et al. BRAF inhibition alleviates immune suppression in murine autochthonous melanoma. Cancer Immunol Res. 2014;2(11):1044–1050. doi:10.1158/2326-6066.CIR-14-0074
35. Ghoreschi K, Laurence A, O’Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228(1):273–287. doi:10.1111/j.1600-065X.2008.00754.x
36. Iamsawat S, Daenthanasanmak A, Voss JH, et al. Stabilization of Foxp3 by targeting JAK2 enhances efficacy of CD8 induced regulatory T cells in the prevention of graft-versus-host disease. J Immunol. 2018;201(9):2812–2823. doi:10.4049/jimmunol.1800793
37. Barbui T, Carobbio A, Finazzi G, et al. Inflammation and thrombosis in essential thrombocythemia and polycythemia vera: different role of C-reactive protein and pentraxin 3. Haematologica. 2011;96(2):315–318. doi:10.3324/haematol.2010.031070
38. Hasselbalch HC, Bjørn ME. MPNs as inflammatory diseases: the evidence, consequences, and perspectives. Mediators Inflamm. 2015;2015:102476. doi:10.1155/2015/102476
39. Fragoulis GE, McInnes IB, Siebert S. JAK-inhibitors. New players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology (Oxford). 2019;58(Suppl1):i43–i54. doi:10.1093/rheumatology/key276
40. Pei H, He L, Shao M, et al. Discovery of a highly selective JAK3 inhibitor for the treatment of rheumatoid arthritis. Sci Rep. 2018;8(1):5273. doi:10.1038/s41598-018-23569-y
41. Fleischmann RM, Damjanov NS, Kivitz AJ, Legedza A, Hoock T, Kinnman N. A randomized, double-blind, placebo-controlled, twelve-week, dose-ranging study of decernotinib, an oral selective JAK-3 inhibitor, as monotherapy in patients with active rheumatoid arthritis. Arthritis Rheumatol. 2015;67(2):334–343. doi:10.1002/art.38949
42. Shimoda K, Kato K, Aoki K, et al. Tyk2 plays a restricted role in IFN alpha signaling, although it is required for IL-12-mediated T cell function. Immunity. 2000;13(4):561–571. doi:10.1016/s1074-7613(00)00055-8
43. Ishizaki M, Akimoto T, Muromoto R, et al. Involvement of tyrosine kinase-2 in both the IL-12/Th1 and IL-23/Th17 axes in vivo. J Immunol. 2011;187(1):181–189. doi:10.4049/jimmunol.1003244
44. Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects [published correction appears in drugs.;77 (8):939][published correction appears in drugs. 2017 Jun 12]. Drugs. 2017;77(5):521–546. doi:10.1007/s40265-017-0701-9
45. Rother N. Disturbed T cell signaling and altered Th17 and regulatory T cell subsets in the pathogenesis of systemic lupus erythematosus. Front Immunol. 2015;6:610. doi:10.3389/fimmu.2015.00610
46. Bussel J, Arnold DM, Grossbard E, et al. Fostamatinib for the treatment of adult persistent and chronic immune thrombocytopenia: results of two Phase 3, randomized, placebo-controlled trials. Am J Hematol. 2018;93(7):921–930. doi:10.1002/ajh.25125
47. Newland A, Lee EJ, McDonald V, Bussel JB. Fostamatinib for persistent/chronic adult immune thrombocytopenia. Immunotherapy. 2018;10(1):9–25. doi:10.2217/imt-2017-0097
48. Keohane C, Kordasti S, Seidl T, et al. JAK inhibition induces silencing of T Helper cytokine secretion and a profound reduction in T regulatory cells. Br J Haematol. 2015;171(1):60–73. doi:10.1111/bjh.13519
49. Sewgobind VD, Quaedackers ME, van der Laan LJ, et al. The Jak inhibitor CP-690550 preserves the function of CD4CD25FoxP3 regulatory T cells and inhibits effector T cells. Am J Transplant. 2010;10(8):1785–1795. doi:10.1111/j.1600-6143.2010.03200.x
50. Meyer A, Wittekind PS, Kotschenreuther K, et al. Regulatory T cell frequencies in patients with rheumatoid arthritis are increased by conventional and biological DMARDs but not by JAK inhibitors. Ann Rheum Dis. 2019:
51. Alzahrani KS, Nadeem A, Ahmad SF, et al. Inhibition of spleen tyrosine kinase attenuates psoriasis-like inflammation in mice through blockade of dendritic cell-Th17 inflammation axis. Biomed Pharmacother. 2019;111:347–358. doi:10.1016/j.biopha.2018.12.060
52. Park JS, Lee J, Lim MA, et al. JAK2-STAT3 blockade by AG490 suppresses autoimmune arthritis in mice via reciprocal regulation of regulatory T cells and Th17 cells. J Immunol. 2014;192(9):4417–4424. doi:10.4049/jimmunol.1300514
53. Tanaka Y, McInnes IB, Taylor PC, et al. Characterization and changes of lymphocyte subsets in baricitinib-treated patients with rheumatoid arthritis: an integrated analysis. Arthritis Rheumatol. 2018;70(12):1923–1932. doi:10.1002/art.40680
54. Jasiecka-Mikolajczyk A, Socha P, Jaroszewski JJ, Maslanka T. Oclacitinib induces Foxp3 expression in CD4+ T cells under activation conditions. J Immunol. 2020;204(1 Supplement):92.
55. Betts BC, Bastian D, Iamsawat S, et al. Targeting JAK2 reduces GVHD and xenograft rejection through regulation of T cell differentiation. Proc Natl Acad Sci U S A. 2018;115(7):1582–1587. doi:10.1073/pnas.1712452115
56. Spoerl S, Mathew NR, Bscheider M, et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood. 2014;123(24):3832–3842. doi:10.1182/blood-2013-12-543736
57. Parampalli Yajnanarayana S, Stübig T, Cornez I, et al. JAK1/2 inhibition impairs T cell function in vitro and in patients with myeloproliferative neoplasms. Br J Haematol. 2015;169(6):824–833. doi:10.1111/bjh.13373
58. Massa M, Rosti V, Campanelli R, Fois G, Barosi G. Rapid and long-lasting decrease of T-regulatory cells in patients with myelofibrosis treated with ruxolitinib. Leukemia. 2014;28(2):449–451. doi:10.1038/leu.2013.296
59. Virtanen T, Haikarainen T, Raivola J, Silvennoinen O. Selective JAKinibs: prospects in inflammatory and autoimmune diseases. BioDrugs. 2019;33(1):15–32. doi:10.1007/s40259-019-00333-w
60. Sigismund S, Avanzato D, Lanzetti L. Emerging functions of the EGFR in cancer. Mol Oncol. 2018;12(1):3–20. doi:10.1002/1878-0261.12155
61. Zaiss DM, van Loosdregt J, Gorlani A, et al. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity. 2013;38(2):275–284. doi:10.1016/j.immuni.2012.09.023
62. Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: beyond discovery and development. Cell. 2019;176(6):1248–1264. doi:10.1016/j.cell.2019.01.021
63. Zhu P, Hu C, Hui K, Jiang X. The role and significance of VEGFR2+ regulatory T cells in tumor immunity. Onco Targets Ther. 2017;10:4315–4319. doi:10.2147/OTT.S142085
64. Farooqi AA, Siddik ZH. Platelet-derived growth factor (PDGF) signalling in cancer: rapidly emerging signalling landscape. Cell Biochem Funct. 2015;33(5):257–265. doi:10.1002/cbf.3120
65. Agrawal S, Ganguly S, Hajian P, Cao JN, Agrawal A. PDGF upregulates CLEC-2 to induce T regulatory cells. Oncotarget. 2015;6(30):28621–28632. doi:10.18632/oncotarget.5765
66. Croce L, Coperchini F, Magri F, Chiovato L, Rotondi M. The multifaceted anti-cancer effects of BRAF-inhibitors. Oncotarget. 2019;10(61):6623–6640. doi:10.18632/oncotarget.27304
67. Shabaneh TB, Molodtsov AK, Steinberg SM, et al. Oncogenic BRAFV600E governs regulatory T-cell recruitment during melanoma tumorigenesis. Cancer Res. 2018;78(17):5038–5049. doi:10.1158/0008-5472.CAN-18-0365
68. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi:10.1038/nature00766
69. Kang ZJ, Liu YF, Xu LZ, et al. The Philadelphia chromosome in leukemogenesis. Chin J Cancer. 2016;35:48. doi:10.1186/s40880-016-0108-0
70. Manlove LS, Berquam-Vrieze KE, Pauken KE, Williams RT, Jenkins MK, Farrar MA. Adaptive immunity to leukemia is inhibited by cross-reactive induced regulatory T cells. J Immunol. 2015;195(8):4028–4037. doi:10.4049/jimmunol.1501291
71. Buckner JH. Mechanisms of impaired regulation by CD4(+)CD25(+)FOXP3(+) regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10(12):849–859. doi:10.1038/nri2889
72. Cai Y, Fleming C, Yan J. New insights of T cells in the pathogenesis of psoriasis. Cell Mol Immunol. 2012;9(4):302–309. doi:10.1038/cmi.2012.15
73. Federici M, Giustizieri ML, Scarponi C, Girolomoni G, Albanesi C. Impaired IFN-gamma-dependent inflammatory responses in human keratinocytes overexpressing the suppressor of cytokine signaling 1. J Immunol. 2002;169(1):434–442. doi:10.4049/jimmunol.169.1.434
74. Morelli M, Scarponi C, Mercurio L, et al. Selective immunomodulation of inflammatory pathways in keratinocytes by the janus kinase (JAK) inhibitor tofacitinib: implications for the employment of JAK-targeting drugs in psoriasis. J Immunol Res. 2018;2018:7897263. doi:10.1155/2018/7897263
75. Jones DS, Jenney AP, Swantek JL, Burke JM, Lauffenburger DA, Sorger PK. Profiling drugs for rheumatoid arthritis that inhibit synovial fibroblast activation. Nat Chem Biol. 2017;13(1):38–45. doi:10.1038/nchembio.2211
76. Vainchenker W, Leroy E, Gilles L, Marty C, Plo I, Constantinescu SN. JAK inhibitors for the treatment of myeloproliferative neoplasms and other disorders. F1000Res. 2018;7:82. doi:10.12688/f1000research.13167.1
77. Liotta F, Gacci M, Frosali F, et al. Frequency of regulatory T cells in peripheral blood and in tumour-infiltrating lymphocytes correlates with poor prognosis in renal cell carcinoma. BJU Int. 2011;107(9):1500–1506. doi:10.1111/j.1464-410X.2010.09555.x
78. Medicines.org.uk; Pfizer Limited; 2021. Inlyta 5 mg film coated tablet; Summary of Product Characteristics. Available from: https://www.medicines.org.uk/emc/product/7948. Accessed 4 March 2021.
79. Medicines.org.uk; Roche Products Limited; 2021. Avastin 25mg/ml concentrate for solution for infusion; Summary of Product Characteristics. Available from: https://www.medicines.org.uk/emc/product/3885. Accessed 4 March 2021.
80. Wijarnpreecha K, Nissaisorakarn P, Sornprom S, et al. Hepatitis C infection and renal cell carcinoma: a systematic review and meta-analysis. World J Gastrointest Pathophysiol. 2016;7(4):314–319. doi:10.4291/wjgp.v7.i4.314
81. Farhadi A, Behzad-Behbahani A, Geramizadeh B, et al. Presence of oncogenic viruses (EBV, HHV-6, BKV and JCV) DNA sequences in renal cell carcinoma. Int J Infect Dis. 2012;16(1):e80. doi:10.1016/j.ijid.2012.05.193
82. Shimakage M, Kawahara K, Harada S, Sasagawa T, Shinka T, Oka T. Expression of Epstein-Barr virus in renal cell carcinoma. Oncol Rep. 2007;18(1):41–46.
83. Heidegger I, Borena W, Pichler R. The role of human papilloma virus in urological malignancies. Anticancer Res. 2015;35(5):2513–2519.
84. Liu C, Wu S, Meng X, et al. Predictive value of peripheral regulatory T cells in non-small cell lung cancer patients undergoing radiotherapy. Oncotarget. 2017;8(26):43427–43438. doi:10.18632/oncotarget.15238
85. Shimizu K, Nakata M, Hirami Y, Yukawa T, Maeda A, Tanemoto K. Tumor-infiltrating Foxp3+ regulatory T cells are correlated with cyclooxygenase-2 expression and are associated with recurrence in resected non-small cell lung cancer. J Thorac Oncol. 2010;5(5):585–590. doi:10.1097/JTO.0b013e3181d60fd7
86. Zhu QG, Zhang SM, Ding XX, He B, Zhang HQ. Driver genes in non-small cell lung cancer: characteristics, detection methods, and targeted therapies. Oncotarget. 2017;8(34):57680–57692. doi:10.18632/oncotarget.17016
87. Liu Y, O’Brien JL, Ajami NJ, et al. Lung tissue microbial profile in lung cancer is distinct from emphysema. Am J Cancer Res. 2018;8(9):1775–1787.
88. O’Mahony C, Scully P, O’Mahony D, et al. Commensal-induced regulatory T cells mediate protection against pathogen-stimulated NF-kappaB activation. PLoS Pathog. 2008;4(8):e1000112. doi:10.1371/journal.ppat.1000112
89. Mertens J, Fabri M, Zingarelli A, et al. Streptococcus pneumoniae serotype 1 capsular polysaccharide induces CD8CD28 regulatory T lymphocytes by TCR crosslinking. PLoS Pathog. 2009;5(9):e1000596. doi:10.1371/journal.ppat.1000596
90. Larsen JM. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology. 2017;151(4):363–374. doi:10.1111/imm.12760
91. Mangalam A, Shahi SK, Luckey D, et al. Human gut-derived commensal bacteria suppress CNS inflammatory and demyelinating disease. Cell Rep. 2017;20(6):1269–1277. doi:10.1016/j.celrep.2017.07.031
92. Robinson LA, Jaing CJ, Pierce Campbell C, et al. Molecular evidence of viral DNA in non-small cell lung cancer and non-neoplastic lung. Br J Cancer. 2016;115(4):497–504. doi:10.1038/bjc.2016.213
93. Kheir F, Zhao M, Strong MJ, et al. Detection of Epstein-Barr virus infection in non-small cell lung cancer. Cancers (Basel). 2019;11(6):759. doi:10.3390/cancers11060759
94. Landtblom AR, Andersson TM, Dickman PW, et al. Risk of infections in patients with myeloproliferative neoplasms-a population-based cohort study of 8363 patients [published online ahead of print, 2020 Jun 16]. Leukemia. 2020. doi:10.1038/s41375-020-0909-7.
95. Ruan S, Tate C, Lee JJ, Ritter T, Kolls JK, Shellito JE. Local delivery of the viral interleukin-10 gene suppresses tissue inflammation in murine Pneumocystis carinii infection. Infect Immun. 2002;70(11):6107–6113. doi:10.1128/iai.70.11.6107-6113.2002
96. Wright TW, Gigliotti F, Finkelstein JN, McBride JT, An CL, Harmsen AG. Immune-mediated inflammation directly impairs pulmonary function, contributing to the pathogenesis of Pneumocystis carinii pneumonia. J Clin Invest. 1999;104(9):1307–1317. doi:10.1172/JCI6688
97. Shrivastava S, TrehanPati N, Patra S, et al. Increased regulatory T cells and impaired functions of circulating CD8 T lymphocytes is associated with viral persistence in Hepatitis B virus-positive newborns. J Viral Hepat. 2013;20(8):582–591. doi:10.1111/jvh.12078
98. Kolata JB, Kühbandner I, Link C, et al. The fall of a dogma? Unexpected high T-cell memory response to Staphylococcus aureus in humans. J Infect Dis. 2015;212(5):830–838. doi:10.1093/infdis/jiv128
99. Frodermann V, Chau TA, Sayedyahossein S, Toth JM, Heinrichs DE, Madrenas J. A modulatory interleukin-10 response to staphylococcal peptidoglycan prevents Th1/Th17 adaptive immunity to Staphylococcus aureus. J Infect Dis. 2011;204(2):253–262. doi:10.1093/infdis/jir276
100. Ferraro A, Buonocore SM, Auquier P, et al. Role and plasticity of Th1 and Th17 responses in immunity to Staphylococcus aureus. Hum Vaccin Immunother. 2019;15(12):2980–2992. doi:10.1080/21645515.2019.1613126
101. Rose WE, Shukla SK, Berti AD, et al. Increased endovascular staphylococcus aureus inoculum is the link between elevated serum interleukin 10 concentrations and mortality in patients with bacteremia [published correction appears in clin infect dis. 2017 Oct 15;65(8):1431–1433]. Clin Infect Dis. 2017;64(10):1406–1412. doi:10.1093/cid/cix157
102. Zhang J, Zhu Z, Zuo X, et al. The role of NTHi colonization and infection in the pathogenesis of neutrophilic asthma. Respir Res. 2020;21(1):170. doi:10.1186/s12931-020-01438-5
103. Essilfie AT, Simpson JL, Horvat JC, et al. Haemophilus influenzae infection drives IL-17-mediated neutrophilic allergic airways disease. PLoS Pathog. 2011;7(10):e1002244. doi:10.1371/journal.ppat.1002244
104. Guan X, Lu Y, Wang G, et al. The role of regulatory T cell in nontypeable haemophilus influenzae-induced acute exacerbation of chronic obstructive pulmonary disease. Mediators Inflamm. 2018;2018:8387150. doi:10.1155/2018/8387150
105. Kim MS, Kim DJ, Na CH, Shin BSA. Study of the changes of T helper 17 cells and regulatory T cells in herpes zoster. Ann Dermatol. 2017;29(5):578–585. doi:10.5021/ad.2017.29.5.578
106. Betts RJ, Prabhu N, Ho AW, et al. Influenza A virus infection results in a robust, antigen-responsive, and widely disseminated Foxp3+ regulatory T cell response. J Virol. 2012;86(5):2817–2825. doi:10.1128/JVI.05685-11
107. Kudva A, Scheller EV, Robinson KM, et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol. 2011;186(3):1666–1674. doi:10.4049/jimmunol.1002194
108. Brereton CF, Sutton CE, Ross PJ, et al. Escherichia coli heat-labile enterotoxin promotes protective Th17 responses against infection by driving innate IL-1 and IL-23 production. J Immunol. 2011;186(10):5896–5906. doi:10.4049/jimmunol.1003789
109. Luo J, Zhang M, Yan B, Zhang K, Chen M, Deng S. Imbalance of Th17 and Treg in peripheral blood mononuclear cells of active tuberculosis patients. Braz J Infect Dis. 2017;21(2):155–161. doi:10.1016/j.bjid.2016.10.011
110. Nikitina IY, Panteleev AV, Kosmiadi GA, et al. Th1, Th17, and Th1Th17 lymphocytes during tuberculosis: th1 lymphocytes predominate and appear as low-differentiated CXCR3+CCR6+ cells in the blood and highly differentiated CXCR3±CCR6- cells in the lungs. J Immunol. 2018;200(6):2090–2103. doi:10.4049/jimmunol.1701424
111. Pai M, Denkinger CM, Kik SV, et al. Gamma interferon release assays for detection of Mycobacterium tuberculosis infection. Clin Microbiol Rev. 2014;27(1):3–20. doi:10.1128/CMR.00034-13
112. Li Q, Li J, Tian J, et al. IL-17 and IFN-γ production in peripheral blood following BCG vaccination and Mycobacterium tuberculosis infection in human. Eur Rev Med Pharmacol Sci. 2012;16(14):2029–2036.
113. Aung H, Toossi Z, McKenna SM, et al. Expression of transforming growth factor-beta but not tumor necrosis factor-alpha, interferon-gamma, and interleukin-4 in granulomatous lung lesions in tuberculosis. Tuber Lung Dis. 2000;80(2):61–67. doi:10.1054/tuld.2000.0235
114. Wu M, Aung H, Hirsch CS, Toossi Z. Inhibition of Mycobacterium tuberculosis-induced signalling by transforming growth factor-β in human mononuclear phagocytes. Scand J Immunol. 2012;75(3):301–304. doi:10.1111/j.1365-3083.2011.02668.x
115. Albillos A, Lario M, Álvarez-mon M. Cirrhosis-associated immune dysfunction: distinctive features and clinical relevance. J Hepatol. 2014;61(6):1385–1396. doi:10.1016/j.jhep.2014.08.010
116. Bentires-Alj M, Paez JG, David FS, et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004;64(24):8816–8820. doi:10.1158/0008-5472.CAN-04-1923
117. Lee IO, Kim JH, Choi YJ, et al. Helicobacter pylori CagA phosphorylation status determines the gp130-activated SHP2/ERK and JAK/STAT signal transduction pathways in gastric epithelial cells. J Biol Chem. 2010;285(21):16042–16050. doi:10.1074/jbc.M110.111054
118. Neumann WL, Coss E, Rugge M, Genta RM. Autoimmune atrophic gastritis–pathogenesis, pathology and management. Nat Rev Gastroenterol Hepatol. 2013;10(9):529–541. doi:10.1038/nrgastro.2013.101
119. Shi WJ, Liu W, Zhou XY, Ye F, Zhang GX. Associations of Helicobacter pylori infection and cytotoxin-associated gene A status with autoimmune thyroid diseases: a meta-analysis. Thyroid. 2013;23(10):1294–1300. doi:10.1089/thy.2012.0630
120. Smyk DS, Koutsoumpas AL, Mytilinaiou MG, Rigopoulou EI, Sakkas LI, Bogdanos DP. Helicobacter pylori and autoimmune disease: cause or bystander. World J Gastroenterol. 2014;20(3):613–629. doi:10.3748/wjg.v20.i3.613
121. Miller WE, Mosialos G, Kieff E, Raab-Traub N. Epstein-Barr virus LMP1 induction of the epidermal growth factor receptor is mediated through a TRAF signaling pathway distinct from NF-kappaB activation. J Virol. 1997;71(1):586–594. doi:10.1128/JVI.71.1.586-594.1997
122. Dawson CW, Laverick L, Morris MA, Tramoutanis G, Young LS. Epstein-Barr virus-encoded LMP1 regulates epithelial cell motility and invasion via the ERK-MAPK pathway. J Virol. 2008;82(7):3654–3664. doi:10.1128/JVI.01888-07
123. Dawson CW, Tramountanis G, Eliopoulos AG, Young LS. Epstein-Barr virus latent membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to promote cell survival and induce actin filament remodeling. J Biol Chem. 2003;278(6):3694–3704. doi:10.1074/jbc.M209840200
124. Vaysberg M, Lambert SL, Krams SM, Martinez OM. Activation of the JAK/STAT pathway in Epstein Barr virus+-associated posttransplant lymphoproliferative disease: role of interferon-gamma. Am J Transplant. 2009;9(10):2292–2302. doi:10.1111/j.1600-6143.2009.02781.x
125. Ando S, Kawada JI, Watanabe T, et al. Tofacitinib induces G1 cell-cycle arrest and inhibits tumor growth in Epstein-Barr virus-associated T and natural killer cell lymphoma cells. Oncotarget. 2016;7(47):76793–76805. doi:10.18632/oncotarget.12529
126. Fujiwara S, Takei M. Epstein‐Barr virus and autoimmune diseases. Clin Exp Neuroimmunol. 2015;6:38–48. doi:10.1111/cen3.12263
127. Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep. 2015;5(1):15179. doi:10.1038/srep15179
128. Ueki IF, Min-Oo G, Kalinowski A, et al. Respiratory virus-induced EGFR activation suppresses IRF1-dependent interferon λ and antiviral defense in airway epithelium. J Exp Med. 2013;210(10):1929–1936. doi:10.1084/jem.20121401
129. Alkharsah KR. VEGF upregulation in viral infections and its possible therapeutic implications. Int J Mol Sci. 2018;19(6):1642. doi:10.3390/ijms19061642
130. Soroceanu L, Akhavan A, Cobbs CS. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature. 2008;455(7211):391–395. doi:10.1038/nature07209
131. DuShane JK, Maginnis MS. Human DNA virus exploitation of the MAPK-ERK cascade. Int J Mol Sci. 2019;20(14):3427. doi:10.3390/ijms20143427
132. Benn J, Schneider RJ. Hepatitis B virus HBx protein activates Ras-GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade. Proc Natl Acad Sci U S A. 1994;91(22):10350–10354. doi:10.1073/pnas.91.22.10350
133. Klein NP, Schneider RJ. Activation of Src family kinases by hepatitis B virus HBx protein and coupled signaling to Ras. Mol Cell Biol. 1997;17(11):6427–6436. doi:10.1128/mcb.17.11.6427
134. Tran TTP, Eichholz K, Amelio P, et al. Humoral immune response to adenovirus induce tolerogenic bystander dendritic cells that promote generation of regulatory T cells. PLoS Pathog. 2018;14(8):e1007127. doi:10.1371/journal.ppat.1007127
135. Gaido CM, Granland C, Laing IA, et al. T-cell responses against rhinovirus species A and C in asthmatic and healthy children. Immun Inflamm Dis. 2018;6(1):143–153. doi:10.1002/iid3.206
136. Tovar-Salazar A, Weinberg A. Understanding the mechanism of action of cytomegalovirus-induced regulatory T cells. Virology. 2020;547:1–6. doi:10.1016/j.virol.2020.05.001
137. Jayaratne HE, Wijeratne D, Fernando S, et al. Regulatory T-cells in acute dengue viral infection. Immunology. 2018;154(1):89–97. doi:10.1111/imm.12863
138. Benson A, Murray S, Divakar P, et al. Microbial infection-induced expansion of effector T cells overcomes the suppressive effects of regulatory T cells via an IL-2 deprivation mechanism. J Immunol. 2012;188(2):800–810. doi:10.4049/jimmunol.1100769
139. Fleming SB. Viral inhibition of the IFN-induced JAK/STAT signalling pathway: development of live attenuated vaccines by mutation of viral-encoded IFN-antagonists. Vaccines (Basel). 2016;4(3):23. doi:10.3390/vaccines4030023
140. Kak G, Raza M, Tiwari BK. Interferon-gamma (IFN-γ): exploring its implications in infectious diseases. Biomol Concepts. 2018;9(1):64–79. doi:10.1515/bmc-2018-0007
141. Gerada C, Campbell TM, Kennedy JJ, et al. Manipulation of the innate immune response by varicella zoster virus. Front Immunol. 2020;11:1. doi:10.3389/fimmu.2020.00001
142. Beekhuizen H, van de Gevel JS. Gamma interferon confers resistance to infection with Staphylococcus aureus in human vascular endothelial cells by cooperative proinflammatory and enhanced intrinsic antibacterial activities. Infect Immun. 2007;75(12):5615–5626. doi:10.1128/IAI.00530-07
143. Norkina O, Dolganiuc A, Shapiro T, Kodys K, Mandrekar P, Szabo G. Acute alcohol activates STAT3, AP-1, and Sp-1 transcription factors via the family of Src kinases to promote IL-10 production in human monocytes. J Leukoc Biol. 2007;82(3):752–762. doi:10.1189/jlb.0207099
144. Psaltopoulou T, Sergentanis TN, Ntanasis-Stathopoulos I, Tzanninis IG, Tsilimigras DI, Dimopoulos MA. Alcohol consumption and risk of hematological malignancies: a meta-analysis of prospective studies. Int J Cancer. 2018;143(3):486–495. doi:10.1002/ijc.31330
145. Rasouli B, Ahlbom A, Andersson T, et al. Alcohol consumption is associated with reduced risk of type 2 diabetes and autoimmune diabetes in adults: results from the Nord-Trøndelag health study. Diabet Med. 2013;30(1):56–64. doi:10.1111/j.1464-5491.2012.03713.x
146. Zeng C, Shi X, Zhang B, et al. The imbalance of Th17/Th1/Tregs in patients with type 2 diabetes: relationship with metabolic factors and complications. J Mol Med (Berl). 2012;90(2):175–186. doi:10.1007/s00109-011-0816-5
147. Farag AGA, Samaka R, Elshafey EN, Shehata WA, El Sherbiny EG, Hammam MA. Immunohistochemical study of janus kinase 1/signal transducer and activator of transcription 3 in psoriasis vulgaris. Clin Cosmet Investig Dermatol. 2019;12:497–508. doi:10.2147/CCID.S202835
148. Gruber CN, Calis JJA, Buta S, et al. Complex autoinflammatory syndrome unveils fundamental principles of JAK1 kinase transcriptional and biochemical function. Immunity. 2020;53(3):672–684.e11. doi:10.1016/j.immuni.2020.07.006
149. Del Bel KL, Ragotte RJ, Saferali A, et al. JAK1 gain-of-function causes an autosomal dominant immune dysregulatory and hypereosinophilic syndrome. J Allergy Clin Immunol. 2017;139(6):2016–2020.e5. doi:10.1016/j.jaci.2016.12.957
150. Arulogun SO, Choong HL, Taylor D, et al. JAK1 somatic mutation in a myeloproliferative neoplasm. Haematologica. 2017;102(8):e324–e327. doi:10.3324/haematol.2017.170266
151. Albacker LA, Wu J, Smith P, et al. Loss of function JAK1 mutations occur at high frequency in cancers with microsatellite instability and are suggestive of immune evasion. PLoS One. 2017;12(11):e0176181. doi:10.1371/journal.pone.0176181
152. Ren Y, Zhang Y, Liu RZ, et al. JAK1 truncating mutations in gynecologic cancer define new role of cancer-associated protein tyrosine kinase aberrations. Sci Rep. 2013;3(1):3042. doi:10.1038/srep03042
153. Hu J, Wu X, Huang P, Teng F, Wang Y, Xue F. The proportion and prognostic significance of T-regulatory cells in patients with gynecological cancers: a systematic review and meta-analysis. J Cancer. 2020;11(11):3340–3348. doi:10.7150/jca.42472
154. Zhang X, Kelaria S, Kerstetter J, Wang J. The functional and prognostic implications of regulatory T cells in colorectal carcinoma. J Gastrointest Oncol. 2015;6(3):307–313. doi:10.3978/j.issn.2078-6891.2015.017
155. Liu X, Zhang Z, Zhao G. Recent advances in the study of regulatory T cells in gastric cancer. Int Immunopharmacol. 2019;73:560–567. doi:10.1016/j.intimp.2019.05.009
156. Dai J, Lu Y, Roca H, et al. Immune mediators in the tumor microenvironment of prostate cancer. Chin J Cancer. 2017;36(1):29. doi:10.1186/s40880-017-0198-3
157. Xu P, Fan W, Zhang Z, et al. The clinicopathological and prognostic implications of FoxP3+ regulatory T cells in patients with colorectal cancer: a meta-analysis. Front Physiol. 2017;8:950. doi:10.3389/fphys.2017.00950
158. Paniagua RT, Sharpe O, Ho PP, et al. Selective tyrosine kinase inhibition by imatinib mesylate for the treatment of autoimmune arthritis. J Clin Invest. 2006;116(10):2633–2642. doi:10.1172/JCI28546
159. Eklund KK, Joensuu H. Treatment of rheumatoid arthritis with imatinib mesylate: clinical improvement in three refractory cases [published correction appears in Ann Med. 2003;35(8):640]. Ann Med. 2003;35(5):362–367. doi:10.1080/07853890310001339
160. Eklund KK, Remitz A, Kautiainen H, Reitamo S, Leirisalo-Repo M. Three months treatment of active spondyloarthritis with imatinib mesylate: an open-label pilot study with six patients. Rheumatology (Oxford). 2006;45(12):1573–1575. doi:10.1093/rheumatology/kel365
161. Kuang YH, Lu Y, Liu YK, et al. Topical Sunitinib ointment alleviates Psoriasis-like inflammation by inhibiting the proliferation and apoptosis of keratinocytes. Eur J Pharmacol. 2018;824:57–63. doi:10.1016/j.ejphar.2018.01.048
162. Ozgen M, Koca SS, Karatas A, et al. Lapatinib ameliorates experimental arthritis in rats. Inflammation. 2015;38(1):252–259. doi:10.1007/s10753-014-0028-6
163. Gore J, Craven KE, Wilson JL, et al. TCGA data and patient-derived orthotopic xenografts highlight pancreatic cancer-associated angiogenesis. Oncotarget. 2015;6(10):7504–7521. doi:10.18632/oncotarget.3233
164. Koblish HK, Hansbury M, Wang L-CS, et al. Novel immunotherapeutic activity of JAK and PI3Kδ inhibitors in a model of pancreatic cancer [abstract]. Cancer Res. 2015;75:1336. doi:10.1158/1538-7445.am2015-1336
165. Zhu Z, Aref AR, Cohoon TJ, et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014;4(4):452–465. doi:10.1158/2159-8290.CD-13-0646
166. Barbie DA, Spira A, Kelly K, et al. Phase 1B study of momelotinib combined with trametinib in metastatic, kirsten rat sarcoma viral oncogene homolog-mutated non-small-cell lung cancer after platinum-based chemotherapy treatment failure. Clin Lung Cancer. 2018;19(6):e853–e859. doi:10.1016/j.cllc.2018.07.004
167. Ng K, Hendifar A, Starodub A, et al. Phase 1 dose-escalation study of momelotinib, a Janus kinase 1/2 inhibitor, combined with gemcitabine and nab-paclitaxel in patients with previously untreated metastatic pancreatic ductal adenocarcinoma. Invest New Drugs. 2019;37(1):159–165. doi:10.1007/s10637-018-0650-5
168. Fogelman D, Cubillo A, García-Alfonso P, et al. Randomized, double-blind, phase two study of ruxolitinib plus regorafenib in patients with relapsed/refractory metastatic colorectal cancer. Cancer Med. 2018;7(11):5382–5393. doi:10.1002/cam4.1703
169. Hurwitz HI, Uppal N, Wagner SA, et al. Randomized, double-blind, phase II study of ruxolitinib or placebo in combination with capecitabine in patients with metastatic pancreatic cancer for whom therapy with gemcitabine has failed. J Clin Oncol. 2015;33(34):4039–4047. doi:10.1200/JCO.2015.61.4578
170. Hurwitz H, Van Cutsem E, Bendell J, et al. Ruxolitinib + capecitabine in advanced/metastatic pancreatic cancer after disease progression/intolerance to first-line therapy: JANUS 1 and 2 randomized phase III studies. Invest New Drugs. 2018;36(4):683–695. doi:10.1007/s10637-018-0580-2
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.