Back to Journals » Journal of Inflammation Research » Volume 12
The Binary Classification Of Chronic Diseases
Authors Elkoshi Z
Received 14 August 2019
Accepted for publication 7 November 2019
Published 16 December 2019 Volume 2019:12 Pages 319—333
DOI https://doi.org/10.2147/JIR.S227279
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Zeev Elkoshi
Taro Pharmaceutical Industries, Haifa Bay, Israel
Correspondence: Zeev Elkoshi Email [email protected]
Abstract: Acute diseases start with an insult and end when insult disappears. If the trauma induces an immune reaction (which happens in most cases), this reaction must be terminated with some type of resolution mechanism, when the cause of the trauma ceases. Chronicity develops if insult is permanent or if the resolution mechanism is defective. Another way to reach disease chronicity is a positive feedback loop, whereby the immune reaction activates an internal, insult-like reaction. A distinction between chronic states characterized by a persistent, low suppressive effect and those characterized by a persistent, high suppressive effect of regulatory T cells (Treg), is proposed. This two-class division represents two ways to reach chronicity: (a) by maintaining inflammatory reaction long after insult disappears (“low Treg”), or (b) by suppressing inflammatory reaction prior to the disappearance of insult (“high Treg”). This two-class division may explain the strong association between certain pathogens and cancer, on one hand, and between several other pathogens and autoimmunity, on the other hand. The weak association between autoimmune diseases and HIV infection and the relatively weak association between autoimmune diseases and cancer may be elucidated as well. In addition, the model rationalizes why immune-modulating drugs, which are effective in cancer, are also effective in “high Treg” viral infections, while corticosteroids, which are generally effective in autoimmune diseases, are also effective in other “low Treg” diseases (such as asthma, atopic dermatitis, and “low Treg” infections) but are not effective in solid malignancies and “high Treg” infections. Moreover, the model expounds why certain bacteria inhibit tumor growth and why these very bacteria induce autoimmune diseases.
Keywords: Treg cells, cancer, autoimmunity, inflammation, chronic diseases, immune-therapy, corticosteroids
Introduction
The role of the immune system is to combat invaders and control diseases. Diseases that are able to manipulate the immune system in the long run, may maintain chronicity.
It should be realized that a self-resolving (acute) inflammation is a tightly controlled reaction of the immune system, to a varying pathological condition. Upon insult initiation, the immune system reacts in an inflammatory mode, combating the invader. This reaction is opposed by the action of anti-inflammatory cytokines, with the result of inflammation resolution. The two arms of the immune system must operate in a timely coordinated manner, and, in proper relation to the changing state of the threat.1
An example of this process was presented by Sagiv et al.2 Mice-induced peritonitis served as a model for self-resolving acute inflammation. In this commonly used model, inflammation reaches a maximum within several hours after it has been triggered (by zymosan administration). This phase, characterized by a flux of neutrophils into the peritoneum, was resolved in most cases within 72 hr post onset. High-density neutrophils (HDNs), the pro-inflammatory neutrophil subpopulation, controlled the process for the first few hours. Low-density neutrophils (LDNs), the anti-inflammatory species, accumulated slowly during this process of acute inflammation, peaking 48 hrs following inflammation triggering.2 This constitutes the peak anti-inflammatory effect of LDNs. In acute self-resolving inflammation, the anti-inflammatory immune reaction follows the course of inflammation in an orchestrated, time-delayed manner.1 Any deviation from this sequence of events may lead to chronicity of the condition.
For example, if during the course of inflammation, an anti-inflammatory effect is not triggered (or is too low), inflammation turns chronic. This is the case with some infections and autoimmune diseases.3 These pathologies are characterized by a lasting, weak effect of regulatory T cells (Treg). Under this condition, the immune system is not suppressed following its arousal (tumor associated neutrophils (TANs) of the N1 type are not switched to the N2 type, HDNs are not switched to LDNs, Th17 are continuously generated) and inflammation or auto-inflammation persist and may damage body tissues.4
The opposite happens if an anti-inflammatory effect persists. This is the case with late-stage cancer and infections characterized by a strong Treg effect.3 A high and continuous TGFβ level within the tumor (in the case of cancer) and in the periphery (in infection and cancer) induces Treg differentiation. This effect was observed in vitro, with or without IL-6 and IL-23 (but was more prominent in the absence of these cytokines).5 This leads to a continuous suppression of the immune system (N2 are not switched back to N1, LDN are not switched back to HDN, Treg cells are continuously differentiated). The immune response weakens for a lengthy period of time, so that cancer and “high Treg” infections are not well controlled, with the result of tumor or pathogen propagation.
During the early stages of cancer, low TGFβ concentrations, which have an anti-tumor effect, inhibit the proliferation of normal cells and induce their apoptosis. At this stage, pre‑cancerous tumor growth is inhibited as well. On the other hand, during late cancer stages, as TGFβ accumulates, it acquires pro‑oncogenic and pro‑metastatic roles.6 Similarly, TANs switch from N1 (anti-tumor) to N2 (pro-tumor) neutrophils as the tumor progresses.7,8 A persistent anti-inflammatory and pro-tumor effect, driven by accumulated TGFβ, hinders cancer eradication. This is another example of a deviation from the sequence of events that characterizes self-resolving inflammation.
The regulation of the immune system by TGFβ is frequently referred to in the literature.9 Generally, TGFβ is described as an immunosuppressor, mediating its effect predominantly by affecting antigen-presenting cells (APCs) and T cells. As far as T cells are concerned, TGFβ inhibits T cell proliferation, activation, and effector functioning (toxicity). TGFβ suppresses the proliferation of IL-2-dependent T cells in vitro,10 by inhibiting IL-2 generation, and it inhibits the activation of T cells by interfering with T cell receptor signaling processes. TGFβ suppresses CD8+ cytotoxic T cells functioning through several mechanisms. It has been shown in vivo that tumors may be eradicated by TGFβ inhibition, which was accompanied by enhanced cytotoxic activity of CD8+ T cells.11 High TGFβ levels characterize cancer and “high Treg” infections.
Autoimmune diseases are characterized by an imbalance between pro-and anti-inflammatory factors. The frequency ratio of Treg cells to Th17 cells (Treg/Th17) is a measure used to assess this imbalance.12 This ratio is expected to decrease in autoimmune diseases, compared to normal control. It is of note that several autoimmune diseases with reduced Tregs’ activity present normal Treg cell frequency.13 In the broad sense, an impaired Treg cells functioning may be due to either: (a) an inadequate number of Treg cells, (b) an impaired suppression mechanism, or (c) the development of T cells that are resistant to the Treg cells suppressive effect.13 One such impaired mechanism is an insufficient secretion of TGFβ by Treg cells. An animal model involving TGFβ knockout mice demonstrated multifocal inflammation lesions that are reminiscent of systemic lupus erythematosus (SLE).14 Moreover, SLE-like autoantibodies were observed in TGFβ deficient mice.15 In fact, an impaired Tregs functioning (related to any of the three modalities) is observed in most, if not all, autoimmune diseases.13 On the other hand, an overexpression of Th17 cell cytokines may also lead to the development of autoimmune diseases. For example, an overexpression of TNFα in mutated mice results in rheumatoid, arthritis-like inflammation.16 Arthritis is also observed in mice with a mutation in the gp130 receptor, which results in enhanced STAT3 activation.17 STAT3 is required in humans for the production of IL-17, and also contributes to the development of Th17 in mouse and man.18
Since T cells frequencies do not always correlate with their activities, the Treg/Th17 cell ratio may not correctly represent the (regulatory/inflammatory) effect of these T cells, and should probably be replaced by the ratio of activities (which are more difficult to assess).
Regulatory T cells may exert opposing effects, in the context of disease. Treg cells may act as foes or friends, depending on whether the immune response is overstimulated or not. If the immune system is not overstimulated, activated Treg cells may promote the disease by down-regulating the immune activity. If the immune reaction is excessive, with the result of collateral tissue damage, activated Treg cells may alleviate this effect by controlling PMN (polymorhonuclear neutrophils) activity.
Based on the above, this work proposes a distinction between “high Treg” and “low Treg” classes of chronic diseases. “High Treg” diseases are defined as the class of diseases characterized by Treg cells’ high anti-inflammatory activity. The activity of Treg cells within the class of “high Treg” diseases promotes the disease by suppressing the immune response. “Low Treg” diseases are defined as the class of diseases that present low anti-inflammatory activity of Treg cells. Although Treg cells drive a positive anti-inflammatory effect within the class of “low Treg” diseases, their activity in the course of “low Treg” diseases is insufficient to control the harmful inflammation, and therefore elicit the chronicity of the condition. Autoimmune diseases are typically “low Treg” diseases.13 A “high Treg” disease improves upon a decreased Treg activity, while a “low Treg” disease improves upon an increased Treg activity. Of note, an anti-inflammatory effect may be exerted by other types of cells, such as granulocytes4 or B cells.3 This article refers solely to the effect of regulatory T cells. It should also be noted that the Treg/Th17 ratio, which represents the balance between pro-and anti-inflammatory effects, may be replaced by a single parameter: Treg or Th17 frequency. This is related to the reciprocal differentiation of Treg and Th17,13 which can be attributed to the effect of IL-2 (which promotes iTreg generation but inhibits Th17 activity); to the inhibitory effect of Treg-produced IL-10 on Th17 activity; to the TEAD transcription factor TAZ, which regulates the reciprocal differentiation of Treg and Th17;19 and to the transcription factor BACH2, which promotes Treg differentiation but suppresses Th17, Th1, and Th2 differentiation.20 In addition, Th17 and Treg lineages may interconvert, under certain inflammatory conditions.12 Therefore, under many circumstances, a high Treg frequency indicates a low Th17 frequency and vice versa.
Due to a similar chronicity mechanism, a strong association is expected between different “high Treg” diseases. Similarly, a strong association is expected between different “low Treg” diseases. On the other hand, a weak association is expected between “high Treg” diseases and “low Treg” diseases. In addition, immune-modulating drugs that are effective in a certain chronic disease are expected to be effective in another chronic disease only if both belong to the same class. Based on the argument above, it is also expected that only “low Treg” microorganisms will exert an anti-tumor effect. Correspondingly, only “low Treg” pathogens are expected to induce autoimmunity. Despite some exceptions, an overwhelming majority of data supports these anticipations. The rest of this work presents these data.
Cancer And “High Treg” Infections Are Strongly Associated
Viral Infections
Many viral infections belong to the “high Treg” class. High serum TGFβ levels were observed in Hepatitis B virus (HBV),21 Hepatitis C virus (HCV),22 human T‐cell leukemia virus type I infection (HTLV-1),23 and Epstein Barr virus (EBV).24 The extent of TGFβ expression in renal transplant patients correlated with sera polyomavirus BK (BKV) viral loads and BKV viremia positivity.25 Liver Treg cell frequency increases in HBV and HCV infections.26
HCV high TGF β levels are correlated with high Treg cell levels: “Treg cell proportions and IL-10 production were significantly elevated in HCV-infected patients, especially for HCV genotype 1b”.27 Expansion in CD39+ CD4+ immunoregulatory T Cells was observed in HTLV-1 infected patients and correlated with severity of neurological disorders.28 High levels of CD4+CD25hi+ Treg cells were recorded in endemic Burkitt’s lymphoma patients, a condition associated with EBV infection.29 Treg cell frequency significantly increased in EBV-associated gastric carcinomas tissues compared to EBV-negative gastric carcinomas tissues.30 Hence, EBV may be classified as a “high Treg” virus, within the context of gastric cancer (and probably other cancers). In addition, Human Papillomavirus (HPV) induced the generation of Treg cells,31 and high levels of Treg cells were associated with polyomavirus.32 Humans co-infected with human herpesvirus 8 and HIV and who developed Kaposi’s sarcoma, demonstrated an increased percentage of Treg cells 1.8 years before they developed Kaposi’s sarcoma. Coinfected humans with a normal Treg cell percentage do not develop Kaposi’s sarcoma.33
Indeed, some cancers are highly related to these viral infections:34
Liver Cancer: Hepatitis B and C viruses are linked to 80% of liver cancer cases.
Adult T-cell Leukemia: Human T lymphotropic virus is linked to almost 100% of adult T-cell Leukaemia cases.
Cervical Cancer: Human Papillomavirus is linked to 100% of cervical cancer cases.
Kaposi Sarcoma: Human Herpesvirus 8 is linked to almost 100% of Kaposi Sarcoma cases.
Merkel Cell Cancer: Merkel cell polyomavirus is linked to almost 100% of Merkel Cell Cancer cases.
Burkitt's Lymphoma and Nasopharyneal Cancer: Epstein Barr virus is linked to 10%–30% of Burkitt’s lymphoma and nasopharyngeal cancer cases.
Bacterial Infections
Stomach Cancer: Eighty percent (80%) of stomach cancer patients reveal H. pylori etiology.34 Th17 cells of the Helicobacter pylori-specific type survive in the gastric mucosa and blood of subjects with a history of Helicobacter pylori infection.35 Although increased IL-17 expression is observed during chronic gastric inflammation, the levels produced are not sufficient to clear the infection.36 Using animal models, Kato et al have shown that dendritic cells derived from bone marrow and infected by H. pylori bacteria skewed the Th17/Treg balance toward a Treg response through a TGFβ-dependent mechanism.37 In addition, gastric biopsies taken from H. pylori-associated gastritis patients indicated an increased number of Treg cells, which correlated with the severity of inflammation and the number of lymphoid follicles.38 Higher TGFβ serum levels were observed in H. pylori-positive gastritis and peptic ulcer patients, compared with those observed in H. pylori-negative patients.39 A high number of gastric Treg cells and high serum TGFβ levels in chronic H. pylori infection contribute to the high association with stomach cancer.
As can be seen, several viruses and bacteria are oncogenic to different cancers, with a prevalence that may reach 100% (with some viruses). This is also true for macro-parasites.34 It must be realized however, that some pathogens are oncogenic, some are oncolytic, and others that are considered neither oncogenic nor oncolytic promote cancer nonetheless by interacting with the immune system,34 as demonstrated above.
Autoimmune Diseases And “Low Treg” Infections Are (moderately) Associated
Viral Infections
As delineated above, the association between viral infections and autoimmune diseases is much less pronounced than their association with cancer, since many viral infections trigger a “high Treg” infection, while autoimmune diseases can generally be considered “low Treg” diseases.13 For example, in spite of the fact that Cytomegalovirus (CMV) was implicated in connection with many autoimmune diseases, a review by Halenius and Hengel40 concludes that “an association of human CMV seroprevalence and autoimmune diseases could not be established”. On the other hand, EBV seems to be involved in the pathogenesis of several autoimmune diseases.41 Salloum et al. revealed a tendency for a linear correlation between EBV DNA levels and serum IL-17A in RA patients. This effect was not observed in non-RA controls.42 Hence, EBV may be regarded as a “low Treg” virus within the context of RA (and probably other autoimmune diseases). This is in contrast to the EBV “high Treg” phenotype within the context of cancer. This exemplifies the plasticity of EBV in the pathogenesis of different chronic diseases, as will be discussed later.
Type 1 diabetes (T1D) is considered an autoimmune disease that involves the destruction of pancreas cells by autoimmune T cells, with or without the involvement of inflammatory cytokines. In the past, diabetes was considered a Th1 disease, but the current notion refers to an involvement of Th1, Th17, and Tfh reactions.43 Tfh is a subset of T helper cells, characterized by an IL-21 signature, that support B cell antibody production. The appellation “Tfh” is derived from their ability to penetrate the B cell follicles of secondary lymphoid tissues, where they form germinal centers in T1D.44 IL-21 counteracts the regulatory effect of Treg cells through the inhibition of IL-2 production.45 There is no evidence that circulating Treg cell numbers are decreased in TID. However, reduced numbers of Treg cells were found in the Langerhans Islets of the pancreas, assessed postmortem in newly diagnosed T1D patients. Other studies have indicated an impaired suppression of immune response by Treg cells isolated from T1D patients (probably due to effector T cell resistance).13Coxsackievirus B (CVB) is a possible cause of type I diabetes.46 It was found that Coxsackievirus B3 directly induced Th17 cell differentiation in patients with acute viral myocarditis, which may imply a reduced number of Treg cells.47
Bacterial Infections
Rheumatic Fever: The group A streptococcus, Streptococcus pyogenes, is linked to rheumatic fever, an autoimmune disease manifested by rheumatic carditis, Sydenham chorea, and pediatric autoimmune neuropsychiatric disorders (a new group of behavioral disorders). Autoantibody T cell responses and molecular mimicry between group A streptococcus, the brain, and the heart contribute to disease pathogenesis.48 Wang et al have shown that group A streptococcal infection induces TGFβ1 and TGFβ1-dependent, predominant Th17 differentiation.49 This is generally indicative of a low TGFβ1 level required for Th17 differentiation. In addition, it was found that memory or effector CD4+ T cells— produced following the nasal priming of mice with S. pyogenes—are not T-regulatory cells (identified by the two markers CD25 and Foxp3) when recirculated through mice spleen.50 Hence, group A streptococcus is probably a “low Treg” pathogen. Similarly, rheumatic heart diseases demonstrate a reduced number of circulating Treg cells.51 Low Treg activity shared by group A streptococcus and rheumatic fever contributes to their association.
Lyme Disease: The cause of Lyme disease is Borrelia burgdorfeii (Bb) bacteria, which is a tick-borne spirochete. Many untreated patients (about 60%) develop arthritis that affects the large joints and can last for several years. Typically, blood from these patients has a high concentration of Bb-specific antibodies, and the joint fluid contains Bb DNA.52 Intensive Th17 immune responses, which may limit early infection, are often developed in these patients.53 This is generally indicative of a low TGFβ level. Patients with chronic Lyme disease demonstrate decreased TGFβ1 synthesis by mononuclear cells, relative to the levels at early stages of the disease.54
Guillain–Barré syndrome (GBS): GBS is a muscle-affecting disease that involves the peripheral nervous system. Anti-glycolipid antibodies have been found in the serum of some of the patients. As a common precursor to GBS, Campylobacter jejuni is the most extensively studied specie among several pathogens associated with the disease.52 The pathogenesis of two GBS subtypes is associated with a short-term decrease in circulating Treg cells.55 Data relating to Campylobacter infection and Th17 or Treg responses is scarce.56
Multiple sclerosis (MS): Impairments in Treg functioning were observed in MS patients.57 Higher levels of the bacteria Chlamydia Pneumoniae were reported in MS patients.52 Benagiano et al found that the expression of several cytokines (IL-1β, IL-6, IL-23, TGF-β, and CCL-20) was upregulated by Chlamydia pneumonia phospholipase D (CpPLD) in monocytes extracted from healthy donors. They conclude that CpPLD promotes Th17 immune response in human atherosclerosis.58 This is generally indicative of “low Treg” bacteria.
Autoimmune Thyroid Diseases (ATDs): A meta-analysis of 7 studies including 862 patients revealed an overall Helicobacter pylori infection association with ATDs (OR 1.92 [CI 1.41–2.61]); the association was significant for Grave’s disease (OR 4.35 [CI 2.48–7.64]) but not for Hashimoto’s thyroiditis (OR 1.45 [CI 0.92–2.26], p=0.11).59 The percentage of Treg cells (CD4+Foxp3+T cells) and Foxp3 mRNA transcription factor was significantly lower in the PBMCs of patients with Grave’s Disease (P<0.05).60 Contrary to the involvement of H. pylori in gastric cancer, where the bacteria skew CD4 cell differentiation towards Treg and drive cancer (“high Treg”) as described above, here the low Treg frequency helps in promoting autoimmunity (a “low Treg” condition). This is an example of H. pylori plasticity, aimed at driving disease pathogenicity.
Parasite Infections
Chagas Heart Disease: About 30% of subjects infected with Trypanosoma cruzi (a protozoan parasite) develop a cardiac inflammation termed Chagas heart disease. During infection, some patients clearly present autoimmune responses, although the role of autoimmunity in the development of Chagas disease is not clear.61 Cai et al found that Th17 cells induce protection against Trypanosoma cruzi infection, both locally (at mucosal tissues) and systemically.62 The protection conferred by Th17 cells is more effective than that conferred by Th1 cells.63 Using a mouse model, it was found that Treg cells are not strongly involved in regulating the response of cytotoxic T cells during acute infection by T. cruzi. In addition, Treg cells were not observed in mice muscles during chronic T. cruzi infection.64 This means either a low frequency or a defective reaction of Treg cells to Trypanosoma cruzi. However, another study in mice points to an immune-suppressive role of Treg cells, which promote the parasite growth but at the same time control early carditis in Chagas disease and thus prolong survival.65
Cystic Fibrosis And “Low Treg” Pseudomonas Aeruginosa Infection
Cystic fibrosis (CF) is an autosomal, recessive genetic disorder that mainly involves the lungs. From an early age, the lungs of CF patients are colonized by bacteria that generate a mucus microenvironment known as biofilm, which impedes the permeability of immune system cells and their cytokines (as well as that of antibiotics) into airways.66 Eighty percent of adult CF patients harbor Pseudomonas aeruginosa. These bacteria elicit an IL-17/IL-23 response, typical to “low Treg” infection, which is associated with the exacerbation of pulmonary inflammation.67 Hector et al. demonstrated Tregs impairment in CF patients with chronic Pseudomonas infection.68 There is evidence suggesting the involvement of autoimmunity in CF. Moreover, the levels of autoantibodies in CF were found to be related to the severity of the disease.69
Taken together, clinical and experimental data generally confirm an association between cancer and “high Treg” infections. This is a strong association in many cases. Autoimmune diseases are generally associated (to a varying degree) with “low Treg” infections.
Cancer And Autoimmune Diseases Are Not Strongly Associated
A continuous inflammation is one of the causes of cancer. In particular, the risk of malignancy at the target organs of autoimmune diseases, such as the guts in celiac disease, the kidneys in lupus nephritis, or the brain in lupus cerebritis, is increased.70 This may be attributed to a local “high Treg” environment, within the systemic “low Treg” surroundings.71 It can be stated that cancers generally develop under “high Treg” conditions. As mentioned throughout this work, high TGFβ levels, the hallmark of “high Treg” chronic maladies, are observed with many types of cancer, especially solid tumors. It is therefore expected that cancer will be more strongly associated with “high Treg” infections compared to autoimmune diseases, which are generally “low Treg” ailments.
A review of the literature published between 2001 and 2011 reveals that the standardized residence ratios (SIR), odds ratios (OR), or hazard ratios of overall cancer risk for most of the 12 autoimmune conditions reported are lower than 2.70 One celiac disease study with a morbidity ratio (MR) of 5.99, one scleroderma study with a SIR value of 4.2, and one Sjogren’s syndrome study with a SIR value of 3.25 are the exceptions. High OR or SIR values may be misleading when the prevalence of cancer in the normal population is low, or when the group analyzed is small. For example, in the celiac disease study with a high MR value, only 1.3% of the celiac patients developed gastric cancer; even though this is a number that is significantly higher than that expected in the normal population, it is still a low number. The scleroderma study with the high SIR value included only 118 patients. In another study with 2040 patients, an SIR value of 1.5 was obtained (see Ref. 70 and the references therein).
Some hematological disorders, especially lymphomas, may present exceptionally high OR values for autoimmune diseases.70 For example, an odds ratio value of 6.5 for autoimmune hemolytic anemia in non-Hodgkin’s lymphoma (NHL) patients was obtained in a large-scale American study.72 Nevertheless, autoimmune hemolytic anemia affected only 87 out of 33, 721 (0.26%) of patients with NHL, i.e., the two conditions are not strongly associated. Moreover, as will be discussed below, the data regarding Treg functioning in hematologic malignancies is conflicting (i.e. some of them may be regarded as “low Treg” diseases).
In a case-control study of 3053 patients with acute myeloid leukemia (AML) and age‐matched population controls, Østgård et al investigated the relation between AML and autoimmune diseases. This association was observed only in patients with previous hematological disorder or those who have undergone chemotherapy (odds ratio=2.0) and not in de novo AML patients. The authors concluded that the development of de novo AML is not affected, or is only marginally affected, by autoimmune disorders.73
Very high OR or SIR values for organ-related malignancies in autoimmune diseases are sometimes reported.70 For example, an Italian study reports an SIR value of 25 for small intestinal cancer, observed before or simultaneously with the diagnosis of celiac disease.70 The original publication reports 5 small bowel cancer cases out of 1968 celiac patients, representing a probability of only 0.25% for small bowel cancer in celiac disease.74 The high SIR value obtained is related to the low value of the expected probability of small bowel cancer (i.e., the probability of small bowel cancer in the healthy population).
The hazard ratio for thyroid cancer in Grave’s disease estimated in a cohort of 5025 Taiwanese patients was 10.4. Still, the incidence rate (per 1000 person-years) was 1.62.75 This means that 1.6 patients out of 1000 patients with Grave’s disease develop thyroid cancer within one year. Assuming a mean chronicity period of 30 years, about 5% of Grave’s disease patients are expected to develop thyroid cancer through life.
It may be concluded that the association between cancer and autoimmune diseases is generally much weaker than the association between cancer and some of the “high Treg” pathogens, which often reaches 100%.
Coinfection/Comorbidity Is More Probable Within Members Of The Same Class
When different pathogens belong to the same class, as defined by Treg activity, they share a common effect on the myeloid and lymphoid arms of the immune system. A “high Treg” pathogen, for example, will induce the generation of anti-inflammatory neutrophil subpopulations, while at the same time it will suppress pro-inflammatory T cell reactions. It is expected therefore, that an infection by a pathogen that relates to one class (“high Treg” or “low Treg”), will increase the probability of a coinfection with other pathogens of the same class. This expectation is verified by clinical data. For example: Both HBV and HTLV-1 are “high Treg” infections.26,28 Indeed, HBV infection was detected in 19.2% and 15.9% of HTLV-1–seropositive indigenous adults admitted to Alice Springs Hospital (ASH) in Central Australia and patients in Northern Australia, respectively. Coinfection with HTLV-1 and HBV has previously been described in South America and southern Japan.76
In tumor tissue specimens taken from patients with squamous cell carcinoma of the larynx, EBV DNA was detected in 27.7% of the samples, 46.3% of which were co-infected with human papillomavirus (HPV).77 This correlates with the fact that EBV (in the context of cancer) and HPV are “high Treg” infections.30,31 In another study of laryngeal, oro-pharyngeal, and oral cavity cancer, the presence of EBV, HPV, or polyoma BK virus was assessed. Coinfection with at least two viruses was detected in 56.2% of the patients.78 All three viruses involved are “high Treg” viruses.30–32
HIV is a “high Treg” pathogen, and Treg frequency in HIV patients correlates with disease severity and viral load.79 Thus, HIV is not expected to be highly associated with autoimmune diseases, which are generally considered “low Treg” diseases.13 Indeed, autoimmune diseases were uncommon (only 0.69%) in 5186 HIV-infected French patients.80
HIV infection, however, does not belong to the discussion on coinfection presented above. HIV infection results in an intense depletion of CD4+ cells, which allows the thriving of many opportunistic pathogens, whether “high Treg” or “low Treg”.
Chronic Diseases That Belong To The Same Class Can Be Treated By The Same Immune-Modulating Drugs
Some drugs that are used to treat chronic diseases harness the immune system for this purpose. Thus, “high Treg” diseases can be treated by a stimulation of the immune response. On the other hand, “low Treg” diseases can be treated by attenuation of the immune response. It appears that an immunotherapy that is effective in one disease may be effective in another disease, only if they belong to the same class. Some examples are the following:
- It has been demonstrated in an animal model that type I interferon suppresses Treg function ing.81 Interferon alpha–2b is indicated for the treatment of hepatitis B and hepatitis C viral infections. In addition, it is indicated for the treatment of different cancers such as hairy cell leukemia, chronic myelogenous leukemia, multiple myeloma, follicular lymphoma, carcinoid tumor, and malignant melanoma. These types of cancer, as well as viral hepatitis B and C, are “high Treg” chronic diseases.
- Nivolumab is a human IgG4, anti-PD-1 monoclonal antibody. The immunogenic checkpoint PD-1 is highly expressed on Treg cells and has an important role in regulating Treg cell activity. Even though an increase or no change in Treg frequency following nivolumab treatment has been reported, some studies have demonstrated a reduced Treg suppressive activity in vitro.82 Nivolumab is indicated for the treatment of several cancers including melanoma, head and neck cancer, and non-small-cell lung cancer. At the same time, nivolumab has shown efficacy in treating chronic hepatitis C virus patients, with a result of a more than 4-log reduction of viral load in some patients.83 Thus, an anti-PD-1 treatment is effective in both cancer and “high Treg” viral infections.
- Corticosteroids (CS) have been used for many years in the treatment of autoimmune diseases.84 Bereshchenko et al.85 report that glucocorticoid-induced leucinezipper drives Treg cell proliferation and enhances Treg signaling. This is one of the mechanisms by which CS exert their anti-inflammatory effect. Being effective in “low Treg” autoimmune diseases, CS are expected to be effective in the treatment of other “low Treg” diseases. On the other hand, CS are not expected to be effective in “high Treg” diseases.
Indeed, the use of CS is recommended in the following infections: pneumocystic pneumonia,86 eye herpes simplex virus (HSV) infection,87 schitosomiasis,88 infectious mononucleosis,89 and sterptococal pharyngitis.90
As the data below demonstrates, these are all “low Treg” infections:
Pneumocystic Pneumonia: Tregs were found to attenuate inflammation in an animal model of the disease.91
Ocular HSV Infection: Tregs dampened the severity of HSV eye inflammation in an experimental mouse model.92
Schisotomiasis: Tregs reduced severity and prolonged survival in a mouse model of the disease.93
Infectious Mononucleosis (IM): “The percentage of Th17/CD4⁺T cells, Th17/Treg, IL-17, and IL-22 contents were significantly elevated in acute IM patients compared to recovery patients and control. Meanwhile, the Treg/CD4⁺T cells, IL-10 and TGF-ß contents were significantly lower”.94
Streptococal Pharyngitis: Group A streptococcus induces a predominant Th17 response in mouse nasal-associated lymphoid tissue.49
In addition, asthma, is commonly treated by inhaled corticosteroids. It has been demonstrated that thymic stromal lymphopoietin suppresses the generation of antigen-specific Tregs.95 An attenuation of thymic stromal lymphopoietin in an asthma mouse model alleviated airway inflammation.96 Therefore, asthma is a “low Treg” disease. This explains the effectiveness of CS in asthma treatment.
Atopic dermatitis is a skin inflammation that mainly affects children. Atopic dermatitis severity is positively related to Th17 cell percentage and Th17/Treg ratio, and negatively related to Treg cell percentage.97 Hence, atopic dermatitis is a “low Treg” condition. Unsurprisingly, topical CS are the mainstay of atopic dermatitis treatment.
Corticosteroids (“Low Treg” Diseases Drugs) Are Not Effective In “High treg” Diseases
Corticosteroids are (categorically) not recommended in the following six out of twenty-six infections listed in Aberdein and Singer’s review on CS use in infections:86 chronic hepatitis B, chronic hepatitis C, severe viral hepatitis, Dengue shock syndrome, cerebral malaria, idiopathic facial nerve (Bell’s) palsy, and cystic fibrosis. Bordetella pertussis, for which only one, small-scale study was retrieved by the authors, was excluded from this list.
The first four infections are “high Treg” viral infections. Symptomatic malaria is associated with high Treg levels.98 The removal of Treg cells induced an immune response against malaria parasite Plasmodium falciparum, in vitro.99 Hence malaria is a “high Treg” disease as well.
A recent Cochrane’s review opposes Aberdine and Singer’s conclusion regarding the ineffectiveness of CS in Bell’s palsy.100 This more recent work found benefit in the use of CS alone over the use of antivirals alone (which were not better than placebo) in treating the disease.100 It should be noted that the ineffectiveness of CS in cystic fibrosis, which is a “low Treg” disease, cannot be explained by the two-class model.
Corticosteroids are generally not effective in solid tumor malignancies, which are, as a whole, “high Treg” diseases.4 However, CS are very effective in treating lymphoid cancer, and were used as such for decades.101 This is not surprising since CS repress the lymphoid immune response (which is the source of pathogenicity in lymphomas). Corticosteroids induce growth arrest and apoptosis in lymphoid tissues through the glucocorticoid receptor.102 This mechanism possibly contributes to their effectiveness in lymphoid cancers and is not necessarily related to the suppression of effector T cell functioning or to the upregulation of Treg functioning by CS. On the other hand, the data on the prognostic significance of regulatory T cells in hematologic malignancies are conflicting, with some studies indicating a “low Treg” profile and others demonstrating a “high Treg” character.103
Only “Low Treg” Bacteria Can Inhibit Malignant Tumor Growth
In recent decades, an inhibitory effect of bacteria on tumor growth has been demonstrated. These include Clostridium novyi, Salmonella typhimurium, Streptococcus pyogenes,104 and Proteus mirabilis.105 Bacillus Calmette–Guérin (BCG) is a live, attenuated strain of Mycobacterium bovis used for tuberculosis vaccination. Since 1977, BCG vaccine has been routinely used as standard care for patients with non-muscle invasive bladder cancer.104 It is currently the only agent approved by the US Food and Drug Administration for primary therapy of this condition.
In accordance with the two-class model, all five bacteria listed above, which demonstrate anti-tumor activity, are pro-inflammatory “low Treg” bacteria:
Clostridium novyi: These bacteria elicit a strong pro-inflammatory effect by the induction of IL-6, G-CSF, MIP-2, and TIMP-1.106
Salmonella Typhimurium: Salmonella infection induces, directly or indirectly, the secretion of many pro-inflammatory cytokines, such as IL-18 (by epithelial cells), 1L-17, and IL-22 (by innate lymphoid cells: neutrophils, gamma delta T cells, innate Th17 cells and αβ T cells), and IFN-γ (by Th1 cells).107
Streptococcus Pyogenes: S. pyogenes intranasal infections mainly generate Th17 cells.108
BCG Vaccine: The major cytokines induced by BCG are IFNγ, IL2, and TNFα secreted by activated CD4+ T cells. All three cytokines are of a pro-inflammatory nature.109
Proteus Miabilis: This gram-negative bacterium, frequently found in urinary tract infections, elicits an immune response which is preferentially pro-inflammatory (even though the anti-inflammatory cytokine, IL-10, has been reported in the urine of infected persons as well).110
“Low treg” Bacteria That Inhibit Tumor Growth Induce Autoimmune Diseases
Adopting the two-class model, it is not surprising that these “low Treg” bacteria, which inhibit cancer, can also trigger autoimmunity: Salmonella species induce reactive arthritis;111 Streptococcus pyogenes induce rheumatic fever;48 Proteus mirabilis induce rheumatoid arthritis;112 Clostridium novyi is not known to induce autoimmunity; however, another strain of the bacteria, Clostridium difficile, is linked to reactive arthritis;113 Mycobacterium bovis is part of the etiology of several autoimmune diseases.114
A Pathogen May Induce Either “High Treg” Or “Low Treg” Responses, Depending On The Host Disease
It must be realized that the induction of a specific regulatory immune response by a pathogen may depend on the host condition. Some pathogens that induce a “high Treg” response in cancer may elicit a “low Treg” response in the context of autoimmune diseases.13 Epstein Barr virus (EBV), which is involved in several cancers, is implicated in autoimmune diseases such as RA, SLE, and MS. In fact, EBNA1, a protein expressed in all latency forms and lytic cycles of EBV, contains three fragments that cross-react with three auto-antigens in RA, SLE, and MS.115 As explained above, EBV may switch from a “high Treg” phenotype in cancer to a “low Treg” phenotype in autoimmune diseases, propagating both diseases. Measles virus has been associated with multiple sclerosis (MS),116 a “low Treg” disease. On the other hand, it has been demonstrated that measles virus induces IL-10-producing CD14+ and CD4+CD25+ cells—a “high Treg” reaction—in peripheral blood mononuclear cells of infected patients not diagnosed with any autoimmune disease.117 It is possible, however, that measles virus transforms into a “low Treg” phenotype in MS, just as EBV does.
Another example is related to herpetic stromal keratitis, a corneal infection caused by herpes virus 1 (HSV-1). Using a murine corneal infection model, a positive correlation between Treg frequency and viral infectivity has been recently observed. On this basis, HSV-1 can be classified as a “high Treg” virus. It has also been found that Tregs promote HSV-1 latency by CD8+ T cell suppression.118 However, stress-induced Treg cells reactivate the latent virus by controlling antiviral CD8+ T cells.118 It seems therefore that the functioning of Treg cells in HSV-1 corneal infection is context-dependent. It is well known that stress induces glucocorticoids secretion (through the HPA axis). Hence, it is possible, that by controlling cytotoxic T cells in the presence of glucocorticoids, Tregs mainly reactivate the latent form of HSV-1, while in their absence, the latency of this virus is established. This also explains the well-documented correlation between psychological and physical stress and the increased risk of developing an active HSV infection.
As noted above, bone marrow dendritic cells infected by H. pylori may skew the Th17/Treg balance toward a Tregs response,37 which may classify H. pylori as a “high Treg” bacterium. This is in accord with the documented association between H. Pylori and (gastric) cancer. However, H. pylori bacteria are also associated with Grave’s disease,60 which is typically a “low Treg” disease.60 As a matter of fact, H. pylori has been linked with several autoimmune diseases.118 Different mechanisms of the H. pylori-induced autoimmunity have been proposed, including molecular mimicry of H. pylori antigens and the ability of H. pylori fragments to induce B cells to produce anti-phospholipid choline, anti-dsDNA antibodies, and IgM rheumatoid factor.119 Whether H. pylori induce cancer or autoimmunity probably depends on the environmental milieu.
Exceptions To The Two-Class Model
Tregs are involved in the impairment of immune control of Mycobacterium tuberculosis.120 Therefore, tuberculous meningitis can be considered a “high Treg” condition, and corticosteroids are not expected to be effective in controlling this disease. However, corticosteroids use is recommended in tuberculous meningitis.91 In addition, the ineffectiveness of CS in cystic fibrosis,88 a “low Treg” disease, cannot be explained by the two-class model.
In another example, chronic myeloid leukemia (CML) seems to be a “high Treg” disease: A negative correlation was observed between Th17 frequency and fusion gene BCR/ABL (a CML marker) frequency in the peripheral blood and bone marrow of CML patients.121 It was also found that the overall survival rate of CML patients with a high peripheral level of CD4+CD25+Foxp3+ Treg was remarkably lower than that of a low Treg group.122 The beneficial effect of CS in the treatment of this “high Treg” disease cannot be elucidated by the model. As explained above, in lymphoid tissues, CS work through glucocorticoid receptors to arrest growth and induce apoptosis,102 a mechanism that is not necessarily related to the anti-inflammatory effect of CS, which is affected by Treg induction and the suppression of effector T cells functioning.
Summary
Two classes of chronicity are discussed in relation to the resolution of inflammation: (a) A “high Treg” class, in which over-active regulatory cells facilitate the persistence of insult (whether malignant cells or pathogens) due to a too-early or too intense suppression of the immune response; and (b) A “low Treg” class, in which an impaired regulatory effect facilitates long-lasting inflammation (and collateral tissue damage), long after the insult disappears or during active autoimmune diseases.
This two-class distinction elucidates the association of some infections with autoimmune diseases and of other infections (mainly viral) with cancer. The high or low probability of a coinfection by different pathogens can also be accounted for by the model. The weak association between autoimmune diseases and HIV infection and the relatively weak association between autoimmune diseases and cancer are explained as well. In addition, different interactions between pathogens, cancer, and autoimmunity are rationalized, as well as the efficacy or inefficacy of immunotherapy by drugs (including corticosteroids) and bacteria, in cancer, infections, and autoimmune diseases. Some exceptions to the model are acknowledged. A summary diagram of the binary model and the conclusions derived by its use is presented in Figure 1.
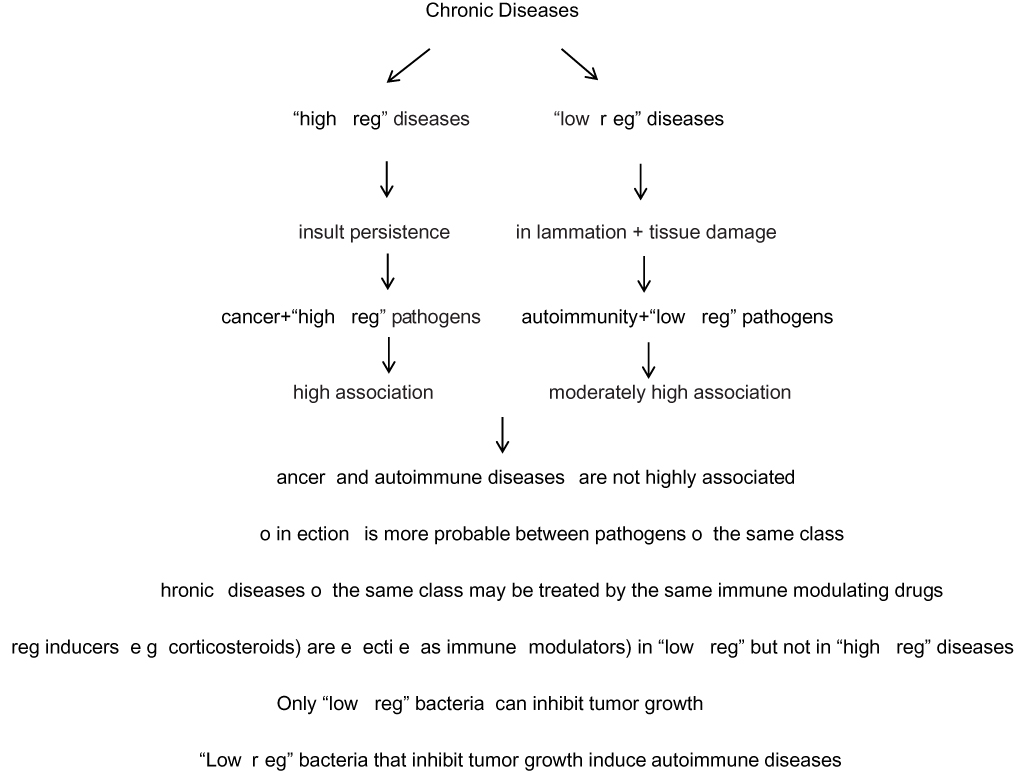 |
Figure 1 A summary diagram of the binary model and the conclusions derived by its use. |
Disclaimer
The views and opinions expressed and/or conclusions drawn in this article are those of the author and do not necessarily reflect those of Taro Pharmaceutical Industries Ltd., its affiliates, directors or employees.
Disclosure
Zeev Elkoshi is employed by Taro Pharmaceutical Industries Ltd and reports personal fees from Taro Pharmaceutical Industries, outside the submitted work. The author reports no other conflicts of interest in this work.
References
1. Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of inflammation: what controls its onset? Front Immunol. 2016;7:160. doi:10.3389/fimmu.2016.00160
2. Sagiv JY, Michaeli J, Assi S, et al. Phenotypic diversity and plasticity in circulating neutrophil subpopulations in cancer. Cell Rep. 2015;10(4):562–573. doi:10.1016/j.celrep.2014.12.039
3. Klinker MW, Lundy SK. Multiple mechanisms of immune suppression by B lymphocytes. Mol Med. 2012;18:123–137. doi:10.2119/molmed.2011.00333
4. Perobelli SM, Silva TG, Bonomo A. Neutrophils plasticity: the regulatory interface in various pathological conditions. In: Khajah M editor. Role of Neutrophils in Disease Pathogenesis. Intech Open Science Open Min d;2017. doi:10.5772/68130.
5. Zhou L, Lopes JE, Chong MM, et al. TGF-beta- induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453(7192):236–240. doi:10.1038/nature06878.
6. Papageorgis P, Stylianopoulos T. Role of TGFβ in regulation of the tumor microenvironment and drug delivery (Review). Int J Oncol. 2015;46(3):933–943. doi:10.3892/ijo.2015.2816.
7. Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. J Clin Invest. 2014;124(12):5466–5480. doi:10.1172/JCI77053.
8. Wu P, Wu D, Ni C, et al. γδT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity. 2014;40(5):785–800. doi:10.1016/j.immuni.2014.
9. Kirkbride KC, Blobe GC. Inhibiting the TGF-beta signalling pathway as a means of cancer immunotherapy. Expert Opin Biol Ther. 2003;3:251–261. doi:10.1517/14712598.3.2.251.
10. Kehrl JH, Wakefield LM, Roberts AB, et al. Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J Exp Med. 1986;163:1037–1050. doi:10.1084/jem.163.5.1037.
11. Dahmani A, Delisle JS. TGF-β in T cell biology:implications for cancer immunotherapy. Cancers (Basel). 2018;10(6):
12. Lee GR. The balance of Th17 versus treg cells in autoimmunity. Int J Mol Sci. 2018;19(3):730. doi:10.3390/ijms19030730.
13. Buckner JH. Mechanisms of impaired regulation by CD4+CD25+FOXP3+regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10(12):849–859. doi:10.1038/nri2889.
14. Shull MM, Ormsby I, Kier AB, et al. Targeted disruption of the mouse transforming growth factor -β 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi:10.1038/359693a0.
15. Dang H, Geiser AG, Letterio JJ, et al. SLE-like autoantibodies and Sjogren’s syndrome-like lymphoproliferation in TGF-β knockout mice. J Immunol. 1995;155:3205–3212.
16. Hashimoto M. Th17 in animal models of rheumatoid arthritis. J Clin Med. 2017;6(7):73. doi:10.3390/jcm6070073.
17. Asquith DL, Miller AM, McInnes IB, Liew FY. Animal models of rheumatoid arthritis. Eur J Immunol. 2009;39(8):2040–2044. doi:10.1002/eji.200939578.
18. Hirahara K, Ghoreschi K, Laurence A, Yang XP, Kanno Y, O’Shea JJ. Signal transduction pathways and transcriptional regulation in Th17 cell differentiation. Cytokine Growth Factor Rev. 2010;21(6):425–434. doi:10.1016/j.cytogfr.2010.10.006.
19. Geng J, Yu S, Zhao H, et al. The transcriptional coactivator TAZ regulates reciprocal differentiation of TH17 cells and Treg cells. Nat Immunol. 2017;18(7):800–812. doi:10.1038/ni.3748.
20. Roychoudhuri R, Hirahara K, Mousavi K, et al. MBACH2 represses effector programs to stabilize T(reg)-mediated immune homeostasis. Nature. 2013;498(7455):506–510. doi:10.1038/nature12199.
21. Khorramdelazad H, Hassanshahi G, Nasiri Ahmadabadi B, Kazemi Arababadi M. High serum levels of TGF-β in Iranians with chronic HBV infection. Hepat Mon. 2012;12(11):e7581. doi:10.5812/hepatmon.7581.
22. Nelson DR, Gonzalez-Peralta RP, Qian K, et al. Transforming growth factor-beta 1 in chronic hepatitis C. J Viral Hepat. 1997;4(1):29–35. doi:10.1046/j.1365-2893.1997.00124.x.
23. Moriuchi M, Moriuchi H. Transforming growth factor‐β enhances human T‐cell leukemia virus type I infection. J Med Virol. 2002;67(3):427–430. doi:10.1002/jmv.10074
24. Xu J, Ahmad A, Jones JF, et al. Elevated serum transforming growth factor beta1 levels in Epstein -Barr virus- associated diseases and their correlation with virus-specific immunoglobulin A (IgA) and IgM. J Virol. 2000;74(5):2443–2446. doi:10.1128/JVI.74.5.2443-2446.2000.
25. Yin WY, Lee MC, Huang HB, Lu MC. Increased gene expression of TGF- in peripheral blood mononuclear cells from renal transplant patients with polyomavirus BK viremia. Clin Transplant. 2016;30(4):393–398. doi:10.1111/ctr.12698.
26. Schmidt J, Blum HE, Thimme R. T-cell responses in hepatitis B and C virus infection: similarities and differences. Emerg Microbes Infect. 2013;2(3):e15. doi:10.1038/emi.2013.14.
27. Hao C, Zhou Y, He Y, et al. Imbalance of regulatory T cells and T helper type 17 cells in patients with chronic hepatitis C. Immunology. 2014;143(4):531–538. doi:10.1111/imm.12330
28. Leal FE, Ndhlovu LC, Hasenkrug AM, et al. Expansion in CD39⁺ CD4⁺ immunoregulatory t cells and rarity of Th17 cells in HTLV-1 infected patients is associated with neurological complications. PLoS Negl Trop Dis. 2013;7(2):e2028. doi:10.1371/journal.pntd.0002028.
29. Futagbi G, Gyan B, Nunoo H, et al. High levels of IL-10 and CD4+CD25hi+ treg cells in endemic burkitt’s lymphoma patients. Biomedicines. 2015;3:224–236. doi:10.3390/biomedicines3030224.
30. Zhang NN, Chen JN, Xiao L, et al. Accumulation mechanisms of CD4(+)CD25(+)FOXP3(+) regulatory T cells in EBV-associated gastric carcinoma. Sci Rep. 2015;20155:18057. doi:10.1038/srep18057.
31. Song D, Li H, Li H, Dai J. Effect of human papillomavirus infection on the immune system and its role in the course of cervical cancer. Oncol Lett. 2015;10(2):600–606. doi:10.3892/ol.2015.3295.
32. Dowlatshahi M, Huang V, Gehad AE, et al. Tumor-specific T cells in human merkel cell carcinomas: a possible role for tregs and T-cell exhaustion in reducing T-cell responses. J Invest Dermatol. 2013;133:1879–1889. doi:10.1038/jid.2013.75.
33. Lepone LM, Rappocciolo G, Piazza PA, Campbell DM, Jenkins FJ, Rinaldo CR. Regulatory T cell effect on CD8+ T cell responses to human herpesvirus 8 infection and development of kaposi’s sarcoma. AIDS Res Hum Retroviruses. 2017;33(7):668–674. doi:10.1089/AID.2016.0155.
34. Jacqueline C, Tasiemski A, Sorci G, et al. Infections and cancer: the “fifty shades of immunity” hypothesis. BMC Cancer. 2017;17:257. doi:10.1186/s12885-017-3234-4.
35. Serelli-Lee V, Ling KL, Ho C, et al. Persistent Helicobacter pylori specific Th17 responses in patients with past H. pylori infection are associated with elevated gastric mucosal IL-1β. PLoS One. 2012;7(6):e39199. doi:10.1371/journal.pone.0039199.
36. Larussa T, Leone I, Suraci E, Imeneo M, Luzza F. Helicobacter pylori and T Helper Cells: mechanisms of immune escape and tolerance. J Immunol Res 2015 2015. Article ID 981328. doi:10.1155/2015/981328.
37. Kao JY, Zhang M, Miller MJ, et al. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology. 2010;138(3):1046–1054. doi:10.1053/j.gastro.2009.11.043.
38. Jang TJ. The number of Foxp3-positivce regulatory T cells is increased in Helicobacter pylori gastritis and gastric cancer. Pathol Res Pract. 2010;206(1):34–38. doi:10.1016/j.prp.2009.07.019.
39. Li N, Xie C, Lu NH. Transforming growth factor-β: an important mediator in Helicobacter pylori-associated pathogenesis. Front Cell Infect Microbiol. 2015;5:77. doi:10.3389/fcimb.2015.00077.
40. Halenius A, Hengel H. Human cytomegalovirus and autoimmune disease. Biomed Res Int. 2014;472978. doi:10.1155/2014/472978.
41. Draborg AH, Duus K, Houen G. Epstein-Barr virus in systemic autoimmune diseases. Clin Dev Immunol. 2013;2013:535738. doi:10.1155/2013/535738.
42. Salloum N, Hussein HM, Jammaz R, Jiche S, Uthman IW, Abdelnoor AM, et al. Epstein-Barr virus DNA modulates regulatory T-cell programming in addition to enhancing interleukin-17A production via Toll-like receptor 9. PLoS One. 2018;13(7):e0200546. doi:10.1371/journal.pone.020054.
43. Walker LSK, von Herrath M. CD4 T cell differentiation in type 1 diabetes. Clin Exp Immunol. 2016;183(1):16–29. doi:10.1111/cei.12672.
44. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol. 2011;29:621–663. doi:10.1146/annurev-immunol-031210-101400.
45. Attridge K, Wang CJ, Wardzinski L, et al. IL-21 inhibits T cell IL-2 production and impairs Treg homeostasis. Blood. 2012;119(20):4656–4664. doi:10.1182/blood-2011-10-388546.
46. Filippi CM, von Herrath MG. Viral trigger for type 1 diabetes. Pros and Cons Diabetes. 2008;57(11):2863–2871. doi:10.2337/db07-1023.
47. Long Q, Liao YH, Xie Y, et al. Coxsackievirus B3 directly induced Th17 cell differentiation by inhibiting Nup98 expression in patients with acute viral myocarditis. Front Cell Infect Microbiol. 2016;6:171. doi:10.3389/fcimb.2016.00171.
48. Cunningham MW. Rheumatic fever, autoimmunity, and molecular mimicry: the streptococcal connection. Int Rev Immunol. 2014;33(4):314–329. doi:10.3109/08830185.2014.917411.
49. Wang B, Dileepan T, Briscoe S, et al. Induction of TGF-β1 and TGF-β1–dependent predominant Th17 differentiation by group A streptococcal infection. Proc Natl Acad Sci U S A. 2010;107(13):5937–5942. doi:10.1073/pnas.0904831107.
50. Costalonga M, Cleary PP, Fischer LA, et al. Intranasal bacteria induce Th1 but not Treg or Th2. Mucosal Immunol. 2009;2(1):85–95. doi:10.1038/mi.2008.67.
51. Mukhopadhyay S, Varma S, Mohan Kumar HN, et al. Circulating level of regulatory T cells in rheumatic heart disease: an observational study. Indian Heart J. 2016;68(3):342–348. doi:10.1016/j.ihj.2015.08.009.
52. Ercolini AM, Miller SD. The role of infections in autoimmune disease. Clin Exp Immunol. 2009;155(1):1–15. doi:10.1111/j.1365-2249.2008.03834.x.
53. Strle K, Sulka KB, Pianta A, et al. T- helper 17 cell cytokine responses in lyme disease correlate with borrelia burgdorferi antibodies during early infection and with autoantibodies late in the illness in patients with antibiotic-refractory lyme arthritis. Clin Infect Dis. 2017;64(7):930–938. doi:10.1093/cid/cix002.
54. Grygorczuk S, Chmielewski T, Zajkowska J, et al. Concentration of TGF-beta1 in the supernatant of peripheral blood mononuclear cells cultures from patients with early disseminated and chronic lyme borreliosis. Adv Med Sci. 2007;52:174–178.
55. Chi LJ, Wang HB, Zhang Y, Wang WZ. Abnormality of circulating CD4(+)CD25(+) regulatory T cell in patients with guillain -barré syndrome. J Neuroimmunol. 2007;192(1–2):206–214. doi:10.1016/j.jneuroim.2007.09.034.
56. Maue AC, Mohawk KL, Giles DK, et al. The polysaccharide capsule of campylobacter jejuni modulates the host immune response. Infect Immun. 2013;81(3):665–672. doi:10.1128/IAI.01008-12.
57. Danikowski KM, Jayaraman S, Prabhakar BS. Regulatory T cells in multiple sclerosis and myasthenia gravis. J Neuroinflammation. 2017;14(1):117. doi:10.1186/s12974-017-0892-8.
58. Benagiano M, Munari F, Ciervo A, et al. Chlamydophila pneumoniae phospholipase D (CpPLD) drives Th17 inflammation in human atherosclerosis. Proc Natl Acad Sci U S A. 2012;109(4):1222–1227. doi:10.1073/pnas.1111833109.
59. Shi WJ, Liu W, Zhou XY, Ye F, Zhang GX. Associations of Helicobacter pylori infection and cytotoxin-associated gene A status with autoimmune thyroid diseases: a meta-analysis. Thyroid. 2013;23(10):1294–1300. doi:10.1089/thy.2012.0630.
60. Li C, Yuan J, Zhu YF, et al. Imbalance of Th17/Treg in different subtypes of autoimmune thyroid diseases. Cell Physiol Biochem. 2016;40(1–2):245–252. doi:10.1159/000452541.
61. Bonney KM, Engman DM. Autoimmune pathogenesis of chagas heart disease. Looking back, looking ahead. Am J Pathol. 2015;185(6):1537–1547. doi:10.1016/j.ajpath.2014.12.023.
62. Cai CW, Blasé JR, Eickhoff CS, Hoft DF. Th17 cells confer both systemic and mucosal protection against Trypanosoma cruzi infection. J Immunol. 2017;198(1 Supplement):
63. Cai CW, Blase JR, Zhang X, Eickhoff CS, Hoft DF. Th17 cells are more protective than Th1 aginst trypanosoma cruzi. PLoS Pathog. 2016;12(10):e1005902. doi:10.1371/journal.ppat.1005902.
64. Kotner J, Tarleton R. EndogenousCD4+ CD25+ regulatory T cells have a limited role in the control of trypanosoma cruzi infection in mice. Infect Immun. 2007;75(2):861–869. doi:10.1128/IAI.01500-06.
65. Flores-García Y, Rosales-Encina JL, Rosales-García VH, Satoskar AR, Talamás-Rohana P. CD4CD25FOXP3 treg cells induced by rSSP4 derived from T. cruzi amastigotes increase parasitemia in an experimental chagas disease model. Biomed Res Int. 2013;2013:632436. doi:10.1155/2013/632436.
66. O’Sullivan BP, Freedman SD. Cyctic fibrosis. Lancet. 2009;373(9678):1891–1904. doi:10.1016/S0140-6736(09)60327-5.
67. Curtis MM, Way SS. Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology. 2009;126(2):177–185. doi:10.1111/j.1365-2567.2008.03017.x.
68. Hector A, Schäfer H, Pöschel S, et al. Regulatory T-cell impairment in cystic fibrosis patients with chronic pseudomonas infection. Am J Respir Crit Care M Ed. 2015;191(8):914–923. doi:10.1164/rccm.201407-1381OC.
69. Skopelja S, Hamilton BJ, Jones JD, et al. The role for neutrophil extracellular traps in cystic fibrosis autoimmunity. JCI Insight. 2016;1(17). doi:10.1172/jci.insight.88912.
70. Franks A, Slansky JE. Multiple associations between a broad spectrum of autoimmune diseases, chronic inflammatory diseases and cancer. Anticancer Res. 2012;32(4):1119–1136.
71. Saxena V, Lienesch DW, Zhou M, et al. Dual roles of immunoregulatory cytokine TGF-beta in the pathogenesis of autoimmunity-mediated organ damage. J Immunol. 2008;180(3):1903–1912. doi:10.4049/jimmunol.180.3.1903.
72. Anderson LA, Gadalla S, Morton LM, et al. Population-based study of autoimmune conditions and the risk of specific lymphoid malignancies. Int J Cancer. 2009;125(2):398–405. doi:10.1002/ijc.24287.
73. Østgård LSG, Nørgaard M, Pedersen L, et al. Autoimmune diseases, infections, use of antibiotics and the risk of acute myeloid leukaemia: a national population -based case-control study. Br J Haematol. 2018;181(2):205–214. doi:10.1111/bjh.15163.
74. Silano M, Volta U, Mecchia AM, et al. Delayed diagnosis of coeliac disease increases cancer risk. BMC Gastroenterol. 2007;7:8. doi:10.1186/1471-230X-7-8.
75. Chen YK, Lin CL, Chang YJ, et al. Cancer risk in patients with graves’ disease: a nationwide cohort study. Thyroid. 2013;23(7):879–884. doi:10.1089/thy.2012.0568.
76. Turpin J, Yurick D, Khoury G, et al. Impact of hepatitis B virus coinfection on human T-lymphotropic virus Type 1 clonality in an Indigenous population of central australia. J Infect Dis. 2018. doi:10.1093/infdis/jiy546.
77. Vazquez-Guillen JM, Palacios-Saucedo GC, Rivera-Morales LG, et al. Infection and coinfection by human papillomavirus, epstein -barr virus and merkel cell polyomavirus in patients with squamous cell carcinoma of the larynx: a retrospective study. Peer J. 2018;6:e5834. doi:10.7717/peerj.5834.
78. Drop B, Strycharz-Dudziak M, Kliszczewska E, Polz-Dacewicz M. Coinfection with Epstein-Barr Virus (EBV), Human Papilloma Virus (HPV) and Polyoma BK Virus (BKPyV) in laryngeal, oropharyngeal and oral cavity cancer. Int J Mol Sci. 2017;18(12):
79. Kleinman AJ, Sivanandham R, Pandrea I, et al. Regulatory T cells as potential targets for HIV cure research. Front Immunol. 2018;9:734. doi:10.3389/fimmu.2018.00734.
80. Virot E, Duclos A, Adelaide L, et al. Autoimmune diseases and HIV infection. A cross-sectional study. Medicine (Baltimore). 2017;96(4):e5769. doi:10.1097/MD.0000000000005769
81. Gangaplara A, Martens C, Dahlstrom E, et al. Type I interferon signaling attenuates regulatory T cell function in viral infection and in the tumor microenvironment. PLoS Pathog. 2018;14(4):e1006985. doi:10.1371/journal.ppat.1006985.
82. Han S, Toker A, Liu ZQ, Ohashi PS. Turning the tide against regulatory T cells. Front Oncol. 2019;9:279. doi:10.3389/fonc.2019.00279.
83. Gardiner D, Lalezari J, Lawitz E, et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PLoS One. 2013;8:e63818. doi:10.1371/journal.pone.0063818.
84. Li P, Zheng Y, Chen X. Drugs for autoimmune inflammatory diseases: from small molecule compounds to anti-TNF biologics. Front Pharmacol. 2017;8:460. doi:10.3389/fphar.2017.00460.
85. Bereshchenko O, Coppo M, Bruscoli S, et al. GILZ promotes production of peripherally induced Treg cells and mediates the crosstalk between glucocorticoids and TGF-b signaling. Cell Rep. 2014;7(2):464–475. doi:10.1016/j.celrep.2014.03.004.
86. Aberdein J, Singer M. Clinical review: a systematic review of corticosteroid use in infections. Crit Care. 2006;10(1):203. doi:10.1186/cc3904.
87. Wilhelmus KR, Gee L, Hauck WW, et al. Herpetic Eye Disease Study. A controlled trial of topical corticosteroids for herpes simplex stromal keratitis. Ophthalmology. 1994;101(12):1883–1895. doi:10.1016/s0161-6420(94)31087-6.
88. Pyrrho AS, Lenzi HL, Ramos JA, et al. Dexamethasone treatment improves morphological and hematological parameters in chronic experimental schistosomiasis. Parasitol Res. 2004;92(6):478–483. doi:10.1007/s00436-004-1078-8.
89. McGee S, Hirschmann J. Use of corticosteroids in treating infectious diseases. Arch Intern Med. 2008;168(10):1034–1046. doi:10.1001/archinte.168.10.1034.
90. Niland ML, Bonsu BK, Nuss KE, Goodman DG. A pilot study of 1 versus 3 days of dexamethasone as add-on therapy in children with streptococcal pharyngitis. Pediatr Infect Dis J. 2006;25(6):477–481. doi:10.1097/01.inf.0000219469.95772.3f.
91. McKinley L, Logar AJ, McAllister F, Zheng M, Steele C, Kolls JK. Regulatory T cells dampen pulmonary inflammation and lung injury in an animal model of pneumocystis pneumonia. J Immunol. 2006;177(9):6215–6226. doi:10.4049/jimmunol.177.9.6215.
92. Suvas S, Azkur AK, Kim BS, Kumaraguru U, Rouse BT. CD4+CD25+ regulatory T cells control the severity of viral immunoinflammatory lesions. J Immunol. 2004;172(7):4123–4132. doi:10.4049/jimmunol.172.7.4123.
93. Hesse M, Piccirillo CA, Belkaid Y, et al. The pathogenesis of schistosomiasis is controlled by cooperating IL-10- producing innate effector and regulatory T cells. J Immunol. 2004;172(5):3157. doi:10.4049/jimmunol.172.5.3157.
94. Gao A, Shao Y. Investigation for the roles of TLR2, TLR9 and Th17/Treg in the pathogenesis of infectious mononucleosis. Int J Clin Exp Med. 2017;10(10):14946–14953.
95. Lei L, Zhang Y, Yao W, Kaplan MH, Zhou B. Thymic stromal lymphopoietin interferes with airway tolerance by suppressing the generation of antigen-specific regulatory T cells. J Immunol. 2011;186(4):2254–2261. doi:10.4049/jimmunol.1002503.
96. Chen YL, Chiang BL. Targeting TSLP with shRNA alleviates airway inflammation and decreases epithelial CCL17 in a murine model of asthma. Mol Ther Nucleic Acids. 2016;5:e316. doi:10.1038/mtna.2016.29.
97. Ma L, Xue HB, Guan XH, et al. The Imbalance of Th17 cells and CD4(+) CD25(high) Foxp3(+) Treg cells in patients with atopic dermatitis. J Eur Acad Dermatol Venereol. 2014;28(8):1079–1086. doi:10.1111/jdv.12288.
98. Frimpong A, Kusi KA, Tornyigah B, Ofori MF, Ndifon W. Characterization of T cell activation and regulation in children with asymptomatic plasmodium falciparum infection. Malar J. 2018;17(1):263. doi:10.1186/s12936-018-2410-6.
99. Walther M, Tongren JE, Andrews L, et al. Upregulation of TGF-beta, FOXP3, and CD4+CD25+ regulatory T cells correlates with more rapid parasite growth in human malaria infection. Immunity. 2005;23(3):287–296. doi:10.1016/j.immuni.2005.08.006.
100. Gagyor I, Madhok VB, Daly F, et al. Antiviral treatment for Bell’s palsy (idiopathic facial paralysis). Cochrane Database Syst Rev. 2015;(11):CD001869. doi:10.1002/14651858.CD001869.pub8.
101. Pufall MA. Glucocorticoids and cancer. Adv Exp Med Biol. 2015;872:315–333. doi:10.1007/978-1-4939-2895-8_14.
102. Herold MJ, McPherson KG, Reichardt HM. Glucocorticoids in T cell apoptosis and function. Cell Mol Life Sci. 2006;63(1):60–72. doi:10.1007/s00018-005-5390-y.
103. D’Arena G, Vitale C, Coscia M, et al. Regulatory T cells and their prognostic relevance in hematologic malignancies. J Immunol Res. 2017;2017:1832968. doi:10.1155/2017/1832968.
104. Linnebacher M, Maletzki C, Klier U, Klar E. Bacterial immunotherapy of gastrointestinal tumors. Langenbecks Arch Surg. 2012;397(4):557–568. doi:10.1007/s00423-011-0892-6.
105. Zhang H, Diao H, Jia L, et al. Proteus mirabilis inhibits cancer growth and pulmonary metastasis in a mouse breast cancer model. PLoS One. 2017;12(12):e0188960. doi:10.1371/journal.pone.0188960.
106. Staedtke V, Roberts NJ, Bai RY, Zhoua S. Clostridium novyi-NT in cancer therapy. Genes Dis. 2016;3(2):144–152. doi:10.1016/j.gendis.2016.01.003.
107. Behnsen J, Perez-Lopez A, Nuccio SP, Raffatellu M. Exploiting host immunity: the Salmonella paradigm. Trends Immunol. 2015;36(2):112–120. doi:10.1016/j.it.2014.12.003.
108. Linehan JL, Dileepan T, Kashem SW, Kaplan DH, Cleary P, Jenkins MK. Generation of Th17 cells in response to in tranasal infection requires TGF-β1 from dendritic cells and IL-6 from CD301b+ dendritic cells. Proc Natl Acad Sci U S A. 2015;112(41):12782–12787. doi:10.1073/pnas.1513532112.
109. Iqbal NT, Hussain R. Non-specific immunity of BCG vaccine: a perspective of BCG immunotherapy. Trials Vaccinol. 2014;3(C):143–149. doi:10.1016/j.trivac.2014.08.002.
110. Armbruster CE, Mobley HLT, Pearson MM. Pathogenesis of proteus mirabilis infection. EcoSal Plus. 2018;8(1). doi:10.1128/ecosalplus.ESP-0009-2017.
111. Soloski MJ, Metcalf ES. Salmonella as an inducer of autoimmunity. EcoSal Plus. 2007;2(2). doi:10.1128/ecosalplus.8.8.13.
112. Erbinger A, Rashid T. Rheumatoid arthritis is caused by a proteus urinary tract infection. Apmis. 2013;122(5). doi:10.1111/apm.12154.
113. Marwat A, Mehmood H, Hussain A, Khan M, Ullah A, Joshi M. Clostridium difficile colitis leading to reactive arthritis: a rare complication associated with a common disease. J Investig Med High Impact Case Rep. 2018;6:2324709618767689. doi:10.1177/2324709618767689.
114. Chai Q, Zhang Y, Liu CH. Mycobacterium tuberculosis: an adaptable pathogen associated with multiple human diseases. Front Cell Infect Microbiol. 2018;8:158. doi:10.3389/fcimb.2018.00158.
115. Ning S. Innate immune modulation in EBV infection. Herpesviridae. 2011;2(1):1. doi:10.1186/2042-4280-2-1.
116. Getts DR, Chastain EM, Terry RL, Miller SD. Virus infection, antiviral immunity, and autoimmunity. Immunol Rev. 2013;255(1):197–209. doi:10.1111/imr.12091.
117. Yu XL, Cheng YM, Shi BS, et al. Measles virus infection in adults induces production of IL-10 and is associated with increased CD4+ CD25+ regulatory T cells. J Immunol. 2008;181(10):7356–7366. doi:10.4049/jimmunol.181.10.7356.
118. Yu W, Geng S, Suo Y, et al. Critical role of regulatory T cells in the latency and stress-induced reactivation of HSV-1. Cell Rep. 2018;25(9):2379–2389.e3. doi:10.1016/j.celrep.2018.10.105.
119. Smyk DS, Koutsoumpas AL, Mytilinaiou MG, Rigopoulou EI, Sakkas LI, Bogdanos DP. Helicobacter pylori and autoimmune disease: cause or bystander. World J Gastroenterol. 2014;20(3):613–629. doi:10.3748/wjg.v20.i3.613.
120. Boer CM, Joosten SA, Ottenhoff THM.Regulatory T-cells at the interface between human host and pathogens in infectious diseases and vaccination. Front Immunol.2015;6:217. doi:10.3389/fimmu.2015.00217. doi:10.3389/fimmu.2015.00217.
121. Chen P, Wang M, Li D, et al. The alteration and clinical significance of Th22/Th17/Th1 cells in patients with chronic m yeloid leukemia. J Immunol Res. 2015;2015:416123. doi:10.1155/2015/416123.
122. Cheng Y, Yang GO, Sun M, Chang K, Long R, Jiang Z. The expression and clinical significance of Treg cells in chronic myeloid leukemia. Biomed Res. 2017;28(21):9188–9192.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.