")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
The association of post-stroke anhedonia with salivary cortisol levels and stroke lesion in hippocampal/parahippocampal region
Authors Terroni L, Amaro Jr E, Iosifescu D , Mattos P, Yamamoto F, Tinone G, Conforto A, Sobreiro M, Guajardo V, De Lucia M, Moreira A, Scaff M, Leite C, Fraguas R
Received 4 September 2014
Accepted for publication 24 October 2014
Published 3 February 2015 Volume 2015:11 Pages 233—242
DOI https://doi.org/10.2147/NDT.S73722
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Luisa Terroni,1 Edson Amaro Jr,2 Dan V Iosifescu,3 Patricia Mattos,4 Fabio I Yamamoto,5 Gisela Tinone,5 Adriana B Conforto,5 Matildes FM Sobreiro,1 Valeri D Guajardo,1 Mara Cristina S De Lucia,7 Ayrton C Moreira,6 Milberto Scaff,5 Claudia C Leite,2 Renerio Fraguas1
1Consultation-Liaison Psychiatry Group, Department and Institute of Psychiatry, Clinical Hospital, University of São Paulo School of Medicine, São Paulo, Brazil; 2Department of Radiology, Clinical Hospital, University of São Paulo School of Medicine, São Paulo, Brazil; 3Mood and Anxiety Disorders Program, Icahn School of Medicine at Mount Sinai, New York, NY, USA; 4Department of Psychiatry, Federal University of São Paulo, São Paulo, Brazil; 5Department of Neurology, Clinical Hospital, University of São Paulo School of Medicine, São Paulo, Brazil; 6Department of Medicine, University of São Paulo, School of Medicine, Ribeirão Preto, Brazil; 7Division of Psychology, Central Institute, Clinical Hospital, University of São Paulo School of Medicine, São Paulo, Brazil
Background: Anhedonia constitutes a coherent construct, with neural correlates and negative clinical impact, independent of depression. However, little is known about the neural correlates of anhedonia in stroke patients. In this study, we investigated the association of post-stroke anhedonia with salivary cortisol levels and stroke location and volume.
Patients and methods: A psychiatrist administered the Structured Clinical Interview for Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition to identify anhedonia in 36 inpatients, without previous depression, consecutively admitted in a neurology clinic in the first month after a first-ever ischemic stroke. Salivary cortisol levels were assessed in the morning, evening, and after a dexamethasone suppression test. We used magnetic resonance imaging and a semi-automated brain morphometry method to assess stroke location, and the MRIcro program according to the Brodmann Map to calculate the lesion volume.
Results: Patients with anhedonia had significantly larger diurnal variation (P-value =0.017) and higher morning levels of salivary cortisol (1,671.9±604.0 ng/dL versus 1,103.9±821.9 ng/dL; P-value =0.022), and greater stroke lesions in the parahippocampal gyrus (Brodmann area 36) compared to those without anhedonia (10.14 voxels; standard deviation ±17.72 versus 0.86 voxels; standard deviation ±4.64; P-value =0.027). The volume of lesion in the parahippocampal gyrus (Brodmann area 36) was associated with diurnal variation of salivary cortisol levels (rho=0.845; P-value =0.034) only in anhedonic patients.
Conclusion: Our findings suggest that anhedonia in stroke patients is associated with the volume of stroke lesion in the parahippocampal gyrus and with dysfunction of the hypothalamic–pituitary–adrenal axis.
Keywords: anhedonia, stroke, glucocorticoids, depression, hippocampus, parahippocampal
Introduction
Anhedonia has been considered a promising endophenotype of depression.1 It is one of the core symptoms of major depressive disorder (MDD),2 one of the specific symptoms of MDD melancholic subtype,2,3 and has been considered particularly relevant for the diagnosis of MDD among the medically ill.4,5 Additionally, anhedonia has been associated with disability independently of sadness,6 poor health status and cognitive symptoms,7 increased major adverse cardiac events,8 lower treatment response in general,9 and specifically with serotonin selective reuptake inhibitors,10 suicidal ideation,11 and increased mortality.8,12 It has its own psychopathological relevance, including the differentiation of anhedonic depression from depression with demoralization13 and from adjustment disorder with depressive mood.14 In addition, it has been related to blunting or to the global inability of reinforcement (ie, negative and positive reinforcement) to alter behavior more than the presence of a diagnosis of depression.15 Its unique importance has also been reported in psychiatric disorders other than depression, including obsessive compulsive disorder,16 eating disorders,17,18 social anhedonia in schizophrenia,19,20 and as a possible predictor for stimulant misuse.21 Anhedonia constitutes a coherent construct, consistent with the new Research Domain Criteria proposed by the National Institutes of Mental Health.22 The Research Domain Criteria aim to identify neurobiological mechanisms underlying clusters of psychiatric symptoms or “domains” of mental function (eg, anhedonia and cognitive deficits) across the traditional psychiatric diagnoses.22,23 Clinical data have indicated that post-stroke depression has imprecise boundaries and better understanding of its psychopathological profile is necessary.24 This approach enhances the relevance of studies investigating the brain morphology and endocrine correlates of anhedonia.25
Neurophysiologically, anhedonia has been associated with reduced connectivity between pregenual anterior cingulate cortex and caudate nucleus, reduced neuronal activity in ventromedial prefrontal cortex, amygdala/ventral striatum,26 and reduced activity in the nucleus accumbens.27 Anhedonia has been associated with vascular depression28 and neuroanatomical alterations that have been related to anhedonia include a greater volume of white matter hyperintensities,29 reduced caudate volume,30 deep white matter lesions, lacunar infarcts in deep white matter, and cortical atrophy.31 Regarding neurotransmitters, antidepressants acting on noradrenaline and not only on serotonin neurotransmission may be relevant to treat anhedonia.32 Neuro-endocrinologically, anhedonia has been related to a dysfunctional feedback between the hypothalamic–pituitary–adrenal (HPA) axis and specific brain regions.33
Of note, increased cortisol levels have been consistently associated with melancholic depression, an MDD subtype where anhedonia is a cardinal marker.34 Clinically, increased cortisol levels have been associated with poor outcomes, increased severity, increased risk for depression,35 poor response to treatment,36,37 and higher relapse of depressive episodes.34,38–40 Neuroanatomical correlates of cortisol that have been reported include reduced hippocampal volume,41 deficits in hippocampus-related neuropsychological functions including verbal and visuospatial memory,42 and dysfunctional hippocampal response to cortisol in depressed women.43 Additionally, depressed patients who are dexamethasone/CRH non-suppressors (a marker of HPA dysfunction) showed metabolic changes in the medial prefrontal cortex (mPFC) and in the hippocampal/parahippocampal brain regions after treatment with antidepressants.44 Recently, the diurnal slope of salivary cortisol has been negatively associated with resting neural activity in the mPFC, hippocampus and parahippocampal gyrus in healthy volunteers; in contrast, anhedonic subjects failed to present an association of salivary cortisol with mPFC activity.33 It should be noted that distorted daily cortisol rhythms may lead to diverse effects on emotional functions depending upon the cellular pathway or part of the network it is disturbing.45 Such effect is influenced by individual regional differences in the sensitivity of the brain determined by differential local epigenetic events.45 In aggregate, these data support the association between anhedonia, HPA dysfunction, and abnormal function in specific brain areas involved in emotional regulation (eg, mPFC and hippocampus/parahippocampal area).
In stroke patients, anhedonia has been associated with increased levels of depression at hospital discharge,46 and cognitive deficits47 including executive dysfunction.48 In contrast with depressive and anxiety symptoms, anhedonia in post-stroke patients has been related to aging and not to a previous history of mood disorders or to the functional status (Barthel Index), supporting its particular brain correlates.49 In rats, anhedonia induced by ischemic stroke has been associated with decreased protein expression of 5-hydroxytryptamine 1A receptors and messenger ribonucleic acid levels in the dentate hippocampal gyrus.50 These changes could be associated with decreased neurogenesis and were reversed by citalopram.50,51 Notwithstanding the abovementioned relevance of anhedonia, it is not known whether its neuroendocrine correlates in stroke patients match the pattern described in MDD.
In post-stroke depression, hypercortisolemia has been associated with lesions in frontal brain areas52–57 and with poor clinical outcome of stroke.55,56,58 Post-stroke hypercortisolemia was a predictor of later depression in some54 but not all studies.53 A recent literature review reported that elevated post-stroke cortisol levels have been associated with dependency, morbidity, and mortality.59 Early studies were conflicting regarding stroke location and depression.60–62 However, a possible explanation for these discrepancies is that depression in acute and inpatients is related to left side lesions, and depression occurring later than 3 months after stroke and in community patients is related to right side lesions.63 Recent studies have attributed the occurrence of depression to disruption of prefrontal subcortical circuits by the stroke lesion.64,65 Recently, post-stroke major depression episode (MDE) has also been associated with stroke lesions in the limbic–cortical–striato–pallido–thalamic41,66,67 circuit including the mPFC, cingulate cortex, hippocampal/parahippocampal region, and amygdala.68 Based on these findings and those relating anhedonia with neuroendocrine and neural dysfunction33 we hypothesized that anhedonia in stroke patients would be associated with cortisol levels and with lesions in mPFC and areas in the hippocampal/parahippocampal region. Therefore, the goal of this study was to investigate the association of anhedonia with salivary cortisol levels and with stroke lesion in the mPFC and hippocampus/parahippocampal areas.
Material and methods
Patients
We screened 326 male and female inpatients, 18 years of age or older, consecutively admitted to the neurology unit of a university hospital with a diagnosis of ischemic stroke between August 2002 and May 2008. Details of the protocol have been previously described.68,69 In short, the diagnosis of stroke was made by a neurologist in accordance with the World Health Organization criteria70 and confirmed by magnetic resonance imaging (MRI). At screening we interviewed patients using the modules for mood episodes, psychotic symptoms, and substance use disorders of the Structured Clinical Interview Patient Version axis I (SCID-I/P) for Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition(DSM-1V) to investigate past and current mood disorders.71 A psychiatrist performed the interview with the patient and a family member/caregiver. A neurologist assessed the daily living activities using the Barthel Index,72 and the stroke severity using the National Institutes of Health Stroke Scale (NIHSS).73 The Barthel Index scores range from 0 (completely dependent) to 100 (completely independent).72 The NIHSS is a graded neurological examination assessing consciousness, eye movements, visual fields, motor and sensory impairments, ataxia, speech, cognition, and inattention and its scores range from 0 to 42 (more severe).73 Both, NIHSS and Barthel Index Brazilian versions have been validated.74
We excluded 253 patients for the following criteria: a) previous history of stroke, infratentorial stroke, greater than 3 weeks interval between stroke occurrence and screening interview, or hemorrhagic transformation of stroke (n=89); b) drug/alcohol dependence, psychoses, delirium, history of MDE, current MDE with pre-stroke onset, dysthymia, or bipolar disorder (n=54); c) aphasia that impeded the interview (n=37); d) neurological diseases or severe clinical condition that impeded the interview (n=22); e) problems during the MRI acquisition (n=19); f) other reasons (n=32).68,69 Of 73 eligible patients, five declined to participate and for 32 we lacked proper cortisol samples, leaving 36 patients for the current analysis.
The 36 patients were evaluated on average within 11.9 days after stroke (standard deviation [SD] ±4.7; range 5–22 days). Patients were free from corticoids, oral contraceptives, and antidepressants. Patients with MDD were only those whose depression had started after the stroke. All patients with MDD were oriented and referred to treatment after the evaluation.
The institutional review board of the Clinics Hospital of the University of São Paulo, School of Medicine, approved the study protocol. Written informed consent was obtained from all participating patients after explanation about the procedures and study.
Anhedonia assessment
Anhedonia, defined as diminished interest or pleasure in response to stimuli previously perceived as rewarding during the premorbid state, was diagnosed by a psychiatrist administering the SCID-I/P for DSM-1V.71 The psychiatrist was blinded for the cortisol and radiological results.
Cortisol measures
Cortisol samples were collected with the supervision of a member of the nursing team or by one of the researchers. Salivary samples (1 mL) were collected in a plastic tube by direct spitting during a 15 minutes period at 9 am and 11 pm. After collecting basal morning and evening samples, 1 mg of dexamethasone was administered orally at 11 pm. The next morning at 9 am, we collected a salivary sample to investigate the inhibitory effect of the dexamethasone. Salivary samples were stored at 4°C and analyses were performed at the neuroendocrine laboratory of the University of Sao Paulo, Ribeirão Preto School of Medicine. After centrifugation at 2,000 rpm, the supernatants were stored at −20°C until assayed. Salivary cortisol measurements were performed by a previously described radioimmunoassay method on 25 μL samples of saliva without previous extraction or chromatography. This method previously demonstrated a good correlation (r=50.95) with plasma free cortisol levels determined by equilibrium dialysis.75 The assay sensitivity was 60 ng/dL.75 The mean intra-assay coefficient of variation was 5.5%. All samples obtained from each subject were analyzed in duplicate in the same assay. The technicians performing cortisol assays were blind to the clinical characteristics of the patients.
MRI protocol, stroke location and volume
All MRIs were acquired on a 1.5-Tesla system (GE-Horizon LX; General Electric Healthcare, WI, USA), using a specific previously described imaging protocol.68 Lesion location and volume quantification were determined using a semi-automated method. Initially, spoiled gradient recalled acquisition in steady state and axial fluid attenuated inversion recovery (FLAIR) acquisitions were both normalized to the Montreal Neurological Institute template.76 We used linear transformation with 12 degrees of freedom and 15 nonlinear interactions implemented in Statistical Parametric Mapping (SPM5, Wellcome Trust for Neuroimaging, London, http://www.fil.ion.ucl.ac.uk/spm/), and based on coordinates referenced in the Talairach and Tornoux Atlas.77 During this process, all images were sampled to 2.3×2.3×2.6 mm. Lesion tracing was performed by a trained psychiatrist (LT), blinded for neurological and cortisol data, using a mouse device to trace the ischemic lesion and analyzing all slices of each FLAIR image using the MRIcro Software (http://www.sph.sc.edu/comd/rorden/mricro.html). All lesions’ tracings from all patients were reviewed by a neuroradiologist, blinded for clinical data and psychiatric diagnoses. The regions of interest were then analyzed automatically using the Brodmann Cytoarchitectonic Atlas placed in the same spaces,78 in order to count the number of voxels within each Brodmann area (BA). The total lesion volume was obtained by multiplying the number of voxels by voxel size in normalized images.
Statistical analysis
Anhedonia was analyzed as a categorical variable (yes/no) according to the specific item on the SCID-I/P for DSM-1V.71 Salivary cortisol levels were analyzed as a continuous variable and by comparing tertiles, as it has been done previously.39 Patients in the highest tertile of cortisol levels were compared to those in the lower two tertiles combined. Patients with cortisol levels below the detection limit of the assay (ie, below 60 ng/dL) were assigned to the inferior tertile. The cut-off for the upper tertile of morning salivary cortisol levels (increased level) was >1.515 ng/dL and for the evening salivary cortisol levels was >575 ng/dL; the cut-off for the upper tertile of morning salivary cortisol levels after dexamethasone supression test was >600 ng/dL. Lesion volume was obtained by determining the voxel-based lesion morphometry in each BA of interest. Lesion volumes were expressed as mean number of voxels and SD (FLAIR voxel size 2.3×2.3×2.6 mm). The a priori defined brain areas for investigation were the mPFC and the BAs 28, 34, 35, and 36 from the hippocampus/parahippocampal region. We estimated the mPFC area by summing the BAs 10, 11, 12, 13, 14, 24, 25, 32, and 47 as previously done.68
Statistical analyses were performed using the Statistical Package for the Social Sciences, version 14 (SPSS Inc., Chicago, IL, USA). Chi-square tests or Fisher’s exact tests were used for categorical data. If not otherwise specified, Student’s t-tests were used for continuous variables; alternatively, the non-parametric Mann–Whitney U-test was used when data did not follow normal distribution according to the Kolmogorov–Smirnov test. The Wilcoxon Signed Rank test was used to compare morning cortisol levels (9 am) with evening cortisol levels (11 pm). We used Spearman correlations to investigate the relationship between diurnal reduction (from morning to evening) in cortisol levels with stroke volumes in the a priori determined brain areas (mPFC and hippocampus/parahippocampal areas), separately in patients with and without anhedonia, in accordance to previous report.33 Results are presented as frequencies or mean ± SD. All statistical tests were two-tailed and alpha was set at 5%.
Results
We identified anhedonia in seven (19.4%) patients and it was significantly associated with new onset post-stroke MDE (Table 1). Salivary cortisol levels were significantly higher in the morning compared to the evening in both anhedonic and non-anhedonic patients (Table 1). Patients with anhedonia had significantly higher mean morning salivary cortisol levels than non-anhedonic patients (Table 1 and Figure 1). Consistent with this, 71.4% of the anhedonic patients had morning salivary cortisol levels in the upper tertile, a rate significantly higher than in non-anhedonic patients (24.1%) (Table 1).
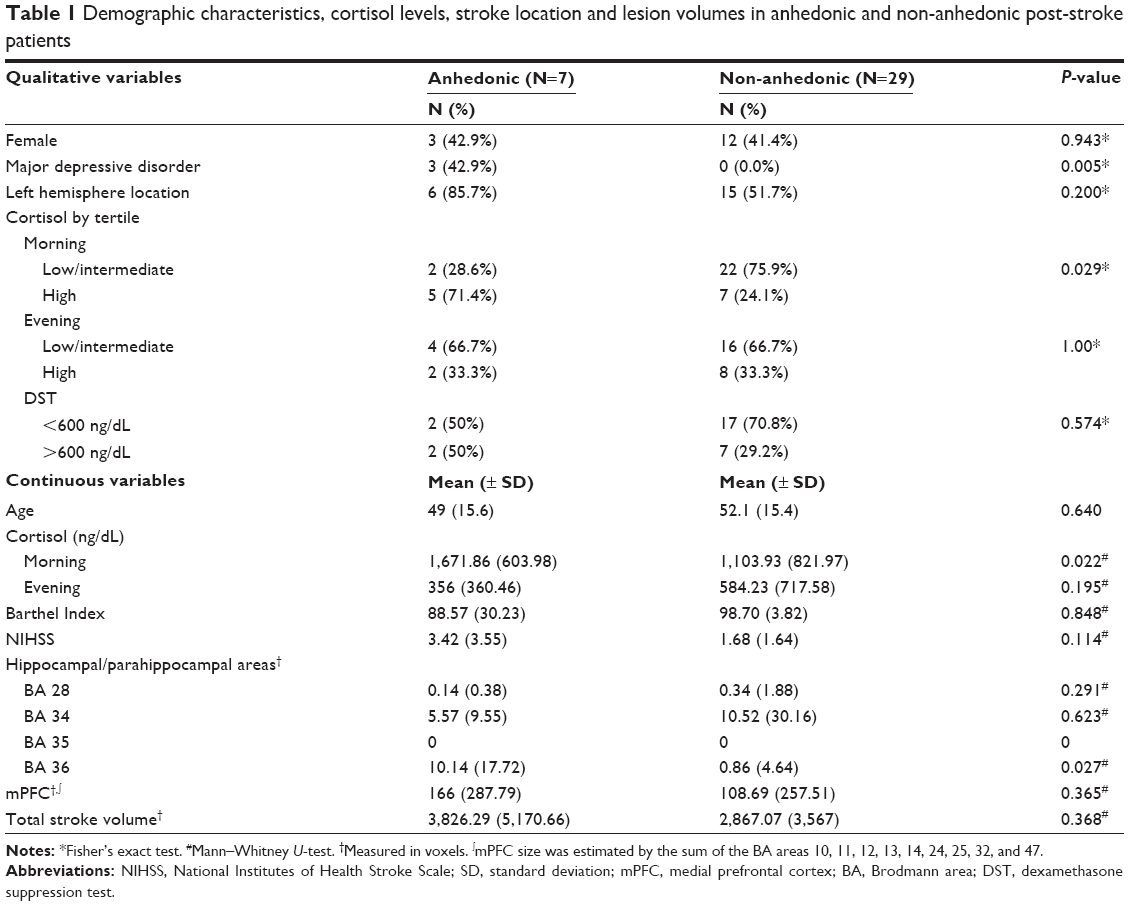 | Table 1 Demographic characteristics, cortisol levels, stroke location and lesion volumes in anhedonic and non-anhedonic post-stroke patients |
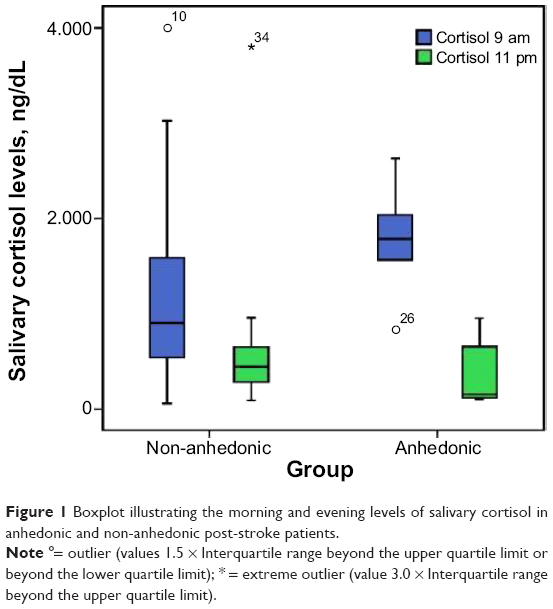 | Figure 1 Boxplot illustrating the morning and evening levels of salivary cortisol in anhedonic and non-anhedonic post-stroke patients. |
Anhedonic patients experienced a larger diurnal decrease in cortisol levels compared to non-anhedonic patients (P-value =0.017, Mann–Whitney U-test).
Anhedonic patients had larger stroke lesion volumes in the parahippocampal gyrus (BA 36) compared to non-anhedonic patients (Table 1; Figure 2). We found no other differences between anhedonic and non-anhedonic patients regarding stroke lesion volumes in the other a priori determined brain areas.
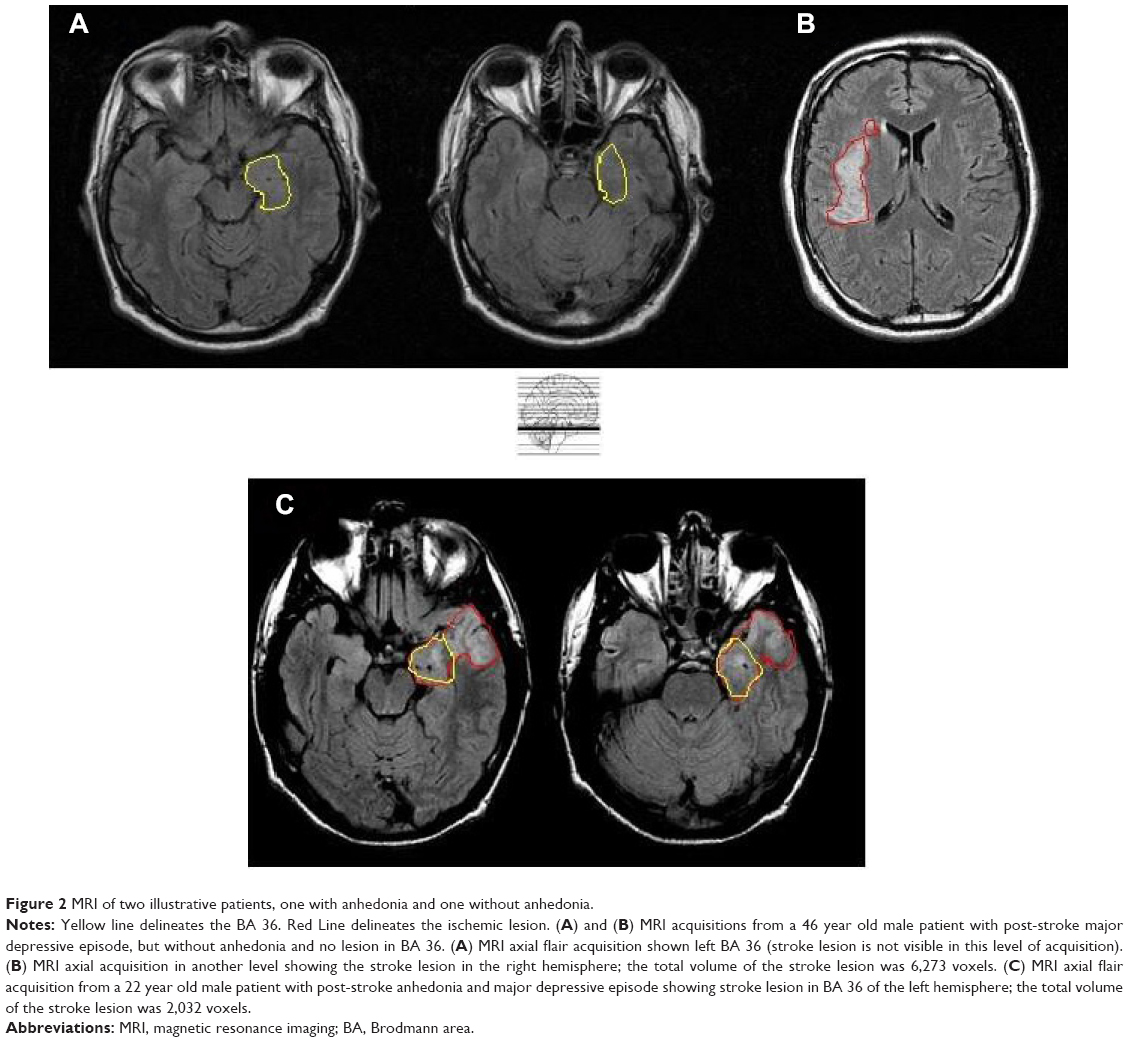 | Figure 2 MRI of two illustrative patients, one with anhedonia and one without anhedonia. |
Stroke lesion volumes in the parahippocampal gyrus (BA 36) were correlated with a larger diurnal decrease of salivary cortisol levels (rho=0.845; P-value =0.034) among anhedonic patients but not among the non-anhedonic ones (rho=0.045; P-value =0.834). No other BAs showed correlation with diurnal variation of salivary cortisol levels.
Discussion
We found that patients experiencing anhedonia within the first month after a first ever ischemic stroke had higher levels of morning salivary cortisol and larger diurnal decrement of salivary cortisol levels than those without anhedonia. Such findings support the existence of specific neuroendocrine correlates of anhedonia, now demonstrated for the first time for post-stroke anhedonia. The association of high morning cortisol levels and anhedonia has been previously reported in other populations, including melancholic depression, a subtype of depression in which anhedonia is a cardinal symptom,34 and in preschool-aged children with maternal history of melancholic depression and anhedonic traits.35 The normal reaction to stress elicits cortisol release.79 However, increased stress has been reported to increase glucocorticoid signaling80 and precipitate neuronal degeneration in the hippocampus,81 further contributing to a dysfunctional stress response. The neural damage caused by the stroke may lead to a dysfunction in the stress response system by disrupting the limbic connectivity and/or deregulating the negative feedback exerted by cortisol secretion. Such dysfunction may explain the association between anhedonia, elevated cortisol levels, and stroke lesion location.
We found that anhedonic patients had an increased volume of stroke lesion in the parahippocampal gyrus (BA 36), an area previously implicated in the abnormal brain connectivity in major depression by neuro functional82 and genetic83 studies. The role of the parahippocampal gyrus in the neural circuitry of anhedonia has previously been supported by studies in bipolar disorder84 and schizophrenia.85 Previous studies also implicate other brain areas in the pathophysiology of anhedonia. In animal models, anhedonia has been related to aberrant hippocampus activity and metabolism,86 while low hippocampus volume in melancholic patients, in which anhedonia is a central feature, has been associated with slower recovery after antidepressant treatment.87 In addition, a catecholamine-depleting diet was reported to induce anhedonia and decreased glucose utilization in the right accumbens.67 From our patients with anhedonia, 85.7% (n=6) had stroke lesion in the left side compared with 51.7% (n=15) from the non-anhedonic ones. Although not significant, such results encourage the investigation of the relationship between left side location and anhedonia, as it has been found for depression in acute post-stroke patients.63
Anhedonic patients, in contrast to those without anhedonia, showed a positive correlation between size of stroke lesion in the parahippocampal gyrus (BA 36) and a larger decrement of the diurnal salivary cortisol levels. Although studies focusing on the relationship of glucocorticoid receptors’ function and parahippocampal gyrus are needed, elevated glucocorticoid levels have been associated with parahippocampal blood flow reduction.88 Hence, it is possible that stroke lesion in the parahippocampal gyrus could impair glucocorticoid receptors’ function leading to an increased morning cortisol level as a compensatory mechanism,89 and consequently a larger decrement of the diurnal levels. Anhedonia could be related to an increased vulnerability to disrupt the modulation of the HPA axis feedback via lesions in the parahippocampal gyrus. Putnam et al reported that health subjects with anhedonia have a dysfunctional feedback between the mPFC and the neuroendocrine HPA axis.33 Using dense-array resting electroencephalography, they found a negative correlation between diurnal salivary cortisol slope and current density in the hippocampal/parahippocampal region for both subjects with and without anhedonia.33 In addition, they found a correlation of diurnal cortisol slope with current density in the mPFC in subjects without anhedonia, but not in subjects with anhedonia. Accordingly, patients with previous vulnerability to anhedonia would have a dysfunctional feedback between mPFC and neuroendocrine regulation. Consequently, it is possible that in anhedonic subjects the HPA axis would depend on feedback from other brain areas, including the hippocampal/parahippocampal region. Thus, stroke lesions in the parahippocampal gyrus (BA 36) in patients with anhedonia would disrupt the feedback of this region to the neuroendocrine HPA axis. Supporting this hypothesis, in our sample, the correlation of salivary cortisol diurnal decrease with the size of lesion in the parahippocampal gyrus (BA 36) was found only in anhedonic patients. Complementary to this, the absence of correlation between size of lesion in parahippocampal gyrus (BA 36) and the decrease in diurnal salivary cortisol levels in patients without anhedonia may suggest that these patients could have the brain HPA axis feedback maintained by the modulation of other brain regions in particular the mPFC as suggested by previous studies.33 In conclusion, our findings indicate that subjects with post-stroke anhedonia may have a dysfunctional feedback between parahippocampal gyrus (BA 36) and the HPA axis.
A consistent line of research has investigated the correlates of apathy in stroke patients. Apathy has a wide array of definitions and conceptualizations usually including diminished interest and motivation among other symptoms.90 The item of the SCID-I/P we have used to assess anhedonia includes two aspects: decrease in motivation and/or reduction in experienced pleasure. Consequently, the symptom “decrease in motivation/interest” included in the definition of anhedonia that we have used, overlaps with some definitions of apathy. Stroke lesions of patients with apathetic depression often did not overlap with lesion of patients with affective depression;91 reinforcing that apathy has a distinct pathophysiology and should not be considered only as a symptom of depression. A recent review concluded that it was not possible to identify a specific stroke lesion location for apathy.90 Actually, post-stroke apathy has been associated with fronto subcortical,92 pontine,93 brainstem and bilateral striatum infarcts.91 The use of diverse definitions of apathy, capturing its distinct aspects may in part explain the diversity of results regarding its stroke location. Similarly, the motivational and consummatory (hedonic response) components of anhedonia have been reported to relay on separate neural systems.25 Thus, assessing specific components of apathy and anhedonia is necessary to address their neural correlates.
Some limitations of our study should be considered. We did not assess the plasma corticotrophin hormone levels. Corticotrophin hormone assessment and several cortisol measurements could give a more detailed and reliable appraisal of HPA function.34 The cross-sectional design of our study restricts conclusions about direction of causality in the association between increased cortisol levels and anhedonia. Although the neuroanatomical areas of interest were determined a priori and literature data support the relationship of the parahippocampal gyrus with anhedonia in non-stroke subjects33 and its deactivation has been reported in paradigms eliciting the reward circuit,94 we did not correct for multiple comparisons. We did not find an association of anhedonia with the mPFC26 and other brain areas previously reported to be involved in the neural basis of anhedonia such as the anterior cingulate cortex and nucleus accumbens.27,95 Similarly to apathy, as commented above, fatigue is common after stroke. Defined as a feeling of lack of energy, weariness, and aversion to effort,96 assessing fatigue in addition to apathy could give a more specific and integrated view of anhedonia. Finally, our sample is relatively young and because of the inclusion and exclusion criteria, patients with more severe stroke have been excluded. Hence, considering the existence of multiple comparisons, the small sample size and small power, our results are better understood as preliminary. Future confirmatory studies with greater sample size allowing the performance of multivariate analysis investigating the influence of total stroke volume, left side location, stroke severity, restriction of daily living activities, the presence of old patients, and more severe stroke cases are warranted.
Conclusion
In conclusion, our data suggest the existence of a relationship between anhedonia with stroke size in the parahippocampal gyrus (area BA 36), with increased morning salivary cortisol levels and increased diurnal cortisol decrement in stroke patients. Our findings contribute to the understanding of post-stroke anhedonia as a result of disruption of neural circuits caused by the stroke lesion and by dysfunctional neuroendocrine feedback of HPA axis associated with that lesion; suggesting that increased morning levels and a larger decrement of diurnal salivary cortisol levels may be a biomarker of anhedonia in stroke patients. Considering the small sample size and absence of correction for multiple comparisons, future studies are necessary to confirm our findings.
Acknowledgments
We thank Mr Bernardo Santos for the statistical analysis, Mr João Ricardo Sato for the essential assistance in the lesion computational analysis of neuroimaging acquisitions, and our colleagues from the Consultation-Liaison Group for the valuable comments and suggestions.
Disclosure
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
Pizzagalli DA. Depression, stress, and anhedonia: toward a synthesis and integrated model. Annu Rev Clin Psychol.2014;10:393–423. | ||
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders Fifth Edition. Arlington, VA, USA: American Psychiatric Publishing; 2013. | ||
Hyett MP, Parker GB, Proudfoot J, Fletcher K. Examining age effects on prototypic melancholic symptoms as a strategy for refining definition of melancholia. J Affect Disord.2008;109(1–2):193–197. | ||
Sibitz I, Berger P, Freidl M, et al. ICD-10 or DSM-IV? Anhedonia, fatigue and depressed mood as screening symptoms for diagnosing a current depressive episode in physically ill patients in general hospital. J Affect Disord.2010;126(1–2):245–251. | ||
Fraguas R Jr, Alves TC. Depressão no Hospital Geral: estudo de 136 casos [Depression in General Hospital: a study of 136 cases]. Rev Assoc Méd Bras.2002;48(3):225–230. Portuguese. | ||
Covinsky KE, Cenzer IS, Yaffe K, O’Brien S, Blazer DG. Dysphoria and Anhedonia as Risk Factors for Disability or Death in Older Persons: Implications for the Assessment of Geriatric Depression. Am J Geriatr Psychiatry.2014;22(6):606–613. | ||
Pelle AJ, Pedersen SS, Erdman RA, et al. Anhedonia is associated with poor health status and more somatic and cognitive symptoms in patients with coronary artery disease. Qual Life Res.2011;20(5):643–651. | ||
Davidson KW, Burg MM, Kronish IM, et al. Association of anhedonia with recurrent major adverse cardiac events and mortality 1 year after acute coronary syndrome. Arch Gen Psychiatry.2010;67(5):480–488. | ||
Spijker J, Bijl RV, de Graaf R, Nolen WA. Determinants of poor 1-year outcome of DSM-III-R major depression in the general population: results of the Netherlands Mental Health Survey and Incidence Study (NEMESIS). Acta Psychiatr Scand.2001;103(2):122–130. | ||
Shelton RC, Tomarken AJ. Can recovery from depression be achieved? Psychiatr Serv.2001;52(11):1469–1478. | ||
Pfeiffer PN, Brandfon S, Garcia E, et al. Predictors of suicidal ideation among depressed Veterans and the interpersonal theory of suicide. J Affect Disord.2014;152(154):277–281. | ||
Furlanetto LM, von Ammon Cavanaugh S, Bueno JR, Creech SD, Powell LH. Association between depressive symptoms and mortality in medical inpatients. Psychosomatics.2000;41(5):426–432. | ||
Clarke DM, issane DW, Trauer T, Smith GC. Demoralization, anhedonia and grief in patients with severe physical illness. World Psychiatry.2005;4(2):96–105. | ||
Zimmerman M, Martinez JH, Dalrymple K, Chelminski I, Young D. “Subthreshold” depression: is the distinction between depressive disorder not otherwise specified and adjustment disorder valid? J Clin Psychiatry.2013;74(5):470–476. | ||
Chase HW, Frank MJ, Michael A, Bullmore ET, Sahakian BJ, Robbins TW. Approach and avoidance learning in patients with major depression and healthy controls: relation to anhedonia. Psychol Med.2010;40(3):433–440. | ||
Abramovitch A, Pizzagalli DA, Reuman L, Wilhelm S. Anhedonia in obsessive-compulsive disorder: Beyond comorbid depression. Psychiatry Res.2014;216(2):223–229. | ||
Tchanturia K, Davies H, Harrison A, Fox JR, Treasure J, Schmidt U. Altered social hedonic processing in eating disorders. Int J Eat Disord.2012;45(8):962–969. | ||
Keating C, Tilbrook AJ, Rossell SL, Enticott PG, Fitzgerald PB. Reward processing in anorexia nervosa. Neuropsychologia.2012;50(5):567–575. | ||
Velthorst E, Meijer C, Investigators G.R.O.U.P. The association between social anhedonia, withdrawal and psychotic experiences in general and high-risk populations. Schizophr Res.2012;138(2–3):290–294. | ||
Goldstein KE, Hazlett EA, New AS, et al. Smaller superior temporal gyrus volume specificity in schizotypal personality disorder. Schizophr Res.2009;112(1–3):14–23. | ||
Leventhal AM, Kahler CW, Ray LA, et al. Anhedonia and amotivation in psychiatric outpatients with fully remitted stimulant use disorder. Am J Addict.2008;17(3):218–223. | ||
Insel TR. Translating scientific opportunity into public health impact: a strategic plan for research on mental illness. Arch Gen Psychiatry.2009;66(2):128–133. | ||
Miller G Psychiatry, Beyond DSM: seeking a brain-based classification of mental illness. Science.2010;327(5972):1437. | ||
da Rocha e Silva CE, Alves Brasil MA, Matos do Nascimento E, de Braganca Pereira B, Andre C. Is poststroke depression a major depression? Cerebrovasc Dis.2013;35(4):385–391. | ||
Treadway MT, Zald DH. Reconsidering anhedonia in depression: lessons from translational neuroscience. Neurosci Biobehav Rev.2011;35(3):537–555. | ||
Keedwell PA, Andrew C, Williams SC, Brammer MJ, Phillips ML. The neural correlates of anhedonia in major depressive disorder. Biol Psychiatry.2005;58(11):843–853. | ||
Wacker J, Dillon DG, Pizzagalli DA. The role of the nucleus accumbens and rostral anterior cingulate cortex in anhedonia: integration of resting EEG, fMRI, and volumetric techniques. Neuroimage.2009;46(1):327–337. | ||
Krishnan KR, Hays JC, Blazer DG. MRI-defined vascular depression. Am J Psychiatry.1997;154(4):497–501. | ||
Lavretsky H, Zheng L, Weiner MW, et al. The MRI brain correlates of depressed mood, anhedonia, apathy, and anergia in older adults with and without cognitive impairment or dementia. Int J Geriatr Psychiatry.2008;23(10):1040–1050. | ||
Pizzagalli DA, Holmes AJ, Dillon DG, et al. Reduced caudate and nucleus accumbens response to rewards in unmedicated individuals with major depressive disorder. Am J Psychiatry.2009;166(6):702–710. | ||
Grool AM, van der Graaf Y, Mali WP, et al. Location and progression of cerebral small-vessel disease and atrophy, and depressive symptom profiles: the Second Manifestations of ARTerial disease (SMART)-Medea study. Psychol Med.2012;42(2):359–370. | ||
Pringle A, McCabe C, Cowen PJ, Harmer CJ. Antidepressant treatment and emotional processing: can we dissociate the roles of serotonin and noradrenaline? J Psychopharmacol.2013;27(8):719–731. | ||
Putnam KM, Pizzagalli DA, Gooding DC, Kalin NH, Davidson RJ. Neural activity and diurnal variation of cortisol: evidence from brain electrical tomography analysis and relevance to anhedonia. Psychophysiology.2008;45(6):886–895. | ||
Stetler C, Miller GE. Depression and hypothalamic-pituitary-adrenal activation: a quantitative summary of four decades of research. Psychosom Med.2011;73(2):114–126. | ||
Dougherty LR, Klein DN, Olino TM, Dyson M, Rose S. Increased waking salivary cortisol and depression risk in preschoolers: the role of maternal history of melancholic depression and early child temperament. J Child Psychol Psychiatry.2009;50(12):1495–1503. | ||
Martiny K, Lunde M, Unden M, Dam H, Bech P. High cortisol awakening response is associated with an impairment of the effect of bright light therapy. Acta Psychiatr Scand.2009;120(3):196–202. | ||
Juruena MF, Pariante CM, Papadopoulos AS, Poon L, Lightman S, Cleare AJ. The role of mineralocorticoid receptor function in treatment-resistant depression. J Psychopharmacol.2013;27(12):1169–1179. | ||
Appelhof BC, Huyser J, Verweij M, et al. Glucocorticoids and relapse of major depression (dexamethasone/corticotropin-releasing hormone test in relation to relapse of major depression). Biol Psychiatry.2006;59(8):696–701. | ||
Brouwer JP, Appelhof BC, van Rossum EF, et al. Prediction of treatment response by HPA-axis and glucocorticoid receptor polymorphisms in major depression. Psychoneuroendocrinology.2006;31(10):1154–1163. | ||
Turkcapar MH, Akdemir A, Orsel SD, et al. The validity of diagnosis of melancholic depression according to different diagnostic systems. J Affect Disord.1999;54(1–2):101–107. | ||
Sheline YI. 3D MRI studies of neuroanatomic changes in unipolar major depression: the role of stress and medical comorbidity. Biol Psychiatry.2000;48(8):791–800. | ||
Hinkelmann K, Moritz S, Botzenhardt J, et al. Cognitive impairment in major depression: association with salivary cortisol. Biol Psychiatry.2009;66(9):879–885. | ||
Abercrombie HC, Jahn AL, Davidson RJ, Kern S, Kirschbaum C, Halverson J. Cortisol’s effects on hippocampal activation in depressed patients are related to alterations in memory formation. J Pychiatr Res.2011;45(1):15–23. | ||
Aihara M, Ida I, Yuuki N, et al. HPA axis dysfunction in unmedicated major depressive disorder and its normalization by pharmacotherapy correlates with alteration of neural activity in prefrontal cortex and limbic/paralimbic regions. Psychiatry Res.2007;155(3):245–256. | ||
Herbert J. Cortisol and depression: three questions for psychiatry. Psychol Med.2013;43(3):449–469. | ||
Sibon I, Lassalle-Lagadec S, Renou P, Swendsen J. Evolution of depression symptoms following stroke: a prospective study using computerized ambulatory monitoring. Cerebrovasc Dis.2012;33(3):280–285. | ||
Farner L, Wagle J, Flekkoy K, et al. Factor analysis of the Montgomery Aasberg Depression Rating Scale in an elderly stroke population. Int J Geriatr Psychiatry.2009;24(11):1209–1216. | ||
Piamarta F, Iurlaro S, Isella V, et al. Unconventional affective symptoms and executive functions after stroke in the elderly. Arch Gerontol Geriatr Suppl.2004;(9):315–323. | ||
Quaranta D, Marra C, Gainotti G. Post-stroke depression: Main phenomenological clusters and their relationships with clinical measures. Behav Neurol.2012;25(4):303–310. | ||
Wang SH, Zhang ZJ, Guo YJ, Teng GJ, Chen BA. Decreased expression of serotonin 1A receptor in the dentate gyrus in association with chronic mild stress: a rat model of post-stroke depression. Psychiatry Res.2009;170(2–3):245–251. | ||
Wang SH, Zhang ZJ, Guo YJ, Sui YX, Sun Y. Involvement of serotonin neurotransmission in hippocampal neurogenesis and behavioral responses in a rat model of post-stroke depression. Pharmacol Biochem Behav.2010;95(1):129–137. | ||
Johansson A, Olsson T, Carlberg B, Karlsson K, Fagerlund M. Hypercortisolism after stroke – partly cytokine-mediated? J Neurol Sci.1997;147(1):43–47. | ||
Olsson T, Astrom M, Eriksson S, Forssell A. Hypercortisolism revealed by the dexamethasone suppression test in patients [corrected] with acute ischemic stroke. Stroke.1989;20(12):1685–1690. | ||
Astrom M, Olsson T, Asplund K. Different linkage of depression to hypercortisolism early versus late after stroke. A 3-year longitudinal study. Stroke.1993;24(1):52–57. | ||
Feibel JH, Hardy PM, Campbell RG, Goldstein MN, Joynt RJ. Prognostic value of the stress response following stroke. JAMA.1977;238(13):1374–1376. | ||
Murros K, Fogelholm R, Kettunen S, Vuorela AL. Serum cortisol and outcome of ischemic brain infarction. J Neurol Sci.1993;116(1):12–17. | ||
Fassbender K, Schmidt R, Mossner R, Daffertshofer M, Hennerici M. Pattern of activation of the hypothalamic-pituitary-adrenal axis in acute stroke. Relation to acute confusional state, extent of brain damage, and clinical outcome. Stroke.1994;25(6):1105–1108. | ||
Olsson T, Marklund N, Gustafson Y, Nasman B. Abnormalities at different levels of the hypothalamic-pituitary-adrenocortical axis early after stroke. Stroke.1992;23(11):1573–1576. | ||
Barugh AJ, Gray P, Shenkin SD, Maclullich AM, Mead GE. Cortisol levels and the severity and outcomes of acute stroke: a systematic review. J Neurol.2014;261(3):533–545. | ||
Carson AJ, MacHale S, Allen K, et al. Depression after stroke and lesion location: a systematic review. Lancet.2000;356(9224):122–126. | ||
Narushima K, Kosier JT, Robinson RG. A reappraisal of poststroke depression, intra- and inter-hemispheric lesion location using meta-analysis. J Neuropsychiatry Clin Neurosci.2003;15(4):422–430. | ||
Yu L, Liu CK, Chen JW, Wang SY, Wu YH, Yu SH. Relationship between post-stroke depression and lesion location: a meta-analysis. Kaohsiung J Med Sci.2004;20(8):372–380. | ||
Bhogal SK, Teasell R, Foley N, Speechley M. Lesion location and poststroke depression: systematic review of the methodological limitations in the literature. Stroke.2004;35(3):794–802. | ||
Vataja R, Pohjasvaara T, Leppavuori A, et al. Magnetic resonance imaging correlates of depression after ischemic stroke. Arch Gen Psychiatry.2001;58(10):925–931. | ||
Vataja R, Leppavuori A, Pohjasvaara T, et al. Poststroke depression and lesion location revisited. J Neuropsychiatry Clin Neurosci.2004;16(2):156–162. | ||
Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct.2008;213(1–2):93–118. | ||
Hasler G, Fromm S, Carlson PJ, et al. Neural response to catecholamine depletion in unmedicated subjects with major depressive disorder in remission and healthy subjects. Arch Gen Psychiatry.2008;65(5):521–531. | ||
Terroni L, Amaro E, Iosifescu DV, et al. Stroke lesion in cortical neural circuits and post-stroke incidence of major depressive episode: a 4-month prospective study. World J Biol Psychiatry.2011;12(7):539–548. | ||
Terroni L, Fraguas R, Lucia M, et al. Importance of retardation and fatigue/interest domains for the diagnosis of major depressive episode after stroke: a four months prospective study. Rev Bras Psiquiatr.2009;31(3):202–207. | ||
[No authors listed]. Stroke – 1989. Recommendations on stroke prevention, diagnosis, and therapy. Report of the WHO Task Force on Stroke and other Cerebrovascular Disorders. Stroke.1989;20(10):1407–1431. | ||
First MB SR, Gibbson M, Williams JBW. Structured clinical interview for axis I DSM-IV disorders (Version 2.0)-patient edition. New York:Biometrics Research Department, New York State Psychiatric Institute;.1995. | ||
Mahoney FI, Barthel DW. Functional Evaluation: The Barthel Index. Md State Med J.1965;14:61–65. | ||
Brott T, Adams HP Jr, Olinger CP, et al. Measurements of acute cerebral infarction: a clinical examination scale. Stroke.1989;20(7):864–870. | ||
Cincura C, Pontes-Neto OM, Neville IS, et al. Validation of the National Institutes of Health Stroke Scale, modified Rankin Scale and Barthel Index in Brazil: the role of cultural adaptation and structured interviewing. Cerebrovasc Dis.2009;27(2):119–122. | ||
Castro M, Elias PC, Quidute AR, Halah FP, Moreira AC. Out-patient screening for Cushing’s syndrome: the sensitivity of the combination of circadian rhythm and overnight dexamethasone suppression salivary cortisol tests. J Clin Endocrinol Metab.1999;84(3):878–882. | ||
Evans AC, Collins DL, Mills SR, Brown ED, Kelly RL, Peters TM. 3D statistical neuroanatomical models from 305 MRI volumes. Proceedings of IEEE-Nuclear Science Symposium and Medical Imaging Conference.October 31 1993-November 6 1993. San Francisco, CA, USA. 1993:1813–1817. | ||
Talairach J, Tornoux P. Co-planar stereotaxic atlas of the human brain. New York: Thieme Medical Publishers Inc.; 1988. | ||
Van Essen DC, Drury HA, Joshi S, Miller MI. Functional and structural mapping of human cerebral cortex: solutions are in the surfaces. Proc Natl Acad Sci U S A.1998;95(3):788–795. | ||
Vaisvaser S, Lin T, Admon R, et al. Neural traces of stress: cortisol related sustained enhancement of amygdala-hippocampal functional connectivity. Front Hum Neurosci.2013;7:313. | ||
Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology.2000;23(5):477–501. | ||
Sapolsky RM. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry.2000;57(10):925–935. | ||
Monkul ES, Silva LA, Narayana S, et al. Abnormal resting state corticolimbic blood flow in depressed unmedicated patients with major depression: a (15)O-H(2)O PET study. Human brain mapping.2012;33(2):272–279. | ||
Montag C, Weber B, Fliessbach K, Elger C, Reuter M. The BDNF Val66Met polymorphism impacts parahippocampal and amygdala volume in healthy humans: incremental support for a genetic risk factor for depression. Psychological medicine.2009;39.(11):1831–1839. | ||
Almeida JR, Mechelli A, Hassel S, Versace A, Kupfer DJ, Phillips ML. Abnormally increased effective connectivity between parahippocampal gyrus and ventromedial prefrontal regions during emotion labeling in bipolar disorder. Psychiatry Res.2009;174(3):195–201. | ||
Crespo-Facorro B, Paradiso S, Andreasen NC, et al. Neural mechanisms of anhedonia in schizophrenia: a PET study of response to unpleasant and pleasant odors. JAMA.2001;286(4):427–435. | ||
Delgado y Palacios R, Campo A, Henningsen K, et al. Magnetic resonance imaging and spectroscopy reveal differential hippocampal changes in anhedonic and resilient subtypes of the chronic mild stress rat model. Biological Psychiatry.2011;70(5):449–457. | ||
Soriano-Mas C, Hernandez-Ribas R, Pujol J, et al. Cross-sectional and longitudinal assessment of structural brain alterations in melancholic depression. Biol Psychiatry.2011;69(4):318–325. | ||
de Quervain DJ, Henke K, Aerni A, et al. Glucocorticoid-induced impairment of declarative memory retrieval is associated with reduced blood flow in the medial temporal lobe. Eur J Neurosci.2003;17(6):1296–1302. | ||
Pariante CM. Risk factors for development of depression and psychosis. Glucocorticoid receptors and pituitary implications for treatment with antidepressant and glucocorticoids. Ann N Y Acad Sci.2009;1179:144–152. | ||
van Dalen JW, Moll van Charante EP, Nederkoorn PJ, van Gool WA, Richard E. Poststroke apathy. Stroke.2013;44(3):851–860. | ||
Murakami T, Hama S, Yamashita H, et al. Neuroanatomic pathways associated with poststroke affective and apathetic depression. Am J Geriatr Psychiatry.2013;21(9):840–847. | ||
Brodaty H, Sachdev PS, Withall A, Altendorf A, Valenzuela MJ, Lorentz L. Frequency and clinical, neuropsychological and neuroimaging correlates of apathy following stroke – the Sydney Stroke Study. Psychol Med.2005;35(12):1707–1716. | ||
Tang WK, Chen YK, Liang HJ, et al. Location of infarcts and apathy in ischemic stroke. Cerebrovasc Dis.2013;35(6):566–571. | ||
Papoiu AD, Nattkemper LA, Sanders KM, et al. Brain’s reward circuits mediate itch relief a functional MRI study of active scratching. PloS One.2013;8(12):e82389. | ||
Gabbay V, Mao X, Klein RG, et al. Anterior cingulate cortex γ-aminobutyric acid in depressed adolescents: relationship to anhedonia. Arch Gen Psychiatry.2012;69(2):139–149. | ||
Mead G, Lynch J, Greig C, Young A, Lewis S, Sharpe M. Evaluation of fatigue scales in stroke patients. Stroke.2007;38(7):2090–2095. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.