")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
The antitumor activity of tumor-homing peptide-modified thermosensitive liposomes containing doxorubicin on MCF-7/ADR: in vitro and in vivo
Authors Wang C, Wang X, Zhong T, Zhao Y, Zhang W, Ren W, Huang D, Zhang S, Guo Y, Yao X, Tang Y, Zhang X , Zhang Q
Received 24 December 2014
Accepted for publication 9 February 2015
Published 19 March 2015 Volume 2015:10(1) Pages 2229—2248
DOI https://doi.org/10.2147/IJN.S79840
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Chao Wang,1* Xin Wang,1* Ting Zhong,1 Yang Zhao,1 Wei-Qiang Zhang,1 Wei Ren,1 Dan Huang,1 Shuang Zhang,1 Yang Guo,1 Xin Yao,1 Yi-Qun Tang,2 Xuan Zhang,1 Qiang Zhang1,3
1Department of Pharmaceutics, School of Pharmaceutical Sciences, Peking University, Beijing, 2Department of Clinical Pharmacy, China Pharmaceutical University, Nanjing, 3State Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Beijing, People’s Republic of China
*These authors contributed equally to this work
Abstract: Clotted plasma proteins are present on the walls of tumor vessels and in tumor stroma. Tumor-homing peptide Cys-Arg-Glu-Lys-Ala (CREKA) could recognize the clotted plasma proteins in tumor vessels. Thermosensitive liposomes could immediately release the encapsulated drug in the vasculature of the heated tumor. In this study, we designed a novel form of targeted thermosensitive liposomes, CREKA-modified lysolipid-containing thermosensitive liposomes (LTSLs), containing doxorubicin (DOX) (DOX-LTSL-CREKA), to investigate the hypothesis that DOX-LTSL-CREKA might target the clotted plasma proteins in tumor vessels as well as tumor stroma and then exhibit burst release of the encapsulated DOX at the heated tumor site. We also hypothesized that the high local drug concentration produced by these thermosensitive liposomes after local hyperthermia treatment will be useful for treatment of multidrug resistance. The multidrug-resistant human breast adenocarcinoma (MCF-7/ADR) cell line was chosen as a tumor cell model, and the targeting and immediate release characteristics of DOX-LTSL-CREKA were investigated in vitro and in vivo. Furthermore, the antitumor activity of DOX-LTSL-CREKA was evaluated in MCF-7/ADR tumor-bearing nude mice in vivo. The targeting effect of the CREKA-modified thermosensitive liposomes on the clotted plasma proteins was confirmed in our in vivo imaging and immunohistochemistry experiments. The burst release of this delivery system was observed in our in vitro temperature-triggered DOX release and flow cytometry analysis and also by confocal microscopy experiments. The antitumor activity of the DOX-LTSL-CREKA was confirmed in tumor-bearing nude mice in vivo. Our findings suggest that the combination of targeting the clotted plasma proteins in the tumor vessel wall as well as tumor stroma by using CREKA peptide and temperature-triggered drug release from liposomes by using thermosensitive liposomes offers an attractive strategy for chemotherapeutic drug delivery to tumors.
Keywords: clotted plasma protein, tumor-homing peptide Cys-Arg-Glu-Lys-Ala (CREKA), lysolipid-containing thermosensitive liposomes, targeting
Introduction
It has been reported that blood clotting takes place on the walls of tumor vessels and in tumor stroma.1–3 This is due to a procoagulant tumor environment and leakage of plasma proteins, including fibrinogen, from the leaky tumor vessels into tissue.4 The expression of highly procoagulant proteins has been suggested to be involved in tumor cell-induced thrombin generation, leading to platelet activation and fibrin clot formation.5 In addition, the presence of fibrin in tumor meshwork is known to be associated with increased microvessel permeability in tumor tissues,1 and fibronectin in tumor stroma is also associated with tumor angiogenesis.2 Therefore, these clotted plasma proteins in tumor vessels as well as in tumor stroma could be an attractive target for antitumor therapy.
The tumor-homing peptide Cys-Arg-Glu-Lys-Ala (CREKA) is a short linear pentapeptide that recognizes clotted plasma proteins and selectively homes in on tumors.6 Interestingly, this tumor-homing peptide has attracted special attention as it homes in on tumors amplifying its own homing activity by stimulating platelet clot formation. The experimental results obtained indicate that CREKA-modified nanoparticles accumulate in tumor vessels, where they induce additional local clotting and thereby produce new binding sites for more nanoparticles in tumor tissues without causing clotting in normal tissues.7–9
Liposomes have been widely used as nanosize carriers in drug delivery systems for tumor therapy.10 It is well known that pegylated liposomes can spontaneously accumulate in solid tumors via an enhanced permeability and retention effect through a passive targeting mechanism.11 In fact, the ability to control and produce a burst release of the encapsulated drug from liposomes would be extremely advantageous and may prove to be an essential step in providing effective levels of drug in the tumor.12 Several approaches for triggered drug release from liposomes have been investigated, such as enzyme-activated,13,14 pH-sensitive,15,16 light-sensitive,17,18 ultrasound-triggered,19–21 and thermosensitive liposomes.22–30
Thermosensitive liposomes can be prepared from phospholipids that exhibit a sharp gel-to-liquid crystalline transition. Since the first developed thermosensitive liposomes using 1,2-dipalmitoyl-sn-glycero-3-phosphatidylcholine (DPPC), various types of thermosensitive liposomes have been prepared using DPPC as a primary membrane component, including the lysolipid-containing thermosensitive liposome (LTSL).31–34 The ability of LTSL to release encapsulated drug was found to be superior to that of conventional DPPC-based thermosensitive liposomes from which the encapsulated drug is released relatively slowly.33–38 Inclusion of ~10 mol% of the lysolipid, such as mono-stearoyl-sn-glycero-3-phosphatidylcholine (MSPC), produced significantly higher membrane permeability, with the highest permeability occurring at phase transition temperature which increased the encapsulated drug release.36
The encapsulated drug could fast release from the thermosensitive liposomes during hyperthermia treatment in the vasculature of tumor tissues site. It produces diffusion into the tumor along a high drug concentration gradient, significantly increasing the concentration of the released drug and its penetration into the tumor resulting in high drug accumulation compared with conventional long-circulating liposomes.12,37 In order to increase the targeting effect, the receptor-mediated targeting of thermosensitive liposomes has been reported.39,40
In the present study, we designed a novel form of targeted thermosensitive liposomes, CREKA-modified LTSLs, containing doxorubicin (DOX) (DOX-LTSL-CREKA). The reason is that CREKA peptide recognizes clotted plasma proteins in tumor vessels as well as tumor stroma and thermosensitive liposomes immediately release the encapsulated drug in the vasculature of the heated tumor. We hypothesized that the DOX-LTSL-CREKA might target the clotted plasma proteins in tumor vessels as well as tumor stroma and then produce burst release of the encapsulated DOX in the heated tumor site. The high local drug concentration produced by thermosensitive liposomes during local hyperthermia treatment will be useful for the treatment of multidrug resistance (MDR) because even extremely resistant experimental MDR cells are killed at high drug concentrations. Therefore, the multidrug-resistant human breast adenocarcinoma (MCF-7/ADR) cell line was chosen as the model tumor cell. The targeting and immediate release characteristics of DOX-LTSL-CREKA were investigated in vitro and in vivo. Furthermore, the antitumor activity of DOX-LTSL-CREKA was evaluated in MCF-7/ADR tumor-bearing nude mice in vivo.
Materials and methods
Materials
DPPC, MSPC, 1,2-distearoyl-sn-glycero-3-phosphatidylethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG-MAL) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Egg phosphatidylcholine (EPC) was provided by Lipoid GmbH (Ludwigshafen, Germany). Cholesterol, sulforhodamine B, and tris base were obtained from Sigma-Aldrich (St Louis, MO, USA). The CREKA peptide was synthesized by Beijing Scilight Biotechnology Co., Ltd. (Beijing, People’s Republic of China). DOX hydrochloride was supplied by Hisun Pharmaceutical Co. Ltd. (Zhejiang, People’s Republic of China). Near-infrared lipophilic carbocyanine dye (cy7) and the fluorescent probe (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate [DiI]) were obtained from Biotium, Inc. (Hayward, CA, USA). Hoechst 33258 and fluorescein isothiocyanate (FITC) goat anti-rabbit secondary antibodies (1:1,000) were purchased from Molecular Probes Inc. (Eugene, OR, USA). Rabbit polyclonal to CD31 (10 μg/mL) and rabbit polyclonal to fibrinogen (10 μg/mL) were obtained from Abcam Inc. (Cambridge, MA, USA). Cell culture media, Roswell Park Memorial Institute 1640, penicillin–streptomycin, fetal bovine serum (FBS), and l-glutamine were obtained from GIBCO Invitrogen Corp. (Carlsbad, CA, USA). All other reagents were of analytical grade.
A MCF-7/ADR cell line and a human breast adenocarcinoma (MCF-7) cell line were obtained from the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (Beijing, People’s Republic of China), and were grown in Roswell Park Memorial Institute 1640 medium with 10% FBS, 1% glutamine, and penicillin–streptomycin at 37°C and 5% CO2.
Female BALB/c nude mice, 6 weeks of age (initially weighing 20–25 g), were supplied by the Experimental Animal Center of Peking University (Beijing, People’s Republic of China). All care and handling of animals were performed with the approval of the Institutional Authority for Laboratory Animal Care of Peking University.
To prepare the tumor-bearing nude mouse model, female BALB/c nude mice were inoculated subcutaneously in the right armpits with 0.1 mL of MCF-7/ADR cell suspension (1×107).
Synthesis of DSPE-PEG-CREKA
DSPE-PEG-CREKA was synthesized from CREKA and DSPE-PEG-MAL in a single step that coupled CREKA to DSPE-PEG using a method previously reported by our laboratory.41,42 In brief, DSPE-PEG-MAL was incubated with CREKA at a 2:1 molar ratio (CREKA:DSPE-PEG-MAL =2:1) in phosphate-buffered saline (PBS) (pH 7.4). This reaction mixture was stirred gently at room temperature for 24 hours under nitrogen gas. The resulting reaction mixture was placed in a dialysis bag (molecular weight cut-off =2,000 Da) and dialyzed against deionized water for 48 hours to remove the unconjugated peptides. The final solution in the dialysis bag was lyophilized and stored at −20°C until required.
Preparation of DOX-LTSL-CREKA
The DOX-loaded CREKA-modified LTSLs (DOX-LTSL-CREKA) were prepared by a thin-film hydration method as described previously.30 Briefly, DPPC, MSPC, DSPE-PEG, and DSPE-PEG-CREKA (86:10:2:2 molar ratio) were dissolved in chloroform. Following evaporation of the solvent in an RE52 rotary evaporator (Shanghai Yarong Biochemistry Instrument Company, People’s Republic of China) in a round-bottomed flask at 45°C for about 40 minutes, a solid film was formed. This film was then flushed with nitrogen gas for 30 minutes and stored overnight in a desiccator to remove any traces of chloroform. After addition of 2 mL of 300 mM citric acid buffer (pH 4.0), the mixture was sonicated in a bath sonicator for 30 minutes. The resulting liposomes were then extruded eleven times through 100 nm polycarbonate membrane filters using a mini-extruder (Avanti Polar Lipids, Inc.). The obtained liposomes were then passed through a Sephadex G-50 column to remove the external citric acid of the liposomes with a 20 mM Hepes buffer solution (HBS, pH 7.4) containing 150 mM NaCl to obtain the blank liposomes. DOX was then loaded into these blank liposomes using a pH-gradient method described previously. Aqueous DOX solution was added to the blank liposomes at a DOX/lipid (w/w) ratio of 1:20. The mixed solution was incubated for 1 hour at 37°C and then passed through a Sephadex G-50 column to remove the un-entrapped DOX with HBS.
For the preparation of unmodified LTSLs containing DOX (DOX-LTSL), an identical procedure was used except that the equivalent molar amount of DSPE-PEG-CREKA was replaced by DSPE-PEG. The sterically stabilized liposomes (SSL) containing DOX (DOX-SSL) were prepared by the same procedures as described above involving EPC, cholesterol, and DSPE-PEG at a molar ratio of 65:30:5, as shown in our published research.41
For preparation of the cy7-loaded liposomes (cy7-LTSL-CREKA and cy7-LTSL) used in the in vivo imaging investigation, the dried lipid film was hydrated with HBS containing cy7, sonicated with a bath-type sonicator, extruded using a mini-extruder, and then passed through a Sephadex G-50 column to remove free cy7.
For preparation of DiI-LTSL-CREKA used in in vivo immunohistochemical analysis, the lipids and DiI were both dissolved in chloroform and dried until a thin film was obtained. The dried lipid film was hydrated with HBS, sonicated in a bath-type sonicator, extruded using a mini-extruder, and then passed through a Sephadex G-50 column to remove the un-entrapped DiI with HBS.
Characterization of DOX-LTSL-CREKA
The particle size and zeta potential of DOX-LTSL-CREKA were measured by photon correlation spectroscopy using a Malvern Zeta Sizer Nano-ZS (Malvern Instruments, Malvern, UK) at 25°C. To determine the liposomal encapsulation efficiency, the final liposomes were passed through a Sephadex G-50 column to remove free DOX. This was followed by disruption of the filtered liposomes with 10% Triton X-100 (v/v), and then the DOX concentration was measured in a spectrofluorimeter (Shimadzu RF-5301 PC). In addition, the same amount of liposomes was treated as above except for passing it through a Sephadex G-50 column to obtain the total concentration of DOX. The encapsulation efficiency was estimated from the following formula:
|

DSC analysis
Differential scanning calorimetry (DSC) analyses were performed using a Thermal Analysis DSC-Q100 differential scanning calorimeter (Thermal Analysis Instruments, New Castle, DE, USA) to analyze the phase transition of DOX-free liposome samples according to the method reported earlier.43 In brief, about 5–10 mg samples of liposomes were encapsulated in flat-bottomed aluminum pans. The thermograms were recorded at a heating rate of 5°C min−1 from 5°C to 90°C using nitrogen as the purging gas.44
In vitro release of DOX from liposomes
The release measurements were performed according to the method previously reported.30 An aliquot of the DOX-loaded liposome dispersion was added to 2 mL of PBS or PBS-containing 50% FBS solution in a quartz cell (final concentration of DOX 1 μg/mL) at a given temperature (37°C–45°C) for various times, and the fluorescence intensity of the solution was monitored using a spectrofluorimeter (Shimadzu RF-5301 PC). The excitation and emission wavelengths were set at 485 nm and 590 nm, respectively. DOX release was measured for 30 minutes, and then the sample at 30 minutes was mixed with 10 μL of 10% (v/v) Triton X-100 as a positive control. The percentage DOX release was calculated as (It − I0)/(I100 − I0) ×100, in which It is the fluorescence at time point t, I0 is the fluorescence at the start of the incubation, and I100 is the fluorescence of the sample 30 minutes after the addition of Triton X-100.
In vitro cytotoxicity of DOX-LTSL-CREKA
MCF-7/ADR cells (6×103 cells per well) were seeded in 96-well plates and grown in medium for 24 hours. The cells were mixed with different amounts of DOX-loaded liposomes. Then, the plates were divided into two groups. For group I, the cells were incubated at 37°C for 24 hours. For group II, the plates were heated immediately by immersing the cell-seeded plates in a water bath at 43°C for 5 minutes, and then, the cells were subsequently incubated at 37°C for 24 hours. After that, the viability of both group I and II was evaluated using an assay with sulforhodamine B.45 Absorbance was measured at 540 nm using a 96-well plate reader (Bio-Rad 680, Bio-Rad Laboratories, Inc., Hercules, CA, USA). The percentage of surviving cells was calculated using the following formula: variability % = (1− A540 nm for treated cells/A540 nm for control cells) ×100, where A540 nm is the absorbance. Each assay was repeated a minimum of three times, with quadruplicate determinations for each dose level. The in vitro cytotoxicity of DOX-LTSL-CREKA was also investigated in MCF-7 cells.
Flow cytometry
MCF-7/ADR cells were seeded at a density of 1×105 cells per well in six-well plates and incubated at 37°C for 24 hours to allow cell attachment. After 24 hours, the medium was replaced with cell culture medium containing DOX-loaded liposomes (40 μg/mL). The plates were divided into three groups. For group I, the cells were incubated at 37°C for 2 hours and washed three times with PBS solution. For group II (heated before incubation treatment), the plates were immediately heated for 5 minutes by immersing them in a water bath at 43°C. Then, the cells were incubated at 37°C for 2 hours and washed three times with PBS solution. For group III (heated after incubation treatment), after a 2-hour incubation, the cells were washed three times with culture medium and then immediately heated for 5 minutes by immersing the cell-seeded plates in a water bath at 43°C. The cells in the two groups were then harvested by trypsinization and centrifuged at 1,000× g for 5 minutes, resuspended in 500 μL of PBS medium, and examined by flow cytometry using an FACScan instrument (Becton Dickinson, San Jose, CA, USA). Cell-associated DOX was excited with an argon laser (488 nm), and fluorescence was detected at 570 nm. The cells not heated for 5 minutes at 43°C (unheated treatment) were also observed using the FACScan instrument.
Confocal microscopy studies
Following incubation of MCF-7/ADR cells in glass-bottomed dishes containing culture medium at 37°C for 24 hours, cell culture media containing DOX-loaded liposomes were added (the final concentration of DOX was 40 μg/mL) to each dish. The dishes were divided into three groups. For group I, the cells were incubated at 37°C for 2 hours. For group II (heated before incubation treatment), the dishes were immediately heated for 5 minutes by immersing the cell-seeded plates in a water bath at 43°C. Then, the cells were incubated at 37°C for 2 hours. For group III (heated after incubation treatment), after a 2-hour incubation, the cells were washed three times with culture medium and then immediately heated for 5 minutes by immersing the cell-seeded plates in a water bath at 43°C. For the above three groups, the medium was then removed, and cells were washed with ice-cold PBS followed by fixing with 4% paraformaldehyde in PBS. After fixing, the cells were processed with Hoechst 33258 for 15 minutes. The fluorescent images of the cells were analyzed using a TCS SP2 confocal microscope (Leica, Germany).
In vivo imaging
Once the tumor masses reached approximately 200 mm3 in volume, the tumor-bearing nude mice received an intravenous (iv) injection, via the tail vein, of physiological saline, cy7-LTSL, or cy7-LTSL-CREKA at a dose of 750 μg/kg. Other tumor-bearing mice received an iv injection of heparin solution (a bolus of 800 units/kg), followed 120 minutes later by cy7-LTSL-CREKA (750 μg/kg), and then they received additional heparin by intraperitoneal injections (a total of 1,000 units/kg) throughout the experiment. All mice were then anesthetized with an intraperitoneal injection of pentobarbital (60 mg/kg), and scanned at 2 hours, 6 hours, 8 hours, and 12 hours using a Kodak In Vivo Imaging System FX PRO (Carestream Health, Inc., Rochester, Minnesota, USA) with an excitation bandpass filter at 711 nm and an emission wavelength of 774 nm. The exposure time was 30 seconds per image. The fluorescent signal intensities were analyzed using Carestream MI SE software. For each near-infrared image, a corresponding X-ray image was taken to identify the anatomical location of the tumor.
Immunohistochemistry
Once the tumor masses reached approximately 200 mm3 in volume, the tumor-bearing nude mice received an iv injection of DiI-LTSL-CREKA at a dose of 200 μg/kg via the tail vein. Then, after 2 hours or 6 hours, the mice in the experiment were sacrificed, and the tumors were harvested. Then, the tumors were frozen in optimal cutting temperature-embedding medium. For immunostaining, tumor sections (6 μm) were first incubated with 1% bovine serum albumin for 1 hour at room temperature followed by incubation with the primary antibody (rabbit polyclonal to CD31 or rabbit polyclonal to fibrinogen) overnight at 4°C. Then, the primary antibodies were detected with FITC goat anti-rabbit secondary antibodies. Nuclei were counterstained with Hoechst 33258 (5 μg/mL). The sections were mounted in Gel Mount™ mounting medium (Biomeda, Foster City, CA, USA) and visualized under a confocal microscope (Leica).
In vivo antitumor activity
Once the tumor volume reached about 150–200 mm3, the tumor-bearing nude mice were randomly assigned to seven groups (six animals per group): group 1 given physiological saline as a control, group 2 given an injection of free DOX (4 mg/kg, iv, q2d ×3), group 3 given an injection of DOX-SSL (4 mg/kg, iv, q2d ×3), groups 4 and 5 were given DOX-LTSL (4 mg/kg, iv, q2d ×3), and groups 6 and 7 were given DOX-LTSL-CREKA (4 mg/kg, iv, q2d ×3). For all administrations, the formulations were given via the tail vein. The total dose of DOX in all treatment groups was 12 mg/kg. Then, 2 hours after drug administration, the mice in groups 5 and 7 were anesthetized by administration of sodium pentobarbital, and the tumor was locally heated at 43°C for 1 hour using a water bath. (The temperature inside the tumor was verified by a thermocouple [HH306 data logger/thermometer, Omega Engineering, Inc., Stamford, CT, USA]) Throughout the study, mice were weighed, and tumors were measured with a caliper twice per week. Tumor volumes were calculated from the formula: V = length (cm) × width (cm2) ×0.5236. Once animals in any group seemed moribund, all animals in the experiment were sacrificed, and the tumors were harvested and weighed. One or two mice in each group were executed, and the tumors were collected for the preparation of histological sections. Terminal Deoxynucleotidyl Transferase Mediated dUTP Nick End Labeling (TUNEL) staining of the paraffin-embedded tissue sections was performed according to the standard protocols provided by the manufacturers.
Statistical analysis
All data are presented as the mean ± standard deviation. One-way analysis of variance was used to determine significance among groups, after which post hoc tests with the Bonferroni correction were used for comparison between individual groups. Statistical significance was established at P<0.05.
Results
Preparation of DOX-LTSL-CREKA
As shown in Figure 1A, CREKA was conjugated to the terminal of PEG through a reaction between the maleimide group of PEG and the cysteine sulfhydryl group of the CREKA peptides through a nucleophilic addition reaction. The results of matrix-assisted laser desorption ionization time-of-flight mass spectrometry indicated that CREKA peptides were successfully conjugated with DSPE-PEG-maleimide (data not shown). The detailed scheme for the preparation and temperature-triggered release characteristic of DOX-LTSL-CREKA is illustrated in Figure 1B and C.
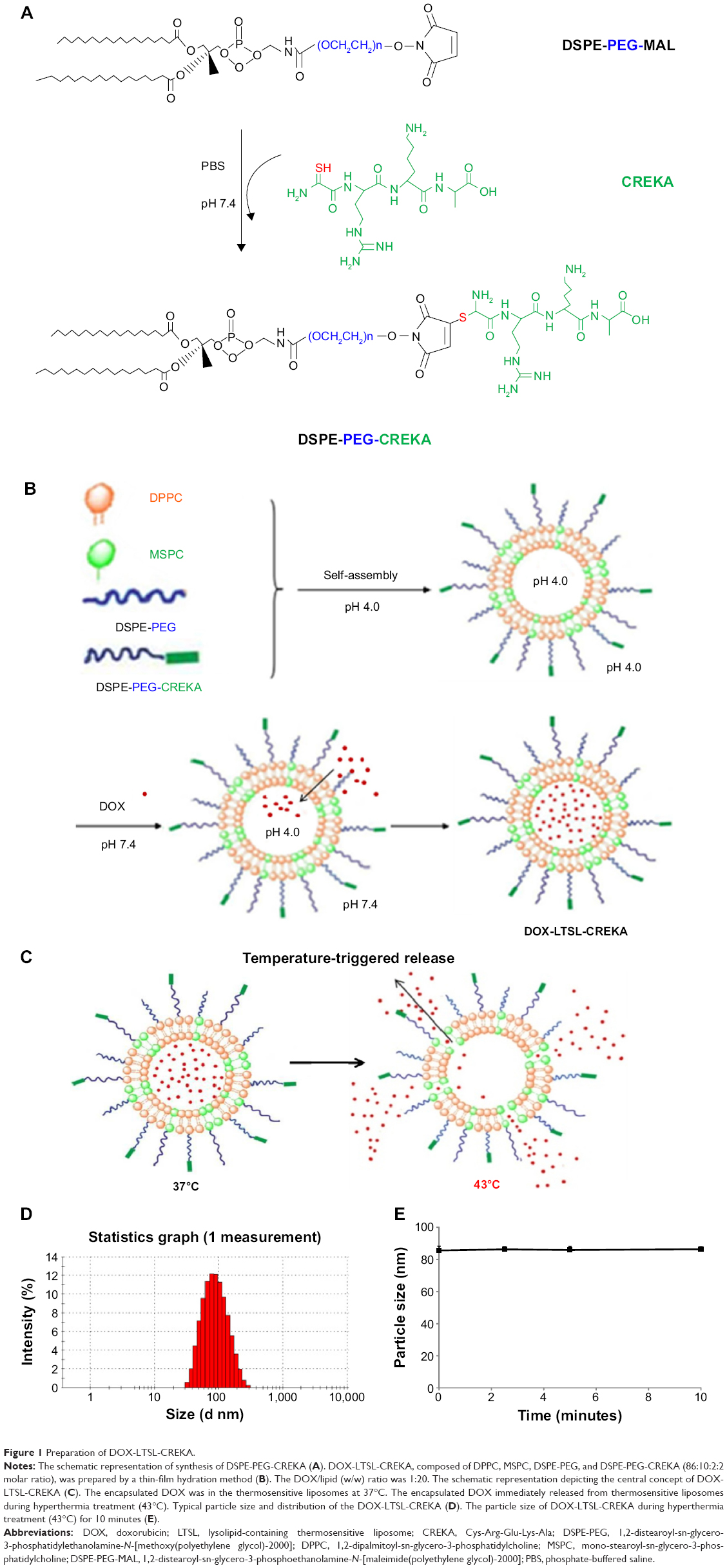 | Figure 1 Preparation of DOX-LTSL-CREKA. |
Characterization of DOX-LTSL-CREKA
The particle size of DOX-LTSL-CREKA was approximately 84±3.4 nm, and the polydispersity was 0.19±0.03. The value of the zeta potential of DOX-LTSL-CREKA was slightly negative −5.96±0.09 mV. The entrapment efficiency of DOX-LTSL-CREKA was 99.03%±0.63% (n=3), as shown in Table 1. A typical particle size and distribution of DOX-LTSL-CREKA is shown in Figure 1D.
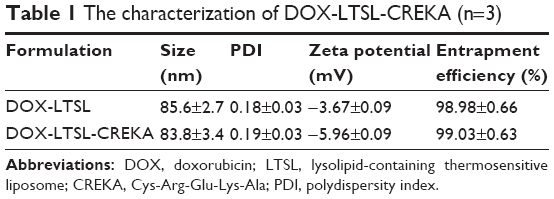 | Table 1 The characterization of DOX-LTSL-CREKA (n=3) |
Transition temperature
The temperature-sensitive transition of LTSL-CREKA, LTSL, and SSL was examined using DSC. Figure S1 shows the DSC curve for LTSL, LTSL-CREKA, and SSL. A large and sharp endotherm was observed centered at 43.0°C for LTSL or 43.7°C for LTSL-CREKA with a low transition temperature at 41.9°C or 42.4°C. In contrast, no phase transition peak was detected for SSL.
Temperature-triggered DOX release
The results of the in vitro release of DOX from DOX-LTSL-CREKA in PBS or PBS-containing 50% FBS are shown in Figure 2. For DOX-LTSL-CREKA groups, the release of DOX exhibited temperature-dependent release kinetics, as shown in Figure 2A and D. Approximately 10%–20% DOX was released from DOX-LTSL-CREKA at 37°C after 30 minutes in PBS (~14%) or PBS-containing 50% FBS (~16%), while almost complete drug release (90%–100%) was observed at 43°C–45°C within 10 minutes. However, the DOX-SSL remained intact and did not release DOX (less than 2%) when the dispersion was heated from 37°C to 45°C, as shown in Figure 2C. In addition, the release of DOX from DOX-LTSL in PBS was similar to that of DOX-LTSL-CREKA (data not shown). The temperature-triggered DOX release from DOX-LTSL-CREKA after 2.5 minutes in PBS or PBS-containing 50% FBS was also observed, as shown in Figure 2B and E. In addition, the release of DOX from DOX-LTSL-CREKA in PBS (about 2%) and PBS-containing 50% FBS (about 4%) at 37°C was almost identical. However, the DOX release from DOX-LTSL-CREKA in PBS-containing 50% FBS at 43°C or 45°C was faster than that in PBS (86% vs 75%, 89% vs 78%). We also investigated the particle size of DOX-LTSL-CREKA in PBS at 43°C for 10 minutes. As shown in Figure 1E, the particle size has no significant change after heating at 43°C for 10 minutes, showing that the structure of DOX-LTSL-CREKA keeps integrity after heat treatment.
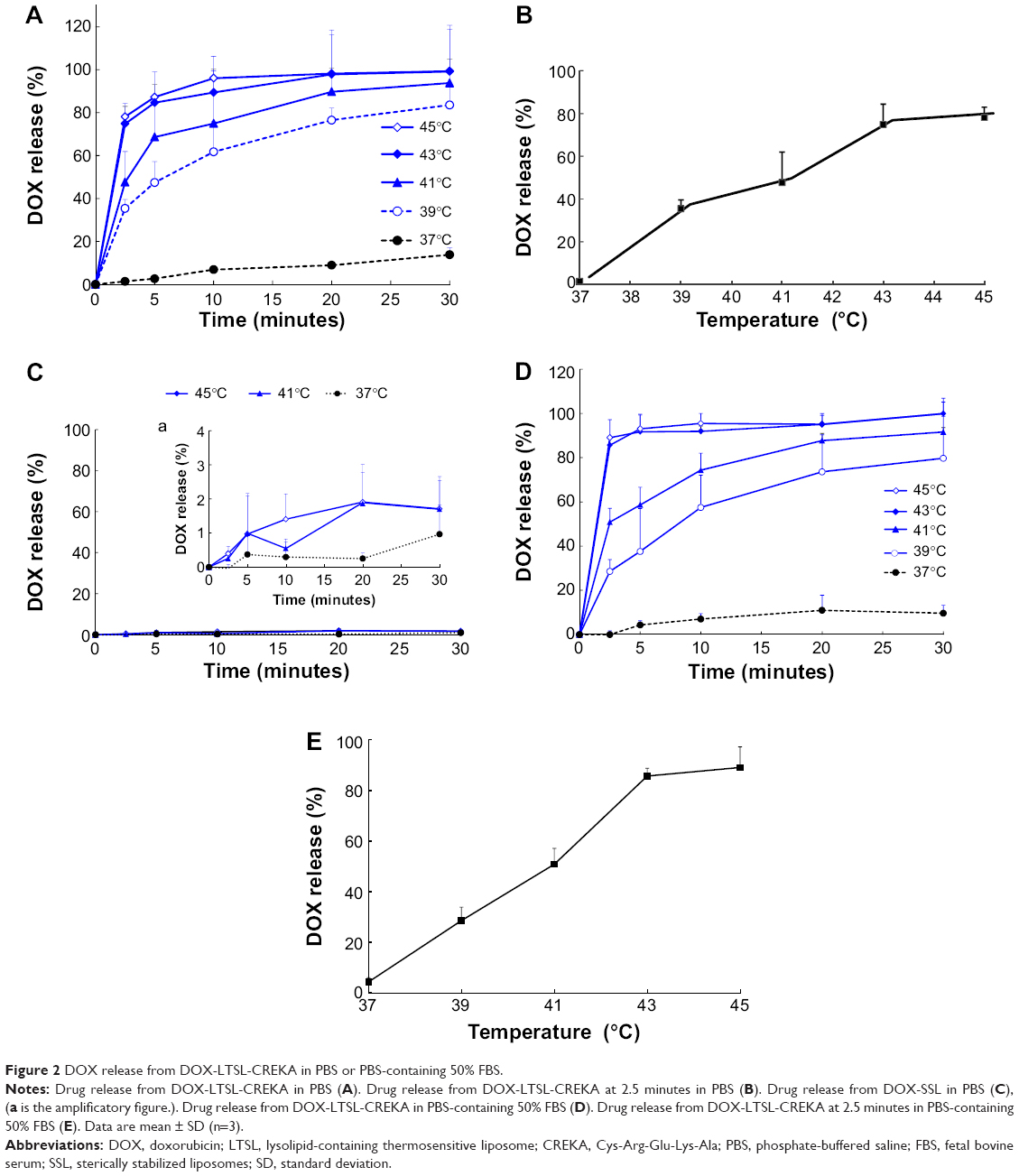 | Figure 2 DOX release from DOX-LTSL-CREKA in PBS or PBS-containing 50% FBS. |
In vitro cytotoxicity of DOX-LTSL-CREKA
The MCF-7/ADR cell line was used to investigate the in vitro cytotoxicity of DOX-LTSL-CREKA, with or without heating at 43°C, in comparison with free DOX. At the unheated condition, we observed that the IC50 values of DOX formulation groups were significantly higher than that of free DOX (about twofold), as shown in Table 2. Meanwhile, the IC50 values among DOX-LTSL-CREKA, DOX-LTSL, and DOX-SSL groups were almost similar at the unheated condition. At the heated condition, the IC50 values of DOX-LTSL-CREKA and DOX-LTSL groups, unlike the DOX-SSL group, were more close to that of free DOX.
 | Table 2 The IC50 (μg/mL) values of DOX-LTSL-CREKA with or without heating at 43°C for 5 minutes in MCF-7/ADR and MCF-7 cells (n=3) |
We also evaluated the in vitro cytotoxicity of DOX-LTSL-CREKA in MCF-7 cell line. The results indicated that MCF-7 is more sensitive to DOX than MCF-7/ADR. As shown in Table 2, at the heated condition, the IC50 values among DOX-LTSL-CREKA, DOX-LTSL, and DOX-SSL groups were almost similar at the unheated condition. At the heated condition, the IC50 values of DOX-LTSL-CREKA and DOX-LTSL groups, unlike the DOX-SSL group, were also more close to that of free DOX.
We suggested that the burst-released DOX from DOX-LTSL-CREKA and DOX-LTSL at heated condition produced more in vitro cytotoxicity in MCF/ADR and MCF-7 cells than that at unheated condition.
Flow cytometry analysis
Flow cytometry was used to quantify the total DOX uptake by MCF-7/ADR cells for DOX-liposome formulations. As shown in Figure 3, the intense fluorescence of DOX from DOX-LTSL-CREKA in the heated before incubation treatment group and the heated after incubation treatment group was about 2.2- and 1.4-fold higher than that from DOX-LTSL-CREKA in the unheated treatment group (Figure 3d and B). However, the cellular DOX level for DOX-SSL in the heated before incubation treatment group and the heated after incubation treatment group was only about 1.2- and 1.1-fold higher than that in the unheated treatment group (Figure 3e and B). In addition, the cellular DOX level for DOX-LTSL-CREKA in the unheated treatment group, the heated before incubation treatment group, and the heated after incubation treatment group was about 1.7-, 3.1-, and 2.1-fold higher than that for DOX-SSL in the unheated treatment group, the heated before incubation treatment group, and the heated after incubation treatment group, respectively (Figure 3a, 2, 3, and B).
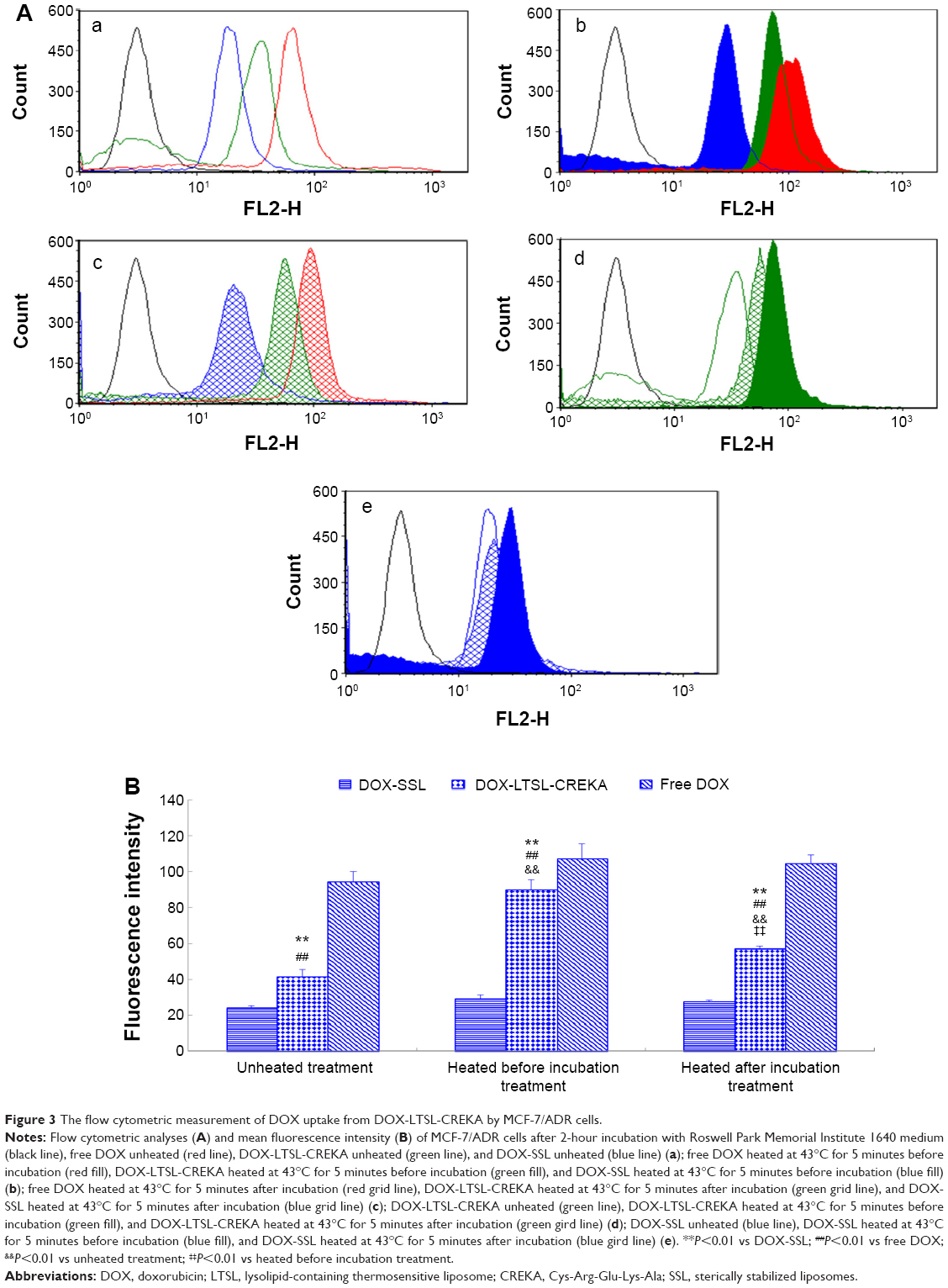 | Figure 3 The flow cytometric measurement of DOX uptake from DOX-LTSL-CREKA by MCF-7/ADR cells. |
Confocal microscopy studies
Figure 4 shows the confocal microscopic images of the MCF-7/ADR cell line after incubation with DOX-liposome formulations. For the DOX-LTSL-CREKA groups, the images showed a more intense fluorescence of DOX in both the heated before incubation treatment and the heated after incubation treatment groups compared with the unheated treatment (Figure 4A). However, for DOX-SSL groups, the intensity of the fluorescence of DOX in both the heated before incubation treatment and the heated after incubation treatment groups was similar to that in the unheated treatment group (Figure 4A). In addition, the intense fluorescence of DOX in the DOX-LTSL-CREKA group in the unheated treatment group, the heated before incubation treatment group, and the heated after incubation treatment group was more marked than that of the DOX-SSL group (Figure 4A).
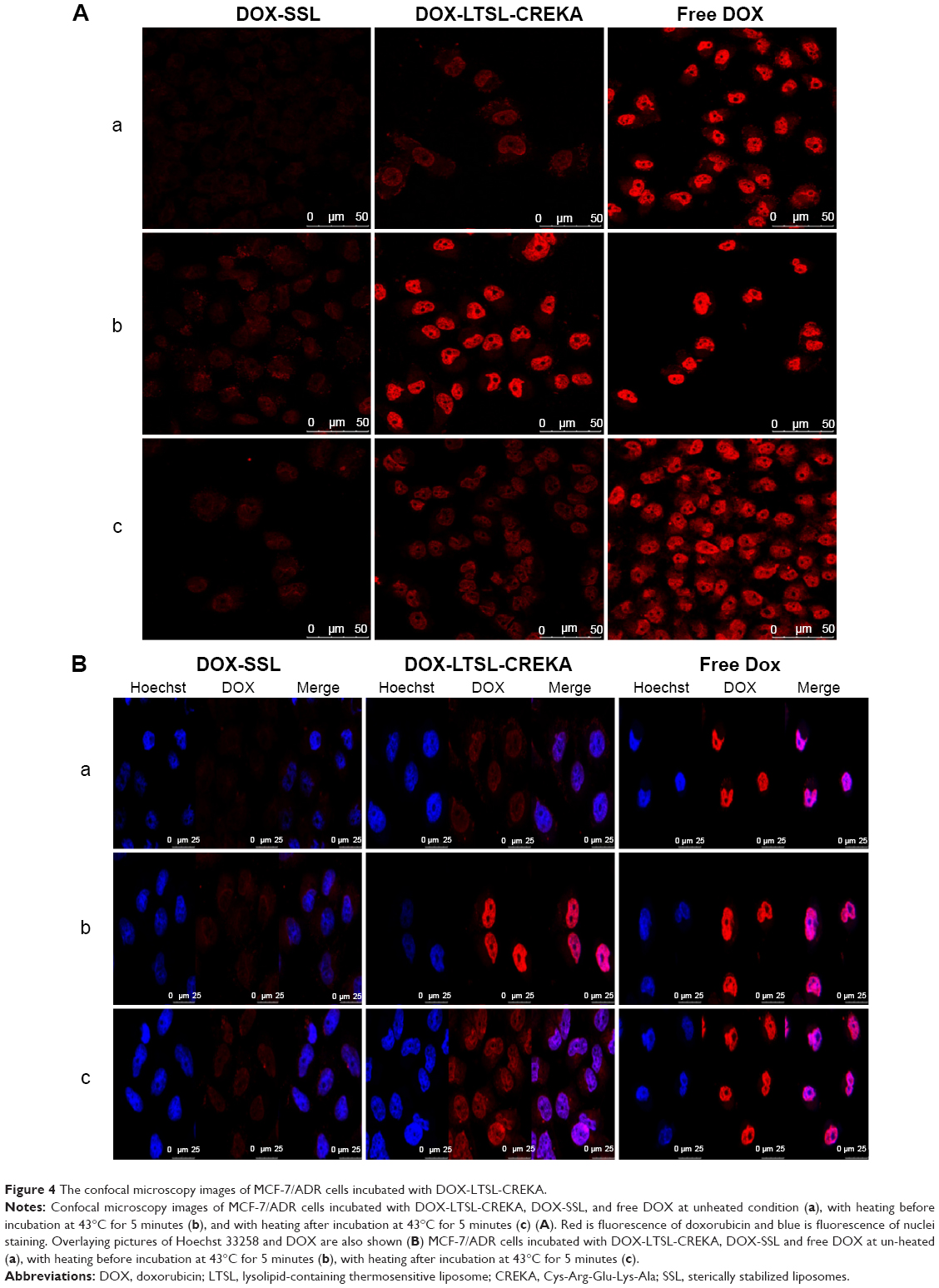 | Figure 4 The confocal microscopy images of MCF-7/ADR cells incubated with DOX-LTSL-CREKA. |
As shown in Figure 4B, the visible red fluorescence of DOX was mainly distributed in the nuclear compartment in the free DOX treatment group. As presented in Figure 4B, MCF-7/ADR cells treated with DOX-SSL and DOX-LTSL-CREKA under unheated conditions displayed intensive and punctate DOX fluorescence. However, the cellular fluorescence of DOX in the DOX-LTSL-CREKA groups was significantly induced by heating the cells at 43°C for 5 minutes, with all the cells displaying intensive fluorescence of the nuclei (Figure 4B).
In vivo imaging
Figure 5 shows the distribution and tumor accumulation of fluorescent cy7 in MCF-7/ADR tumor-bearing mice. The cy7 fluorescence signal at the tumor site was stronger for the mice treated with cy7-LTSL-CREKA than those treated with cy7-LTSL at all observed time points (Figure 5B and C). When the mice received an injection of heparin, a strong clotting inhibitor, the cy7 fluorescence signal induced by cy7-LTSL-CREKA at the tumor site was significantly reduced compared with that in the group treated with cy7-LTSL-CREKA alone and was almost identical at all the observed time points (Figure 5D).
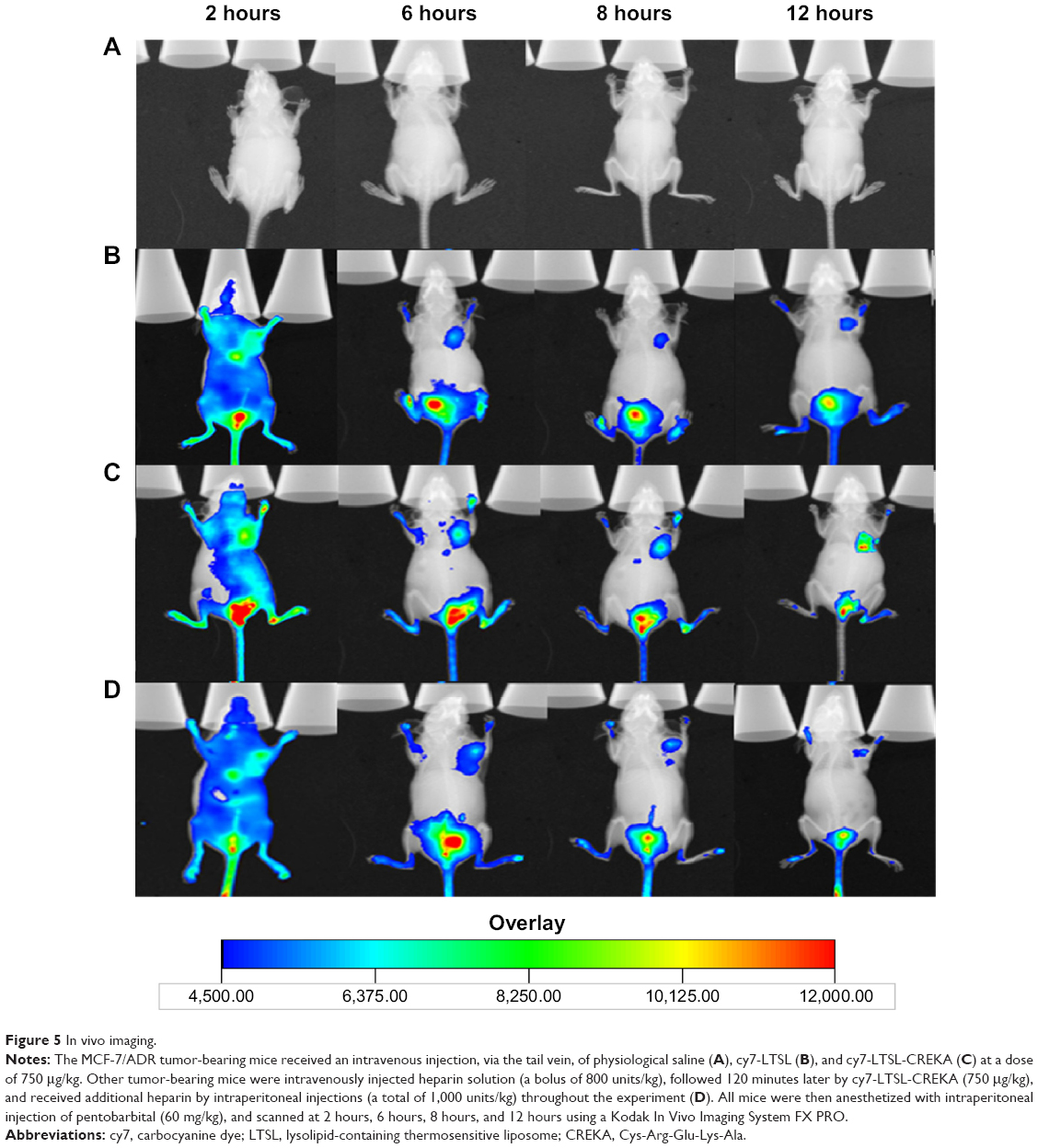 | Figure 5 In vivo imaging. |
Immunohistochemistry study
As shown in Figure 6A, the blood vessels stained with CD31 exhibited a green fluorescence, while those stained with DiI exhibited a red fluorescence. DiI-LTSL-CREKA mostly accumulated in the tumor blood vessels as shown by its co-localization with CD31 staining (Figure 6A). Some of the blood vessels appeared to have their lumens filled with a red fluorescent mass. In Figure 6B, clotted plasma proteins stained with antifebrin (ogen) exhibited a green fluorescence, and those stained with DiI exhibited a red fluorescence. DiI-LTSL-CREKA was also bound to the clotted plasma proteins. We also observed that the red fluorescence intensity of DiI-LTSL-CREKA accumulated in the tumor blood vessels or bound to the clotted plasma proteins at 6 hours was higher than that at 2 hours.
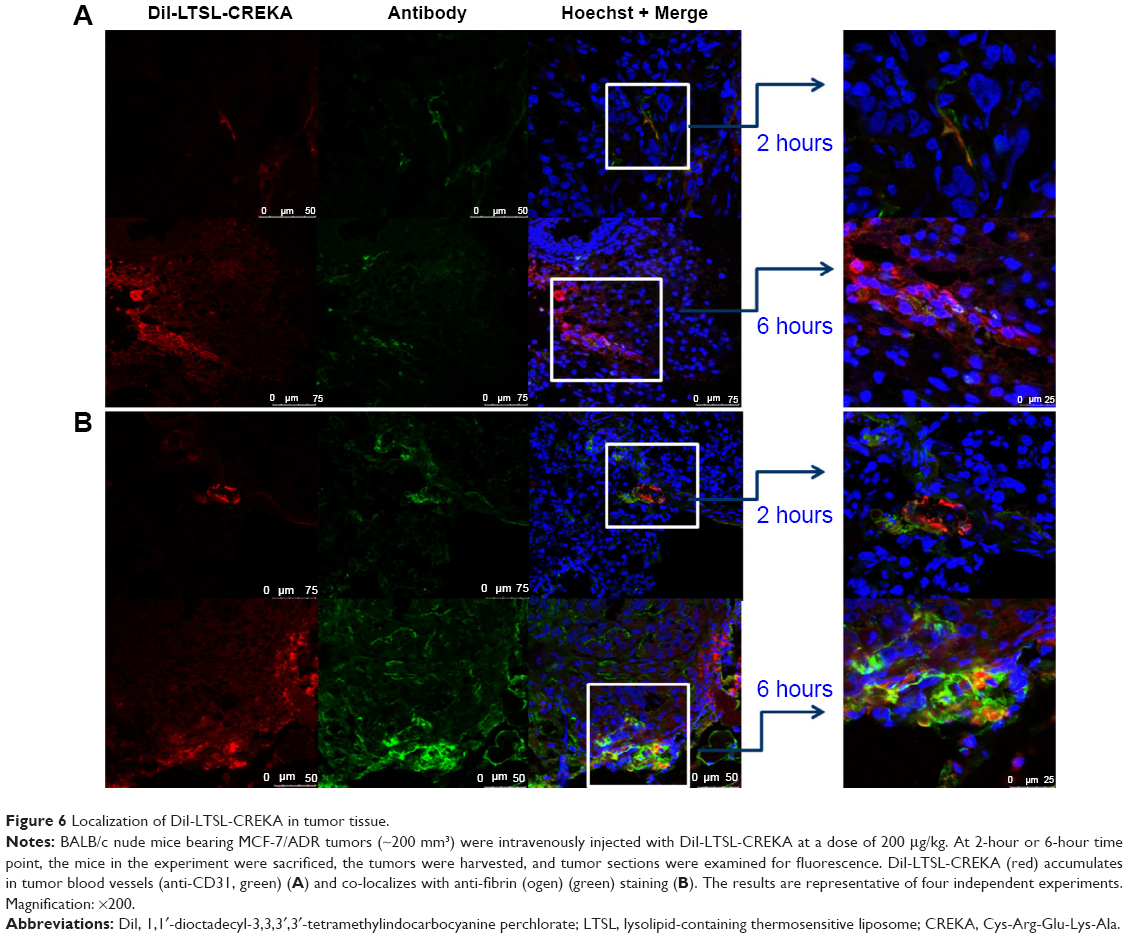 | Figure 6 Localization of DiI-LTSL-CREKA in tumor tissue. |
The antitumor effect of DOX formulations was evaluated in MCF-7/ADR tumor-bearing mice after cell implantation. As shown in Figure 7A, the tumor growth was significantly inhibited in all DOX-liposome treatment groups compared with the control group given physiological saline (P<0.01), but the effect varied. The antitumor activity of DOX-LTSL was similar to that of DOX-SSL (P>0.05). The groups given DOX-LTSL-CREKA, with or without heating, showed significant inhibition in the growth of MCF-7/ADR tumors compared with the DOX-LTSL, DOX-SSL, and free DOX treatment groups (P<0.01). In addition, the antitumor activity in the heated treatment groups (DOX-LTSL-CREKA/Heated and DOX-LTSL/Heated) was significantly higher than that in unheated treatment groups (DOX-LTSL-CREKA and DOX-LTSL) (P<0.01). The highest antitumor activity among the DOX-liposome treatment groups was observed in the DOX-LTSL-CREKA/Heated treatment group. The mean tumor size at day 25 after implantation in the free DOX, DOX-SSL, DOX-LTSL, DOX-LTSL/Heated, DOX-LTSL-CREKA, and DOX-LTSL-CREKA/Heated groups was 2,053±524 mm3, 1,688±339 mm3, 1,789±141 mm3, 1,164±243 mm3, 1,092±287 mm3, and 521±223 mm3, respectively, compared with 3,572±1,167 mm3 in the control group (P<0.01). The corresponding tumor growth inhibition in the free DOX, DOX-SSL, DOX-LTSL, DOX-LTSL/Heated, DOX-LTSL-CREKA, and DOX-LTSL-CREKA/Heated treated groups compared with the control group was 42.5%, 52.7%, 49.9%, 67.4%, 69.4%, and 85.4%, respectively.
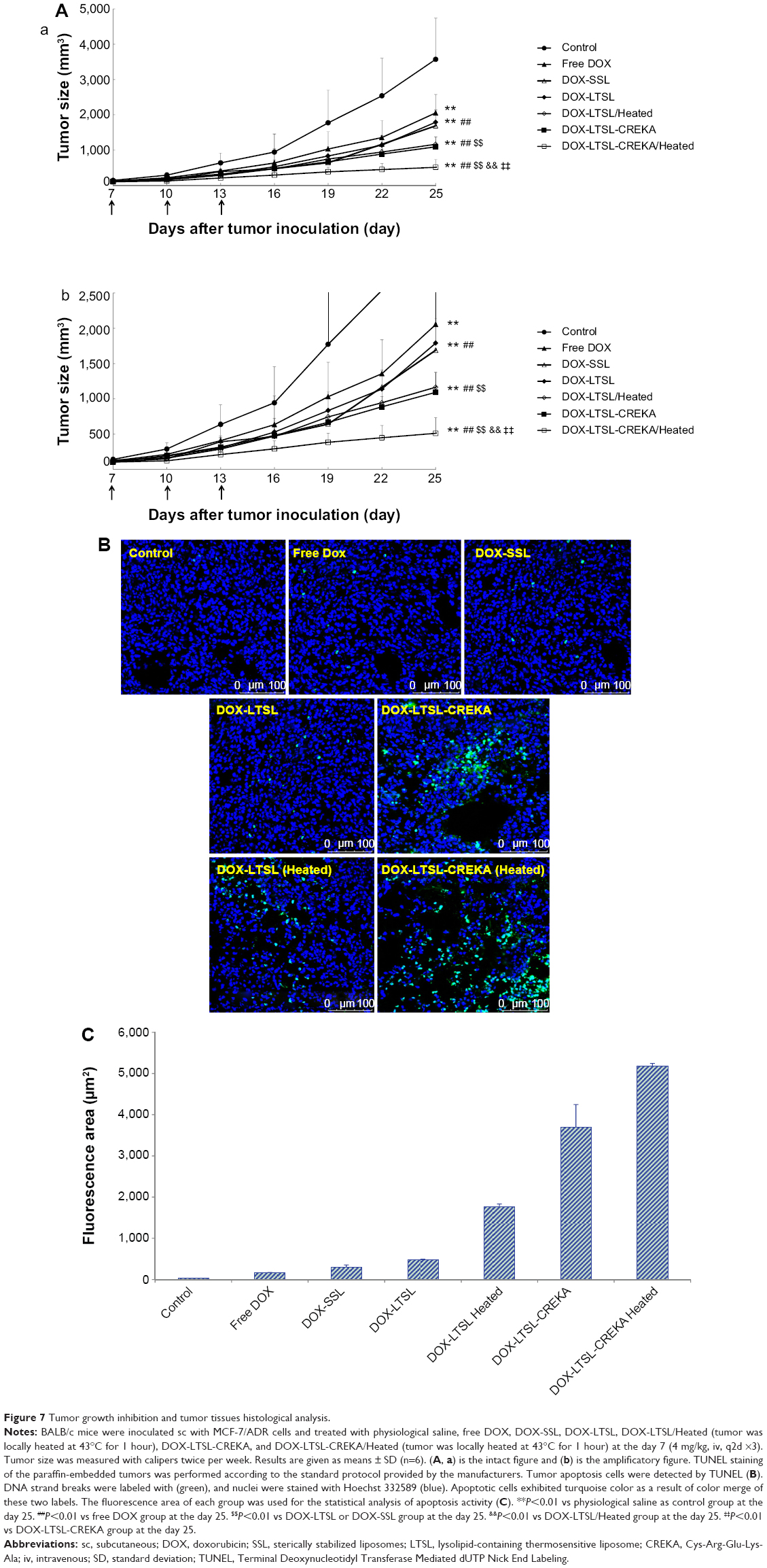 | Figure 7 Tumor growth inhibition and tumor tissues histological analysis. |
The tumor cell apoptosis by TUNEL analysis staining of tumor tissue sections was also evaluated. As shown in Figure 7B, tumors from the DOX-LTSL-CREKA/Heated treated group exhibited more advanced cell apoptosis compared with the control and other DOX treatment groups. In addition, the activity in the heated treatment groups (DOX-LTSL-CREKA/Heated and DOX-LTSL/Heated) was significantly higher than that in unheated treatment groups (DOX-LTSL-CREKA and DOX-LTSL) (P<0.01). The calculated results are shown in Figure 7C.
Discussion
In the present study, we prepared novel CREKA-modified thermosensitive liposomes containing DOX (DOX-LTSL-CREKA). We hypothesized that the DOX-LTSL-CREKA will specifically target the clotted plasma proteins in the tumor vessel wall as well as tumor stroma. Once this drug carrier system has undergone sufficient tumor accumulation, the drug could be burst released from the carrier by applying hyperthermia to render it bioavailable. The targeting and burst release characteristics of this delivery system were investigated in vitro and in vivo. The antitumor activity of DOX-LTSL-CREKA was confirmed in MCF-7/ADR tumor-bearing nude mice in vivo.
It has been reported that specific clotted plasma proteins were present in tumor vessel walls independent of tumor cell species. Therefore, these clotted plasma proteins are regarded as targeting sites for drug delivery systems. The homing peptide CREKA can specifically bind to the clotted plasma proteins in tumor vessels. More importantly, this binding induces additional local clotting and thereby produces new binding sites. This amplification system that enhanced homing of the nanoparticles in a mouse tumor model, without causing clotting or other obvious side effects elsewhere in the body, has been reported. In addition, these clotted plasma proteins are also present in tumor stroma, being a procoagulant tumor environment, and seepage of plasma proteins (such as fibrinogen) takes place from the leaky tumor vessels into the tissue. In the present study, the targeting effect of CREKA was confirmed by our in vivo imaging experiment. The self-amplified binding accumulation produced by CREKA was also confirmed. It has been reported that DOX can damage both endothelial and tumor cells.46–48 The death of endothelial cells might directly cause intravascular blood coagulation.49 Therefore, we suggest that the CREKA-binding sites might increase along with the DOX action.
The MDR to chemotherapeutic agents is a major challenge for those involved in cancer treatment.50 One of the main players in MDR mechanisms is a drug efflux transport protein, P-glycoprotein (P-gp), which transports drugs out of tumor cells. A large number of P-gp inhibitors have been developed; however, clinical trials using these inhibitors have shown limited success, largely due to the lack of specificity and efficacy.51 Until now, many attempts have been made to reverse MDR and hence restore the sensitivity of resistant cancer cells to multiple anticancer drugs. It has been reported that MDR tumor cells could be eventually killed upon exposure to tenfold to 100-fold higher drug concentrations than that to sensitive tumor cells.52 Normally, nano-carrier systems release drug slowly; thus, tumor cells may not be exposed to concentrations high enough to result in cell death.53 However, stimuli-responsive nano-carrier systems could trigger drug release in the target tissue upon hyperthermia treatment/temperature change, pH change, or other stimuli.54 It has been reported that hyperthermia combined with chemotherapy is playing an increasing role in the treatment of multidrug-resistant tumors in vivo.55 In addition, combining targeted drug delivery with hyperthermia has also been explored to overcome MDR.56 The combination of tumor vascular targeting and temperature-triggered drug release from liposomes has also been reported.40 Its potential therapeutic efficacy may be due to slowing the transit time of liposomes in the tumor vasculature to improve drug release and improving total drug accumulation in the tumor.40 In the present research, we investigated the high local drug concentration produced by DOX-LTSL-CREKA during local hyperthermia treatment for the treatment of MDR.
Our results of the in vitro cytotoxicity of DOX-LTSL-CREKA with heating indicated that the temperature-triggered released DOX was almost complete producing a similar in vitro cytotoxicity to that of free DOX in MCF-7/ADR cells and MCF-7 cells. In addition, the release of DOX from DOX-LTSL-CREKA which had been absorbed into MCF-7/ADR cells was also triggered by temperature, indicating that burst release was still exhibited when it entered the tumor cells. Our in vivo antitumor results indicate that the highest antitumor activity among the DOX-liposome treatment groups was observed in the DOX-LTSL-CREKA/Heated treatment group, showing that the combination of targeting the clotted plasma proteins in the tumor vessel wall as well as tumor stroma and temperature-triggered drug release from liposomes is an attractive strategy for drug delivery to tumors.
Conclusion
In summary, we designed and prepared novel CREKA-modified thermosensitive liposomes containing DOX (DOX-LTSL-CREKA). The targeting effect of the CREKA-modified thermosensitive liposomes on the clotted plasma proteins was confirmed in our in vivo imaging and immunohistochemistry experiments. The burst release of this delivery system was observed in our in vitro temperature-triggered DOX release and flow cytometry analysis and confocal microscopy experiments. The antitumor activity of the DOX-LTSL-CREKA was confirmed in tumor-bearing nude mice in vivo. Considering all our findings, we suggest that the combination of targeting the clotted plasma proteins in the tumor vessel wall as well as tumor stroma by using CREKA peptide and temperature-triggered drug release from liposomes by using thermosensitive liposomes is an attractive strategy for chemotherapeutic drug delivery to tumors.
Acknowledgments
The authors gratefully acknowledge the financial support from the Specialized Research Fund for the Doctoral Program of Higher Education (20120001110012) and the National Basic Research Program of China (973 Program 2013CB932501) and National Natural Science Foundation of China (number 81172992).
Disclosure
The authors report no conflicts of interest in this work.
References
Dvorak HF, Senger DR, Dvorak AM, Harvey VS, McDonagh J. Regulation of extravascular coagulation by microvascular permeability. Science. 1985;227:1059–1061. | ||
Neri D, Carnemolla B, Nissim A, et al. Targeting by affinity-matured recombinant antibody fragments of an angiogenesis associated fibronectin isoform. Nat Biotechnol. 1997;15:1271–1275. | ||
Ye F, Wu X, Jeong EK, et al. A peptide targeted contrast agent specific to fibrin-fibronectin complexes for cancer molecular imaging with MRI. Bioconjug Chem. 2008;19:2300–2303. | ||
Abe K, Shoji M, Chen J, et al. Regulation of vascular endothelial growth factor production and angiogenesis by the cytoplasmic tail of tissue factor. Proc Natl Acad Sci U S A. 1999;96:8663–8668. | ||
Ahmad S, Ansari AA. Therapeutic roles of heparin anticoagulants in cancer and related disorders. Med Chem. 2011;7:504–517. | ||
Simberg D, Duza T, Park JH, et al. Biomimetic amplification of nanoparticle homing to tumors. Proc Natl Acad Sci U S A. 2007;104:932–936. | ||
Agemy L, Sugahara KN, Kotamraju VR, et al. Nanoparticle-induced vascular blockade in human prostate cancer. Blood. 2010;116:2847–2856. | ||
Karmali PP, Kotamraju VR, Kastantin M, et al. Targeting of albumin-embedded paclitaxel nanoparticles to tumors. Nanomedicine. 2009;5:73–82. | ||
Kruse AM, Meenach SA, Anderson KW, Hilt JZ. Synthesis and characterization of CREKA-conjugated iron oxide nanoparticles for hyperthermia applications. Acta Biomater. 2014;10:2622–2629. | ||
Slingerland M, Guchelaar HJ, Gelderblom H. Liposomal drug formulations in cancer therapy: 15 years along the road. Drug Discov Today. 2012;17:160–166. | ||
Yuan F, Dellian M, Fukumura D, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55:3752–3756. | ||
Needham D, Anyarambhatla G, Kong G, Dewhirst MW. A new temperature-sensitive liposome for use with mild hyperthermia: characterization and testing in a human tumor xenograft model. Cancer Res. 2000;60:1197–1201. | ||
Meers P. Enzyme-activated targeting of liposomes. Adv Drug Deliv Rev. 2001;53:265–272. | ||
Pedersen PJ, Adolph SK, Subramanian AK, et al. Liposomal formulation of retinoids designed for enzyme triggered release. J Med Chem. 2010;53:3782–3792. | ||
Hatakeyama H, Murata M, Sato Y, et al. The systemic administration of an anti-miRNA oligonucleotide encapsulated pH-sensitive liposome results in reduced level of hepatic microRNA-122 in mice. J Control Release. 2014;173:43–50. | ||
Yuba E, Tajima N, Yoshizaki Y, Harada A, Hayashi H, Kono K. Dextran derivative-based pH-sensitive liposomes for cancer immunotherapy. Biomaterials. 2014;35:3091–3101. | ||
Alvarez-Lorenzo C, Bromberg L, Concheiro A. Light-sensitive intelligent drug delivery systems. Photochem Photobiol. 2009;85:848–860. | ||
Seo HJ, Kim JC. Light-sensitive liposomes containing coumarin-proteinoid conjugate. J Nanosci Nanotechnol. 2012;12:4044–4050. | ||
Ibsen S, Benchimol M, Simberg D, Schutt C, Steiner J, Esener S. A novel nested liposome drug delivery vehicle capable of ultrasound triggered release of its payload. J Control Release. 2011;155:358–366. | ||
Sutton JT, Raymond JL, Verleye MC, Pyne-Geithman GJ, Holland CK. Pulsed ultrasound enhances the delivery of nitric oxide from bubble liposomes to ex vivo porcine carotid tissue. Int J Nanomedicine. 2014;9:4671–4683. | ||
Cool SK, Geers B, Roels S, et al. Coupling of drug containing liposomes to microbubbles improves ultrasound triggered drug delivery in mice. J Control Release. 2013;172:885–893. | ||
Kneidl B, Peller M, Winter G, Lindner LH, Hossann M. Thermosensitive liposomal drug delivery systems: state of the art review. Int J Nanomedicine. 2014;9:4387–4398. | ||
Li L, ten Hagen TL, Haeri A, et al. A novel two-step mild hyperthermia for advanced liposomal chemotherapy. J Control Release. 2014;174:202–208. | ||
Iyer AK, Singh A, Ganta S, Amiji MM. Role of integrated cancer nanomedicine in overcoming drug resistance. Adv Drug Deliv Rev. 2013;65:1784–1802. | ||
Li L, ten Hagen TL, Schipper D, et al. Triggered content release from optimized stealth thermosensitive liposomes using mild hyperthermia. J Control Release. 2010;143:274–279. | ||
Chen Q, Tong S, Dewhirst MW, Yuan F. Targeting tumor microvessels using doxorubicin encapsulated in a novel thermosensitive liposome. Mol Cancer Ther. 2004;3:1311–1317. | ||
Tagami T, May JP, Ernsting MJ, Li SD. A thermosensitive liposome prepared with a Cu2−gradient demonstrates improved pharmacokinetics, drug delivery and antitumor efficacy. J Control Release. 2012;161:142–149. | ||
Tagami T, Ernsting MJ, Li SD. Optimization of a novel and improved thermosensitive liposome formulated with DPPC and a Brij surfactant using a robust in vitro system. J Control Release. 2011;154:290–297. | ||
Agarwal A, Mackey MA, El-Sayed MA, Bellamkonda RV. Remote triggered release of doxorubicin in tumors by synergistic application of thermosensitive liposomes and gold nanorods. ACS Nano. 2011;5:4919–4926. | ||
Tagami T, Ernsting MJ, Li SD. Efficient tumor regression by a single and low dose treatment with a novel and enhanced formulation of thermosensitive liposomal doxorubicin. J Control Release. 2011;152:303–309. | ||
Weinstein JN, Magin RL, Yatvin MB, Zaharko DS. Liposomes and local hyperthermia: selective delivery of methotrexate to heated tumors. Science. 1979;204:188–191. | ||
Yatvin MB, Weinstein JN, Dennis WH, Blumenthal R. Design of liposomes for enhanced local release of drugs by hyperthermia. Science. 1978;202:1290–1293. | ||
Mills JK, Needham D. Lysolipid incorporation in dipalmitoylphosphatidylcholine bilayer membranes enhances the ion permeability and drug release rates at the membrane phase transition. Biochim Biophys Acta. 2005;1716:77–96. | ||
Poon RT, Borys N. Lyso-thermosensitive liposomal doxorubicin: a novel approach to enhance efficacy of thermal ablation of liver cancer. Expert Opin Pharmacother. 2009;10:333–343. | ||
Lindner LH, Hossann M, Vogeser M, et al. Dual role of hexadecylphosphocholine (miltefosine) in thermosensitive liposomes: active ingredient and mediator of drug release. J Control Release. 2008;125:112–120. | ||
Landon CD, Park JY, Needham D, Dewhirst MW. Nanoscale drug delivery and hyperthermia: the materials design and preclinical and clinical testing of low temperature-sensitive liposomes used in combination with mild hyperthermia in the treatment of local cancer. Open Nanomed J. 2011;3:38–64. | ||
Kong G, Anyarambhatla G, Petros WP, et al. Efficacy of liposomes and hyperthermia in a human tumor xenograft model: importance of triggered drug release. Cancer Res. 2000;60:6950–6957. | ||
Banno B, Ickenstein LM, Chiu GN, et al. The functional roles of poly(ethylene glycol)-lipid and lysolipid in the drug retention and release from lysolipid-containing thermosensitive liposomes in vitro and in vivo. J Pharm Sci. 2010;99:2295–2308. | ||
Smith B, Lyakhov I, Loomis K, et al. Hyperthermia-triggered intracellular delivery of anticancer agent to HER2(+) cells by HER2-specific affibody (ZHER2-GS-Cys)-conjugated thermosensitive liposomes (HER2(+) affisomes). J Control Release. 2011;153:187–194. | ||
Negussie AH, Miller JL, Reddy G, Drake SK, Wood BJ, Dreher MR. Synthesis and in vitro evaluation of cyclic NGR peptide targeted thermally sensitive liposome. J Control Release. 2010;143:265–273. | ||
Yu KF, Zhang WQ, Luo LM, et al. The antitumor activity of a doxorubicin loaded, iRGD-modified sterically-stabilized liposome on B16-F10 melanoma cells: in vitro and in vivo evaluation. Int J Nanomedicine. 2013;8:2473–2485. | ||
Du R, Zhong T, Zhang WQ, et al. Antitumor effect of iRGD-modified liposomes containing conjugated linoleic acid-paclitaxel (CLA-PTX) on B16-F10 melanoma. Int J Nanomedicine. 2014;9:3091–3105. | ||
Yudina A, de Smet M, Lepetit-Coiffé M, et al. Ultrasound-mediated intracellular drug delivery using microbubbles and temperature-sensitive liposomes. J Control Release. 2011;155:442–448. | ||
Kono K, Ozawa T, Yoshida T, et al. Highly temperature-sensitive liposomes based on a thermosensitive block copolymer for tumor-specific chemotherapy. Biomaterials. 2010;31:7096–7105. | ||
Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–1116. | ||
Wang S, Kotamraju S, Konorev E, Kalivendi S, Joseph J, Kalyanaraman B. Activation of nuclear factor-kappaB during doxorubicin-induced apoptosis in endothelial cells and myocytes is pro-apoptotic: the role of hydrogen peroxide. Biochem J. 2002;367:729–740. | ||
Pastorino F, Brignole C, Marimpietri D, et al. Vascular damage and anti-angiogenic effects of tumor vessel-targeted liposomal chemotherapy. Cancer Res. 2003;63:7400–7409. | ||
Zhang L, Yu D, Hicklin DJ, Hannay JA, Ellis LM, Pollock RE. Combined anti-fetal liver kinase 1 monoclonal antibody and continuous low-dose doxorubicin inhibits angiogenesis and growth of human soft tissue sarcoma xenografts by induction of endothelial cell apoptosis. Cancer Res. 2002;62:2034–2042. | ||
Caine GJ, Stonelake PS, Lip GY, Kehoe ST. The hypercoagulable state of malignancy: pathogenesis and current debate. Neoplasia. 2002;4:465–473. | ||
Ullah MF. Cancer multidrug resistance (MDR): a major impediment to effective chemotherapy. Asian Pac J Cancer Prev. 2008;9:1–6. | ||
Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. | ||
Kim D, Lee ES, Oh KT, Gao ZG, Bae YH. Doxorubicin-loaded polymeric micelle overcomes multidrug resistance of cancer by double-targeting folate receptor and early endosomal pH. Small. 2008;4:2043–2050. | ||
Manzoor AA, Lindner LH, Landon CD, et al. Overcoming limitations in nanoparticle drug delivery: triggered, intravascular release to improve drug penetration into tumors. Cancer Res. 2012;72:5566–5575. | ||
Kapse-Mistry S, Govender T, Srivastava R, Yergeri M. Nanodrug delivery in reversing multidrug resistance in cancer cells. Front Pharmacol. 2014;5:159. | ||
Ren Y, Zhang H, Chen B, et al. Multifunctional magnetic Fe3O4 nanoparticles combined with chemotherapy and hyperthermia to overcome multidrug resistance. Int J Nanomedicine. 2012;7:2261–2269. | ||
Gaber MH. Modulation of doxorubicin resistance in multidrug-resistance cells by targeted liposomes combined with hyperthermia. J Biochem Mol Biol Biophys. 2002;6:309–314. |
Supplementary material
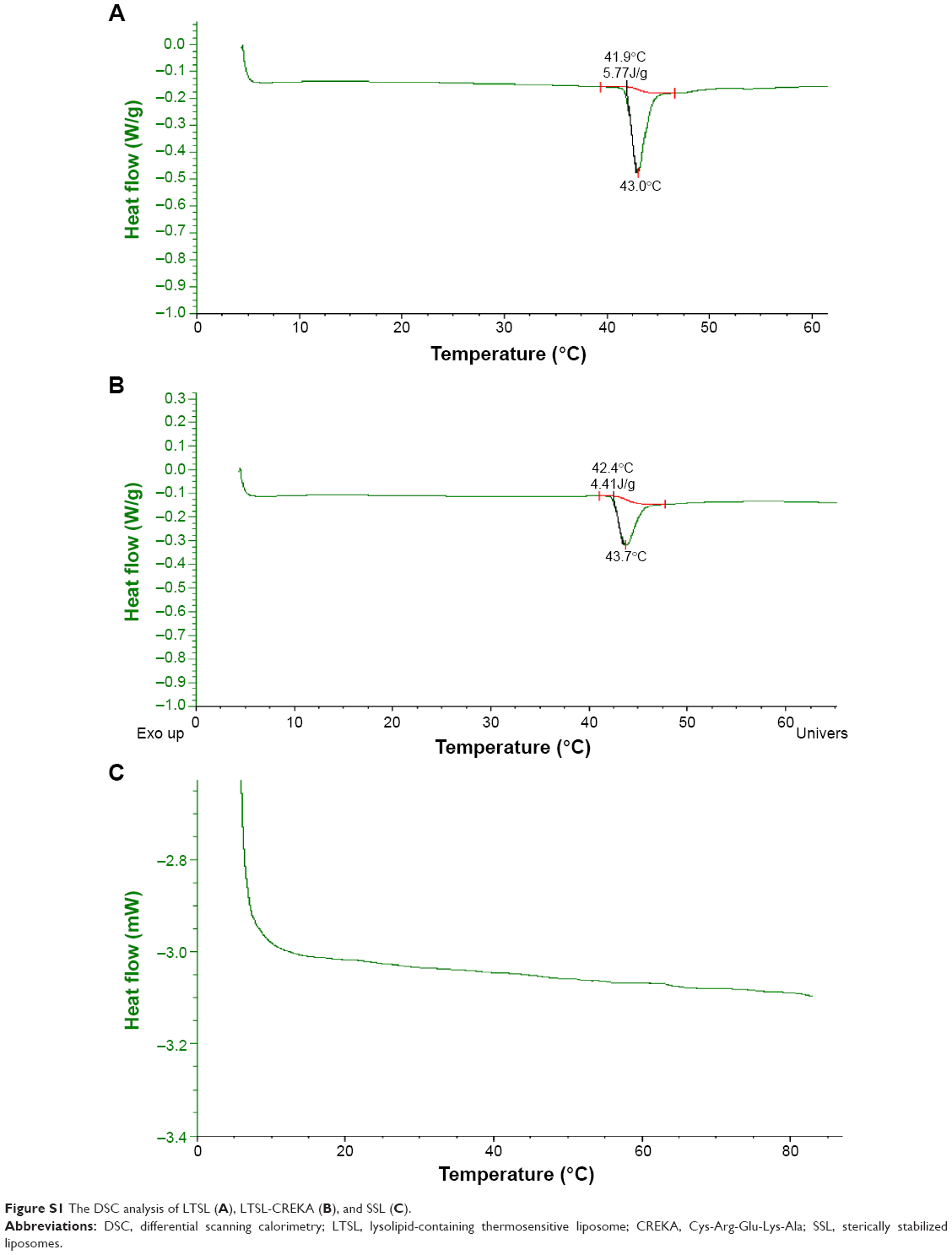 | Figure S1 The DSC analysis of LTSL (A), LTSL-CREKA (B), and SSL (C). |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.