")
Back to Journals » Drug Design, Development and Therapy » Volume 17
The Anti-Inflammatory Mediator 17(R)-Resolvin D1 Attenuates Pressure Overload-Induced Cardiac Hypertrophy and Fibrosis
Authors Wang M, Pan W, Wei C, Liu J, Zhang J, Yu J, Zhao M, Xu S, Ye J, Wang Z, Ye D, Feng Y, Xu Y, Wan J
Received 13 June 2023
Accepted for publication 22 September 2023
Published 11 October 2023 Volume 2023:17 Pages 3073—3083
DOI https://doi.org/10.2147/DDDT.S421894
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Menglong Wang,1– 3,* Wei Pan,1– 3,* Cheng Wei,1– 3,* Jianfang Liu,1– 3 Jishou Zhang,1– 3 Junping Yu,1– 3 Mengmeng Zhao,1– 3 Shuwan Xu,1– 3 Jing Ye,1– 3 Zhen Wang,1– 3 Di Ye,1– 3 Yongqi Feng,1– 3 Yao Xu,1– 3 Jun Wan1– 3
1Department of Cardiology, Renmin Hospital of Wuhan University, Wuhan, 430060, People’s Republic of China; 2Cardiovascular Research Institute, Wuhan University, Wuhan, 430060, People’s Republic of China; 3Hubei Key Laboratory of Cardiology, Wuhan, 430060, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jun Wan; Yao Xu, Department of Cardiology, Renmin Hospital of Wuhan University, 238 Jiefang Road, Wuhan, 430060, People’s Republic of China, Tel +86-27-88041911, Fax +86-27-88042293, Email [email protected]; [email protected]
Background: Increased inflammation contributes to pressure overload-induced myocardial remodeling. 17(R)-Resolvin D1 (17(R)-RvD1), a potent lipid mediator derived from docosahexaenoic acid, possesses anti-inflammatory and pro-resolving properties. However, the association between 17(R)-RvD1 and pressure overload-induced cardiac hypertrophy remains unclear.
Methods: Transverse aortic constriction (TAC) surgery was performed to establish a cardiac hypertrophy model. C57BL/6J mice were randomly assigned to the Sham, TAC and TAC+17(R)-RvD1 groups. 17(R)-RvD1 was injected (2 μg/kg, i.p.) before TAC surgery and once every other day after surgery for 4 weeks. The same volume of saline was injected into the mice in both Sham group and TAC group. Then, cardiac function was evaluated and heart tissues were collected for biological analysis.
Results: 17(R)-RvD1 treatment attenuated TAC-induced increase in left ventricular diameter and decrease in left ventricular contractility, mitigated increased cardiomyocyte cross-sectional area, and downregulated the expression of hypertrophic genes. Besides, 17(R)-RvD1 attenuated myocardial fibrosis, as indicated by the decreased LV collagen volume and expression of fibrotic genes. In addition, 17(R)-RvD1 ameliorated the inflammatory response in cardiac tissue, as illustrated by the decreased infiltration of CD68+ macrophages and reduced production of pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6. 17(R)-RvD1 treatment significantly suppressed the activation of NLRP3 inflammasome after TAC surgery, which might be responsible for the attenuation of inflammation in cardiac tissue.
Conclusion: 17(R)-RvD1 attenuated pressure overload-induced cardiac hypertrophy and fibrosis, and the possible mechanism may be associated with the inhibition of NLRP3 inflammasome. 17(R)-RvD1 may serve as a potential drug for the treatment of cardiac hypertrophy.
Keywords: inflammation resolution, resolvin D1, cardiac hypertrophy, NLRP3 inflammasome, transverse aortic constriction
Introduction
Heart failure (HF), which is the severe and final stage of heart diseases, such as coronary heart disease and hypertension, is currently a widespread concern and a crucial risk factor for cardiac disease-related mortality.1 It is clear that cardiac remodeling is the main mechanism underlying the occurrence and development of HF, and cardiac hypertrophy is one of its main manifestations.2,3 As a compensatory response of the myocardium to long-term pressure or volume overload, cardiac hypertrophy is generally characterized by cardiomyocyte enlargement and increased extracellular matrix deposition.4,5 Under conditions of sustained pressure or volume overload, the progression of cardiac remodeling is accompanied by the decompensation of cardiac hypertrophy, interstitial fibrosis, inflammatory infiltration, and metabolic disorders4 and will ultimately develop into end-stages.3,6 Despite advances in treatment, the mortality rate associated with HF remains high. Therefore, it is essential to discover specific molecules and signaling pathways that block cardiac hypertrophy to further identify potential strategies for preventing myocardial hypertrophy.
Resolvins are naturally occurring lipid-derived pro-resolving mediators that function in the resolution phase of inflammation.7,8 Immune responsive resolvin D1 (RvD1) is a potent anti-inflammatory molecule with pro-resolving activities.9 17(R)-RvD1 is the epimer of RvD1, and it exhibits equivalent functionality to RvD1.10 It has been demonstrated that RvD1 can improve left ventricular (LV) function post-myocardial infarction (MI), reduce neutrophil recruitment to the LV, decrease collagen deposition, and promote macrophage clearance post-MI.11 Additionally, it has been shown that RvD1 decreases the activation of the nuclear factor κB (NF-κB) pathway in a rat model of the vascular artery and smooth muscle inflammation.12 Our previous study also showed that RvD1 protects against sepsis-induced cardiac injury in mice by attenuating the infiltration of pro-inflammatory M1 macrophages and the expression of inflammatory cytokines in the heart by suppressing the NK-κB and mitogen-activated protein kinase (MAPK) signaling pathways.13 A recent study showed that the administration of RvD1 attenuated angiotensin II (Ang II) induced cardiac hypertrophy and perivascular fibrosis but without exploring the underlying mechanisms.14 Despite there are several studies providing evidence of the role of RvD1 on acute cardiac damage, the role of 17(R)-RvD1 in pressure overload-induced cardiac remodeling is still unclear, prohibiting the understanding of any potential mechanism for the effect of RvD1 on chronic myocardial remodeling. Therefore, in this study, we evaluated the protective role of 17(R)-RvD1 in pressure overload-induced cardiac remodeling and further explored the potential mechanisms.
Materials and Methods
Reagents
17(R)-RvD1 (7,8,17-trihydroxydocosa-4,9,11,13,15,19-hexanoic acid) was purchased from Cayman Chemical (Ann Arbor, MI, United States).
Animals
All of the animal care and experimental procedures conformed to the Guidelines for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH Publication, revised 2011) and the protocol was developed according to ARRIVE guidelines 2.0.15 This study was approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University (Wuhan, China). Male C57BL/6 mice (8 weeks old, weighing 23–25 g) were supplied by Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China). The animals were maintained on a 12/12-light/dark cycle with free access to regular rodent chow and tap water for 1 week before the experiments. The mice were randomly assigned to three groups: the Sham group, the Transverse aortic constriction (TAC) group, and TAC+17(R)-RvD1 group. A minimally invasive transverse aortic constriction (TAC) technique in mice without endotracheal intubation and ventilator ventilation. Briefly, after animal preparation, the skin at the midline position of the neck was opened and the aortic arch was separated subsequently under a dissection microscope. The 27G needle was used as a spacer and then the aortic arch was ligated with 6/0 monofilament suture. The sham group underwent the same procedure but without ligation. After the operation, the mice were placed in a pre-warmed cage for recovery and post-operative monitoring. Buprenorphine (0.05–0.1 mg/kg s.c. every 12 h) was injected after mice regained consciousness to relieve pain. Each mouse received a daily subcutaneous injection of 50,000 U of penicillin for infection prophylaxis from day 1 to day 3 after the operation. The animals were sent back to a 12-h light/dark cycle room after full recovery. In the TAC+17(R)-RvD1 group, 17(R)-RvD1 (Cayman Chemical, Ann Arbor, Michigan) was injected (2 μg/kg, i.p.) once, 30 min prior to the TAC surgery and then administrated once every other day after the surgery until the mice were sacrificed. The dose and dosing schema were selected based on prior published work.13,16 The same volume of saline was injected into the mice in both the Sham group and the TAC group. Four weeks after TAC surgery, echocardiography was performed, and then, the mice were euthanized by carbon dioxide asphyxiation. The heart weight of mice was collected for the ratio of heart weight (HW)/body weight (BW) and HW/tibia length (TL). The left cardiac tissues were collected for detailed analyses.
Echocardiography
Echocardiography was performed on the mice as described previously.17 Briefly, mice were anesthetized using 1.5–2.0% isoflurane. Heart rates and images were acquired using a VINNO 6 device (VINNO Technology, Suzhou, China) with a linear probe working at a frequency of 23 MHz. The left ventricular end-systolic diameter (LVESd) and left ventricular end-diastolic diameter (LVEDd) were measured, parameters were given in millimeters, and the left ventricle ejection fraction (EF) and fractional shortening (FS) were calculated using the LVESd and LVEDd values.
Histological Analysis and Immunohistochemistry
Hearts were isolated and arrested in a 10% KCl solution. After fixation with 4% paraformaldehyde for 5 days, the hearts were embedded in paraffin and sliced into approximately 5-µm-thick sections. Subsequently, the hearts were stained with hematoxylin and eosin (H&E) for histopathology and Picrosirius Red (PSR) for collagen deposition analysis. The sections were then visualized by light microscopy. The cross-sectional area (CSA) from randomly selected fields (40 myocytes for each group) was measured using a quantitative digital image analysis system (Image-Pro Plus, version 6.0). The LV collagen volume fraction was calculated from the PSR slides as the positive area stained by PSR/the total tissue area. For immunohistochemistry, The heart sections were heated using the pressure cooker method for antigen retrieval, incubated with anti-CD68 antibodies (ab955, Abcam), incubated with goat anti-rabbit EnVision™+/horseradish peroxidase (HRP) reagent, and stained using a DAB detection kit. The number of CD68+ cells was quantified using Image-Pro Plus and the percentage of positive cells was calculated as follows: [(positive cells) / (total cell number)] × 100%.
Immunofluorescence
The paraffin sections were generated as described above. Immunofluorescence staining was used to confirm the expression of nucleotide-binding oligomerization domain-like receptor 3 (NLRP3) in the heart. After a 5-min high-pressure antigen retrieval process (sodium citrate buffer, 100×, pH 6.0), the sections were incubated in PBS containing 10% fetal bovine serum for 60 min and subsequently incubated overnight at 4 °C with the anti-NLRP3 primary antibody (GB11300, Servicebio, China). The sections were then washed with PBS and incubated with the appropriate secondary antibodies for 1 hour at 37 °C. Fluorescein isothiocyanate-conjugated wheat germ agglutinin (WGA) staining was utilized for the detection of cell membranes. The nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI). Images were all obtained with a fluorescence microscope (Olympus Dx51) and DP2-BSW software (Version 2.2), and the images were analyzed with Image-Pro Plus (Version 6.0) in a blinded manner.
Western Blotting
Protein was extracted from left ventricle tissue and the protein concentrations of all the samples were normalized before analysis. Procedures were performed as previously described.13 The cell lysates (50 μg) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and subsequently transferred to Immobilon-FL polyvinylidene fluoride (PVDF) membranes (Millipore, Beijing, China). A gel transfer device (Invitrogen) was used, and the membranes were incubated with different primary antibodies, including antibodies against GAPDH (#2118, Cell Signaling), P-Smad2/3 (abs130992, absin), T-Smad2/3 (#8685, Cell Signaling), ASC (SC-514414, Santa Cruz Biotechnology), IL-1β (sc-52012, Santa Cruz Biotechnology), and c-caspase-1 (sc-56036, Santa Cruz Biotechnology). Incubation with the secondary antibodies, either goat anti-rabbit IgG (#7074, Cell Signaling) or goat anti-mouse IgG (#7076; Cell Signaling), was performed for 60 min. The blots were scanned by a two-color infrared imaging system (Odyssey; LICOR) to quantify protein expression. Specific protein expression levels were normalized to those of GAPDH.
Quantitative Real-Time PCR
Total RNA was extracted from frozen heart tissues with TRIzol reagent (Invitrogen Life Technologies, United States). Two micrograms of RNA from each sample were reverse-transcribed into cDNA using oligo(dT) primers and a Transcriptor First Strand cDNA Synthesis kit (Roche, Germany). LightCycler 480 SYBR Green Master Mix (Roche, Germany) was used to quantify the PCR products. The mRNA levels of the target genes were analyzed with the 2−ΔΔCt method, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the internal control. The PCR cycling conditions were as follows: initial activation at 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The sequences of the target gene-specific primers used for qRT-PCR are shown in Table 1.
 |
Table 1 Sequences for the Primers Used in the qRT-PCR Experiments |
Statistical Analysis
All the data were analyzed by SPSS (version 24.0) and are presented as the means ± standard errors of the mean (SEMs). To compare the differences among groups, a one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test was conducted if the dataset exhibited a normal distribution, otherwise Kruskal–Wallis H-test followed by Dunnett’s post hoc test was utilized. A level of P<0.05 was considered to be statistically significant.
Results
17(R)-Resolvin D1 Treatment Attenuates Cardiac Dysfunction and Hypertrophy in Response to Pressure Overload
Figure 1A depicts the experimental protocol. TAC induced cardiac dysfunction and hypertrophy, as illustrated by a significantly higher left ventricular (LV) diameter (TAC vs Sham, LVEDd, 4.56±0.07 mm vs 3.68±0.18 mm; LVESd, 3.86±0.13 mm vs 2.36±0.13 mm) and lower left ventricular contractility (TAC vs Sham, EF%, 35.20±4.79 vs 70.20±2.56; FS%, 14.60±2.38 vs 35.80±2.36), and this effect was reversed by the administration of 17(R)-RvD1 (TAC vs TAC+17(R)-RvD1, LVEDd, 4.56±0.07 mm vs 4.42±0.15 mm; LVESd, 3.86±0.13 mm vs 3.26±0.13 mm; EF%, 35.20±4.79 vs 57.40±2.59; FS%, 14.60±2.38 vs 26.30±1.42) (Figure 1B–E).
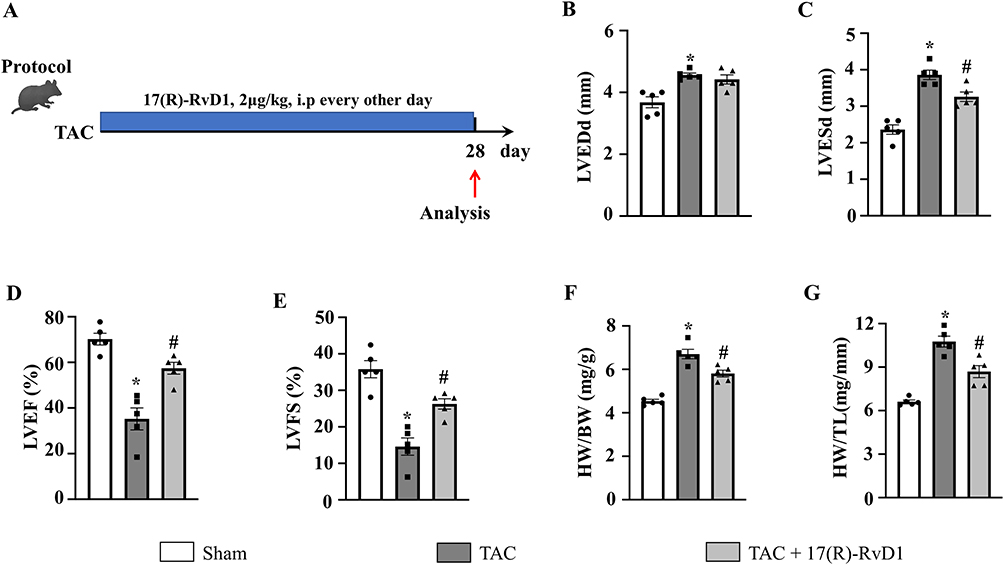 |
Figure 1 17(R)-Resolvin D1 treatment ameliorates pressure overload-induced cardiac dysfunction in mice. Protocol (A). Mice were subjected to either transverse aortic constriction (TAC) or a sham operation. 2 μg/kg 17(R)-RvD1 (Cayman ChemicalAnn, Arbor, Michigan) or saline was injected intraperitoneally before TAC and once every other day for 4 weeks until harvest. The left ventricular end-diastolic diameter (LVEDd) (B) and left ventricular end-systolic diameter (LVESd) (C) were measured by echocardiography in each group (n=5). The left ventricle ejection fraction (LVEF) and fractional shortening (LVFS) were calculated using the LVEDd and LVESd values (D and E). Heart weight (HW)/body weight (BW) (F) and HW/tibia length (TL) (G) ratios in the indicated group (n=5). The data were analyzed using One-way ANOVA followed by Tukey’s post hoc test. A level of P<0.05 was considered to be statistically significant. Data are presented as the mean ± SEMs. *P < 0.05 compared with the Sham group, #P < 0.05 compared with the TAC group. |
Compared to the mice in the Sham group, mice in the TAC group also exhibited increased HW/BW and HW/TL ratios (Figure 1F and G) and an increased cardiomyocyte CSA, as indicated by the morphology of H&E and WGA staining (Figure 2A–D). Consistently, we found an upregulation of hypertrophic markers, including atrial natriuretic peptide (ANP), B-type natriuretic peptide (BNP), and β-myosin heavy chain (β-MHC), induced by TAC surgery (Figure 2E–G). In contrast, 17(R)-RvD1 treatment notably mitigated these changes, except for the changes in the levels of β-MHC. These results suggested that 17(R)-RvD1 treatment attenuated pressure overload-induced cardiac dysfunction and cardiac hypertrophy.
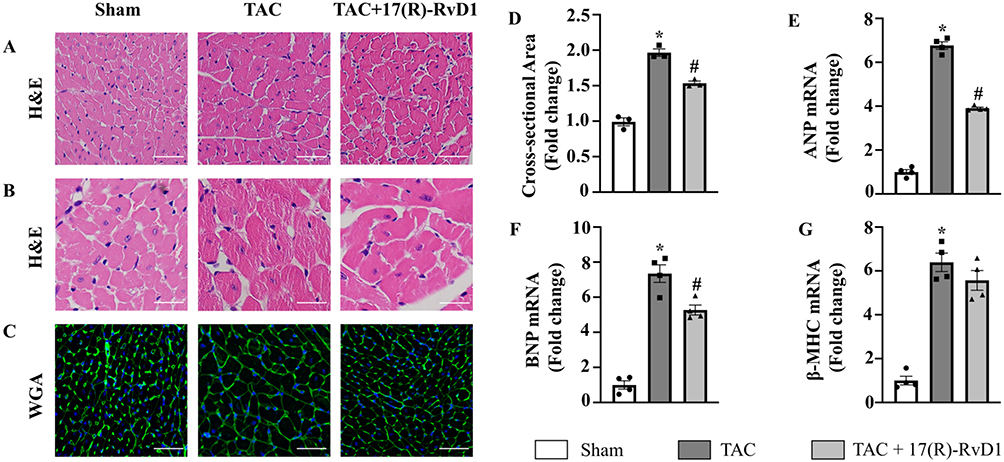 |
Figure 2 17(R)-Resolvin D1 treatment improves cardiac hypertrophy induced by pressure overload. Histological analyses of the hematoxylin-eosin (HE) staining (A and B), wheat germ agglutinin (WGA) staining (C) and quantification of the average cross-sectional areas (D) in each group (n=3) (Scale Bar, 50μm). Expression of transcripts of hypertrophic biomarkers ANP (E), BNP (F), β-MHC (G) in each group, as determined by RT-PCR analysis (n=4). The data were analyzed using One-way ANOVA followed by Tukey’s post hoc test. Data are presented as the mean ± SEMs. *P < 0.05 compared with the Sham group, #P < 0.05 compared with the TAC group. |
17(R)-Resolvin D1 Treatment Improves Cardiac Fibrosis Induced by Pressure Overload
Four weeks after TAC surgery, the mice in the TAC group developed significantly increased interstitial and perivascular fibrosis (Figure 3A–C). The expression of the fibrotic markers Collagen type I alpha 1 chain (Col1a1), Collagen type III alpha 1 chain (Col3a1), transforming growth factor-β1 (TGFβ1), and connective tissue growth factor (CTGF) was markedly increased in the TAC group, while 17(R)-RvD1 treatment significantly attenuated the cardiac fibrosis induced by TAC surgery (Figure 3D–G).
 |
Figure 3 17(R)-Resolvin D1 treatment improves cardiac fibrosis in mice after TAC surgery. PSR staining of histological sections of left ventricles (A) and statistical results of LV collagen volume (B and C) (n=3) (Scale Bar, 50μm). The mRNA expressions of collagen I (D), collagen III (E), transforming growth factor (TGF)-β1 (F), and connective tissue growth factor (CTGF) (G), in the myocardium were obtained from the indicated groups using RT-PCR analysis (n=4). Protein expressions of P-Smad2/3 and T-Smad2/3 were tested by Western blot analysis. GAPDH was used as an internal control (H) and quantification of the p-Smad2/3 level in the indicated groups (n=3) (I). The data were analyzed using One-way ANOVA followed by Tukey’s post hoc test. Data are presented as the mean ± SEMs. *P < 0.05 compared with the Sham group, #P < 0.05 compared with the TAC group. |
To further elucidate the molecular mechanisms by which 17(R)-RvD1 inhibits fibrosis, we examined the Smad cascade. As expected, TAC induced significant activation of reactive Smad 2/3, and the increased phosphorylation of Smad2/3 was suppressed by 17(R)-RvD1 treatment (Figure 3H and I.) Therefore, our results suggested that 17(R)-RvD1 negatively regulates the extent of cardiac fibrosis induced by pressure overload, possibly by blocking the Smad cascade.
17(R)-Resolvin D1 Reduces the Infiltration of Macrophages Induced by Pressure Overload in Cardiac Tissue
To confirm the effects of 17(R)-RvD1 treatment on inflammatory cells, the infiltration of macrophages into the heart was detected by immunohistochemistry. Compared to the Sham group, the infiltration of CD68+ macrophages was significantly increased in the TAC group, while 17(R)-RvD1 treatment obviously attenuated the increased infiltration of CD68+ macrophages. These results indicated that 17(R)-RvD1 attenuated the cardiac inflammatory response by reducing the infiltration of CD68+ macrophages into the heart (Figure 4A and B).
 |
Figure 4 17(R)-Resolvin D1 reduces inflammatory cytokine production and the infiltration of inflammatory cells induced by pressure overload in cardiac tissue. Infiltration of CD68+ macrophages (A and B) in the heart in different groups (n=3) (Scale Bar, 40μm), the black arrowheads indicate CD68-positive cells. The mRNA levels of pro-inflammatory cytokines tumor necrosis factor-α (TNF-α) (C), interleukin (IL)-1β (D), and IL-6 (E) were assessed in cardiac tissue (n=4). The data were analyzed using One-way ANOVA followed by Tukey’s post hoc test. Data are presented as the mean ± SEMs. *P < 0.05 compared with the Sham group, #P < 0.05 compared with the TAC group. |
17(R)-Resolvin D1 Decreases the Inflammatory Cytokine Production Induced by Pressure Overload in Cardiac Tissue
We further analyzed the mRNA expression of inflammatory cytokines, including TNF-α, IL-1β, and IL-6, in the heart tissues of the three groups. Compared to that in the Sham group, increased expression of inflammatory cytokines was observed in the TAC group. However, the increased expression of inflammatory cytokines was notably attenuated by treatment with 17(R)-RvD1 (Figure 4C–E).
NLRP3 Signaling Plays an Important Role in the Beneficial Effect of 17(R)-RvD1 on Pressure Overload-Induced Hypertrophy
Previous studies have suggested that the NLRP3 inflammasome may play a crucial role in regulating inflammatory cytokines.18 We further examined the activation of the NLRP3 inflammasome. As shown in Figure 5A, the staining results showed that NLRP3 was mainly expressed in the cytoplasm of cardiomyocytes, and the expression level of NLRP3 was upregulated 4 weeks after TAC surgery, which was significantly downregulated after 17(R)-RvD1 treatment.
 |
Figure 5 17(R)-Resolvin D1 treatment suppressed the activation of the NLRP3 signalling pathway in the heart. NLRP3 was assessed by immunofluorescence in the LV tissue of each group (n=3) (Scale Bar, 40μm) (A). Protein expressions of apoptosis-associated speck-like protein containing a CARD (ASC), c-caspase-1, procaspase-1, IL-1β, and pro-IL-1β in myocardial tissue were tested by Western blot analysis (B), GAPDH was used as internal control and quantification of the ASC, c-caspase-1, IL-1β level in the indicated groups were analyzed (C–E) (n=4). The data were analyzed using One-way ANOVA followed by Tukey’s post hoc test. Data are presented as the mean ± SEMs. *P < 0.05 compared with the Sham group, #P < 0.05 compared with the TAC group. |
To further investigate the molecular mechanism underlying the protective effects of 17(R)-RvD1 treatment on myocardial remodeling, we evaluated the activation of the NLRP3 pathway via Western blotting. As expected, our results revealed that TAC surgery dramatically increased the expression levels of apoptosis-associated speck-like protein containing a CARD (ASC), c-caspase-1, and IL-1β in myocardial tissue, and 17(R)-RvD1 treatment markedly reversed these changes (Figure 5B–E).
Discussion
In this study, we clearly revealed that 17(R)-RvD1 could attenuate pressure overload-induced cardiac hypertrophy and fibrosis. Mechanistically, 17(R)-RvD1 inhibited pressure overload-induced macrophage infiltration and inflammatory cytokine secretion, and NLRP3 signaling may be responsible for the attenuation of inflammation and cardiac hypertrophy.
Cardiac hypertrophy is a complex, multi-factor, dynamic process. The main pathological changes of myocardial hypertrophy include increased myocardial cell volume, increased intracellular protein synthesis, and increased extracellular matrix and collagen deposition.4,19 Similarly, fibroblast proliferation and increased intracellular matrix protein synthesis are also partly responsible for the deposition of extracellular matrix and collagen and the increase in ventricular wall stiffness, which contribute to cardiac dysfunction in many cardiac pathophysiologic conditions.20,21 In addition to the role of cardiomyocytes and fibroblasts, inflammation is also associated with the occurrence and development of cardiac hypertrophy. Studies have shown that the administration of exogenous TNF-α, IL-1β, and IL-6 or the overexpression of pro-inflammatory cytokines can promote myocardial hypertrophy and fibrosis, causing cardiac insufficiency.22–24 Consistently, the absence of TNF-α and IL-6 can inhibit myocardial hypertrophy and interstitial fibrosis, thereby improving the cardiac dysfunction induced by pressure overload.25–27 In addition, as the main source of inflammatory factors, immune cells are also involved in the progression of cardiac hypertrophy.28,29 Therefore, the inflammation response was a promising target to improve myocardial remodeling.
Specialized pro-resolving lipid mediators (SPMs) are a series of small molecular substances that were discovered in recent years and exert significant anti-inflammatory and pro-resolving effects;30 these molecules are metabolized and synthesized by omega-3 polyunsaturated fatty acids in the body.31 Resolvin D1 (RvD1), a bioactive lipid mediator that is synthesized from docosahexaenoic acid, is a member of the SPM family, and it has been reported to reduce the production of pro-inflammatory cytokines and the infiltration of immune cells in inflamed tissues and exert protective effects in ischaemic injury models, asthma models, and acute lung injury models.11,30,32
We are aware that there are several relevant studies that analyze the protective effect of RvD1 on cardiac remodeling in different models. One study suggested RvD1 could prevent cardiac remodeling via reducing cardiac hypertrophy, cardiac fibrosis, and the serum levels of Interferon γ (IFN-γ) and IL-1β, with the participation of N-formyl peptide receptor 2 (FPR2),33 but limited to a murine model of early chronic Chagas disease caused by the protozoan Trypanosoma cruzi. Another study reported that RvD1 is capable of attenuating Ang II–induced chronic cardiac inflammation and cardiac hypertrophy, but without exploring the underlying mechanisms.14 In addition, RvD1 was reported to prevent atrial fibrillation-promoting right atrial remodeling in an animal model of right heart disease, suggesting the potential mechanism may be partially associated with suppressing inflammatory changes and fibrotic/electrical remodeling.34 And the in vitro study showed that RvD1 could prevent all Ang II effects in rat cardiac fibroblasts via reducing the intracellular Ca2+ increase.35 However, the research content of the above two studies is fundamentally different from that of this study. One study found that RvD1 could stimulate the pro-resolving marker Arginase 1 (Arg-1) in both WT and 12/15-lipoxygenase (LOX)−/− macrophages and diminished expression of cyclooxygenase-2 (COX-2) but upregulated 5-LOX expression in 12/15-LOX−/− cardiac fibroblast, suggesting RvD1 are responsive in determining macrophages phenotype and cardiac fibroblast plasticity,36 consistent with our result. As expected, this study shows that TAC surgery exacerbated cardiac hypertrophy and fibrosis, increased the infiltration of CD68+ macrophages and increased the expression of pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6; however, 17(R)-RvD1 treatment attenuated pressure overload-induced cardiac hypertrophy, fibrosis, and dysfunction. Moreover, 17(R)-RvD1 reduced macrophage infiltration and attenuated the increase in pro-inflammatory cytokine production, which is consistent with previous works.13,37
The NLRP3 signaling pathway is generally considered an important driving force of the activation of the cardiac inflammatory response.38–40 Studies suggest that the NLRP3 inflammasome pathway is involved in the pro-inflammatory response of pressure load-induced myocardial remodeling.18 NLRP3 inflammasome is upregulated in pressure overload-induced myocardial remodeling, the inhibition of the NLRP3 signaling pathway could attenuate inflammatory lesions, decrease fibroblast proliferation and collagen deposition, and improve cardiac mechanical function.41 Another study suggests that caspase-1 inhibitor could significantly inhibit the increase in mean cardiomyocyte surface area and reduce the fibrotic area in the myocardium after TAC.42 In addition, our recent work found that the administration of selective NLRP3 inhibitor MCC950 from two weeks after TAC surgery could reverse pressure overload-induced pathological cardiac remodeling, indicating the clinical therapeutic effects of NLRP3 inhibitor.43 In this study, our results suggest that 17(R)-RvD1 treatment could inhibit the activation of NLRP3 signaling pathways, which may contribute to the cardio-protective effects of 17(R)-RvD1.
The possible mechanisms underlying the inhibition of NLRP3 inflammasome 17(R)-RvD1 were not clarified, which is a limitation of this study. One possible mechanism is that RvD1 interferes with the production of mitochondrial reactive oxygen species (ROS) in cardiomyocytes. Mitochondrial ROS are involved in the activation of the NLRP3 signaling pathway, and RvD1 could decrease the levels of ROS via activation of the Nrf2/Keap1 pathway, which indicates that RvD1 may participate in the regulation of the NLRP3 signaling pathways by mediating the reduction in the levels of mitochondrial ROS.44,45 In addition, we cannot exclude the fact that other mechanisms are involved in the 17(R)-RvD1-mediated attenuation of pressure overload-induced cardiac hypertrophy. More studies are required to clarify the detailed molecular relationship between pro-resolving lipid mediators and NLRP3 inflammasome.
Conclusion
In conclusion, we demonstrated that 17(R)-RvD1 could attenuate pressure overload-induced cardiac hypertrophy and fibrosis, and the possible mechanism may be associated with the inhibition of NLRP3 inflammasome. 17(R)-RvD1 may serve as a potential drug for the treatment of cardiac remodeling.
Data Sharing Statement
The datasets supporting the conclusions of this article are included within the article.
Ethics Approval and Consent to Participate
This study was approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University (Wuhan, China).
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by grants from National Natural Science Foundation of China (NO. 82270454 and NO.82070436) and National College Students’ innovation and entrepreneurship training (NO.202110486083).
Disclosure
The authors declare no conflict of interest in relation to this paper. Each author takes responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation.
References
1. Tanai E, Frantz S. Pathophysiology of heart failure. Compr Physiol. 2015;6(1):187–214. doi:10.1002/cphy.c140055
2. Gibb AA, Hill BG. Metabolic coordination of physiological and pathological cardiac remodeling. Circ Res. 2018;123(1):107–128. doi:10.1161/CIRCRESAHA.118.312017
3. Tham YK, Bernardo BC, Ooi JYY, et al. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets. Arch Toxicol. 2015;89(9):1401–1438. doi:10.1007/s00204-015-1477-x
4. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15(7):387–407. doi:10.1038/s41569-018-0007-y
5. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245–262. doi:10.1016/j.yjmcc.2016.06.001
6. Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011;8(1):30–41. doi:10.1038/nrcardio.2010.165
7. Krishnamoorthy S, Recchiuti A, Chiang N, et al. Resolvin D1 receptor stereoselectivity and regulation of inflammation and proresolving microRNAs. Am J Pathol. 2012;180(5):2018–2027. doi:10.1016/j.ajpath.2012.01.028
8. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J Clin Invest. 2018;128(7):2657–2669. doi:10.1172/JCI97943
9. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8(5):349–361. doi:10.1038/nri2294
10. Schmitz Nunes V, Rogerio AP, Abrahao O. Insights into the activation mechanism of the ALX/FPR2 receptor. J Phys Chem Lett. 2020;11(21):8952–8957. doi:10.1021/acs.jpclett.0c02052
11. Kain V, Ingle KA, Colas RA, et al. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J Mol Cell Cardiol. 2015;84:24–35. doi:10.1016/j.yjmcc.2015.04.003
12. Wu B, Mottola G, Chatterjee A, et al. Perivascular delivery of resolvin D1 inhibits neointimal hyperplasia in a rat model of arterial injury. J Vasc Surg. 2017;65(1):207–217 e3. doi:10.1016/j.jvs.2016.01.030
13. Wang M, Liu M, Zhang J, et al. Resolvin D1 protects against sepsis-induced cardiac injury in mice. Biofactors. 2020;46(5):766–776. doi:10.1002/biof.1668
14. Olivares-Silva F, De Gregorio N, Espitia-Corredor J, et al. Resolvin-D1 attenuation of angiotensin II-induced cardiac inflammation in mice is associated with prevention of cardiac remodeling and hypertension. Biochim Biophys Acta Mol Basis Dis. 2021;1867(12):166241. doi:10.1016/j.bbadis.2021.166241
15. Percie du Sert N, Hurst V, Ahluwalia A, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol. 2020;18(7):e3000410. doi:10.1371/journal.pbio.3000410
16. Hellmann J, Tang Y, Kosuri M, et al. Resolvin D1 decreases adipose tissue macrophage accumulation and improves insulin sensitivity in obese-diabetic mice. FASEB J. 2011;25(7):2399–2407. doi:10.1096/fj.10-178657
17. Wang M, Zhang J, Yin Z, et al. Microglia-mediated neuroimmune response regulates cardiac remodeling after myocardial infarction. J Am Heart Assoc. 2023;12(12):e029053. doi:10.1161/jaha.122.029053
18. Suetomi T, Willeford A, Brand CS, et al. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca 2+ /calmodulin-dependent protein kinase II δ signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation. 2018;138(22):2530–2544. doi:10.1161/CIRCULATIONAHA.118.034621
19. Zheng L, Han P, Liu J, et al. Role of copper in regression of cardiac hypertrophy. Pharmacol Ther. 2015;148:66–84. doi:10.1016/j.pharmthera.2014.11.014
20. MacKenna D, Summerour SR, Villarreal FJ. Role of mechanical factors in modulating cardiac fibroblast function and extracellular matrix synthesis. Cardiovasc Res. 2000;46(2):257–263. doi:10.1016/s0008-6363(00)00030-4
21. Ma ZG, Yuan Y-P, Wu H-M, et al. Cardiac fibrosis: new insights into the pathogenesis. Int J Biol Sci. 2018;14(12):1645–1657. doi:10.7150/ijbs.28103
22. Kubota T, McTiernan CF, Frye CS, et al. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ Res. 1997;81(4):627–635. doi:10.1161/01.res.81.4.627
23. Van Tassell BW, Arena RA, Toldo S, et al. Enhanced interleukin-1 activity contributes to exercise intolerance in patients with systolic heart failure. PLoS One. 2012;7(3):e33438. doi:10.1371/journal.pone.0033438
24. Melendez GC, McLarty JL, Levick SP, et al. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. 2010;56(2):225–231. doi:10.1161/HYPERTENSIONAHA.109.148635
25. Sun M, Chen M, Dawood F, et al. Tumor necrosis factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation. 2007;115(11):1398–1407. doi:10.1161/CIRCULATIONAHA.106.643585
26. Zhao L, Cheng G, Jin R, et al. Deletion of interleukin-6 attenuates pressure overload-induced left ventricular hypertrophy and dysfunction. Circ Res. 2016;118(12):1918–1929. doi:10.1161/circresaha.116.308688
27. Lai NC, Gao MH, Tang E, et al. Pressure overload-induced cardiac remodeling and dysfunction in the absence of interleukin 6 in mice. Lab Invest. 2012;92(11):1518–1526. doi:10.1038/labinvest.2012.97
28. Frieler RA, Mortensen RM. Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation. 2015;131(11):1019–1030. doi:10.1161/CIRCULATIONAHA.114.008788
29. Wang Z, Xu Y, Wang M, et al. TRPA1 inhibition ameliorates pressure overload-induced cardiac hypertrophy and fibrosis in mice. EBioMedicine. 2018;36:54–62. doi:10.1016/j.ebiom.2018.08.022
30. Anderson P, Delgado M. Endogenous anti-inflammatory neuropeptides and pro-resolving lipid mediators: a new therapeutic approach for immune disorders. J Cell Mol Med. 2008;12(5B):1830–1847. doi:10.1111/j.1582-4934.2008.00387.x
31. Fredman G, Spite M. Specialized pro-resolving mediators in cardiovascular diseases. Mol Aspects Med. 2017;58:65–71. doi:10.1016/j.mam.2017.02.003
32. Levy BD. Resolvin D1 and resolvin E1 promote the resolution of allergic airway inflammation via shared and distinct molecular counter-regulatory pathways. Front Immunol. 2012;3:390. doi:10.3389/fimmu.2012.00390
33. Carrillo I, Rabelo RAN, Barbosa C, et al. Aspirin-triggered resolvin D1 reduces parasitic cardiac load by decreasing inflammation in a murine model of early chronic Chagas disease. PLoS Negl Trop Dis. 2021;15(11):e0009978. doi:10.1371/journal.pntd.0009978
34. Hiram R, Xiong F, Naud P, et al. The inflammation-resolution promoting molecule resolvin-D1 prevents atrial proarrhythmic remodelling in experimental right heart disease. Cardiovasc Res. 2021;117(7):1776–1789. doi:10.1093/cvr/cvaa186
35. Salas-Hernández A, Ruz-Cortés F, Bruggendieck F, et al. Resolvin D1 reduces expression and secretion of cytokines and monocyte adhesion triggered by angiotensin II, in rat cardiac fibroblasts. Biomed Pharmacother. 2021;141:111947. doi:10.1016/j.biopha.2021.111947
36. Kain V, Halade GV. Immune responsive resolvin D1 programs peritoneal macrophages and cardiac fibroblast phenotypes in diversified metabolic microenvironment. J Cell Physiol. 2019;234(4):3910–3920. doi:10.1002/jcp.27165
37. Yang D, Liu H-Q, Liu F-Y, et al. Critical roles of macrophages in pressure overload-induced cardiac remodeling. J Mol Med. 2021;99(1):33–46. doi:10.1007/s00109-020-02002-w
38. Gao R, Shi H, Chang S, et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces myocardial fibrosis and improves cardiac remodeling in a mouse model of myocardial infarction. Int Immunopharmacol. 2019;74:105575. doi:10.1016/j.intimp.2019.04.022
39. Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361. doi:10.1038/nature08938
40. Toldo S, Marchetti C, Mauro AG, et al. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int J Cardiol. 2016;209:215–220. doi:10.1016/j.ijcard.2016.02.043
41. Li R, Lu K, Wang Y, et al. Triptolide attenuates pressure overload-induced myocardial remodeling in mice via the inhibition of NLRP3 inflammasome expression. Biochem Biophys Res Commun. 2017;485(1):69–75. doi:10.1016/j.bbrc.2017.02.021
42. Yue R, Zheng Z, Luo Y, et al. NLRP3-mediated pyroptosis aggravates pressure overload-induced cardiac hypertrophy, fibrosis, and dysfunction in mice: cardioprotective role of irisin. Cell Death Discov. 2021;7(1):50. doi:10.1038/s41420-021-00434-y
43. Zhao M, Zhang J, Xu Y, et al. Selective inhibition of NLRP3 inflammasome reverses pressure overload-induced pathological cardiac remodeling by attenuating hypertrophy, fibrosis, and inflammation. Int Immunopharmacol. 2021;99:108046. doi:10.1016/j.intimp.2021.108046
44. Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10(3):210–215. doi:10.1038/nri2725
45. Posso SV, Quesnot N, Moraes JA, et al. AT-RVD1 repairs mouse lung after cigarette smoke-induced emphysema via downregulation of oxidative stress by NRF2/KEAP1 pathway. Int Immunopharmacol. 2018;56:330–338. doi:10.1016/j.intimp.2018.01.045
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.