")
Back to Journals » Cancer Management and Research » Volume 12
The Anti-Apoptotic Role of EBV-LMP1 in Lymphoma Cells
Authors Zeng M, Chen Y, Jia X, Liu Y
Received 29 April 2020
Accepted for publication 18 August 2020
Published 22 September 2020 Volume 2020:12 Pages 8801—8811
DOI https://doi.org/10.2147/CMAR.S260583
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Mei Zeng,1,* Yuhua Chen,2,* Xintao Jia,2 Yan Liu2
1Pathology Teaching and Research Section, Xiangyang Polytechnic, Xiangyang 441021, Hubei, People’s Republic of China; 2Department of Pathology, Xiangyang Central Hospital, Affiliated Hospital of Hubei University of Arts and Science, Xiangyang 441021, Hubei, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xintao Jia; Yan Liu
Department of Pathology, Xiangyang Central Hospital, Affiliated Hospital of Hubei University of Arts and Science, No. 136, Jingzhou Street, Xiangcheng County, Xiangyang 441021, Hubei, People’s Republic of China
Tel/Fax +86-0710-3524082
Email [email protected]; [email protected]
Background: Epstein-Barr virus (EBV) has been indicated in the development of some tumors, including lymphoma. However, the potential role of latent membrane protein 1 (LMP1) encoded by EBV in the tumorigenesis of lymphoma remains debated. Herein, we examined the function of LMP1 in lymphoma.
Methods: The expression of LMP1 was downregulated or upregulated in EBV negative cell line SNT-8 and positive cell line KHYG-1, respectively. Subsequently, the cell viability, apoptosis, as well as the expression patterns of p53, mouse double minute 2 (MDM2), B-cell CLL/lymphoma 2 (Bcl-2) and NF-κB were evaluated. Next, the binding relationship between MDM2 and p53 along with p53 ubiquitination in cells was tested by Western blot and co-immunoprecipitation. Finally, the effects of LMP1 on lymphoma cell growth through p53, Bcl-2 and NF-κB pathways were verified by functional rescue experiments.
Results: Overexpression of LMP1 promoted KHYG-1 cell growth and inhibited cell apoptosis. Moreover, LMP1 upregulation significantly enhanced the activation of NF-κB pathway, thus increasing MDM2 binding to p53, leading to p53 ubiquitination and degradation as well as Bcl-2 expression enhancement. Further inhibition of the NF-κB pathway or Bcl-2 expression significantly weakened the promotive role of LMP1 in the growth of KHYG-1 cells.
Conclusion: EBV-LMP1 promoted the p53 ubiquitination and degradation by activating NF-κB signaling pathway and the following binding of MDM2 and p53 in cells to enhance Bcl-2 expression, thus promoting the growth of lymphoma cells and inhibiting cell apoptosis.
Keywords: EBV-LMP1, p53, MDM2, ubiquitination and degradation, lymphadenoma, apoptosis
Introduction
In adolescents, lymphoma is among the most frequent-occurring tumors (20%), and cases of Hodgkin lymphoma was almost as twice as non-Hodgkin lymphoma by 2020 in the United states.1 Epstein-Barr virus (EBV) comprised of a linear double-stranded DNA molecule that encodes over 85 genes, is highly associated with different subtypes of lymphoma.2 Initially discovered due to its correlation with Burkitt lymphoma, the pathogenesis of EBV-related lymphomas is considered as an intricate interplay between different viral gene expression profiles and cellular genetic changes.3 Even though chemotherapy is effective enough for EBV-related malignancies, up to half of survivors of Hodgkin lymphoma die from complications caused by chemotherapy, secondary malignancies, or relapse.4 Therefore, it is of great significance to investigate the molecular basis of the progression of lymphoma, which may be beneficial to develop better strategical options to prevent and treat lymphoma.
There are three latent membrane proteins (LMP)-1-2A that are essential for the survival and transformation of infected cells into eternally proliferating cells.5 LMP1, a 60–66 kDa fully phosphorylated transmembrane glycoprotein consists of 386 amino acid (aa) residues.6 The cytoplasmic fragment of LMP-1 contains three important domains which induce several pathways involving the nuclear factor κB (NF-κB) pathway.7 Moreover, delivery of LMP1 recombinant constructs into human large cell lung carcinoma (with p53 deleted gene) has proposed that the carboxyl-terminus activating regions of LMP1 repressed the transactivation of p53, and p53-dependent DNA repair and transcription was inhibited through the NF-κB pathway.8 Enhanced interactions with its regulator murine double minute (MDM2) is one of the possible mechanisms whereby the functions of the p53 protein can be altered in tumor cells.9 Meanwhile, the decline in p53 expression is accomplished by MDM2-mediated p53 ubiquitination as well as proteasomal degradation via the ubiquitin proteolytic system and through MDM2-mediated suppression of p53 transactivation.10 Intrinsic apoptosis is a kind of response to intracellular cell death stimuli, which is modulated by the connection between the B-cell lymphoma 2 (Bcl-2) family and the membrane interactions.11 As a consequence, we set to evaluate the function of LMP1 in mediating cell apoptosis in lymphoma in the present study. Based on the results, the detailed mechanism was further examined. Moreover, the effect of LMP1 on the activation of NF-κB pathway and MDM2-mediated p53 ubiquitination was explored. The purpose of the current work is to clarify the mechanistic connection between LMP1 and p53 ubiquitination in EBV-positive or EBV-negative lymphoma cells as well as their implications in apoptosis.
Materials and Methods
Cell Culture and Treatment
EBV positive cell line SNT-8 (RRID: CVCL_A677) and EBV negative cell line KHYG-1 (RRID: CVCL_2976) were purchased from Cell Bank (https://cellbank.nibiohn.go.jp/english/). SNT-8 and KHYG-1 cells were cultured in Roswell Park Memorial Institute-1640 medium supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL), streptomycin (100 g/mL) and recombinant human interleukin-2 (IL-2, 100 U/mL).
SNT-8 cells in good growth condition were delivered with short hairpin RNA (shRNA) targeting EBV-LMP1 and the corresponding scramble shRNA (GenePharma, Shanghai, China). KHYG-1 cells in good growth conditions were infected with EBV-LMP1 lentiviral overexpression vector (GenePharma, Shanghai, China) with empty vector Lv-NC as a control. After cell transfection or infection, Western blot and RT-qPCR were used to detect LMP1 expression in cells to determine transfection and infection efficiency. Cells were collected after 48 h of transfection or 24 h of infection for subsequent experiments.
5-Ethynyl-2ʹ-Deoxyuridine (EdU) Assay
EdU labelling was conducted using the Click-iTEdU kit (Invitrogen Inc., Carlsbad, CA, USA) according to the manufacturer’s instructions. Briefly, cells were exposed to 50 μM EdU for a period of 2 h and fixed with 4% formaldehyde. Cells were then treated with 2 mg/mL glycine to neutralize formaldehyde and permeated with 0.5% Triton X-100. Lastly, the cells were reacted with 100 μL 1 × Apollo reaction mixture for 30 min and then with 100 μL 4ʹ,6-diamidino-2-phenylindole (5 μg/mL). Images were obtained using an Olympus IX-71 inverted microscope (Tokyo, Japan). The percentage of EdU positive cells was calculated by dividing the number of EdU positive cells by the number of cells stained by Hoechst.
Cell Proliferation Assay
KHYG-1 and SNT-8 cells were plated into a 24-well plate at 2 × 104 cells/well and incubated at 37°C in a 5% CO2 incubator for 24 h. The cells were then detached with 0.25% ethylenediaminetetracetic acid-trypsin (Solarbio, Beijing, China), centrifuged at 1000 rpm for 5 min and then resuspended with F12 medium. Next, the carboxyfluorescein succinimidyl ester (CFSE) solution (Thermo, shanghai, china) was added for a 15-min incubation at 37°C. After the incubation, FBS was used to terminate the reaction. The cells were resuspended again with F12 medium. Finally, cell viability was detected by a flow cytometer (Beckman Coulter, Inc., Brea, CA, USA).
Colony Formation Assay
SNT-8 and KHYG-1 cells in logarithmic growth period were plated in a six-well plate at 200 cells/well. After 14 days, colonies formed were fixing with 1% paraformaldehyde and stained with 1% crystal violet (Sigma-Aldrich Chemical Company, St Louis, MO, USA).
Cell Cycle Detection
KHYG-1 and SNT-8 cells were fixed with 70% ice ethanol and placed overnight in a 4°C refrigerator. After washing and centrifugation, the cells were resuspended in 500 mL FxCycle propidium iodide (PI)/RNase staining solution, then filtered using nylon meshes and incubated at room temperature without light for 30 min. The cell cycle distribution was detected using a FACS-Aria flow cytometer (BD Biosciences, San Jose, CA, USA).
Apoptosis Detection
To measure cell cycle distribution, SNT-8 and KHYG-1 cells were incubated for 48 h, then fixed with 75% ethanol (Sigma), and incubated with RNase A and PI solution for a period of 30 min at 37°C. To perform apoptosis analysis, cells were resuspended in binding buffer after 48 h of culture and then stained with Annexin V-fluorescein isothiocyanate (FITC) and PI (Beyotime, Shanghai, China) in the dark.
Western Blot
Total protein was extracted from cultured cells using radioimmunoprecipitation assay lysis buffer containing protease inhibitors, and protein concentrations were determined by bicinchoninic acid assay protein assay (Beyotime, Shanghai, China). A total of 20 μg protein lysates was subjected to a sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrically transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). The membranes were then sealed with 5% skim milk for 2 h, incubated overnight with specific primary antibodies (Table 1) and with secondary antibodies for 2 h. Visualization of proteins was performed using the enhanced chemiluminescence solution (Beyotime) with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the normalizer.
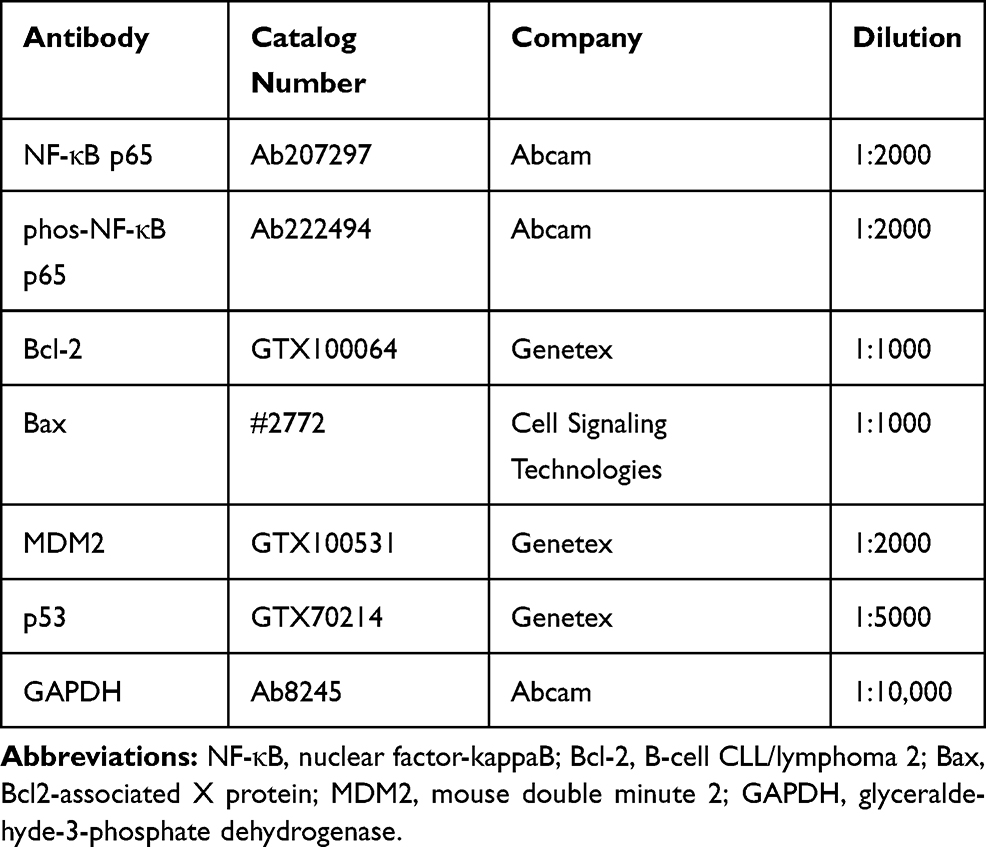 |
Table 1 The Primary Antibodies Used in the Western Blot |
Co-Immunoprecipitation (Co-IP)
Co-IP was performed using the Co-IP kit (Wanlei, Shenyang, Liaoning, China). In short, cells in the petri dishes were lysed in 500 μL lysis buffer. The proteins were isolated from SNT-8 and KHYG-1 cells, incubated at 4°C for a period of 30 min and centrifuged to separate the lysate. The total protein was quantified and diluted to 1 μg/μL. A total of 500 μL lysate was incubated with antibody (2 μg) at 4°C for 12 h with 60 μL protein A beads at room temperature for 2 h. Finally, the target antigen was washed with loading buffer and prepared for Western blot analysis.
p53 Ubiquitination Detection
KHYG-1 and SNT-8 cells were transfected with pCDNA3.1–3 × HA-UB expression vectors (Miaolingbio, Wuhan, Hubei, China). After 24 h, the cells were treated with 10 mM MG132 for 3 h and then lysed. Ubiquitinated p53 was immunoprecipitated with the antibody against p53 and analyzed by Western blot with anti-HA.
Animal Experiments
All animals were maintained and used according to strict guidelines established by the Animal Ethics Committee of Xiangyang Central Hospital (Approval number: XYCH/IAEC/2018/39). Twenty BALB/c nude mice (4 weeks old, male) from Shanghai Animal Experiment Center (Shanghai, China) were allowed to acclimatize for 1 week in the animal facility before any intervention was initiated. KHYG-1 cells expressing LMP1 (Lv-LMP1), control (Lv-NC) or SNT-9 cells stably expressing shLMP1 or Scramble shRNA (2 × 106 cells) were subcutaneously injected into the right armpit of nude mice (n = 5). The tumor volume was monitored every 7 days and calculated using the following formula: volume (mm3) = length × width2 × 0.5. Five weeks after injection, mice were euthanized, and tumors were collected for weight and immunohistochemical analysis.
Immunohistochemical Analysis
The sections were fixed at room temperature with 4% paraformaldehyde (30,525–89-4, Aladdin, Shanghai, China) for 48 h, embedded in paraffin, and cut into 4-μm thickness sections. Sections were then dewaxed in gradient concentrations of xylene (1330–20-7, Aladdin) and ethanol. Afterwards, the sections were heated in a citrate buffer (10 mM, pH = 6.0) below boiling temperature for a period of 10 min for antigen retrieval. Next, the sections were probed with the primary antibody against Ki-67 (AF1738, 1:400, Beyotime) and diluted in immunostaining blocking buffer (P0102, Beyotime) for 4°C for 2 h. Subsequently, the sections were washed two times at ambient temperature with washing buffer (P0106, Beyotime) for 15 min and subjected to a 30-min incubation with SignalStain® Boost immunohistochemistry detection reagent (horseradish peroxidase-labelled, 8114S, Cell Signaling Technologies, Beverly, MA, USA) at room temperature. Finally, sections were observed under an inverted microscope (Olympus IX-71, Tokyo, Japan).
Statistical Analysis
Statistical analysis was executed by the SPSS version 22.0 (SPSS Inc, Chicago, IL, USA). All values are displayed as mean values ± standard deviation (SD) of three individual experiments. Statistical differences were determined using unpaired t-test, one-way or two-way analysis of variance (ANOVA), followed by Tukey’s multiple comparisons test. All tests were two-tailed, and statistical significance was considered to be reached when p < 0.05.
Results
LMP1 Promotes Lymphoma Cell Proliferation and Inhibits Apoptosis
To expound the specific role of LMP1 in lymphoma cells, we transfected SNT-8 cells with shRNAs targeting LMP1 (shLMP1) or Scramble (Scr) and infected KHYG-1 cells with Lv-LMP1 (LMP1) or Lv-NC. EdU staining revealed that the number of EdU positive cells was increased significantly after overexpression of LMP1 (p < 0.05, Figure 1A). Moreover, after CFSE staining, the cell proliferation was detected by flow cytometric analysis, and it was also revealed that increasing the expression of LMP1 in cells significantly promoted cell proliferation (p < 0.05, Figure 1B). The colony formation assay was applied to detect the number of colonies formed by cells, and the number of colonies formed was increased significantly by overexpression of LMP1 in KHYG-1 cells (p < 0.01, Figure 1C). We then used flow cytometry to detect cell cycle distribution and apoptosis and found that after overexpression of LMP1, cell cycle progression was significantly promoted (p < 0.05, Figure 1D), and the number of apoptosis was decreased significantly (p < 0.05, Figure 1E). Nevertheless, knocking down the expression of LMP1 in EBV positive SNT-8 cells led to declines in cell proliferation, cell cycle arrest in G0/G1 stages, and also promotion in apoptosis level (all p < 0.05, Figure 1A–E).
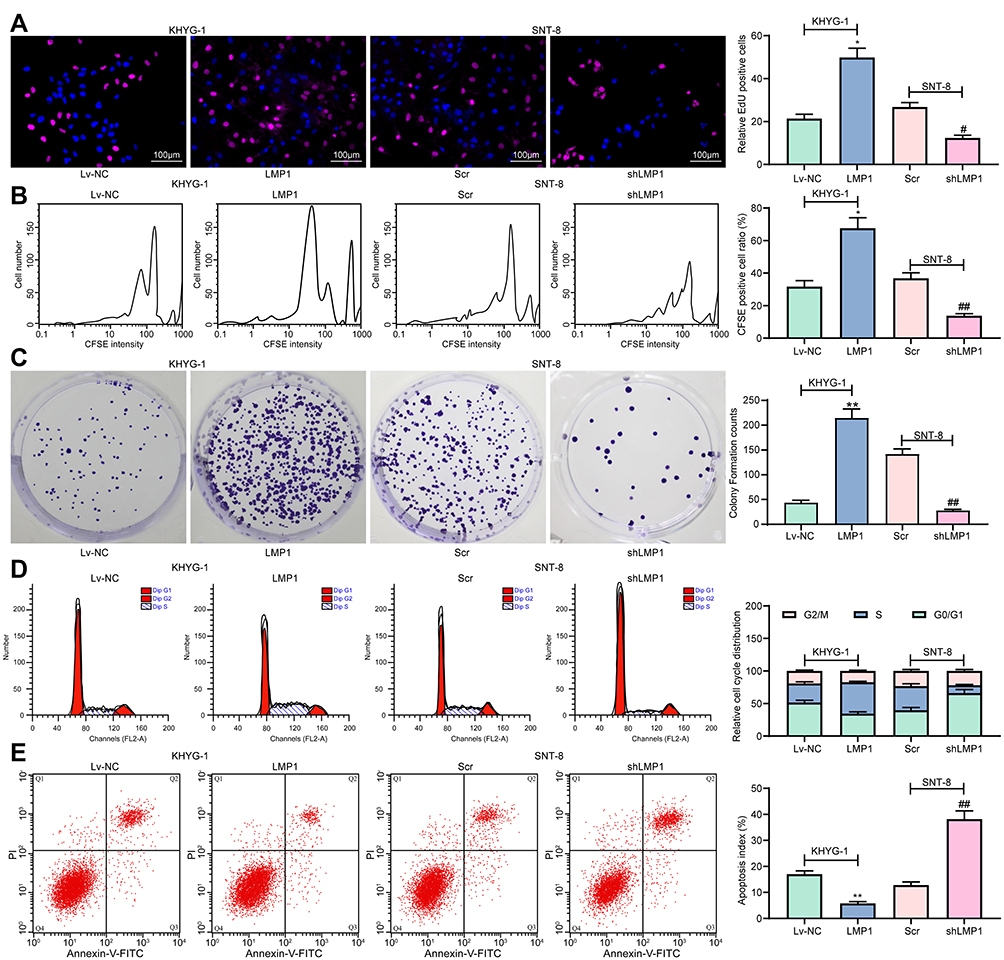 |
Figure 1 LMP1 promotes lymphoma cell proliferation and inhibits apoptosis. (A) The proliferation ability of KHYG-1 and SNT-8 cells evaluated by EdU staining; (B) Cell viability after CFSE staining determined by flow cytometry; (C) The number of colonies formed by cells detected by the colony formation assay; (D) Cell cycle distribution detected by flow cytometry; (E) Cell apoptosis level assessed by flow cytometry. The data was performed as means ± SD from three independent experiments. One-way ANOVA was applied to compare the mean of samples among multiple groups, followed by Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01 vs. the Lv-NC group; #p < 0.05, ##p < 0.01 vs. the Scr group. |
LMP1 Enhances NF-κB Activation to Facilitate MDM2-Mediated p53 Protein Degradation
To determine the effect of LMP1 on lymphoma cells, we used Western blot to detect phosphorylation levels of NF-κB signaling pathway in KHYG-1 and SNT-8 cells. Overexpression of LMP1 significantly promoted phosphorylation levels of NF-κB p65, decreased p53 and Bax expression as well as the ratio of Bax/Bcl-2, and elevated MDM2 and Bcl-2 expression. The downregulation of LMP1 in SNT-8 cells significantly inhibited the extent of NF-κB p65 phosphorylation, elevated the expression of p53 and Bax, along with the ratio of Bax/Bcl-2, while repressed the expression of MDM2 and Bcl-2 (p < 0.05, Figure 2A). Therefore, we suspected that LMP1 promoted MDM2-mediated p53 ubiquitination by potentiating the NF-κB signaling pathway to promote lymphoma cell growth. Afterwards, we conducted Co-IP experiments to detect the binding relation of MDM2 to p53 in KYHG-1 and SNT-8 cells. Overexpression of LMP1 expedited the interaction of p53 and MDM2 in KYHG-1 cells, but the binding of p53 to MDM2 in cells was significantly reduced in SNT-8 cells (Figure 2B). Subsequently, we detected p53 ubiquitination in KHYG-1 and SNT-8 cells and found that the p53 ubiquitination was notably increased in KHYG-1 cells, while the p53 ubiquitination was remarkably inhibited after knockdown of LMP1 in the cells (Figure 2C).
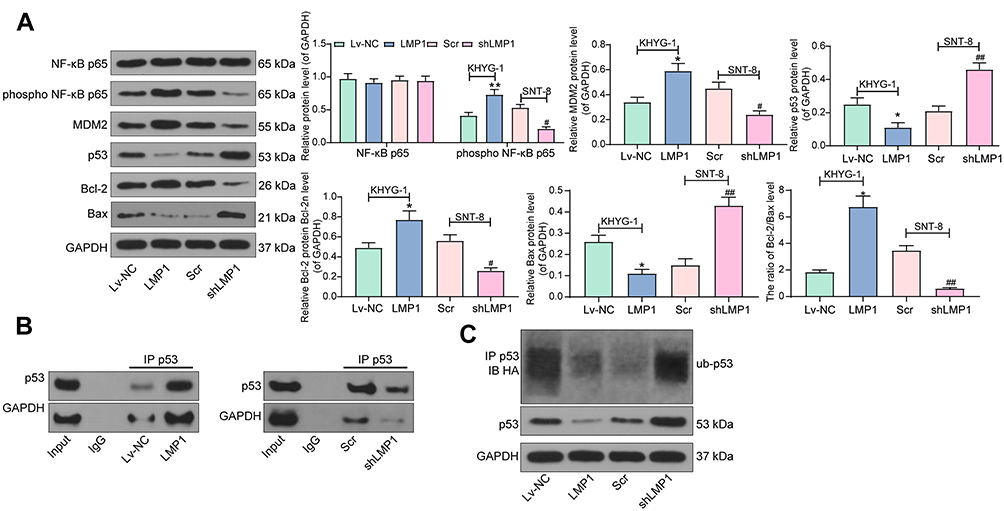 |
Figure 2 LMP1 activates the NF-κB pathway to induce MDM2-mediated p53 protein degradation. (A) NF-κB p65 phosphorylation levels and p53, MDM2, Bax and Bcl-2 protein expression in cells detected by Western blot (one-way ANOVA was used to compare the mean of samples among multiple groups, followed by Tukey’s multiple comparisons test); (B) MDM2 and p53 binding in KHYG-1 and SNT-8 cells detected by Co-IP; (C) Ubiquitination of p53 in KHYG-1 and SNT-8 cells detected by Co-IP. The data was performed as means ± SD of three independent experiments. *p < 0.05, **p < 0.01 vs. the Lv-NC group; #p < 0.05, ##p < 0.01 vs. the Scr group. |
A NF-κB Inhibitor Blocks the Supporting Role of LMP1 for Lymphoma Cell Growth and p53 Degradation
We subsequently added a NF-κB-specific inhibitor Flaconitine to KHYG-1 cells overexpressing LMP1 and a NF-κB-specific agonist to SNT-8 cells with LMP1 knockdown. We observed that specific inhibition of NF-κB activation in KHYG-1 cells significantly reduced the number of EdU (p < 0.01) and CFSE-positive cells (p < 0.05, Figure 3A and B) and promoted apoptosis and G0/G1 cell cycle arresting (both p < 0.05, Figure 3C and D). While specific activation of the NF-κB pathway in SNT-8 cells with poor expression of LMP1 significantly increased cell viability, inhibited apoptosis and promoted cell cycle progression (all p < 0.05). Moreover, we found that p53 ubiquitination decreased significantly in KHYG-1 cells and increased significantly in SNT-8 cells (Figure 3E), suggesting that p53 ubiquitination was associated with NF-κB pathway activation.
 |
Figure 3 The NF-κB pathway inhibitor abrogates the promotive role of LMP1 for lymphoma cell growth and p53 degradation. (A) The proliferation ability of KHYG-1 and SNT-8 cells evaluated by EdU staining; (B) Cell viability after CFSE staining determined by flow cytometry; (C) Cell cycle distribution detected by flow cytometry; (D) Cell apoptosis level assessed by flow cytometry; (E) Ubiquitination of p53 in KHYG-1 and SNT-8 cells detected by Co-IP. The data was performed as means ± SD of three independent experiments. One-way ANOVA was used to compare the mean of samples among multiple groups, followed by Tukey’s multiple comparisons test. The data was performed as means ± SD of three independent experiments. *p < 0.05, **p < 0.01 vs. the LMP1 + DMSO group; #p < 0.05, ##p < 0.01 vs. the shLMP1 + DMSO group. |
MG132, a Proteasome Blocker, Weakens LMP1 Action on Lymphoma Cell Growth and p53 Degradation
Significant inhibition of p53 ubiquitination level was observed in KHYG-1 cells overexpression LMP1 following the addition of MG132 (Figure 4A). Subsequently, we tested the proliferation ability of KHYG-1 cells, which displayed that the number of EdU positive cells (p < 0.01) and CFSE positive cells (p < 0.05) was decreased significantly, while apoptosis was increased remarkably (p < 0.01), in concomitant with an enhancement in the cell number in the G0/G1 phase and a decline in the cell number in the S phase (p < 0.05, Figure 4B–E).
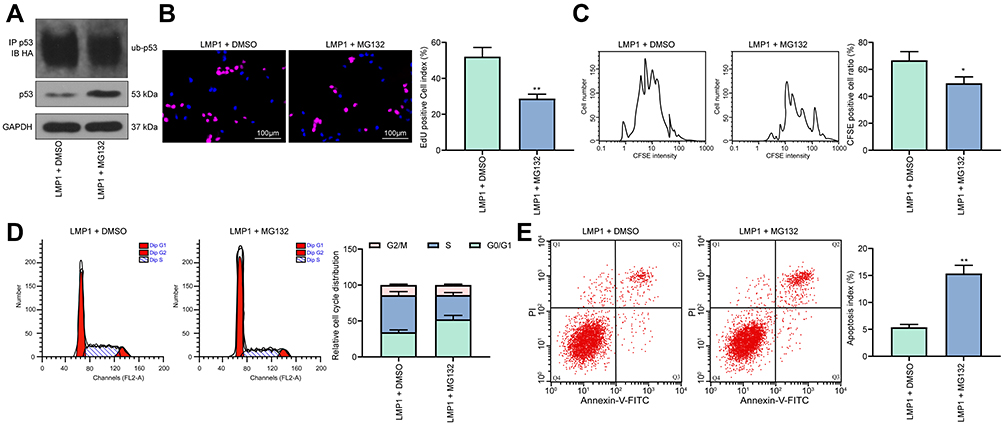 |
Figure 4 MG132 significantly attenuates the action of LMP1 on lymphoma cell growth and p53 degradation. (A) Ubiquitination of p53 in KHYG-1 cells detected by Co-IP; (B) The proliferation ability of KHYG-1 cells evaluated by EdU staining; (C) Cell viability after CFSE staining determined by flow cytometry; (D) Cell cycle distribution detected by flow cytometry; (E) Cell apoptosis level assessed by flow cytometry. The data was performed as means ± SD of three independent experiments. Unpaired t-test was applied to compare the mean of samples between groups. The data was performed as means ± SD of three independent experiments. *p < 0.05, **p < 0.01 vs. the LMP1 + DMSO group. |
Specific Activation of p53 or Inhibition of Bcl-2 Inhibits LMP1 Action on Lymphoma Cell Apoptosis
To further verify our conjecture, we added a Bcl-2-specific inhibitor TW-37 to KHYG-1 cells overexpressing LMP1 and a p53-specific inhibitor Pifithrin-β to SNT-8 cells with LMP1 knockdown. We used Western blot to detect Bcl-2 and p53 expression in those cells to determine the success of delivery (p < 0.05, Figure 5A). Also, we monitored that after specific inhibition of Bcl-2 expression, the proliferation activity of KHYG-1 cells was significantly decreased, and the level of apoptosis was significantly promoted (all p < 0.05). However, after inhibition of p53 expression in SNT-8 cells, cell proliferation activity was significantly enhanced, and apoptosis level was significantly inhibited (all p < 0.05, Figure 5B–E).
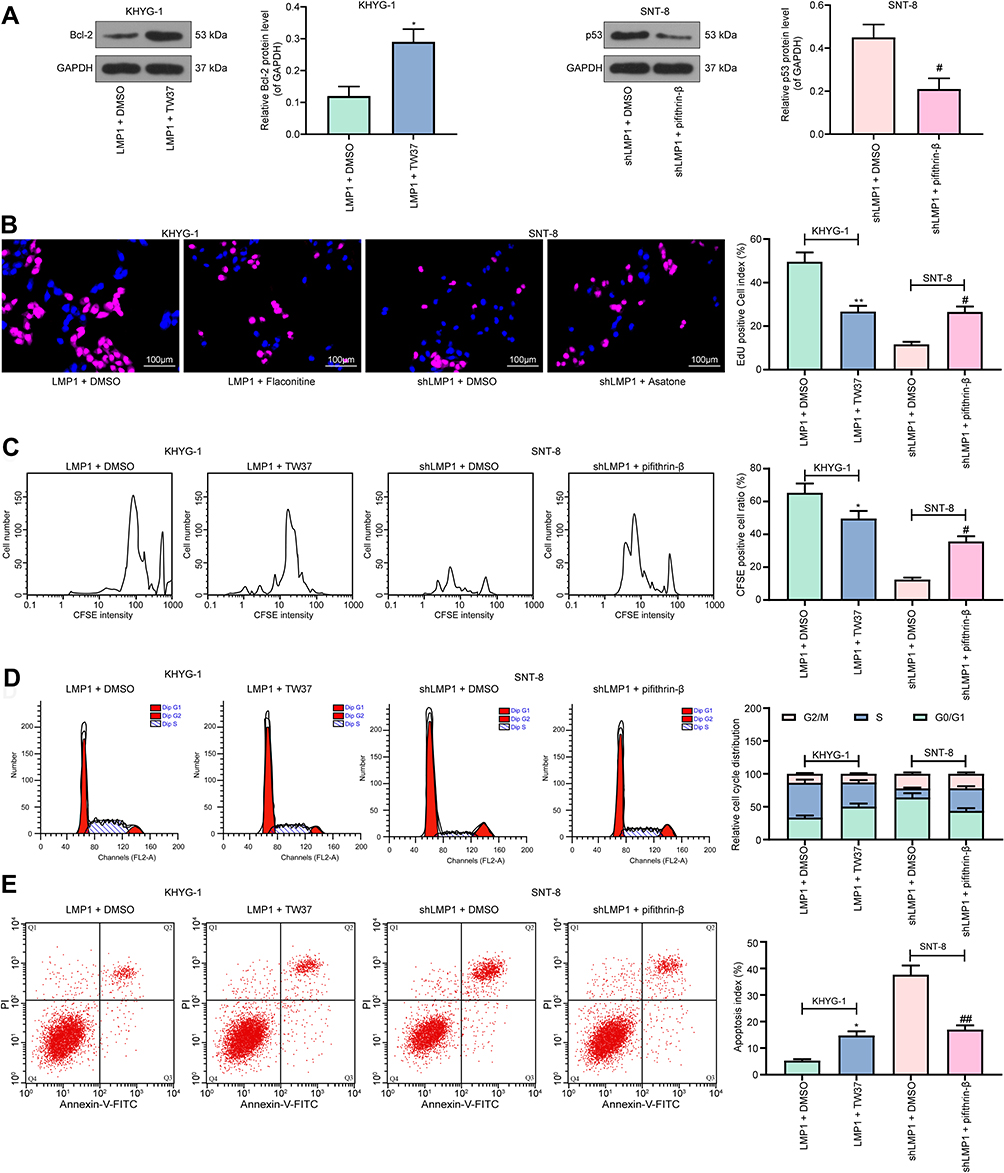 |
Figure 5 Specific activation of p53 or inhibition of Bcl-2 inhibits the LMP1 action on lymphoma cell viability and apoptosis. (A) Bcl-2 expression in KHYG-1 cells and p53 expression in SNT-8 cells determined by Western blot; (B) The proliferation ability of KHYG-1 and SNT-8 cells evaluated by EdU staining; (C) Cell viability after CFSE staining determined by flow cytometry; (D) Cell cycle distribution detected by flow cytometry; (E) Cell apoptosis level assessed by flow cytometry. The data was performed as means ± SD of three independent experiments. One-way ANOVA was used to compare the mean of samples among multiple groups, followed by Tukey’s multiple comparisons test. *p < 0.05, **p < 0.01 vs. the LMP1 + DMSO group; #p < 0.05, ##p < 0.01 vs. the shLMP1 + DMSO group. |
LMP1 Accelerates the Growth of Lymphoma in vivo
After that, we examined the effects of LMP1 on the growth of lymphoma cells in vivo. The growth rate of tumors from mice injected with KHYG-1 cells overexpressing LMP1 was increased significantly and the weight of transplanted tumor was elevated significantly as well (all p < 0.05, Figure 6A–C). By contrast, the growth rate of tumors from mice injected with SNT-8 cells with LMP1 knockdown was significantly inhibited, and the weight of tumor was significantly reduced. Furthermore, we detected the protein expression of KI67 in tumor by immunohistochemistry, and found that overexpression LMP1 increased KI67 expression, while inhibition of LMP1 expression decreased KI67 expression (all p < 0.01, Figure 6D).
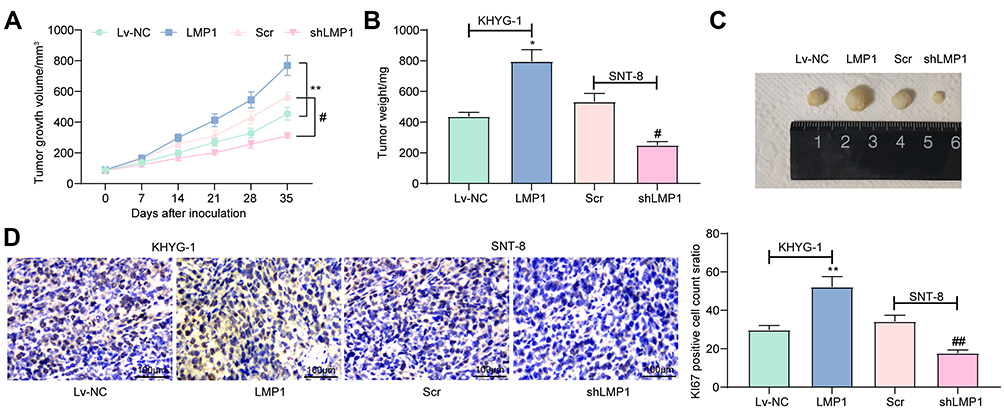 |
Figure 6 LMP1 potentiates the growth of lymphoma in nude mice. (A) The tumor growth volume (cm3) (two-way ANOVA was used to compare the mean of samples among multiple groups, followed by Tukey’s multiple comparisons test); (B) Tumor weight (mg) of nude mice (one-way ANOVA was used to compare the mean of samples among multiple groups, followed by Tukey’s multiple comparisons test); (C) The representative images of tumors in each group; (D) Immunohistochemical staining and statistical analysis of KI67 expression in tumors (one-way ANOVA was used to compare the mean of samples among multiple groups, followed by Tukey’s multiple comparisons test). *p < 0.05, **p < 0.01 vs. the Lv-NC group; #p < 0.05, ##p < 0.01 vs. the Scr group. n = 5. |
Discussion
EBV, the first human virus associated with oncogenesis, was originally identified in 1964 in a Burkitt lymphoma cell line and disturbs over 90% of the global population.12 Moreover, EBV-encoded LMP1 has been indicated to promote cell cycle progression and repress apoptosis via the induction of the NF-κB pathway.13 However, the precise mechanism of its tumor-facilitating effect remains unclear. In the present investigation, we first determined that LMP1 augmented the proliferation of lymphoma cells in vitro and in vivo. Additionally, LMP1 overexpression activated NF-κB pathway to enhance p53 protein degradation in an MDM2-dependent manner and to promote Bcl-2 expression. These data suggested pro-proliferative and anti-apoptotic roles for LMP1 in lymphoma.
Initially, the plasma expression of EBV-LMP1 was notably elevated in patients with metastatic nasopharyngeal carcinoma (NPC) relative to non-metastatic patients, and EBV-LMP1 was tightly linked to tumor stage and node stage for metastatic NPC patients.14 More specifically, LMP1 has been suggested to mimic human CD40 receptor to activate several pathways and to lead to the progression of disorders associated with EBV.15 For instance, LMP1 upregulated the expression and activity of DNA methyltransferases 1 and promoted its mitochondrial translocation, contributing to metabolic reprogramming in NPC.16 Besides, EBV-encoded LMP1 suppressed the repair of DNA double-strand breaks by repressing the phosphorylation and activity of DNA-dependent protein kinase to elevate radio-resistance in NPC.17 While in the current study, we performed EdU, colony formation and flow cytometric assays to reveal that LMP1 overexpression in EBV negative KHYG-1 cells promoted lymphoma cell proliferation and prevented apoptosis, while LMP1 knockdown resulted in opposite trends in EBV positive SNT-8 cells. In line with our findings, LMP1 upregulation promoted the growth of CNE-2 cells relative to LMP2A overexpression, and LMP1 knockout markedly repressed EBV proliferation in CNE-2 cells.18 As a consequence, we sought to probe the underlying mechanism.
Previously, Kim et al found that LMP1-activated V-set and Ig domain-containing 4 expression is modulated through the NF-κB pathway.19 Moreover, LMP1 encouraged miR-146a expression via the NF-κB pathway in EBV-associated cells, hence manipulating cell phenotypes.20 Similarly, we observed that the NF-κB pathway is involved in the mediation of MDM2-mediated degradation of p53 and cell viability enhancement caused by LMP1, supporting by lowered number in EdU- and CFSE-positive cells, facilitated cell apoptosis as well as diminished p53 ubiquitination levels in KHYG-1 cells treated with Flaconitine in the presence of LMP1. LMP1 was also established to confer cancer stem-like cell phenotype to NPC cells by impairing the p53-modulated apoptosis pathway.21 Furthermore, IKKβ could stabilize MDM2, thus resulting in degradation of GADD45α in HepG2 cells in an ubiquitination-dependent manner.22 Casein kinase-2 inhibitors reduced the association between MDM2 and p53 and blocked p53 ubiquitination to enhance p53 levels.23 ZCCHC10 bound to and stabilized p53 by impairing the binding relation between p53 and MDM2, and the p53 inhibitor pifithrin-α weakened the impacts of ZCCHC10 restoration on p53 pathway, cell cycle and apoptosis in lung cancer.24 Our data demonstrated that specific inhibition of p53 by pifithrin-β reversed the influence of LMP1 knockdown on cell viability and apoptosis. Additionally, MG132, a proteasome blocker, and suppression of Bcl-2 inhibited the function of LMP1 in cell growth. Klenke et al found that TW-37 treatment resulted in enhanced apoptosis and diminished proliferation rates in neuroblastoma cell lines,25 which was in line with our findings. Apoptosis in cancer cells is highly related to the activity of ubiquitin-proteasome signaling, and MG132 was monitored to stimulate cell apoptosis through generating reactive oxygen species or altering the expression of transcription factors and cell cycle proteins.26 Also, MG132 contributed to a significant decline in HepG2 cell viability.27 Consistently, MG132 was displayed in our observations to reduce the EdU- and CFSE-positive cells and enhance cell apoptosis in KHYG-1 cells overexpressing LMP1.
Conclusion
Altogether, the results from our assays reveal that LMP1 acts as an oncogene in lymphoma and interacts with NF-κB to coregulate the MDM2/p53/Bcl-2 axis to regulate the cell viability, apoptosis as well as cell cycle progression. As a consequence, LMP1 knockdown may become a novel molecular target for the exploration of novel treatment approaches for lymphoma. Nevertheless, more detailed molecular analyses are necessary to explore the probable role of the existing axis in lymphoma, bringing possibility in novel approaches for the management and treatment of the tumor.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflict of interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30.
2. AbuSalah MAH, Gan SH, Al-Hatamleh MAI, Irekeola AA, Shueb RH, Yean Yean C. Recent advances in diagnostic approaches for Epstein-Barr virus. Pathogens. 2020;9(3):226.
3. Shannon-Lowe C, Rickinson AB, Bell AI. Epstein-Barr virus-associated lymphomas. Philos Trans R Soc Lond B Biol Sci. 2017;372(1732):20160271.
4. Cohen JI, Fauci AS, Varmus H, Nabel GJ. Epstein-Barr virus: an important vaccine target for cancer prevention. Sci Transl Med. 2011;3(107):107fs107.
5. Ayee R, Ofori MEO, Wright E, Quaye O. Epstein-Barr virus associated lymphomas and epithelia cancers in humans. J Cancer. 2020;11(7):1737–1750.
6. Ye D, Zhu J, Zhao Q, et al. LMP1 up-regulates calreticulin to induce epithelial-mesenchymal transition via TGF-beta/Smad3/NRP1 pathway in nasopharyngeal carcinoma cells. J Cancer. 2020;11(5):1257–1269.
7. Sun L, Zhao Y, Shi H, Ma C, Wei L. LMP-1 induces survivin expression to inhibit cell apoptosis through the NF-kappaB and PI3K/Akt signaling pathways in nasal NK/T-cell lymphoma. Oncol Rep. 2015;33(5):2253–2260.
8. Chatterjee K, Das P, Chattopadhyay NR, Mal S, Choudhuri T. The interplay between Epstein-Bar virus (EBV) with the p53 and its homologs during EBV associated malignancies. Heliyon. 2019;5(11):e02624.
9. Pujals A, Favre L, Pioche-Durieu C, et al. Constitutive autophagy contributes to resistance to TP53-mediated apoptosis in Epstein-Barr virus-positive latency III B-cell lymphoproliferations. Autophagy. 2015;11(12):2275–2287.
10. Egorova O, Lau HH, McGraphery K, Sheng Y. Mdm2 and MdmX RING domains play distinct roles in the regulation of p53 responses: a comparative study of Mdm2 and MdmX RING domains in U2OS cells. Int J Mol Sci. 2020;21(4):1309.
11. Banjara S, Suraweera CD, Hinds MG, Kvansakul M. The Bcl-2 family: ancient origins, conserved structures, and divergent mechanisms. Biomolecules. 2020;10(1):128.
12. Geng L, Wang X. Epstein-Barr virus-associated lymphoproliferative disorders: experimental and clinical developments. Int J Clin Exp Med. 2015;8(9):14656–14671.
13. Kucuk C, Wang J, Xiang Y, You H. Epigenetic aberrations in natural killer/T-cell lymphoma: diagnostic, prognostic and therapeutic implications. Ther Adv Med Oncol. 2020;12:1758835919900856.
14. Sun L, Wang Y, Shi J, Zhu W, Wang X. Association of plasma Epstein-Barr virus LMP1 and EBER1 with circulating tumor cells and the metastasis of nasopharyngeal carcinoma. Pathol Oncol Res. 2019;1–9.
15. Zhang Y, Zhang W, Liu W, Liu H, Zhang Y, Luo B. Epstein-Barr virus miRNA-BART16 modulates cell proliferation by targeting LMP1. Virus Res. 2018;256:38–44.
16. Luo X, Hong L, Cheng C, et al. DNMT1 mediates metabolic reprogramming induced by Epstein-Barr virus latent membrane protein 1 and reversed by grifolin in nasopharyngeal carcinoma. Cell Death Dis. 2018;9(6):619.
17. Lu J, Tang M, Li H, et al. EBV-LMP1 suppresses the DNA damage response through DNA-PK/AMPK signaling to promote radioresistance in nasopharyngeal carcinoma. Cancer Lett. 2016;380(1):191–200.
18. Huo H, Hu G. CRISPR/Cas9-mediated LMP1 knockout inhibits Epstein-Barr virus infection and nasopharyngeal carcinoma cell growth. Infect Agent Cancer. 2019;14:30.
19. Kim SM, Oh SW, Park SH, Hur DY, Hong SW, Han SY. Epstein-Barr virus-encoded latent membrane protein 1 induces epithelial to mesenchymal transition by inducing V-set Ig domain containing 4 (VSIG4) expression via NF-kB in renal tubular epithelial HK-2 cells. Biochem Biophys Res Commun. 2017;492(3):316–322.
20. Wang W, Zhang Y, Liu W, et al. LMP1-miR-146a-CXCR4 axis regulates cell proliferation, apoptosis and metastasis. Virus Res. 2019;270:197654.
21. Yang CF, Peng LX, Huang TJ, et al. Cancer stem-like cell characteristics induced by EB virus-encoded LMP1 contribute to radioresistance in nasopharyngeal carcinoma by suppressing the p53-mediated apoptosis pathway. Cancer Lett. 2014;344(2):260–271.
22. Hu Y, Jin R, Gao M, et al. Transcriptional repression of IKKbeta by p53 in arsenite-induced GADD45alpha accumulation and apoptosis. Oncogene. 2019;38(5):731–746.
23. Dixit D, Sharma V, Ghosh S, Mehta VS, Sen E. Inhibition of casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumor necrosis factor (TNFalpha)-induced apoptosis through SIRT1 inhibition. Cell Death Dis. 2012;3:e271.
24. Ning Y, Hui N, Qing B, et al. ZCCHC10 suppresses lung cancer progression and cisplatin resistance by attenuating MDM2-mediated p53 ubiquitination and degradation. Cell Death Dis. 2019;10(6):414.
25. Klenke S, Akdeli N, Stelmach P, Heukamp L, Schulte JH, Bachmann HS. The small molecule Bcl-2/Mcl-1 inhibitor TW-37 shows single-agent cytotoxicity in neuroblastoma cell lines. BMC Cancer. 2019;19(1):243.
26. Guo N, Peng Z. MG132, a proteasome inhibitor, induces apoptosis in tumor cells. Asia Pac J Clin Oncol. 2013;9(1):6–11.
27. Ozgun GS, Ozgun E. The cytotoxic concentration of rosmarinic acid increases MG132-induced cytotoxicity, proteasome inhibition, autophagy, cellular stresses, and apoptosis in HepG2 cells. Hum Exp Toxicol. 2020;39(4):514–523.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.