")
Back to Journals » Journal of Pain Research » Volume 16
The American Society of Pain and Neuroscience (ASPN) Guidelines for Radiofrequency Ablative Procedures in Patients with Implanted Devices
Authors Sowder T, Sayed D , Concannon T, Pew SH, Strand NH , Abd-Elsayed A, Wie CS, Gomez Ramos DE, Raslan AM, Deer TR
Received 2 May 2023
Accepted for publication 26 October 2023
Published 3 November 2023 Volume 2023:16 Pages 3693—3706
DOI https://doi.org/10.2147/JPR.S419594
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Michael Schatman
Timothy Sowder,1 Dawood Sayed,1 Tyler Concannon,1 Scott H Pew,2 Natalie H Strand,2 Alaa Abd-Elsayed,3 Christopher S Wie,2 Daniel E Gomez Ramos,4 Ahmed M Raslan,5 Timothy R Deer6
1Department of Anesthesiology, University of Kansas Medical Center, Kansas City, KS, USA; 2Department of Anesthesiology, Mayo Clinic, Phoenix, AZ, USA; 3Department of Anesthesiology, University of Wisconsin, Madison, WI, USA; 4Department of Neurology, Mayo Clinic, Phoenix, AZ, USA; 5Department of Neurological Surgery, Oregon Health and Science University, Portland, OR, USA; 6The Spine and Nerve Center of the Virginias, Charleston, WV, USA
Correspondence: Timothy Sowder, Department of Anesthesiology, University of Kansas Medical Center, 3901 Rainbow Boulevard, Mail Stop 1034, Kansas City, KS, 66160, USA, Tel +1-913-588-6670, Fax +1-913-588-3365, Email [email protected]
Abstract: Radiofrequency ablation (RFA) is a treatment modality used in interventional pain management to treat several conditions including chronic neck or back pain, sacroiliac joint pain, major joint pain, and pain from sites that can be isolated to a sensory nerve amenable to RFA. The goals of such procedures are to reduce pain, improve function, delay need for surgical intervention, and reduce pain medication consumption. As applications for RFA expand through novel techniques and nerve targets, there is concern with how RFA may impact patients with implanted medical devices. Specifically, the electrical currents used in RFA produce electromagnetic interference, which can result in unintentional energy transfer to implanted devices. This may also interfere with device function or cause damage to the device itself. As the number of patients with implanted devices increases, it is imperative to establish guidelines for the management of implanted devices during RFA procedures. This review aims to establish guidelines to assist physicians in the preoperative, intraoperative, and postoperative management of implanted devices in patients undergoing procedures using radiofrequency energy. Here, we provide physicians with background knowledge and a summary of current evidence to allow safe utilization of RFA treatment in patients with implanted devices such as cardiac implantable electronic devices, spinal cord stimulators, intrathecal pumps, and deep brain stimulators. While these guidelines are intended to be comprehensive, each patient should be assessed on an individual basis to optimize outcomes.
Keywords: radiofrequency ablation, implanted medical devices, cardiac implantable electronic device, spinal cord stimulator, intrathecal pump, deep brain stimulator
Introduction
Radiofrequency Ablation
Radiofrequency ablation (RFA) is a treatment modality used in multiple specialties from treatment of cardiac arrhythmias to targeted destruction of tumors. It is used in interventional pain management to treat several conditions including chronic neck or back pain, sacroiliac joint pain, major joint pain, and pain from sites that can be isolated to a sensory nerve amenable to RFA. Conventional or thermal RFA uses a needle that delivers continuous high-voltage current to produce a heat lesion on a pain-transmitting nerve. The tip of the needle is heated to approximately 80 degrees Celsius, and the resulting lesion disrupts the pain signals to the brain. Pulsed radiofrequency utilizes short bursts of high-voltage current with intermittent pauses resulting in lower temperatures of approximately 42 degrees Celsius. The proposed mechanism of action is thought to be modulation of pain signals rather than thermal destruction of the nerve. The goals of such procedures are to reduce pain, improve function, delay or eliminate the need for surgical intervention, and reduce pain medication requirements. Contraindications to RFA are few but include site specific use of anticoagulants, site infection, and the inability of the patient to consent or desire not to proceed with the procedure. The electrical currents used in RFA produce electromagnetic interference (EMI) which can rarely result in unintentional energy transfer to implanted devices. This could theoretically interfere with device function or damage the device. As a result of this potential interference and the growing number of implanted devices, it is imperative to establish guidelines for the management of these devices during RFA procedures.
Problem
Implanted devices are becoming more prevalent as technologies improve and average lifespan continues to grow. Applications for RFA are also expanding with novel techniques and nerve targets. This increases the potential use of RFA in patients with implanted devices.
Goal
The primary objective of this review is to update previous recommendations and provide current guidelines to assist physicians and affiliated healthcare professionals in the preoperative, intraoperative, and postoperative management of implanted devices in patients undergoing procedures using radiofrequency energy.1–3 In addition, we hope to shape procedures and policies of institutions to improve patient safety in this population.
Application of Guidelines
These guidelines aim to provide physicians with background knowledge and a summary of current evidence to allow safe utilization of RFA treatment in patients with implanted devices. While these guidelines are intended to be comprehensive, each patient should be assessed on an individual basis to optimize outcomes.
Methods
We have prepared a clinical guideline on management of implanted devices in patients undergoing RFA procedures. Data sources included relevant literature identified through searches of PubMed as well as manufacturer-provided information. Bibliographies of included primary and review articles were also searched for additional relevant literature. Search terms included relevant key words and phrases such as radiofrequency ablation, electromagnetic interference, cardiac implantable electronic devices, pacemaker, deep brain stimulator, spinal cord stimulator, and intrathecal pump. Guidelines were then developed by a multidisciplinary physician group with input from experts in the fields of electrophysiology, interventional pain management, and neurosurgery.
Quality Assessment, Evidence Ranking, and Level of Certainty
Identified peer-reviewed literature was critiqued using the United States Preventive Services Task Force (USPSTF) criteria for quality of evidence, with modifications for neuromodulation studies (Table 1).4 After USPSTF letter grading was assigned, the panel then assigned the “level of certainty regarding benefit” as described in Table 2.
 |
Table 1 Quality of Evidence Ranking Using United States Preventative Services Task Force Criteria Modified for Neuromodulation |
 |
Table 2 Levels of Certainty Regarding Net Benefit |
Results
Cardiac Implantable Electronic Devices
Background
Cardiac implantable electronic devices (CIEDs) include pacemakers, implanted cardioverter-defibrillators (ICDs), and cardiac resynchronization therapy devices (CRTDs). Each of these devices have specific functions which need to be clearly identified and managed. Pacemakers are used to manage cardiac arrhythmias and conduction abnormalities. In heart failure patients, ICDs provide immediate shock therapy when non-perfusing tachyarrhythmias are detected, and CRTDs provide pacing for dyssynchronous ventricular activation to optimize cardiac output.5 CIEDs are becoming more common as technological advances have broadened indications. Increased device complexity makes it even more important that providers have adequate knowledge to manage these devices.
Components
CIEDs are comprised of a pulse generator attached to electrodes or leads. The pulse generator is typically implanted in the infraclavicular region of the anterior chest wall and connected to transvenously placed leads anchored in the myocardium. The electrodes may function in sensing, pacing, defibrillating, or some combination depending on the device type. Sensing electrodes detect both intrinsic and extrinsic electrical currents. Output is then delivered in the form of pacing or defibrillation based on input from the sensing electrode.5
Effect of EMI on CIEDs
EMI from an extrinsic source, including RFA treatments, may be sensed and trigger a response. In pacemakers, sensed electrical current will inhibit the device from delivering additional current to avoid cardiac dyssynchrony and “R on T” phenomenon. Inhibition of therapy can result in asystole in a pacemaker-dependent patient. In ICDs, sensed electrical current may be interpreted as a non-perfusing tachyarrhythmia which will elicit delivery of cardioversion therapy. Less common problems that may be caused by EMI are device reset, pulse generator damage, and lead-tissue interface damage. To avoid interference, some manufacturers recommend a minimum distance of 15cm between sources of EMI and CIEDs.6 Manufacturer recommendations should be confirmed for each device. In anatomical terms, EMI below the umbilicus (correlates with L3–4) is unlikely to cause significant interference.
Preoperative Evaluation
Preoperative evaluation lays the foundation for safe intra- and post-operative outcomes. Management should be individualized to each patient based on the device and planned procedure. Information that should be obtained includes device type (including manufacturer and model), indication, most recent device interrogation, pacemaker dependency, programming, and response to magnet placement. This information may be obtained by patient history, review of records, manufacturer ID card, or requested from the managing CIED team. Direct communication and planning with the CIED team should be considered in high-risk patients (above the umbilicus, presence of both ICD and PPM, pacemaker dependency, etc) or if there are any questions regarding device management. Ideally, device interrogation should be performed within the last six months for ICDs and 12 months for pacemakers.7 A physical exam should be considered in high-risk patients to assess IPG location. This will help determine the distance from the planned RFA and identify any potential issues with magnet placement.
Preoperative Management
If EMI is located below the umbilicus, no changes to the device are needed. If EMI is going to be located above the umbilicus, recommendations from the managing CIED team should be obtained or the following algorithm should be considered (Figure 1). In general, magnet placement alters pacemaker function to asynchronous mode or suspends the anti-tachycardia function of the ICD. In patients with combined ICD-PPM, a magnet will only suspend the anti-tachycardia function of the ICD without affecting the function of the pacemaker. If the patient is pacemaker-dependent and/or has an ICD, the device must be managed with either placement of a magnet or reprogramming of the device. Placing a magnet is sufficient in pacemaker-dependent patients without an ICD and in non-pacemaker-dependent patients with an ICD. If the patient has an ICD and is also pacemaker-dependent, the device will need to be reprogrammed as a magnet will only disable anti-tachycardia function without providing asynchronous pacing. Positioning may present a challenge for management of CIEDs as patients are often in the prone position for applications of RFA in interventional pain. If the magnet will be difficult to keep in place over the generator due to body habitus, chest roll, operating table configuration, or any other reason, consider contacting the device representative to reprogram the device prior to positioning. If using a magnet at any time during surgery, the magnet must remain in place the entire time. Once the magnet is removed, the CIED reverts to its baseline settings.5
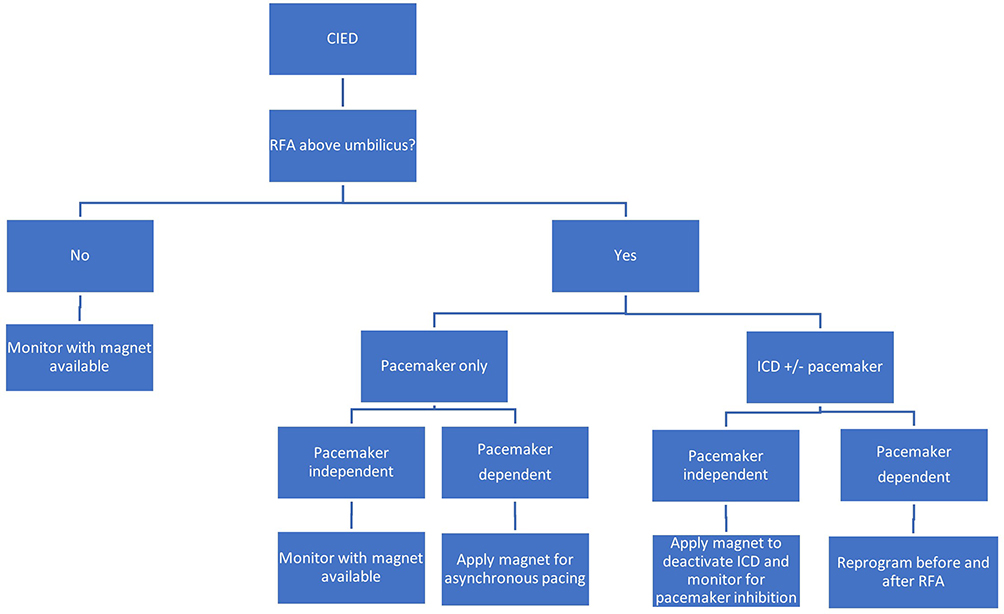 |
Figure 1 CIED Management Algorithm. |
Intraoperative Management
Patients should be monitored with standard ASA monitors including ECG (in pacing mode, if available), blood pressure, ETCO2, and pulse oximetry with plethysmography. A magnet as well as transcutaneous pacing and defibrillation pads should be available for all patients. Care should be taken to place the cautery pad away from the CIED in a manner to direct the EMI path away from the CIED. This may be achieved using bipolar RFA, which has been used safely in patients with CIEDs.8 Time of EMI exposure should be limited to the minimal amount needed to achieve the desired clinical outcome.9
Postoperative Management
Postoperatively, patients with a CIED should have continuous cardiac rate monitoring until device settings are restored. Back-up pacing and cardioversion-defibrillation equipment should remain immediately available.10 If a magnet was used, programmed settings typically resume with magnet removal. If the device was reprogrammed for the procedure, preprocedural settings will need to be restored by a device representative or CIED team member. An immediate post-operative interrogation of a CIED may not be required but should be considered if RFA occurred above the umbilicus. CIED interrogation should be performed if:
- CIED was reprogrammed pre- or intraoperatively.
- Significant intraoperative events occurred including arrhythmias requiring transcutaneous pacing, delivery of anti-tachycardia therapy (by CIED or external pads), cardiac arrest, etc.
- There was concern for CIED malfunction.9,10
Recommendations: Application of guidelines and risk mitigation strategies in patients with CIEDs undergoing RFA will likely improve patient safety and adverse events. Evidence is limited to case reports and additional high-quality studies are needed. Grade B, moderate level of certainty (Table 3).
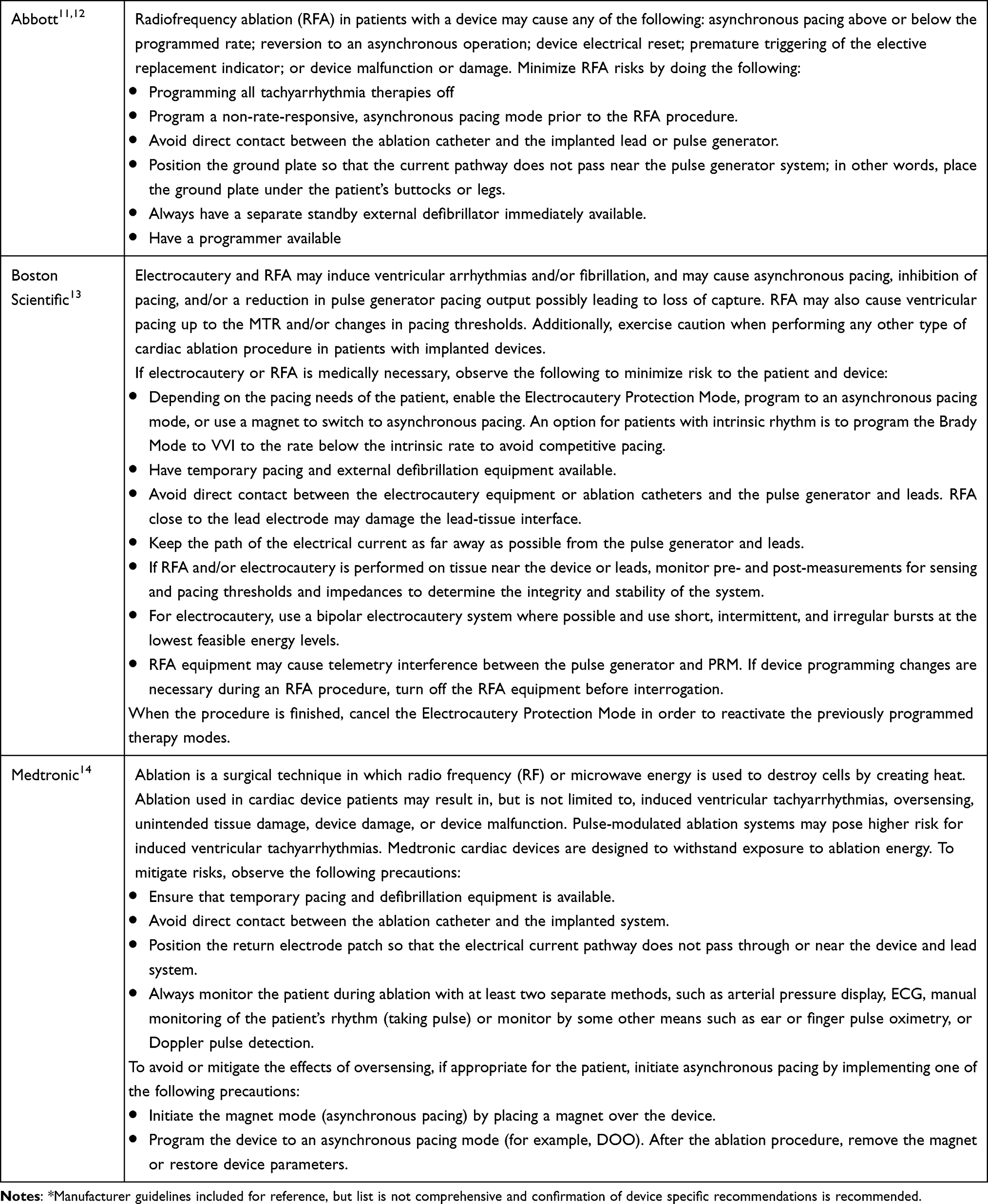 |
Table 3 CIED Manufacturer Recommendations* |
Spinal Cord Stimulators
Background
Spinal cord stimulators (SCS) have been used for chronic pain treatment since 1967.15,16 The mechanism of action is explained mostly by Gate Control Theory, which proposes that non-painful stimuli “close” the gate, prevent transmission of painful afferent signals via wide-dynamic range neurons to the brain, and attenuate perception of pain.16 Other subsequent theories proposed include dorsal horn GABAergic inhibitory interneurons, supraspinal involvement, and vasculature involvement in ischemic pain. SCS devices are typically utilized in the setting of chronic axial or appendicular neuropathic pain, radicular pain, complex regional pain syndrome (CRPS), and failed back surgery syndrome, all of which are approved indications by the Food and Drug Administration (FDA).17 Additional indications exist including chronic intractable angina, peripheral vascular disease, postherpetic neuralgia, central pain from multiple sclerosis (MS), visceral pain, and painful spasms of atypical stiff limb syndrome.
Components
Traditional SCS devices use an array of electrodes that are implanted in the epidural space targeting the dorsal columns, specifically stimulating the A-beta fibers. The electrode at its distal portion is placed over the dorsal column. The proximal segment is connected to an internal pulse generator (IPG), which administers energy to the electrodes.18 It should be noted that this device is only permanently implanted following a successful lead trial with more than 50% pain reduction in addition to patient-reported satisfaction.18
Implantation of permanent SCS leads can be performed either via an interlaminar approach or laminectomy (typically when paddle leads are utilized). For the interlaminar approach, the entry point is several levels below the targeted spinal level for lead placement. Anchoring of the leads occurs at the point of epidural access. If using a laminectomy to place a paddle lead, the lead would be anchored directly over or very close to the intended paddle site. Proximal ends of the leads are tunneled beneath the skin and connected to the IPG, which is located subcutaneously, often in the flank or abdomen.18
Effect of EMI on Spinal Cord Stimulators
During use of a monopolar RFA needle, current travels through the patient and arrives to the ground electrode/panel, which is typically placed on the contralateral side of the patient’s body. Increased risk of damage or injury may occur if the RFA needle is near enough to the IPG, SCS lead, or any tissue near the leads.17 In contrast, it has been reported that use of a bipolar RFA needle, which uses one active electrode and one ground electrode with a shorter inter-distance, has decreased risk since it provides the opportunity for precise and predictive ablation of the target.19
Preoperative Evaluation and Management
The first task in preoperative evaluation of patients with implanted SCS devices is to determine the location of the device and any specific neurostimulator recommendations regarding EMI. Some devices have a surgery mode. Others do not, and the device needs to simply be turned off. Clinicians should also understand the difference between an implantable device being set in the “off” versus “surgery” mode. In certain devices, surgery mode creates greater protection of the IPG by releasing a small amount of electrical current to protect the lead from becoming a ground for the RFA. Informed consent should also include risks of EMI to the IPG, as the most severe risk includes damage requiring reimplantation of a new IPG. In addition, patient education is recommended on the need for frequent communication during the procedure regarding any abnormal sensation around the electrodes or IPG. Symptoms could include pain, paresthesia, or muscle activation.
Intraoperative Management
There is a risk of SCS devices being affected by EMI created during RFA. RFA uses radio waves to create current. This causes ions in the tissue to move, which then creates friction and the ultimate heating of tissues. Therefore, an electromagnetic field at the exposed tip of the needle is generated. The primary goal is to reduce the amount of EMI from the radiofrequency cannula to the IPG and stimulator leads. This can be done by reducing the distance between the radiofrequency cannula and the grounding pad, ensuring that it is less than the distance between the needle and IPG and/or leads, if possible. Specifically, Abbott (Abbott Laboratories, Chicago, IL, USA) has recommended that the IPG be placed in surgery mode and the bipolar setting utilized. Other medical device companies do not have these specific recommendations. In summary, bipolar ablation and keeping the grounding pad as close to the ablation site as possible will create the least amount of EMI.
Recommendations for intraoperative monitoring during the RFA procedure are primarily clinical. There have been reported cases of RFA causing interference with patients with implanted neuromodulating devices.20 These were both with dorsal root ganglion stimulation using monopolar lumbar radiofrequency (motor activation) and cervical SCS with ablation of the cervical spine (pain and paresthesia in hands). In addition, it is recommended that patients have minimal sedation so there is adequate communication to the proceduralist. It is also advised that sensory and motor testing start at 0.1V with a slow increase and continuous patient communication. During RFA lesioning, the patient should be directed to report any abnormal sensations as discussed pre-procedurally. Although there are concerns regarding activation of the stimulator device during the procedure, the main concern is permanent loss of therapy secondary to EMI.
Postoperative Management
Following the procedure, the stimulator should be turned back on, or surgery mode should be turned off. Ideally, the functioning of the SCS device should be confirmed immediately after the procedure.
Recommendations: Application of guidelines and risk mitigation strategies in patients with SCS devices will likely improve patient safety and adverse events. Evidence is limited to case reports and additional high-quality studies are needed. Grade B, low level of certainty (Table 4).
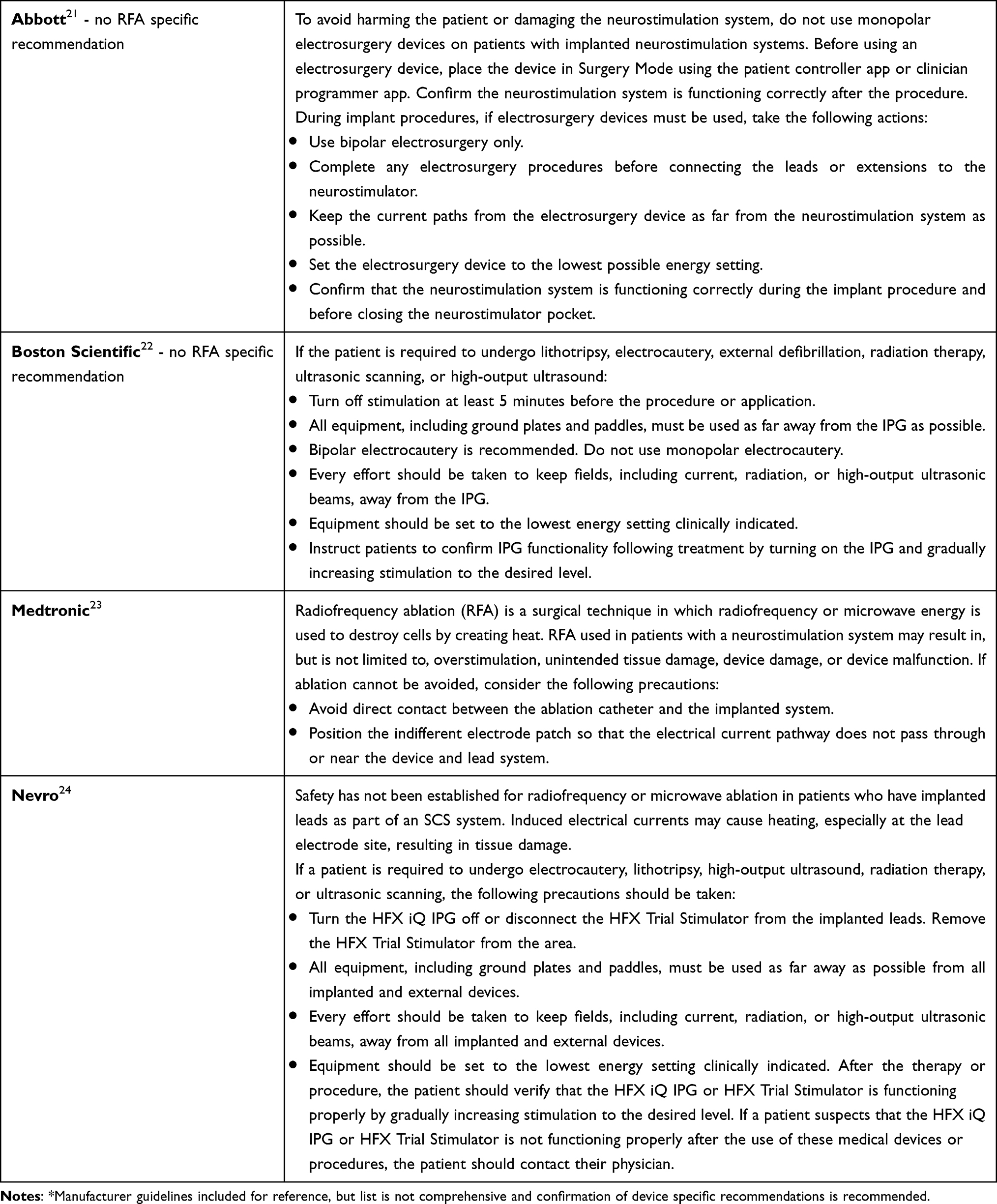 |
Table 4 SCS Manufacturer Recommendations* |
Intrathecal Pumps
Background
Delivering intrathecal medication experimentally in humans dates to as early as 1898, with it gaining more widespread use clinically after 1979 for obstetric analgesia.25,26 In 1981, an implantable intrathecal opioid delivery device was demonstrated for use in chronic pain secondary to malignancy.27 Currently in the US, morphine sulfate and ziconotide are on-label for intrathecal infusions in the setting of chronic intractable pain. Baclofen is on-label for intrathecal infusion for the management of severe spasticity. Morphine, because of its low cost, support in literature, duration of action, and ease of use, remains the most common medication used in intraspinal analgesia globally; however, other off-label agents are frequently used in intraspinal therapy.28 In 2007, the International Neuromodulation Society organized a group of experts to evaluate evidence and create a Polyanalgesic Consensus Conference (PACC) to guide practice for intrathecal drug delivery which continues to update their guidelines.29
Components
An intrathecal pump consists of two components: a reservoir/pump and a catheter. The pump is anchored in the pump pocket using a suture loop on the outside of the pump, and the drug to be infused is stored in the pump reservoir. The two most common reservoir sizes are 20 mL and 40 mL.30 The intrathecal catheter connects to the pump catheter port. After the drug is put in the reservoir, the drug moves from the pump reservoir, through the pump tubing, catheter port, catheter, and then to the infusion site. Typically, the manufacturer and model of pump are recorded on a radiopaque identifier visible on X-ray.
Location
Limited data exist regarding location or best placement level of the catheter tip. Guidelines recommend the catheter be centered in the spinal dermatome associated with the pain generator.29 Thus, the location of catheter and pump will vary from patient to patient, although placing the pump in the lower quadrant of the abdomen is most common.28
Effect of EMI on Device
Safety has not been established for radiofrequency or microwave ablation patients who have an ITP.30 Published data is limited to two case reports in which patients with an ITP underwent RFA; however, the authors did not specifically discuss concerns with the procedure in patients with an IT pump.31,32 Strong sources of EMI can undesirably interact with the pump. This can include heating of the implanted pump that results in system damage and/or changes in pump operation or flow rate resulting in patient injury from tissue heating, reoccurrence of underlying symptoms, or significant or fatal drug underdose or overdose.30
Preoperative Evaluation and Management
Prior to RFA or any procedure that might interfere with an ITP, the provider should determine age of the device, the last time the pump was interrogated, current pump medication(s), and when the patient is due for refill or pump change.33 Most pumps last 5–7 years and require a refill every 1–6 months, with this interval contingent on drug delivery rate and reservoir volume.34 When known, it is recommended to review the manufacturer’s guidelines for the specific device used. Consultation with the provider responsible for pump management is encouraged as this will help diminish possible complications. If unable to communicate prior to the procedure, it is important to have contact information for the managing physician as well as a representative from the device’s manufacturer company should any complications occur during the procedure.28
Intraoperative Management
If the anesthetic plan or post-procedural pain control involves opioids, vigilance must be taken to prevent inappropriate titration in patients receiving intrathecal opioids as administered opioids may induce overdose.35 If the ITP is delivering baclofen, the addition of opioids for sedation or pain control may have an exaggerated response due to the synergistic nature of the drugs.36 The patient’s pre-procedural baseline status and hemodynamics should be documented as any significant variation may indicate overdose or withdrawal, which may represent a medical emergency.
Patient positioning as well as location of RFA needs to be considered as these variables can affect the pump or catheter. Most ITPs are implanted in the lower quadrant of the abdomen and are preferably positioned to avoid contact with the patient’s waist or beltline as well as the pelvic girdle and ribs. For obese patients, the device may be implanted in the flank region to allow greater comfort while avoiding difficulties inherently found with a large abdominal pannus.28 To decrease catheter damage or malfunction, avoid positions including twisting, excessive bending, or stretching that might obstruct, kink, dislodge, or damage the catheter.37
As previously discussed, safety is not established for patents with ITP undergoing RFA. If RFA is going to be performed in patients with an ITP, it is prudent to be cognizant of the catheter and pump location in relation to the ablation location while maintaining the greatest distant possible between the radiofrequency cannula and ITP components.
Postoperative Management
Given the various potential sources of device malfunction, it is essential to recognize the signs and symptoms of overdose or withdrawal. If device malfunction is questioned, a provider experienced with ITP management should be consulted and the ITP should be interrogated. There is no literature or guidelines which recommend the routine or prophylactic interrogation of ITP status post RFA where device malfunction is not suspected.
Recommendations: Application of guidelines and risk mitigation strategies in patients with ITP devices will likely improve patient safety and adverse events. Evidence is limited to case reports and additional, high-quality studies are needed. Grade B, low level of certainty (Table 5).
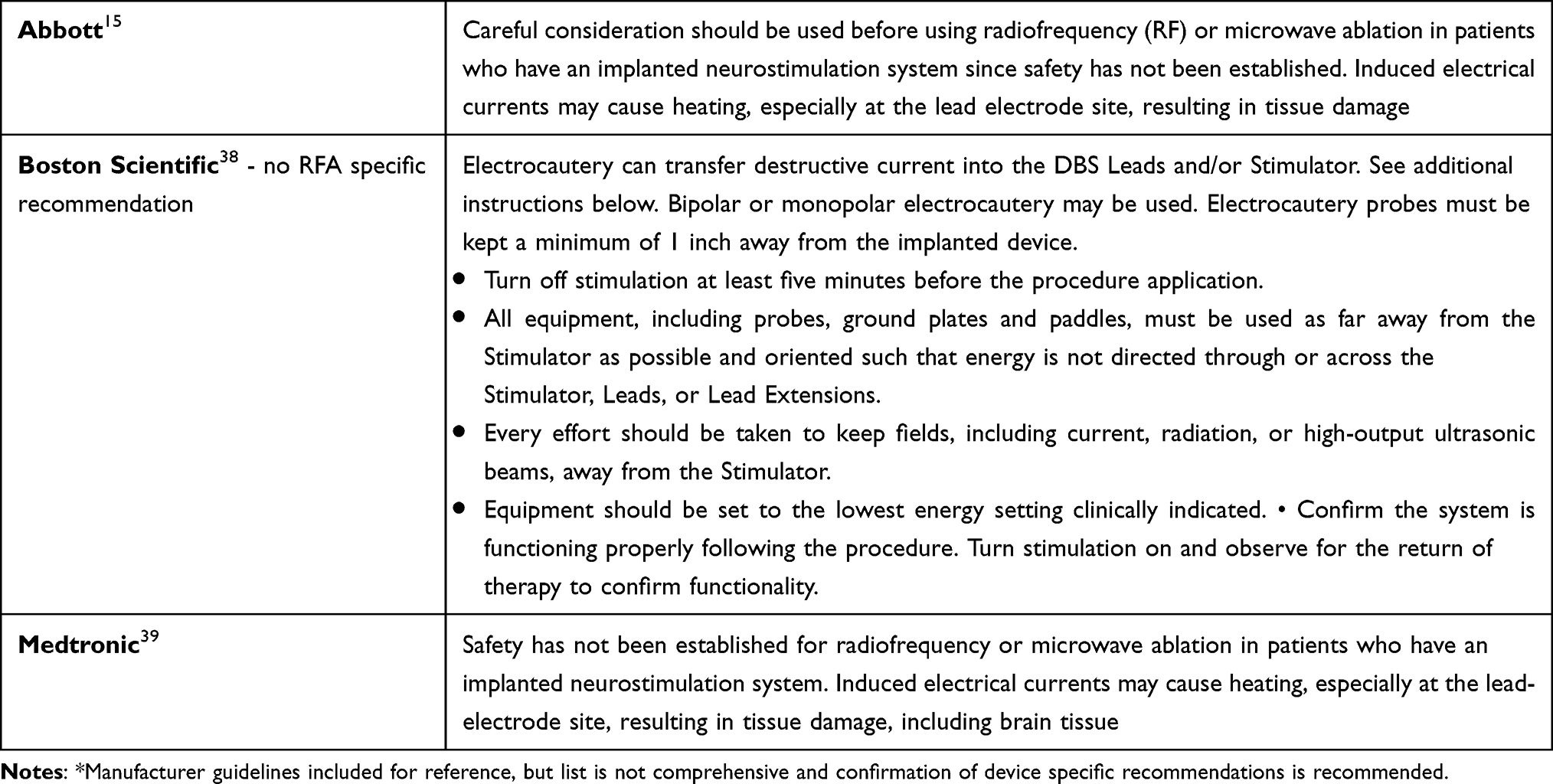 |
Table 5 DBS Manufacturer Recommendations* |
Deep Brain Stimulators (DBS)
Background
Stimulation of cortical structures dates back as far as the 1930s. Initially employed using modified pacemakers, deep brain stimulation was developed to treat chronic pain and later applied to tremor disorders. The mechanisms underpinning the therapeutic effects of DBS remain unknown; however, neurophysiological, neurochemical, neurovascular, neurogenic, and neuro-oscillations all play a role. Therapy can be individualized to each patient using different combinations of amplitude, frequency, pulse width, and electrode configuration.40
Indications
DBS has been approved to treat a variety of conditions including Parkinson’s disease, dystonia, essential tremors, chronic pain, intractable focal epilepsy, and certain neuropsychiatric conditions such as Tourette’s syndrome, depression, or obsessive-compulsive disorder. Several experimental indications are being tested such as obesity, PTSD, drug abuse, and addiction.40
Components
DBS technology involves placement of one or two leads with four (4CH) or eight (8CH) channel contacts into a specific and predetermined brain target. The 4CH leads have four cylindrical electrodes that can deliver stimulation in all directions. The 8CH lead has two cylindrical electrodes, similar to the 4CH lead, and has two central contacts consisting of three segments that can be activated independently to concentrate stimulation in one direction. This theoretically reduces the current needed to produce meaningful therapeutic effects.41 The electrodes are typically placed within the basal ganglia structures, specifically the subthalamic nucleus. The leads are connected to an insulated extension wire that is tunneled to an implanted pulse generator. The IPG is commonly implanted in the infraclavicular region, though location may vary based on surgeon and patient preference. IPGs are either single cell or rechargeable. Newer models allow for recording and sensing brain signals while delivering stimulation.
Effect of EMI on Device
EMI from RFA treatments may be transmitted to implanted DBS devices. Delivery of excess electrical energy through the leads can cause neurolytic changes resulting in severe injury or death.42 Patients may describe “jolting” or “shocking” in cases of increased stimulation. EMI can cause system damage or reset requiring reprogramming or surgical replacement.
Preoperative Evaluation and Management
DBS devices have become more complex in recent years, offering multiple program settings and therapies. Prior to proceeding with the RFA procedure, information should be obtained regarding the system such as indication for implantation, device manufacturer, and IPG location.43 Another important piece of information to delineate is severity of symptoms should the device be turned off, as this may preclude patients from undergoing the procedure from a safety perspective. It is also important to have detailed discussion with patients outlining risks to device and damage resulting in the need for IPG/lead revision.
Intraoperative Management
Intraoperatively, risk to the DBS system is present during RFA. This risk is present during the activation of the radio waves, resulting in a current to heat and ablate the targeted nerve. The risk in this scenario is primarily through inappropriate activation of the stimulator as well as generating EMI that damages the IPG, rendering the entire system nonfunctional. First, the system should be turned off or programmed to surgery mode prior to performing the procedure to minimize EMI. Second, the grounding pad should be placed as near as possible to the RFA needle to minimize the dispersion of electrical currents. Bipolar RFA is also a preferable treatment option, if available.43
Postoperative Management
Immediately after the ablative procedure, the DBS system should be powered on and returned to prior settings. It is also recommended that the device be interrogated in the immediate postoperative period by either a DBS device representative or a physician/advanced practice provider with experience involving the device.43
Recommendations: Application of guidelines and risk mitigation strategies in patients with DBS devices will likely improve patient safety and adverse events. Evidence is limited to case reports and additional, high-quality studies are needed. Grade B, low level of certainty.
Conclusions
Considering an aging population, the dichotomy of both osteoarthritic disease and cardiovascular or central nervous system disease in the same patient is strikingly likely. Considering this, the need for both RFA ablation and implantable cardiac, neuro, or other devices will become more common in all settings. Adherence and attention to this guidance will improve safety and efficacy, thus leading to improved care.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
Dr Timothy Deer reports grants, personal fees from Abbott, during the conduct of the study; personal fees, Consultant, Stock Options, Research from Vertos, personal fees, Consultant, Stock Options from SpineThera, personal fees, Consultant, Stock Options, Research from Saluda, personal fees, Consultant, Stock Options from Nalu, Consultant, Stock Options from Cornerloc, personal fees, Consultant, Stock Options from Ethos, personal fees, Consultant, Stock Options, Research from SPR Therapeutics, personal fees from Medtronic, personal fees, consultant, Research from Boston Scientific, personal fees, Consultant, Stock Options, Research from PainTeq, personal fees from Tissue Tech, personal fees, Consultant, Stock Options from Spinal Simplicity, personal fees from Biotronik, Research from Mainstay, Research from Avanos, outside the submitted work; In addition, Dr Timothy Deer has a patent Abbott pending to pending The authors do not have any financial disclosures or other conflicts of interest to report.
References
1. Cohen SP, Bhaskar A, Bhatia A, et al. Consensus practice guidelines on interventions for lumbar facet joint pain from a multispecialty, international working group. Reg Anesth Pain Med. 2020;45(6):424–467. doi:10.1136/rapm-2019-101243
2. Hurley RW, Adams MCB, Barad M, et al. Consensus practice guidelines on interventions for cervical spine (facet) joint pain from a multispecialty international working group. Pain Med. 2021;22(11):2443–2524.
3. Smith CC, McCormick ZL, Mattie R, MacVicar J, Duszynski B, Stojanovic MP. The effectiveness of lumbar transforaminal injection of steroid for the treatment of radicular pain: a comprehensive review of the published data. Pain Med. 2020;21(3):472–487. doi:10.1093/pm/pnz160
4. Grade definitions after July 2012. United States Preventive Services Taskforce; 2018. Available from: https://www.uspreventiveservicestaskforce.org/uspstf/about-uspstf/methods-and-processes/grade-definitions#july2012.
5. Crossley GH, Poole JE, Rozner MA, et al. The Heart Rhythm Society (HRS)/American Society of Anesthesiologists (ASA) Expert Consensus Statement on the perioperative management of patients with implantable defibrillators, pacemakers and arrhythmia monitors: facilities and patient management this document was developed as a joint project with the American Society of Anesthesiologists (ASA), and in collaboration with the American Heart Association (AHA), and the Society of Thoracic Surgeons (STS). Heart Rhythm. 2011;8(7):1114–1154. doi:10.1016/j.hrthm.2010.12.023
6. Medtronic. Magnet operation: CRHF technical services standard letter; 2016.
7. Friedrich J, Itano EM, Lynn RR. Management of cardiac implantable electrical devices in patients undergoing radiofrequency ablation for spine pain: physician survey and review of guidelines. Pain Physician. 2020;23(4):E335–e342. doi:10.36076/ppj.2020/23/E335
8. Hanna R, Abd-Elsayed A. Review of the safety of bipolar radiofrequency ablation in patients with chronic pain with implantable cardiac rhythm management devices. Pain Physician. 2021;24(2):E169–e176.
9. American Society of Anesthesiologists Task Force on Perioperative Management of Patients with Cardiac Rhythm Management Devices and others. Practice advisory for the perioperative management of patients with cardiac implantable electronic devices: pacemakers and implantable cardioverter-defibrillators 2020: an updated report by the American Society of anesthesiologists task force on perioperative management of patients with cardiac implantable electronic devices: Erratum. Anesthesiology. 2020;132(4):938. doi:10.1097/ALN.0000000000003217
10. Neelankavil J, Thompson A, Mahajan A. Managing cardiovascular implantable electronic devices (CIEDs) during perioperative care. Anesth Patient Saf Found Newsl. 2013;28(2):29–35.
11. Quadra Assura MP CRT-D. Abbott; 2021. Available from: https://www.cardiovascular.abbott/us/en/hcp/products/cardiac-rhythm-management/cardiac-resynchronization-therapy/quadra-assura.html.
12. Pulse generator and cardiac resynchronization therapy. St. Jude Medical; 2021. Available from: https://manuals.sjm.com/search-form?re=north-america&cc=us&ln=en&ct=professional&cat=382d7298-7dc3-4301-a2fd-0c2739c21e4e&seg=59897b67-1f73-4b0d-9689-fe31a212e062&ipp=10&Page=1.
13. Physicians technical manual: pacemaker. Boston Scientific. 2018. Available from: https://www.bostonscientific.com/content/dam/elabeling/crm/359248-002_multi_PTM_en-US_S.pdf.
14. Medical procedure and EMI warnings and precautions for pacemakers. Medtronic Manuals: region; 2016. Available from: https://manuals.medtronic.com/manuals/main/.
15. Chapman KB, Schirripa F, Yousef T, Deygoo J, van Helmond N. Lumbar radiofrequency ablation interfering with S1 dorsal root ganglion stimulation systems: experience from two cases. Pain Pract. 2020;20(7):780–786. doi:10.1111/papr.12901
16. Daroff R, Jankovic J, Mazziotta JC, Pomeroy SL. Bradley’s Neurology in Clinical Practice. Elsevier; 2021.
17. Abdullah N, Muir C, Eldrige JS, Pingree MJ, Hagedorn JM. Peri-procedural management of implanted spinal cord stimulators in patients undergoing radiofrequency ablation: a case report and manufacturer-specific recommendations. Pain Pract. 2020;20(4):405–411.
18. Kanakarajan S. Radiofrequency techniques in pain management. Anaesth Intensive Care Med. 2013;14(12):543–545. doi:10.1016/j.mpaic.2013.09.011
19. Harned ME, Gish B, Zuelzer A, Grider JS. Anesthetic considerations and perioperative management of spinal cord stimulators: literature review and initial recommendations. Pain Physician. 2017;20(4):319–329. doi:10.36076/ppj.2017.329
20. Jeon HY, Shin JW, Kim DH, Suh JH, Leem JG. Spinal cord stimulator malfunction caused by radiofrequency neuroablation -A case report. Korean J Anesthesiol. 2010;59(Suppl(Suppl)):S226–228.
21. Abbott proclaim 3660 clinician manual. Abbott proclaim 3660 pulse generator clinician manual; 2023.Available from: https://all-guidesbox.com/manual/1481772/abbott-proclaim-3660-clinician-manual-80.html.
22. Boston scientific wavewriter alpha and wavewriter alpha prime systems information for prescribers; 2021. https://www.bostonscientific.com/content/dam/elabeling/nm/92619387-01_Rev_B_WW_%20Alpha_System_IFP_en-US_s.pdf.
23. Medtronic pain therapy: intellisTM, VantaTM, SequentiaTM LT, neurostimulation systems for pain therapy. Medtronic Manuals: Region. Available from: https://manuals.medtronic.com/manuals/main/.
24. Nevro information for prescribers. 2022. Available from: https://s28.q4cdn.com/260621474/files/doc_downloads/2022/12/16/10001223-Information-For-Prescribers-Rev-B_Final.pdf.
25. Miller RD. Miller’s Anesthesia.
26. Alper MH. Intrathecal morphine: a new method of obstetric analgesia? Anesthesiology. 1979;51(5):378–379. doi:10.1097/00000542-197911000-00002
27. Onofrio BM, Yaksh TL, Arnold PG. Continuous low-dose intrathecal morphine administration in the treatment of chronic pain of malignant origin. Mayo Clin Proc. 1981;56(8):516–520.
28. Nadherny W, Anderson B, Abd-Elsayed A. Perioperative and periprocedural care of patients with intrathecal pump therapy. Neuromodulation. 2019;22(7):775–780. doi:10.1111/ner.12880
29. Deer TR, Pope JE, Hayek SM, et al. The Polyanalgesic Consensus Conference (PACC): recommendations on intrathecal drug infusion systems best practices and guidelines. Neuromodulation. 2017;20(2):96–132.
30. Medtronic. Synchromed®, IsoMed®: Implantable Infusion Systems. Minneapolis, MN: Medtronic; 2017.
31. Samuelson CG, Shah R, Arul-Kumar S. Cooled radiofrequency ablation in combination with kyphoplasty for treating primary and metastatic osteolytic disease [11689]. Neuromodulation. 2017;20(7):e276–e277.
32. Bruel B, Viswanathan A. Neurodestruction and neuromodulation for the treatment of intractable pain from radiation induced brachial plexopathy. Neuromodulation. 2011;14(6):544.
33. Whitney P-S, Sturgess J. Anaesthetic considerations for patients with neurosurgical implants. BJA Education. 2015;16(7):230–235. doi:10.1093/bjaed/mkv049
34. Bolash R, Udeh B, Saweris Y, et al. Longevity and cost of implantable intrathecal drug delivery systems for chronic pain management: a retrospective analysis of 365 patients. Neuromodulation. 2015;18(2):
35. Naumann C, Erdine S, Koulousakis A, Van Buyten JP, Schuchard M. Drug adverse events and system complications of intrathecal opioid delivery for pain: origins, detection, manifestations, and management. Neuromodulation. 1999;2(2):92–107. doi:10.1046/j.1525-1403.1999.00092.x
36. Slonimski M, Abram SE, Zuniga RE. Intrathecal baclofen in pain management. Reg Anesth Pain Med. 2004;29(3):269–276. doi:10.1097/00115550-200405000-00014
37. Prager J, Deer T, Levy R, et al. Best practices for intrathecal drug delivery for pain. Neuromodulation. 2014;17(4):
38. Vercise Safety Information: Boston scientific. Available from: https://www.bostonscientific.com/en-US/products/deep-brain-stimulation-systems/vercise-tm-dbs/vercise_indications_safety_and_warnings.html.
39. Medtronic deep brain stimulation therapy implanted neurostimulators. Medtronic. Available from: https://www.medtronic.com/us-en/healthcare-professionals/therapies-procedures/neurological/deep-brain-stimulation.html.
40. Gardner J. A history of deep brain stimulation: technological innovation and the role of clinical assessment tools. Soc Stud Sci. 2013;43(5):707–728. doi:10.1177/0306312713483678
41. Rahimpour S, Kiyani M, Hodges SE, Turner DA. Deep brain stimulation and electromagnetic interference. Clin Neurol Neurosurg. 2021;203:106577. doi:10.1016/j.clineuro.2021.106577
42. Nutt JG, Anderson VC, Peacock JH, Hammerstad JP, Burchiel KJ. DBS and diathermy interaction induces severe CNS damage. Neurology. 2001;56(10):1384–1386. doi:10.1212/WNL.56.10.1384
43. Yeoh TY, Manninen P, Kalia SK, Venkatraghavan L. Anesthesia considerations for patients with an implanted deep brain stimulator undergoing surgery: a review and update. Can J Anaesth. 2017;64(3):308–319. doi:10.1007/s12630-016-0794-8
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.