Back to Journals » Journal of Multidisciplinary Healthcare » Volume 15
The Added Value of a Multidisciplinary Clinic for Systemic Autoinflammatory Diseases
Authors Zinterl C, Costa-Reis P, Esteves IC, Marques JG ![]() , Sousa AB, Fonseca JE, Oliveira Ramos F
, Sousa AB, Fonseca JE, Oliveira Ramos F
Received 16 December 2021
Accepted for publication 5 April 2022
Published 4 May 2022 Volume 2022:15 Pages 999—1010
DOI https://doi.org/10.2147/JMDH.S351546
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Carolina Zinterl,1– 3 Patrícia Costa-Reis,1,4,5 Isabel Castro Esteves,5 José Gonçalo Marques,5 Ana Berta Sousa,5,6 João Eurico Fonseca,2,3 Filipa Oliveira Ramos1– 3
1Pediatric Rheumatology Unit, Hospital de Santa Maria, Centro Hospitalar Universitário Lisboa Norte, Centro Académico de Medicina de Lisboa, Lisbon, Portugal; 2Rheumatology Research Unit, Instituto de Medicina Molecular, Faculdade de Medicina, Universidade de Lisboa, Centro Académico de Medicina de Lisboa, Lisbon, Portugal; 3Rheumatology Department, Hospital de Santa Maria, Centro Hospitalar Universitário Lisboa Norte, Centro Académico de Medicina de Lisboa, Lisbon, Portugal; 4Faculdade de Medicina, Universidade de Lisboa, Centro Académico de Medicina de Lisboa, Lisbon, Portugal; 5Pediatrics Department, Hospital de Santa Maria, Centro Hospitalar Universitário Lisboa Norte, Centro Académico de Medicina de Lisboa, Lisbon, Portugal; 6Department of Basic Immunology, Faculty of Medicine, Universidade de Lisboa, Centro Académico de Medicina de Lisboa, Lisbon, Portugal
Correspondence: Carolina Zinterl, Serviço de Reumatologia, Centro Hospitalar Lisboa Norte, Hospital de Santa Maria, EPE, R. Prof. Egas Moniz, Lisboa, 1700, Portugal, Tel +351 217805139, Email [email protected]
Background: Systemic autoinflammatory diseases (SAID) are characterized by inappropriate activation of the innate immune system and include not only monogenic periodic fever syndromes but also multifactorial conditions. As SAID are rare and represent a diagnostic challenge, a multidisciplinary approach is important to ensure successful diagnosis and adequate follow-up of these patients.
Objective: To describe the organization of our multidisciplinary SAID clinic and to characterize our clinical experience, highlighting the benefits of multidisciplinary team management.
Methods: Our SAID clinic takes place monthly and is managed by pediatric rheumatologists closely collaborating with pediatricians specialized in infectious diseases and immunodeficiencies and one medical geneticist. Patients’ data are systematically incorporated in the Rheumatic Diseases Portuguese Register (Reuma.pt). Biological samples are stored in a biobank. We describe our clinical experience based on SAID patients registered into Reuma.pt/SAID between July 2011 and June 2020.
Results: We have registered 176 patients, with a median age of disease onset of 3.1 ± 4.4 years and median age at disease diagnosis of 4.7 ± 4.0 years. Most patients were diagnosed with periodic fever, aphthous stomatitis, pharyngitis, adenitis syndrome (PFAPA) (n=133), 20 with undefined SAID (uSAID) and 13 with monogenic SAID, including familial Mediterranean fever (FMF) (n=5), tumor necrosis factor receptor-associated periodic syndrome (TRAPS) (n=1), cryopyrin-associated periodic disease (CAPS) (n=1), and hyperimmunoglobulin D syndrome/mevalonate kinase deficiency (HIDS/MKD) (n=2). A genetic test was performed in 31 patients (18%), and in 26% of these a mutation responsible for the phenotype was found. Thirty-four patients (19%) achieved remission.
Conclusion: FMF was the most common monogenic SAID and the percentage of patients with an identified causal mutation was low. A structured electronic clinical record coupled with a biobank and a multidisciplinary approach are crucial to ensure successful diagnosis and adequate follow-up of these patients.
Keywords: pediatrics, rheumatology, systemic autoinflammatory syndromes
Introduction
Systemic autoinflammatory diseases (SAID) are characterized by inappropriate activation of the innate immune system, classically including periodic fever syndromes, but also autoinflammatory disorders that do not present with fever as a major manifestation. Except for periodic fever with aphthous stomatitis, pharyngitis, and adenitis (PFAPA syndrome), SAID are rare.1–4
SAID most frequently have their onset in childhood and the differential diagnosis often represents a challenge.5 The diagnosis of monogenic SAID is challenging due to genetic heterogeneity and phenotypic overlap.3,6,7 Manifestations include fever, rash, serositis, arthritis, meningitis, and uveitis among others.3,6,8 In other SAIDs, such as PFAPA syndrome, multiple genes might be involved and there is no single genetic cause.9 Due to their extreme rarity and complexity, and because of their demanding management, achieving a specific SAID diagnosis is often very difficult. Patients and their families can face difficulties accessing the appropriate clinical services and undergo long, stressful, and inconclusive diagnostic journeys. The use of Next Generation Sequencing (NGS) technologies has proven to be a successful strategy to answer these unmet medical needs by inducing a marked acceleration in rare disease gene discovery and effective mutation screening, thus improving genetic SAID diagnosis, prompting further clinical assessments, providing insight into biological mechanisms, and increasing therapeutic opportunities. Indeed, NGS was revolutionary for SAID diagnosis, since it allowed the simultaneous analysis of different genes and the detection of somatic mosaicism, as demonstrated in patients presenting a typical clinical phenotype, but negative for germ-line mutations. However, in some other cases, namely in the so-called undefined SAIDs (uSAID), the NGS based gene panel sequencing results can be inconclusive or even misleading. Furthermore, a proportion of uSAID are considered multifactorial diseases, and thus particularly challenging as they are inherently complex.
As multidisciplinary patient care combines expertise from different medical areas and provides the most differentiated care, it has been acknowledged as the most adequate model in the management of complex chronic disease, as are the SAID.10,11
The aim of this experience-based manuscript is to highlight the benefits of multidisciplinary team management of patients with SAID, describing the general functioning of our SAID outpatient clinic, coupled with the Rheumatic Diseases Portuguese Register (Reuma.pt)/SAID and with the institutional Biobank. It was also an opportunity to retrospectively review the demographics, clinical entities, and treatment of all patients with SAID followed at the clinic.
This study was approved by the Scientific Committee of Reuma.pt and by the Ethics Committee of Lisbon Academic Medical Center. Reuma.pt was approved by the National Data Protection Authority and by the local ethics committees of the participating centers. The study was conducted according to the Declaration of Helsinki.
Materials and Methods
Systemic Autoinflammatory Diseases Outpatient Clinic
Since October 2012, the Rheumatology Department of Centro Hospitalar Universitário Lisboa Norte (CHLN) has a dedicated multidisciplinary clinic for patients with SAID. The CHLN-SAID clinic takes place monthly and is managed by two pediatric rheumatologists, two pediatricians specialized in Infectious Diseases and Immunodeficiencies and one medical geneticist. It is a multidisciplinary clinic, in which these medical specialties collaborate closely, sharing the care of these patients. The whole team regularly discusses cases of suspected SAID to define the best approach to genetic diagnosis and to make treatment decisions. Focus areas recognized as crucial in the development of the CHLN-SAID clinic are depicted in Table 1.
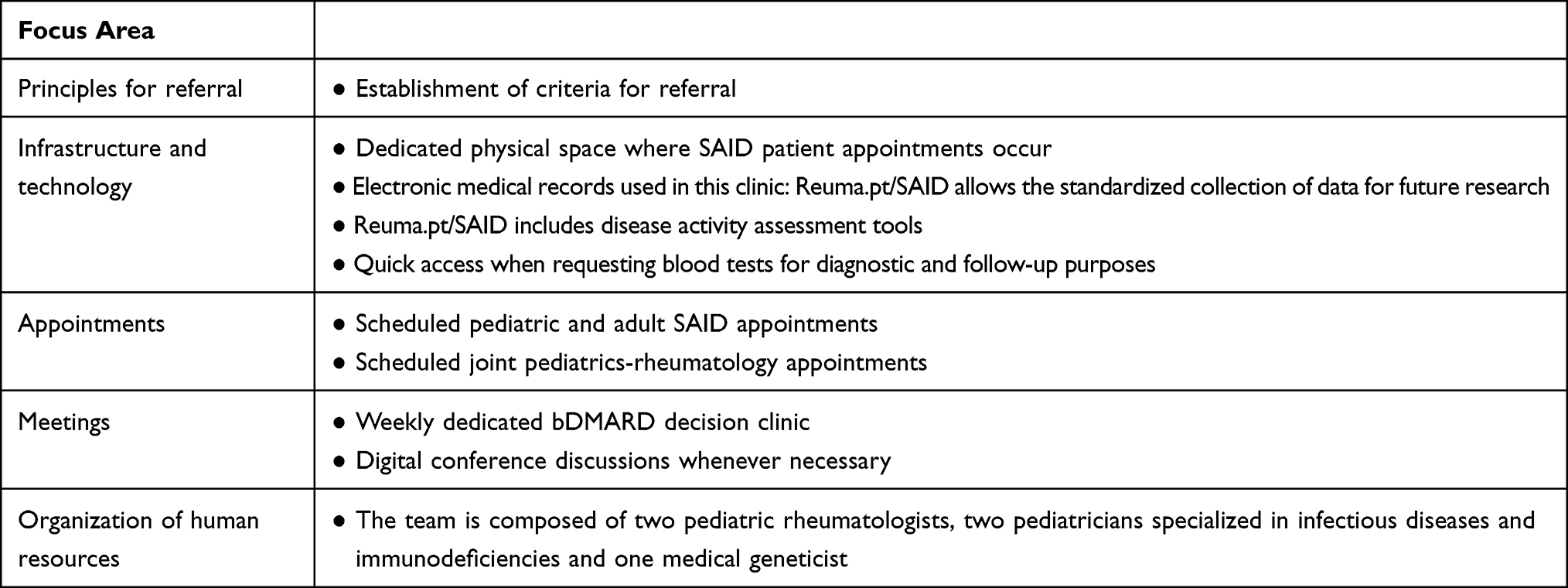 |
Table 1 Focus Areas of the CHLN-SAID Clinic |
Assessment Tools
Data from every patient with a confirmed diagnosis of SAID is registered into the Reuma.pt/SAID module. Registered patients are required to sign an informed consent. Reuma.pt was created in June 2008, and has since been an essential tool in the clinical practice of Portuguese rheumatologists, allowing the structured follow-up of patients with various systemic rheumatic diseases.12,13
The database information security was approved by the Portuguese National Commission for Data Protection and the ethics committees of the participating institutions. All identifiable data is encrypted, and an individual password is attributed to physicians, only allowing access to data related to their center.
In September 2013, a module specifically developed for SAIDs was introduced in Reuma.pt and has since been used by Portuguese rheumatologists and pediatricians.
The Reuma.pt/SAID module guides the clinical appointment as it can be used as an electronic clinical record. Data captured by this registry are specific for SAID and include: 1) clinical data, including the diagnosis, symptom onset, clinical manifestations, fever characterization, genetic testing, classification criteria, family history, comorbidities, and source of referral (Supplementary Figures 1–4); 2) disease activity assessment, specifically the auto-inflammatory diseases activity index (AIDAI) (Supplementary Figure 5 and 6); 3) complementary diagnostic exams, including laboratory screening with serum amyloid A (Supplementary Figure 7); 4) vaccination status and tuberculosis screening; 5) therapy and adverse events (Supplementary Figure 8). Data is collected directly from patients attending regular outpatient appointments and is captured immediately into Reuma.pt or, in selected conditions, obtained from clinical notes. Participants are not asked to attend extra visits or interviews.
With this tool we can collect detailed clinical data, using an electronic clinical record linked to a SQL server database, with input screens specifically developed for autoinflammatory diseases, integrated on Reuma.pt.12
Categorical variables are described as frequencies and percentages and continuous variables as median ± interquartile range (IQR).
Genetic Testing in SAID
Biological samples from all SAID patients (whole blood, serum, plasma, DNA, RNA) are stored in Biobanco-IMM, Lisbon Academic Medical Centre, since July 2013, authorized by the Ethics Committee of Lisbon Academic Medical Centre and the Portuguese commission for data protection.14 Informed consent and assent are requested.
A biobank is crucial to adequately collect and preserve biological samples from SAID patients for long periods of time, so they can be used for research, or to later update the diagnosis whenever novel SAIDs are described.
The genetic characterization of these patients is provided by the analysis of one or more SAID-related genes.5,14,15 This analysis is performed by a commercial laboratory with specific software either by Sanger sequencing for single genes, such as MEFV, TNFRSF1A, and MVK or by NGS for multigene panels associated with recurrent fever syndromes, which include the following genes: ADAR, AP1S3, ASAH1, CARD14, DDX58, ELANE, HAX1, IFIH1, IL10RA, IL10RB, IL1RN, IL36RN, LPIN2, MEFV, MVK, NLRC4, NLRP1, NLRP12, NLRP3, NOD2, OTULIN, PLCG2, PSMB8, PSTPIP1, RBCK1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, SLC29A3, TMEM173, TNFAIP3, TNFRSF11A, TNFRSF1A, TREX1. This workflow allows us to detect already described as well as not yet reported genetic variants. If the identified variants are pathogenic or probably pathogenic, we propose family studies according to the underlying transmission model. This approach allows us to identify affected individuals who will profit from special assistance, and to refer carriers at risk of having affected offspring to genetic counselling. In contrast, if the variant is of uncertain significance, segregation studies are made to help clarify its clinical importance, although they cannot be used for clinical guidance.
If a diagnosis is still not established after NGS for multigene panels, especially if it will affect the therapeutic decision, we proceed to whole exome sequencing in centers with experience in SAID with dedicated bioinformatic scientists. We are currently making efforts to collaborate with international centers where somatic mutations associated with SAID are being investigated.
Results
Clinical Characteristics
The most frequent subtype of SAID was PFAPA (n=133, 76%), followed by uSAID (n=20, 11%). Figure 1 shows diagnoses found in adult and pediatric patients followed at our clinic. Consanguinity was present in 2 families, and 87 patients had a positive family history, especially Hyperimmunoglobulin D Syndrome/Mevalonate Kinase Deficiency (HIDS/MKD) and PFAPA patients, with a family history of recurrent tonsillitis in all HIDS/MKD patients (n=2, 100%) and in approximately half of the PFAPA patients (n=72, 54%).
 |
Figure 1 Diagnoses of adult and pediatric patients with SAID followed at the CHLN-SAID clinic. Abbreviations: uSAID, undefined systemic autoinflammatory disease; NLRP12, NLRP12-related autoinflammatory disease; HIDS/MKD, hyperimmunoglobulin D syndrome/mevalonate-kinase deficiency; CAMPS, CARD14-mediated psoriasis; DADA2, deficiency of adenosine deaminase 2; TRAPS, tumor necrosis factor receptor associated periodic syndrome; CAPS, cryopyrin associated periodic syndromes; FMF, familial Mediterranean fever; SAPHO, synovitis, acne, pustulosis, hyperostosis, osteitis syndrome; CNO, chronic recurrent multifocal osteomyelitis; PFAPA, periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis syndrome. |
The median of the delay between disease onset and diagnosis was 2.3 ± 2.8 years.
Genetic testing was performed in 32 (18%) patients, and in 9 (28%) of them a mutation was found, as shown in Table 2.
 | 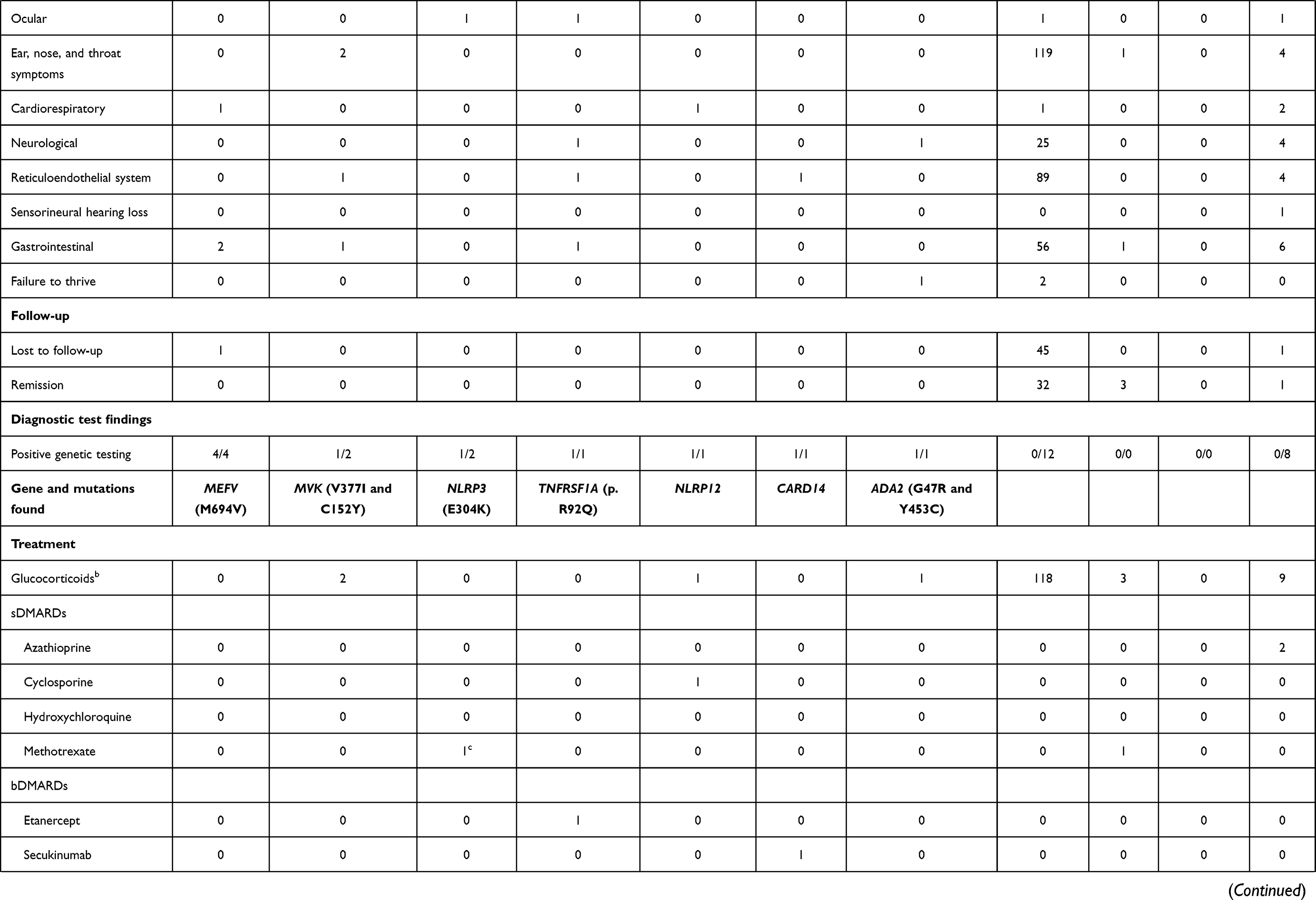 |  |
Table 2 Demographics, Clinical Manifestations, Diagnostic Test Findings and Treatment of the Patients Followed at the CHLN-SAID Clinic |
Forty-seven patients were lost to follow-up (27%), 36 (20%) patients were discharged from our clinic due to sustained remission and 4 (2%) were transferred to their hometown hospital.
No deaths were reported.
Table 2 details clinical characteristics for each SAID sub-type.
Monogenic Autoinflammatory Diseases
In total, 13 (13/176, 7%) patients were diagnosed with a monogenic autoinflammatory disease, 9 (9/13, 69%) of whom had genetic results consistent with the clinical diagnosis. The median age of diagnosis was 4.0 ± 7.4 years with a median time gap between symptom onset and diagnosis of 3.9 ± 16.5 years. Mutations in the most relevant genes were not found in one patient (1/13; 8%), two patients (2/13; 15%) were not genetically tested, and one patient (1/13; 8%) still awaits genetic results. Clinical manifestations of these patients are reported in Table 2. Although the AIDAI score16 is included in the Reuma.pt/SAID module, the results are not specified due to the low number of cases it applies to.
The diagnosis of FMF was made in 5 patients (5/176; 3%) – 3 children and 2 adults. Four of these patients carried the Met694Val mutation of MEFV, in two of them (father and daughter) an autosomal dominant pattern of inheritance was present; another patient had compound heterozygosity for the MEFV mutation (M694V and R202Q). One patient was Turkish and the other four Portuguese. No cases of consanguinity were found.
One adult patient (1/176; 1%) presented with Tumor Necrosis Factor Receptor-Associated Periodic Syndrome (TRAPS), and this patient was positive for the low-penetrance p.R92Q variant. The patient’s father also had this variant and symptoms of TRAPS.
Our registry included two adult patients (2/176; 1%) diagnosed with CAPS: one patient with Familial Cold Autoinflammatory Syndrome (FCAS), and one patient with Chronic Infantile Neurological Cutaneous and Articular (CINCA). The FCAS patient had no identifiable pathogenic or likely pathogenic variant in the NLRP3 gene, but the CINCA patient carried the NLRP3 mutation E304K.
Two children (2/176; 1%) diagnosed with HIDS/MKD were registered. One of them had compound heterozygosity for two mutations in the MVK gene – V377I and C152Y. The genetic results of the second patient were still unavailable. A family history suggestive of HIDS/MKD was absent.
One adult patient (1/176; 1%) with Deficiency of Adenosine Deaminase 2 (DADA2) with two heterozygous pathogenic variants in the ADA2 gene, namely G47R and Y453C, was included. This patient presented with painful erythematous subcutaneous nodules, asymmetric polyarthritis, livedo reticularis, digital necrosis, involvement of the central and peripheral nervous system, as well as gastrointestinal bleeding.
There was also one child (1/176; 1%) with NLRP12-related SAID (NLRP12-rSAID) and one (1/176; 1%) with CARD14 mediated psoriasis (CAMPS) with heterozygosity for a variant of exon 3 on CARD14 (c277A>G) with unknown clinical significance.
Multifactorial SAID
PFAPA was diagnosed in 133 patients (133/176; 76%). The median age at disease onset was 2.6 ± 3.0 years with 107 (80%) patients first showing symptoms before age 5. No case of consanguinity was identified, and 57 (57/133, 43%) patients reported a family history of recurrent tonsillitis. Remission occurred after a median of disease duration of 6.8 ± 5.1 years. Twelve (12/133; 9%) patients were screened for genetic defects by Sanger sequencing for genes MEFV, TNFRSF1A, and MVK, because the phenotype did not exactly fit into a specific monogenic syndrome – no mutations were found. Apart from the typical manifestations of PFAPA, there were cases of abdominal pain (49/133; 37%), headache (25/133; 19%), myalgia (21/133; 16%), arthralgia (12/133; 9%), maculopapular rash (4/133; 3%), failure to thrive (2/133; 2%), splenomegaly (1/133; 1%) and conjunctivitis (1/133; 1%).
Six patients (6/176; 3%) with Chronic nonbacterial osteomyelitis (CNO) were registered and four adult patients (4/176; 2%) were diagnosed with Synovitis–Acne–Pustulosis–Hyperostosis–Osteitis (SAPHO) syndrome, with a median age at disease onset of 12.5 ± 3.4 and 35.8 ± 19.3 years, respectively.
Undefined SAID
20 patients (20/176; 11%) – 17 children and 3 adults – were enrolled in the category of uSAID because they had clinical manifestations consistent with SAID after exclusion of either (1) other defined inflammatory conditions (ie rheumatic diseases, inflammatory bowel diseases) or other mimicking conditions (ie hematological diseases, tumors, immune deficiencies) and (2) defined autoinflammatory diseases.
Median age at which first manifestations occurred was 3.8 ± 6.7 years. Eight patients (8/20; 42%) were screened for at least one autoinflammatory disease-associated gene – in most of them by Sanger sequencing of MEFV, TNFRSF1A and MVK and in the remainder by multigene panels –, but no mutations were found. In the past, if there was strong suspicion of a specific monogenic disease, our center first proceeded to genetic screening of MEFV, TNFRSF1A and MVK. Nowadays all patients are screened with a multigene panel.
Treatment and Outcomes
For PFAPA, the most prescribed therapeutic regimen was a single dose of glucocorticoid (1mg/kg) at the beginning of a crisis. Colchicine was also prescribed to one patient due to shortening of intercritical periods and aphthae but had to be interrupted after 1 month because of gastrointestinal intolerance. Of twenty-seven PFAPA patients (27/133; 20%) who underwent tonsillectomy, twenty (20/133, 27%) achieved complete remission, three (3/133, 2%) achieved incomplete response (fever without pharyngitis) and no response was seen in four (4/133, 3%) of them.
For three of the FMF patients (3/5; 60%), the prescribed drug was colchicine 1 mg/day which led to symptom remission.
Glucocorticoids were also used in all patients with HIDS/MKD (2/2; 100%), 9 patients with uSAID (9/20; 45%), in the NLRP12-rSAID and DADA2 patients, as well as in 3 CNO patients (3/6; 50%). Three CNO patients (3/6; 50%) were treated with bisphosphonates.
As detailed in Table 2, five patients (5/176; 3%) were on synthetic disease-modifying anti-rheumatic drugs (sDMARDs) and five patients (5/176; 3%) were treated with biologic disease-modifying anti-rheumatic drugs (bDMARDs).
Three patients with CNO (3/6; 50%) and one with uSAID (1/20, 5%) also achieved sustained remission.
Discussion
Most of our SAID cases were PFAPA. When comparing our registry to other studies, the demographic and clinical characteristics are similar, including gender distribution, age of disease onset, age at diagnosis and occurrence of abdominal pain, found in almost 70% of our patients.17
Out of 48 patients who were lost to follow-up, 45 were PFAPA cases, likely due to resolution of symptoms – expected in PFAPA18,19 –, after which patients simply stopped attending visits.
Frequently, the only clinical manifestation in young infants with SAID was recurrent fever. Genetic testing was not immediately performed in these patients, since most of them have a benign clinical course and respond to prednisolone, eventually developing criteria for PFAPA. Once the diagnosis of PFAPA is established, each patient is redirected to their individual doctor.
Of the 13 patients with the diagnosis of a monogenic SAID, FMF was the most frequent one, which is in line with other studies.14,20
Regarding genetic diagnosis, one HIDS/MKD patient carried the V377I variant in the MVK gene, which is the most common, and it was associated with a mild phenotype and some residual MVK activity. As is the case in our patient, this variant is present in a compound heterozygous state in most HIDS/MKD patients.14,21
The TRAPS patient carried the R92Q variant in the TNFRSF1A gene, which is a common variant in Caucasian populations but not undoubtedly considered to be a true mutation – it could represent a low‐penetrance variant, a functional polymorphism, or a susceptibility factor for inflammation, as it is associated with adult-onset and milder clinical features, as well as lower risk of amyloidosis.22,23
In the case of our patient, however, clinical manifestations suggestive of TRAPS started at 1 year of age.
Four of the monogenic SAID patients had non confirmatory genetic testing, which is expectable in up to 60% of patients with suspected SAID, despite implementation of NGS into routine diagnostic practice of SAID.24,25 This is probably due to the limited number of tested genes, unknown gene mutations that may not be detected by the available molecular technology, genetic heterogeneity and/or a complex mode of inheritance.20,26 Also, misdiagnosis cannot be excluded as these diseases have very similar phenotypes, which makes their clinical diagnosis difficult. It is important to continue to improve the efficiency of SAID diagnosis based on clear criteria to guide genetic screening.5,25
Regarding CAPS, it is well documented that some patients with a classical phenotype of this SAID may not have mutations in NLRP3 gene, which can be explained by mosaicism.15,27,28 Interestingly, dominant inheritance is evident in about 75% of patients with Muckle-Wells-syndrome and FCAS, whereas CINCA is usually due to de novo mutations and somatic mosaicism occurring during fetal growth, which may explain late onset of the disease.29–33
All mutations found in our patients are already registered as pathogenic in the Infevers database (https://infevers.umai-montpellier.fr/web/).34
Offering clinical expertise with a fully dedicated and multidisciplinary team is potentiated by the Reuma.pt/SAID module that guides the clinical appointment, minimizing medical error. This tool presents two simultaneous advantages: 1) it improves medical practice, and 2) it provides the center with precious, accurate and comprehensive clinical data on all patients, allowing high quality clinical and translational research.
Furthermore, our CHLN-SAID clinic enables a smooth transition from childhood to adulthood in terms of medical care, which is guaranteed by the rheumatologists of the team that continue to follow-up these patients throughout adulthood.34
The strength of this retrospective analysis is the structured data collection based on the Reuma.pt/SAID registry. Limitations of our work are the relatively short period of functioning of this clinic as well as some missing data in our registry. Another limitation is the fact that genetic testing was performed by different commercial laboratories using different gene panels, which may have underdiagnosed some genetic variants (especially when only the MEFV, TNFRSF1A and MVK trio was analyzed). However, we are currently working on a standardized multigene panel that will be used for all future patients followed at our clinic. Defining performance indicators and evaluating them regularly will improve the multidisciplinary team functioning.
A formalized and structured multidisciplinary clinic is vital to increase the chances of successful management of patients with SAID, and thus improve their prognosis. Maintaining this structure demands coordination, literature update, teamwork, and institutional willingness to support activities provided by a higher physician-to-patient ratio. This latter aspect has been recognized as a barrier to the development of this type of service.35,36 However, establishing a multidisciplinary clinic dedicated to SAID allows to gather expertise and broaden medical knowledge. In addition, this structure helps to settle patient pathways for diagnosis and follow-up, promoting systematic monitoring and the use of treatment protocols, crucial for the management of these infrequent diseases.37 This improves quality of care and opens research avenues, ultimately increasing cost-effectiveness. The goal of this paper was to share our experience with the organization of a SAID multidisciplinary clinic, coupled with an electronic registry and a biobank.
Conclusion
In our clinic FMF was the most common monogenic condition and overall, the percentage of patients with an identified causal mutation was low.
SAID are a continuously expanding spectrum of diseases and represent a diagnostic challenge. A dedicated multidisciplinary clinic, where geneticists, pediatricians, rheumatologists, specialists in Infectious Diseases and Immunodeficiencies collaborate closely, improves diagnostic accuracy, builds clinical expertise, and allows for close monitoring of patients with these rare diseases.
The use of a structured electronic clinical record as an integrating part of our clinical activity, linked to a biobank, has been crucial for research, genetic characterization, and healthcare planning.
Abbreviations
ADA2, adenosine deaminase 2 gene; AIDAI, auto-inflammatory diseases activity index; bDMARDs, biological disease-modifying antirheumatic drugs; CAMPS, CARD14 mediated psoriasis; CAPS, cryopyrin-associated periodic disease; CARD14, caspase recruitment domain-containing protein 14 gene; CHLN, Centro Hospitalar Universitário Lisboa Norte; CINCA, chronic infantile neurological cutaneous and articular syndrome; CNO, chronic non-bacterial osteomyelitis syndrome; DADA2, deficiency of adenosine deaminase 2 syndrome; FCAS, familial cold autoinflammatory syndrome; FMF, familial Mediterranean fever; HIDS/MKD, hyperimmunoglobulin D syndrome/mevalonate kinase deficiency; IQR, interquartile range; MEFV, Mediterranean fever gene; MVK, mevalonate-kinase gene; NGS, next generation sequencing; NLRP3, NOD-like receptor 3 gene; NLRP12, NOD-like receptor 12 gene; NLRP12-rAID, NLRP12-related autoinflammatory disease; N/A, not applicable; PFAPA, periodic fever, aphthous stomatitis, pharyngitis, adenitis syndrome; Reuma.pt, rheumatic diseases Portuguese register; SAID, systemic autoinflammatory diseases; SAPHO, synovitis–acne–pustulosis–hyperostosis–osteitis syndrome; sDMARDs synthetic disease-modifying antirheumatic drugs; TNFRSF1A, tumor necrosis factor receptor superfamily member 1A gene; TRAPS, tumor necrosis factor receptor-associated periodic syndrome; uSAID, undefined SAID.
Disclosure
Dr Carolina Zinterl reports grants from Novartis, during the conduct of the study. Prof. Dr. Patrícia Costa-Reis reports personal fees from Kyowa Kirin, outside the submitted work. The authors report no other conflicts of interest in this work. The abstract of this paper was presented at the 23rd Portuguese Rheumatology Congress as a poster presentation with interim findings. The poster’s abstract was published in “Poster Abstracts” in Acta reumatologica portuguesa: https://www.arprheumatology.com/archive_detail.php?id=237.38 This project had the financial contribution of Novartis.
References
1. Thomas KT, Feder JHM, Lawton AR, Edwards KM. Periodic fever syndrome in children. J Pediatr. 1999;135(1):15–21. doi:10.1016/S0022-3476(99)70321-5
2. Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell. 2010;140(6):784–790. doi:10.1016/j.cell.2010.03.002
3. Russo RA, Brogan PA. Monogenic autoinflammatory diseases. Rheumatology. 2014;53(11):1927–1939. doi:10.1093/rheumatology/keu170
4. Toplak N, Frenkel J, Ozen S, et al. An international registry on autoinflammatory diseases: the Eurofever experience. Ann Rheum Dis. 2012;71(7):1177–1182. doi:10.1136/annrheumdis-2011-200549
5. Rowczenio DM, Lachmann HJ. How to prescribe a genetic test for the diagnosis of autoinflammatory diseases? La Presse Méd. 2019;48(1):e49–e59. doi:10.1016/j.lpm.2018.08.015
6. Kallinich T, Gattorno M, Grattan C, et al. Unexplained recurrent fever: when is autoinflammation the explanation? Allergy. 2013;68(3):285–296. doi:10.1111/all.12084
7. Gattorno M, Hofer M, Federici S, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis. 2019;78(8):1025–1032. doi:10.1136/annrheumdis-2019-215048
8. De Jesus AA, Goldbach-Mansky R. Monogenic autoinflammatory diseases: concept and clinical manifestations. Clin Immunol. 2013;147(3):155–174. doi:10.1016/j.clim.2013.03.016
9. Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol. 2009;27:621–668. doi:10.1146/annurev.immunol.25.022106.141627
10. Ugwumadu L, Chakrabarti R, Williams-Brown E, et al. The role of the multidisciplinary team in the management of deep infiltrating endometriosis. Gynecol Surg. 2017;14(1):1–4. doi:10.1186/s10397-017-1018-0
11. Cobo-Ibáñez T, Villaverde V, Seoane-Mato D, et al. Multidisciplinary dermatology–rheumatology management for patients with moderate-to-severe psoriasis and psoriatic arthritis: a systematic review. Rheumatol Int. 2016;36(2):221–229. doi:10.1007/s00296-015-3377-z
12. Canhao H, Faustino A, Martins F, Fonseca JE, Nero P, Branco JC. Reuma. pt-the rheumatic diseases Portuguese register. Acta Reumatol Port. 2011;36(1):45–56.
13. Santos MJ, Canhão H, Faustino A, Fonseca JE. Reuma. pt–a case study. Acta Médica Portuguesa. 2016;29(2):83–84. doi:10.20344/amp.7243
14. Martorana D, Bonatti F, Mozzoni P, Vaglio A, Percesepe A. Monogenic autoinflammatory diseases with Mendelian inheritance: genes, mutations, and genotype/phenotype correlations. Front Immunol. 2017;8(344). doi:10.3389/fimmu.2017.00344
15. Welzel T, Kuemmerle-Deschner JB. Diagnosis and management of the cryopyrin-associated periodic syndromes (CAPS): what do we know today? J Clin Med. 2021;10(1):128. doi:10.3390/jcm10010128
16. Piram M, Koné-Paut I, Lachmann HJ, et al. Validation of the auto-inflammatory diseases activity index (AIDAI) for hereditary recurrent fever syndromes. Ann Rheum Dis. 2014;73(12):2168–2173. doi:10.1136/annrheumdis-2013-203666
17. Vanoni F, Caorsi R, Aeby S, et al. Towards a new set of classification criteria for PFAPA syndrome. Pediatric Rheumatol. 2018;16(1):1–4. doi:10.1186/s12969-018-0277-2
18. Wekell P. Periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis syndrome–PFAPA syndrome. La Presse Médicale. 2019;48(1):e77–e87. doi:10.1016/j.lpm.2018.08.016
19. Georgin-Lavialle S, Fayand A, Rodrigues F, Bachmeyer C, Savey L, Grateau G. Autoinflammatory diseases: state of the art. La Presse Méd. 2019;48(1):e25–e48. doi:10.1016/j.lpm.2018.12.003
20. Yao Q, Lacbawan F, Li J. Adult autoinflammatory disease frequency and our diagnostic experience in an adult autoinflammatory clinic.
21. van der Hilst JC, Bodar EJ, Barron KS, et al. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine. 2008;87(6):301–310. doi:10.1097/MD.0b013e318190cfb7
22. Grandemange S, Cabasson S, Sarrabay G, et al. Clinical dose effect and functional consequences of R92Q in two families presenting with a TRAPS/PFAPA‐like phenotype. Mol Genet Genomic Med. 2017;5(2):110–116. doi:10.1002/mgg3.229
23. Ravet N, Rouaghe S, Dode C, et al. Clinical significance of P46L and R92Q substitutions in the tumour necrosis factor superfamily 1A gene. Ann Rheum Dis. 2006;65(9):1158–1162. doi:10.1136/ard.2005.048611
24. Savic S, Dickie LJ, Battellino M, McDermott MF. Familial Mediterranean fever and related periodic fever syndromes/autoinflammatory diseases. Curr Opin Rheumatol. 2012;24(1):103–112. doi:10.1097/BOR.0b013e32834dd2d5
25. Karacan İ, Balamir A, Uğurlu S, et al. Diagnostic utility of a targeted next-generation sequencing gene panel in the clinical suspicion of systemic autoinflammatory diseases: a multi-center study. Rheumatol Int. 2019;39(5):911–919. doi:10.1007/s00296-019-04252-5
26. Federici S, Sormani MP, Ozen S, et al. Evidence-based provisional clinical classification criteria for autoinflammatory periodic fevers. Ann Rheum Dis. 2015;74(5):799–805. doi:10.1136/annrheumdis-2014-206580
27. Stoffels M, Kastner DL, Dogs O. New tricks: monogenic autoinflammatory disease unleashed. Annu Rev Genomics Hum Genet. 2016;17(1):245–272. doi:10.1146/annurev-genom-090413-025334
28. Tanaka N, Izawa K, Saito MK, et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum. 2011;63(11):3625–3632. doi:10.1002/art.30512
29. Rowczenio DM, Gomes SM, Aróstegui JI, et al. Late-onset cryopyrin-associated periodic syndromes caused by somatic NLRP3 mosaicism—UK single center experience. Front Immunol. 2017;8:1410. doi:10.3389/fimmu.2017.01410
30. Saito M, Nishikomori R, Kambe N, et al. Disease-associated CIAS1 mutations induce monocyte death, revealing low-level mosaicism in mutation-negative cryopyrin-associated periodic syndrome patients. Blood. 2008;111(4):2132–2141. doi:10.1182/blood-2007-06-094201
31. Nakagawa K, Gonzalez-Roca E, Souto A, et al. Somatic NLRP3 mosaicism in Muckle-Wells syndrome. A genetic mechanism shared by different phenotypes of cryopyrin-associated periodic syndromes. Ann Rheum Dis. 2015;74(3):603–610. doi:10.1136/annrheumdis-2013-204361
32. Kuemmerle-Deschner JB, Lohse P, Koetter I, et al. NLRP3 E311K mutation in a large family with Muckle-Wells syndrome-description of a heterogeneous phenotype and response to treatment. Arthritis Res Ther. 2011;13(6):1–9. doi:10.1186/ar3526
33. Mensa-Vilaró A, García-Morato MB, de la Calle-martin O, et al. Unexpected relevant role of gene mosaicism in patients with primary immunodeficiency diseases. J Allergy Clin Immunol. 2019;143(1):359–368. doi:10.1016/j.jaci.2018.09.009
34. Foster HE, Minden K, Clemente D, et al. EULAR/PReS standards and recommendations for the transitional care of young people with juvenile-onset rheumatic diseases. Ann Rheum Dis. 2017;76(4):639–646. doi:10.1136/annrheumdis-2016-210112
35. Singh-Grewal D. Multidisciplinary paediatric rheumatology services in Australia and New Zealand. Med J Aust. 2017;206(2):96–97. doi:10.5694/mja16.00710
36. Leal I, Romão VC, Mano S, et al. A non-infectious uveitis multidisciplinary clinic in a tertiary referral center: clinical impact and added value. J Multidiscip Healthc. 2021;14:695. doi:10.2147/JMDH.S292981
37. Rosell L, Wihl J, Hagberg O, Ohlsson B, Nilbert M. Function, information, and contributions: an evaluation of national multidisciplinary team meetings for rare cancers. Rare Tumors. 2019;11:2036361319841696. doi:10.1177/2036361319841696
38. Zinterl C, Reis PC, Esteves IC, et al. The experience of a multidisciplinary clinic for systemic autoinflammatory diseases. Acta Reumatol Port. 2021;46(4):104.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.