")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 18
The Activation of M1 Macrophages is Associated with the JNK-m6A-p38 Axis in Chronic Obstructive Pulmonary Disease
Authors Hu T, Pang N , Li Z , Xu D , Jing J, Li F , Ding J, Wang J , Jiang M
Received 23 May 2023
Accepted for publication 29 September 2023
Published 6 October 2023 Volume 2023:18 Pages 2195—2206
DOI https://doi.org/10.2147/COPD.S420471
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Min Zhang
Tingting Hu,1,* Nannan Pang,2,* Zheng Li,1 Dan Xu,1 Jing Jing,1 Fengsen Li,1 Jianbing Ding,3 Jing Wang,1 Min Jiang1
1Xinjiang Laboratory of Respiratory Disease Research, Traditional Chinese Medicine Hospital Affiliated to Xinjiang Medical University, Urumqi, 830000, People’s Republic of China; 2CAS Key Laboratory of Bio-Medical Diagnostics, Suzhou Institute of Biomedical Engineering and Technology, Chinese Academy of Sciences, Suzhou, 215163, People’s Republic of China; 3Department of Immunology, College of Basic Medicine, Xinjiang Medical University, Urumqi, 830000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jing Wang; Min Jiang, Xinjiang Laboratory of Respiratory Disease Research, Traditional Chinese Medicine Hospital Affiliated to Xinjiang Medical University, Urumqi, People’s Republic of China, Tel +86-13999908413 ; +86-13999162873, Email [email protected]; [email protected]
Background: Excessive activation of M1 macrophages affects the chronic inflammatory response of the airways and leads to the development of chronic obstructive pulmonary disease (COPD). Therefore, it needs to be closely monitored and investigated. MAPK signaling pathway is involved in the activation of M1 macrophages, and N6-methyladenosine (m6A) is involved in the pathogenesis of COPD. However, it is unknown whether activation of the MAPK signaling pathway is mediated by m6A in M1 macrophages in COPD.
Methods: The GEO data were analyzed using bioinformatics techniques to assess the differences between COPD and healthy individuals in the levels of M1 macrophages, their secreted cytokines, and m6A regulators. The MAPK signaling pathway was significantly enriched in the list of differentially regulated genes between COPD and healthy individuals. We further analyzed the correlation between M1 macrophages, m6A, and the MAPK signaling pathway. Next, blood samples from COPD and healthy individuals were collected and analyzed by using flow cytometry, ELISA, and RT-PCR. Western blotting was performed using CSE-induced THP-1 cells. COPD and healthy mice were used for Me-RIP sequencing and flow cytometry experiments. Validation of the results of the above bioinformatics analysis by molecular biology experiments and sequencing techniques.
Results: We found that GEO data and blood specimens from COPD patients showed increased M1 macrophages, higher levels of IL-6 and TNF-α, and higher mRNA expression of key mediators of the MAPK signaling pathway (p38, ERK, and JNK). Western blotting showed increased expression of p38, ERK, and JNK in the CSE group. In contrast, the expression of m6A regulators was low. Also, M1 macrophages in COPD mice were hyperactivated and had reduced m6A modifications of p38, ERK, and JNK compared with control.
Conclusion: m6A may be involved in M1 macrophage hyperactivation by regulating the MAPK signaling pathway, thereby influencing the development of COPD.
Keywords: chronic obstructive pulmonary disease, COPD, N6-methyladenosine, m6A, M1 macrophages, mitogen-activated protein kinase signaling pathway, MAPK signaling pathway
Introduction
Chronic obstructive pulmonary disease (COPD) is a progressive disease that results in symptoms such as shortness of breath, coughing, and mucus overproduction. The incidence of the disease has increased significantly since 1980 and is now the third leading cause of death after cardiovascular diseases and stroke.1 The progression of COPD is characterized by intense chronic inflammation of the lungs.2,3 Macrophages play a role in the development of COPD, especially M1 macrophages.4,5 Activated M1 macrophages are involved in the development of COPD by releasing various cytokines and chemokines such as interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α). The mitogen-activated protein kinase (MAPK) is a family of protein kinases, and they are involved in the development of COPD by affecting M1 macrophage activation and the production of inflammatory cytokines.6–8
The role of epigenetic regulation in inflammation is emerging as an important area of research. The N6-methyladenosine(m6A) is the most common epigenetic modification in eukaryotic RNA and represents the methylation of the 6th nitrogen atom of adenine in the RNA molecule.9 The methylation of m6A is primarily regulated by three families of enzymes: m6A methyltransferase (writer), m6A demethylase (eraser), and m6A RNA-binding proteins. In inflammatory diseases, the knockdown of m6A methyltransferase-like 3 (METTL3) and m6A RNA-binding protein YTH domain-containing family protein 2 (YTHDF2) promoted the upregulation of p38, ERK, and JNK in M1 macrophages.10,11 Recently, Huang et al found that m6A regulators play a role in the pathogenesis of COPD by analyzing data of small airway tissues from the GEO database.12,13 The expression of m6A regulatory factors was significantly associated with key genes involved in the development of COPD.12
In this study, we explored the potential contribution of M1 macrophages and the related m6A modifications in an animal model of COPD and clinical samples from COPD patients through an integrated bioinformatics approach and molecular biology experiments.
Materials and Methods
Subject Characteristics
We included outpatients and inpatients from the Traditional Chinese Medicine Hospital affiliated with Xinjiang Medical University between January and September 2022. All patients were diagnosed with COPD according to the Global Initiative on Chronic Obstructive Pulmonary Disease (GOLD): FEV1/FVC after the use of bronchodilators < 70% and predicted FEV1 < 80% of patients. Patients with bronchial asthma, bronchiectasis, pulmonary fibrosis, lung cancer, and tuberculosis were excluded. A total of 21 patients with COPD were obtained according to the above criteria. According to the GOLD grading method, the samples were categorized into 3 disease stages. At the same time, we recruited 21 healthy controls. The characteristics of the samples are shown in Table 1. More sample characteristics can be found in the Excel file called Sample Information. This study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Xinjiang Uygur Autonomous Region Hospital of Traditional Chinese Medicine (Ethical approval number: 2022XE0109). Written informed consent was obtained from each patient.
 |
Table 1 The Sample Information |
Animal Experiments
Six-week-old male C57 mice (Xinjiang Medical University Animal Center) were used. Mice were kept on a 12 h light/12 h dark cycle at 23 ± 1°C with 50 ± 10% relative humidity under specific-pathogen-free circumstances, fed a non-purified diet, and provided with water ad libitum at the animal facility of Xinjiang Medical University. The present study was approved by the Xinjiang Medical University Institutional Review Board and Committee on Animal Experimentation (License number: SYXK-2018–0003) and conducted in compliance with the University’s guidelines for the care and use of laboratory animals.
Mice were acclimatized for 1 week before beginning the experiments. Mice were divided into two subgroups (n = 10 for each subgroup). Mice in the COPD group were exposed to smoke from four cigarettes without filters via a smoking machine as described previously.14 Mice were placed in a 60×40 × 30 cm3 smoke box for 30 min, four times per day. Mice were exposed to smoke 5 days per week for 14 weeks.15 Each cigarette yields 10 mg tar, 1.0 mg nicotine, and 11 mg CO. The total particulate matter concentration of 361.3 ± 49.2 mg/m3/day.
Cell Culture and Treatment
A human monocyte leukemia cell line (THP-1) was obtained from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences. The cells were cultured at a density of 5×105 cells/mL in RPMI 1640 (Gibco, USA) medium supplemented with 10% FBS (Gibco, USA) and 1% penicillin/streptomycin solution (Gibco, USA) at 37°C in a 5% CO2 incubator. THP-1 monocytes were cultured in 6-well plates treated with 100 nM phorbol 12-myristate 13-acetate (PMA, Sigma-Aldrich, USA) for 24 h to transform into adherent macrophages.16
THP-1 cells were induced with 2% cigarette smoke extract (CSE) for 24 h.17 After cigarette smoke extract exposure, non-adherent cells were discarded and adherent cells were incubated with 500 μL cold PBS for 5 min, then harvested by scraping the adherent cells with a plastic scraper into 1.5 mL tubes before centrifugation at 427 RCF at 4°C. The supernatant was removed and the cell pellet was stored at –80°C for protein expression analysis.
Flow Cytometry
A total of 2 mL of peripheral blood was drawn from the median elbow vein of the enrolled subjects and 100 μL of this blood was used for flow cytometry. At the same time, a total of 200 μL of peripheral blood was drawn from the tail end of C57 mice and 100 μL was used for flow cytometry. The gating strategy for human peripheral blood was as follows: SSC and CD14 were used to set a gate to circle the macrophage population, then CD68 and CD14 were utilized to set another gate to circle the macrophage population, and CD14 and CD86 were used to circle the M1 macrophages.18 The gating strategy for peripheral blood of C57 mice was as follows: SSC and CD45 were used to set a gate to circle the macrophage population, then SSC and F4/80 were utilized to circle the macrophage population again and F4/80 and CD11C were used to locate the M1 macrophages.19,20 A total of 100 µL of peripheral blood was inserted into the flow tube and 1 uL of antibody was added to the negative control sample without any antibody. The following antibodies were used: PE/Dazzle 594 anti-human CD14 (Clone 63D3, BioLegend), Alexa Fluor 488 anti-human CD86 (Clone 2331 (FUN-1), BD), PE/Cyanine7 anti-human CD68 (Clone Y1/82A (RUO), BD), PE/Dazzle 594 anti-mouse F4/80 (Clone BMB, BioLegend), PE/Cyanine5 anti-mouse CD11c (Clone N418, BioLegend) and FITC anti-mouse CD45 (Clone 30-F11, BioLegend). The red blood cell residue was dissolved with 2 mL of red blood cell lysate (BD Bioscience). After 10 min, PBS solution was added to terminate the lysis of erythrocyte residues. Finally, centrifugation at 427 RCF for 5 min was used to elute all dissolved residues, morphological particles, and soluble proteins. The stained cells were analyzed by Beckman and DXFLEX flow cytometry and the results were processed by Kaluza software. Approximately 50,000 cells were collected from each sample and M1 macrophages were assessed as a percentage of total macrophages. At the same time, we have supplemented the detailed experimental method evaluating human peripheral blood flow cytometry, subject information and other missing data.
Detection of Cytokines by ELISA
The serum was collected from blood samples of COPD and healthy individuals. The levels of TNF-α and IL-6 were detected using an automated chemistry analyzer. The experiment was performed according to the instructions provided by the manufacturers (Jianglai, China).
RNA Extraction and Quantitative Polymerase Chain Reaction Analysis
The total RNA was extracted using Trizol (Invitrogen, USA) in accordance with the manufacturer’s protocol. We used the TaqMan RNA-to-CT 1-Step Kit from Thermo Fisher (METTL3, METTL14, and YTHDF1) and PrimeScript RT Reagent Kit (p38, ERK, and JNK) from Takara. Real-time quantitative PCR was performed on an ABI 7500 fast real-time PCR system (Thermo Fisher Scientific, USA) to determine the target RNA expression levels. The PCR thermal cycling parameters of TaqMan RNA-to-CT 1-Step Kit were as follows: 48°C for 15 min, 95°C for 10 min, 40 cycles of 95°C for 15 s and 60°C for 1 min. The PCR thermal cycling parameters of Prime Script RT Reagent Kit were as follows: 95°C for 3 min, 40 cycles of 60°C for 30 s, and 72°C for 10 s. The relative quantity of the target gene was calculated with the 2−ΔΔCt method and normalized GAPDH. The expression levels of METTL3, METTL14, and YTHDF1 were assessed by TaqMan technology. Their primer sequence has not been listed because of intellectual property protection. However, other primer sequences are shown in Table 2.
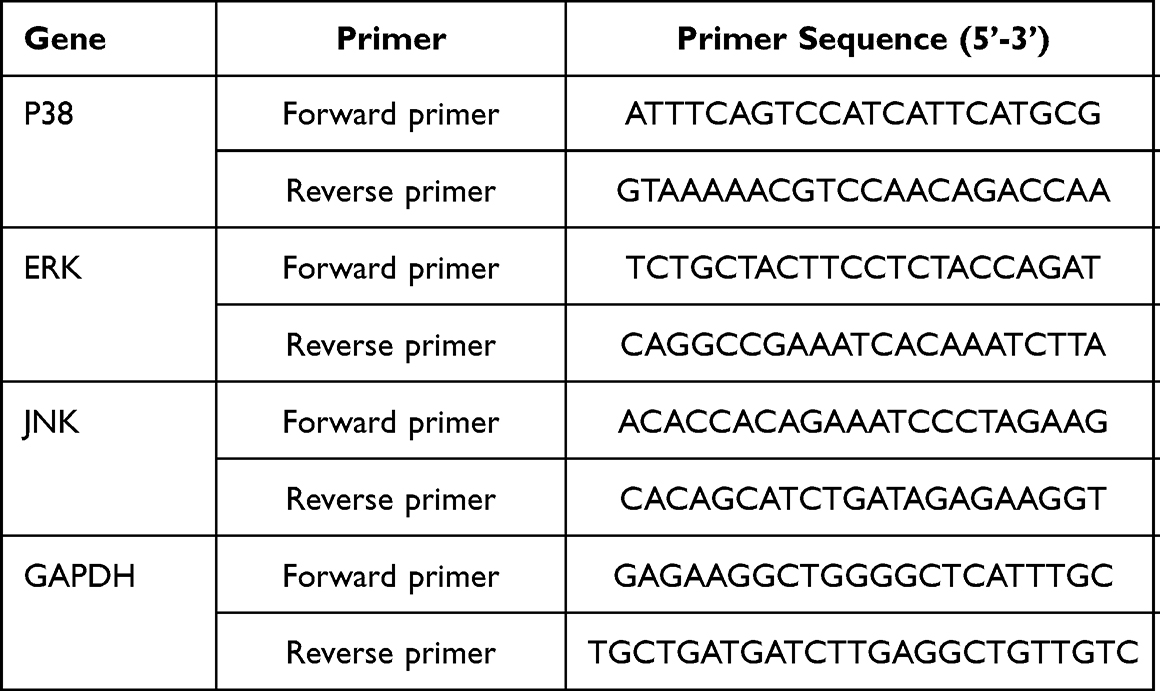 |
Table 2 Primers of Reverse Transcription PCR Analysis for Genes |
Western Blotting
The THP-1 cells were lysed by cold RIPA buffer (87787, Thermo Fisher Scientific, USA). Total protein lysates (25 mg/lane) were separated via 12% SDS-PAGE and then transferred to polyvinylidene fluoride membranes. The membranes were subsequently blocked with 5% nonfat milk in Tris-Buffered Saline and Tween-20 (TBST, maintaining 20 mM Tris-HCl, 0.15 M NaCl, 0.05% Tween-20, pH 7.5) for 2 h at room temperature and further incubated overnight with rabbit anti-p38 (ab170099, Abcam, USA), rabbit anti- phosphorylated -p38 (ab195049, Abcam, USA), rabbit anti-JNK (ab179461, Abcam, USA), rabbit anti- phosphorylated -JNK (ba124956, Abcam, USA), mouse anti- ERK (BSM-33337M, BIOSS, USA), rabbit anti- phosphorylated -ERK (bs-1522R, BIOSS, USA), and mouse anti-GAPDH antibodies (ab8245, Abcam, USA) at 4°C according to the manufacturer’s instructions. After washing with Tris-Buffered Saline and Tween 20 (TBST, Beyotime Biotech. Shanghai, China) three times, the membranes were incubated with the secondary antibody (150113,150077, Abcam, USA) and finally processed using enhanced chemiluminescence (ECL) reaction kit (Cell Signaling Technology, USA) and quantification was performed using Image J.
Data Collection and Pre-Processing
The data used in this study included lung and tracheal tissue samples. The lung tissues included 98 COPD samples and 91 healthy samples, while tracheal tissue included 23 COPD samples and 112 healthy samples. The data were retained in the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The sequence number of lung tissues was GSE57148, and the sequence number of tracheal tissues was GSE20257. All the data were analyzed through the R software and the limma package (version 3.40.6).
MeRIP-Seq
The mRNA m6A was sequenced by MeRIP-seq at Novogene (Beijing, China). Briefly, a total of 300 µg RNAs was extracted from lung tissues. The integrity and concentration of extracted RNAs were detected using an Agilent 2100 bioanalyzer (Agilent) and simpliNano spectrophotometer (GE Healthcare), respectively. Fragmented mRNA (~100 nt) was incubated for 2 h at 4°C with the anti-m6A polyclonal antibody (Synaptic Systems) in the immunoprecipitation experiment. Then, immunoprecipitated mRNAs or Input were used for library construction with the NEB Next Ultra RNA library preparation kit for Illumina (New England Biolabs). The library preparations were sequenced on an Illumina Novaseq or HiSeq platform with a paired-end read length of 150 bp according to the standard protocols. The sequencing was carried out with 3 independent biological replicates. The data have been uploaded to NCBI’s BioProject (PRJNA853736).
Enrichment of M1 Macrophages and Their Surface-Specific Proteins
A single sample gene set enrichment analysis (ssGSEA) was used to determine the immune cell types of the dataset.21 The expression of each gene across all the samples was normalized using the Z-score. The genes were then ranked in descending order based on the Z-score (mean of z-scores) for each sample (or set of samples). Associations were represented by the normalized enrichment score (NES). Immune cell types were considered enriched in a sample or set of the samples when FDR (q value) ≤ 10%. After determining the immune cell scores in all the samples, the differences in M1 macrophages and their surface characteristic proteins between COPD and healthy individuals were analyzed using the method of two sample t-tests.
Identification of COPD Genes and Gene-Enriched Pathways
The differentially expressed genes of lung and tracheal tissues between COPD and healthy individuals were identified by using the limma package (version 3.40.6). The KEGG enrichment analysis was performed in the ENRICHR database.
Analysis of m6A Regulator Changes in COPD and Healthy Individuals
A total of 18 broadly recognized m6A RNA methylated regulators were retrieved from the published literature.9,22 Then, their mRNA expression data were obtained from the lung and the tracheal tissues, respectively. Thereafter, differential expression analysis of 18 m6A RNA methylation regulators between COPD and healthy individuals was performed using the t-test. The LASSO regression was used for feature selection and dimension reduction to excrete unimportant m6A regulatory factors. M6A regulators were evaluated for their discriminatory properties against COPD and healthy samples using the ROC curve analysis.
Correlation Analysis
Pearson’s analysis was used to analyze the correlation of the m6A regulator, M1 macrophages, and the MAPK signaling pathway between the two groups. p < 0.05 was considered statistically significant.
Statistical Analysis
Data from bioinformatics were statistically analyzed using R software (version 3.6.2), and p < 0.05 was considered statistically significant. Gene expression levels of the samples were statistically analyzed using the Student’s t-test of GraphPad Prism 8 (GraphPad Software Inc, San Diego, CA, USA), and p < 0.05 was considered statistically significant.
Results
M1 Macrophages, Inflammatory Factors and MAPK Signaling Were Involved in COPD
By analyzing the sequencing data from the GEO database, we found that the number of M1 macrophages and the levels of their surface-specific proteins (IL-1β, TLR2, TLR4 and CD86) were significantly higher in lung and tracheal tissue samples of COPD patients compared to healthy controls (Figure 1A and B). Activation of M1 macrophages was closely associated with the activity of TNF-α and IL-6 (Fig. S1). We further explored the differentially expressed genes (DEGs) between COPD and healthy samples. A total of 4638 DEGs were obtained from lung tissues and 1788 DEGs from tracheal tissues. A total of 768 DEGs were in common between lung and tracheal tissues (Figure 2A and B). These 768 DEGs were subjected to enrichment analysis, with the top-ranked genes pertaining to the MAPK signaling pathway (Figure 2C and D). The correlation analysis between M1 macrophages and the MAPK signaling pathway showed a positive correlation in lung and tracheal tissues (Fig. S2). These results suggest that the MAPK signaling pathway may play a role in COPD by activating the inflammatory response of M1 macrophages. In COPD patients, we found a significant decrease in lung function, an increased number of CD86+ and CD14+ M1 macrophage-like cells and an augmented secretion of IL-6 and TNF-α (Figure 3A and B, Fig. S3 and Table 2). RT-PCR results showed that mRNA expression levels of p38, ERK, and JNK were significantly higher in blood specimens from COPD patients compared with healthy controls (Figure 3C). Western blotting confirmed that CSE induced significantly higher levels of p38, ERK, and JNK protein expression in THP-1 cells compared to controls (Figure 3D).
 |
Figure 1 The proportion of activated M1 macrophages and the level of secreted cytokines in COPD were analyzed by using GEO data. (A) Violin plots showing different immune cells in lung and tracheal tissues of COPD patients and healthy controls. (B) Box plots showing different inflammatory cytokines in lung and airway tissues of COPD patients and healthy controls (*p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 compared with healthy controls). |
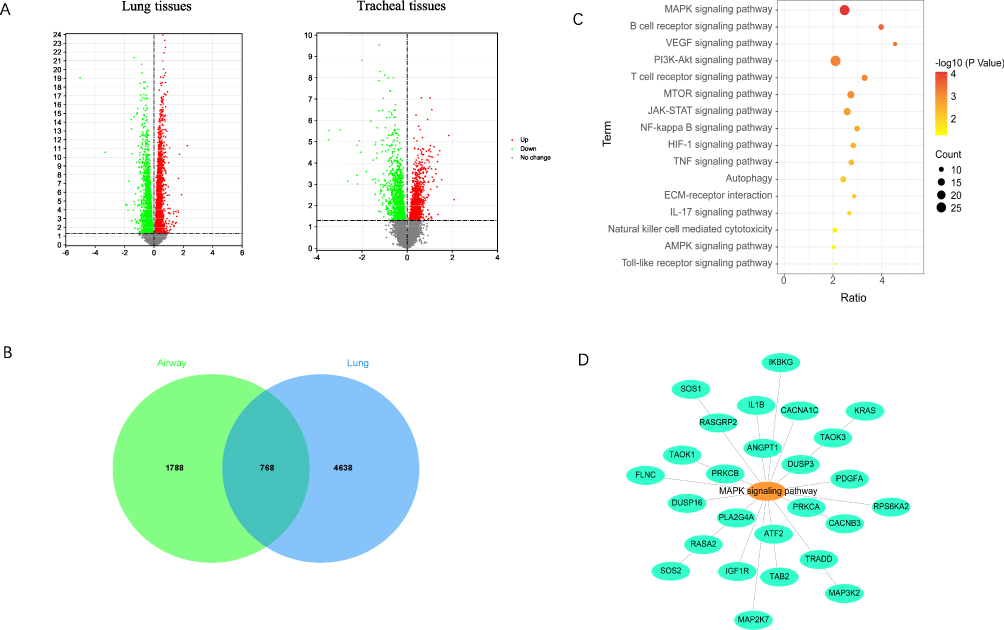 |
Figure 2 The relationship between the MAPK signaling pathway and M1 macrophages in COPD was analyzed by using GEO data. (A) Volcano plots showing the differentially expressed genes in lung and tracheal tissues of COPD patients and healthy controls. (B and C) Differentially expressed genes in lung and tracheal tissues and enriched pathways for co-differentially expressed genes. (D) Differentially expressed genes related to the MAPK signaling pathway in lung and tracheal tissues. |
 |
Figure 3 Increased M1 macrophage activity during COPD is associated with elevated expression levels of key factors involved in the MAPK signaling pathway. (A and B) Flow cytometry and ELISA results of 21 blood samples from COPD patients and healthy controls. (C) RT-PCR results of 8 blood samples each from COPD patients and healthy controls. (D) Western blotting results of CSE-induced THP-1 cells (4 replicates each in control and CSE groups). ***p < 0.001 compared with healthy controls. |
Methylation of mRNA for Key Factors of the MAPK Signaling Pathway May Activate M1 Macrophages and Be Involved in COPD Development
Analysis of DEGs showed that levels of several m6A regulators were significantly different between COPD patients and healthy controls. In lung tissues of COPD patients, nine genes were upregulated and three genes were downregulated. In the tracheal tissues of COPD patients, seven genes were downregulated (Figure 4A). In lung tissues, the levels of METTL14 were increased whereas the levels of METTL3 and YTHDF1 were decreased. In tracheal tissues, the levels of METTL3 were decreased. We further analyzed the association of m6A regulators with COPD by feature selection and dimension reduction techniques. We found that 13 m6A regulators in lung tissues and 10 m6A regulators in tracheal tissues were associated with COPD (Figure 4B and C). According to the least absolute shrinkage and selection operator (LASSO) regression, we analyzed the ROC curve of risk model through the scoring formula 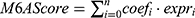 . The results showed that the AUC was greater than 85% in both lung and tracheal tissues, indicating that m6A modifiers correctly classified COPD and healthy samples (Figure 4D). By using RT-PCR, we found that the expression of METTL14 and YTHDF1 was significantly decreased in blood samples of COPD patients compared to healthy controls. In contrast, the expression levels of METTL3 were not significantly different (Figure 4E). Validation indicates that m6A is involved in COPD.
. The results showed that the AUC was greater than 85% in both lung and tracheal tissues, indicating that m6A modifiers correctly classified COPD and healthy samples (Figure 4D). By using RT-PCR, we found that the expression of METTL14 and YTHDF1 was significantly decreased in blood samples of COPD patients compared to healthy controls. In contrast, the expression levels of METTL3 were not significantly different (Figure 4E). Validation indicates that m6A is involved in COPD.
 |
Figure 4 Involvement of m6A regulators in COPD. (A) Box plots were obtained by analyzing the GEO data, which shows the transcriptomic differences of 18 m6A regulators in COPD and healthy samples. (B) Least Absolute Shrinkage and Selection Operator (LASSO) coefficient curves for the 18 m6A regulators in the GEO dataset. (C) 10-fold cross-validation in LASSO regression adjusting for parameter selection in GEO data. The partial likelihood bias is compared to log(λ), with λ being the tuning parameter. The partial likelihood bias values are shown in the figure, the error bars represent the standard error (SE). The vertical dashed lines indicate the optimal values using the minimum and (1-SE) criteria. (D) ROC curves and AUC values were used to assess the ability of m6A modulators in the GEO data to discriminate between COPD and healthy samples. (E) RT-PCR results of 8 blood samples each from COPD patients and healthy controls. *p < 0.05, compared with healthy controls. ns, p=0.2357. |
Correlation analysis showed that m6A regulators were negatively associated with M1 macrophages and the MAPK signaling pathway in lung and tracheal tissues of COPD patients (Figure 5A and B). We performed Me-RIP sequencing analysis on mouse lung tissues, showing that 740 upregulated genes were significantly associated with m6A modifications and 1373 downregulated genes were associated with m6A modifications in COPD samples compared with the healthy controls. By using the KEGG enrichment analysis, we showed that many differentially methylated mRNAs were enriched in the MAPK signaling pathway (Figure 6A and B). The m6A mRNA modifications of p38, ERK, and JNK were significantly reduced in the lung tissues of COPD mice compared with controls (Figure 6C). The proportion of M1 macrophages in the blood of COPD mice was significantly increased, which was consistent with the results in the peripheral blood of patients with COPD (Figure 6D).
 |
Figure 5 Correlations between m6A regulators, the MAPK signaling pathway, and M1 macrophages were analyzed by using GEO data. (A) Correlation between m6A regulators and M1 macrophages in lung and tracheal tissues. (B) Correlation between m6A regulators and the MAPK signaling pathway in lung and tracheal tissues. |
 |
Figure 6 Decreased mRNA methylation modifications of key factors of the MAPK signaling pathway. (A) MeRIP-seq results showing volcano maps of differentially expressed genes altered by m6A regulatory factors in lung tissues of COPD mice and healthy mice. (B) MeRIP-seq results show 20 pathways enriched for genes differentially regulated by m6A regulators in lung tissues of COPD mice and healthy mice. (C) MeRIP-seq results show the IGV track of the regional distribution of mRNA modifications associated with key factors of the MAPK signaling pathway. (D) Flow cytometry results of blood samples obtained from mice. ***p < 0.001 compared with healthy controls. |
Discussion
Our analysis of GEO data and RT-PCR results from the peripheral blood of COPD patients revealed that m6A modifications were significantly decreased in COPD patients compared with healthy controls. The m6A modifications in M1 macrophages might play a role in COPD pathogenesis. The activation of M1 macrophages involves a variety of pathways, including the MAPK signaling pathway.23 In inflammatory conditions, the MAPK signaling pathway over-activates M1 macrophages while stimulating the secretion of large amounts of inflammatory cytokines, which may contribute to the development of COPD.24–27 The expression of p38, ERK, and JNK is influenced by a variety of factors, with m6A RNA methylation being one of the most common post-transcriptional modifications documented in a variety of inflammatory diseases.28 Consistent with our findings, m6A regulators may be involved in the inflammatory response induced by M1 macrophage hyperactivation by regulating the MAPK signaling pathway, thereby participating in the development of COPD.10,29,30
Previous studies found that reducing the m6A regulatory enzymes METTL3 and YTHDF2 promoted the mRNA expression of p38, ERK, and JNK, thus activating the inflammatory response of macrophages.10,11 By analyzing GEO data, peripheral blood flow cytometry, and ELISA results from COPD patients, we found that activated M1 macrophages and released inflammatory cytokines were significantly increased in COPD patients. The mRNA expression of p38, ERK, and JNK was found to be significantly up-regulated in the peripheral blood of COPD patients, meanwhile, the protein expression levels of p38, ERK, and JNK were significantly up-regulated in CSE-induced THP-1 cells. Overall, these findings suggest that p38, ERK, and JNK in M1 macrophages can effectively participate in the pathologic process of COPD.
Writers and erasers synergistically regulate the dynamic and reversible modification of m6A, whereas readers perform biological functions by recognizing m6A modification sites. Writers, erasers and readers collaborate to adapt the level of m6A methylation to the overall state of the organism.31 The m6A-modified, methyltransferase-dependent, writers are a complex consisting of three core components: METTL3, methyltransferase-like 14 (METTL14), and the cofactor Wilms tumor 1-associated protein (WTAP). Erasers can mediate m6A demethylation modifications, including Fat Mass and Obesity-associated (FTO) proteins and ALKB homolog 5 (ALKBH5). Recognition of m6A modifications is mediated by m6A-binding proteins. The main reader proteins identified to date include YTH domain-containing proteins, such as YTHDF1. A decrease in writers, an increase in erasers and a decrease in readers may ultimately result in a reduction of the m6A modifier. RT-PCR findings showed that gene expression of the m6A regulatory enzymes METTL14 and YTHDF1 was dramatically down-regulated in the peripheral blood of patients with COPD, with a decrease in METTL14 resulting in the inability to form the writers’ complex. The decrease in YTHDF1 resulted in the unrecognized or reduced recognition of m6A modifications, ultimately in the reduction of the m6A modification. MeRIP-seq results indicated that m6A modifications of p38, ERK and JNK were markedly reduced, pointing out that m6A modifications exerted a negative regulatory effect on the MAPK signaling pathway.
In conclusion, we found that reduced levels of m6A modification of p38, ERK and JNK may increase their expression, thereby over-activating M1 macrophages and promoting the secretion of inflammatory cytokines, which are ultimately involved in the development of COPD. We hypothesized that reduced levels of m6A modifications of p38, ERK, and JNK might be associated with reduced METTL14 and YTHDF1. We speculate that the activity of the METTL14-m6A-YTHDF1 axis can regulate the modification of key factors of the MAPK signaling pathway, upregulating the mRNA expression of p38, ERK, and JNK in COPD. We propose a novel mechanism of macrophage m6A-RNA methylation in patients with COPD. This suggests that m6A can be involved in the pathogenesis of COPD through the over-activation of M1 macrophages, thus providing a potential therapeutic target for the treatment of COPD.
Our study is also associated with limitations. Firstly, we did not clearly demonstrate that the increase in IL-6 and TNF-α was directly caused by activated M1 macrophages. Secondly, we found altered expression levels of methyltransferases, methylated reading proteins, and key factors of the MAPK signaling pathway in clinical samples of COPD. Even so, the precise mechanisms by which m6A regulators could regulate the activation of M1 macrophages through the MAPK signaling pathway were not discussed in detail. In addition, our study was characterized by a limited sample size. In the future, novel studies are needed to validate these findings.
Conclusion
Our results revealed that reduced levels of m6A modifications of p38, ERK and JNK may be associated with M1 macrophage hyperactivation, ultimately leading to an augmented expression of inflammatory factors involved in COPD. This study provides a new direction for screening potential therapeutic targets for COPD.
Ethical Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Xinjiang Uygur Autonomous Region Chinese Medicine Hospital. The informed consent obtained from patients included the consent to have their individual details (eg age, gender, ethnicity) published.
Acknowledgments
We acknowledge TopEdit LLC for the linguistic editing and proofreading during the preparation of this manuscript.
Authors´ Contribution
Jing Wang conceived and designed the experiments. Tingting Hu, Min Jiang, Zheng Li, Dan Xu, Jing Jing, Fengsen Li and Jianbing Ding performed the experiments. Tingting Hu analyzed the data. Jing Wang, Tingting Hu, Min Jiang, Dan Xu collected the funds. Tingting Hu and Min Jiang wrote the manuscript. Jing Wang revised the manuscript. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was jointly supported by the Natural Science Foundation of Xinjiang Uygur Autonomous Region (Project No: 2022D01C176), the Natural Science Foundation of Xinjiang Uygur Autonomous Region (project No: 2022D01E28), Xinjiang Key Laboratory of Pulmonary Disease Research,The Fourth Affiliated Hospital of Xinjiang Medical University (Project No: ZYYHX202104), Xinjiang Uygur Autonomous Region Key Laboratory Open Subjects (Project No: 2022D04022, 2021D04023), the National Natural Science Foundation Regional Fund Project (Project No: 82160844).
Disclosure
The authors report no conflicts of interest in relation to this work.
References
1. Soriano JB, Kendrick PJ, Paulson KR. Prevalence and attributable health burden of chronic respiratory diseases, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Respir Med. 2020;8(6):585–596. doi:10.1016/S2213-2600(20)30105-3
2. Tetley TD. Macrophages and the pathogenesis of COPD. Chest. 2002;121(5 Suppl):156s–159s. doi:10.1378/chest.121.5_suppl.156S
3. Wang Y, Xu J, Meng Y, Adcock IM, Yao X. Role of inflammatory cells in airway remodeling in COPD. Int J Chron Obstruct Pulmon Dis. 2018;13:3341–3348. doi:10.2147/COPD.S176122
4. Lea SR, Reynolds SL, Kaur M, et al. The effects of repeated Toll-like receptors 2 and 4 stimulation in COPD alveolar macrophages. Int J Chron Obstruct Pulmon Dis. 2018;13:771–780. doi:10.2147/COPD.S97071
5. Lee JW, Chun W, Lee HJ, et al. The role of macrophages in the development of acute and chronic inflammatory lung diseases. Cells. 2021;10(4):897.
6. Subhashini CPS, Dash D, Paul BN, Singh R. Intranasal curcumin ameliorates airway inflammation and obstruction by regulating MAPKinase activation (p38, Erk and JNK) and prostaglandin D2 release in murine model of asthma. Int Immunopharmacol. 2016;31:200–206. doi:10.1016/j.intimp.2015.12.025
7. Liu L, Guo H, Song A, et al. Progranulin inhibits LPS-induced macrophage M1 polarization via NF-кB and MAPK pathways. BMC Immunol. 2020;21(1):32. doi:10.1186/s12865-020-00355-y
8. Gu W, Song L, Li XM, Wang D, Guo XJ, Xu WG. Mesenchymal stem cells alleviate airway inflammation and emphysema in COPD through down-regulation of cyclooxygenase-2 via p38 and ERK MAPK pathways. Sci Rep. 2015;5(1):8733. doi:10.1038/srep08733
9. Luo J, Xu T, Sun K. N6-methyladenosine RNA modification in inflammation: roles, mechanisms, and applications. Front Cell Dev Biol. 2021;9:670711. doi:10.3389/fcell.2021.670711
10. Zhang Y, Gu X, Li D, Cai L, Xu Q. METTL3 regulates osteoblast differentiation and inflammatory response via Smad signaling and MAPK signaling. Int J Mol Sci. 2019;21(1):199. doi:10.3390/ijms21010199
11. Fang C, He M, Li D, Xu Q. YTHDF2 mediates LPS-induced osteoclastogenesis and inflammatory response via the NF-κB and MAPK signaling pathways. Cell Signal. 2021;85:110060. doi:10.1016/j.cellsig.2021.110060
12. Huang X, Lv D, Yang X, Li M, Zhang H. m6A RNA methylation regulators could contribute to the occurrence of chronic obstructive pulmonary disease. J Cell Mol Med. 2020;24(21):12706–12715. doi:10.1111/jcmm.15848
13. Guo X, Lin Y, Lin Y, et al. PM2.5 induces pulmonary microvascular injury in COPD via METTL16-mediated m6A modification. Environ Pollut. 2022;303:119115. doi:10.1016/j.envpol.2022.119115
14. Jiang M, Li Z, Zhang F, et al. Butyrate inhibits iILC2-mediated lung inflammation via lung-gut axis in chronic obstructive pulmonary disease (COPD). BMC Pulm Med. 2023;23(1):163. doi:10.1186/s12890-023-02438-z
15. Mustra Rakic J, Liu C, Veeramachaneni S, et al. Lycopene inhibits smoke-induced chronic obstructive pulmonary disease and lung carcinogenesis by modulating reverse cholesterol transport in Ferrets. Cancer Prev Res. 2019;12(7):421–432. doi:10.1158/1940-6207.CAPR-19-0063
16. Zhao W, Ma L, Cai C, Gong X. Caffeine inhibits NLRP3 inflammasome activation by suppressing MAPK/NF-κB and A2aR signaling in LPS-induced THP-1 macrophages. Int J Biol Sci. 2019;15(8):1571–1581. doi:10.7150/ijbs.34211
17. Buscetta M, Di Vincenzo S, Miele M, Badami E, Pace E, Cipollina C. Cigarette smoke inhibits the NLRP3 inflammasome and leads to caspase-1 activation via the TLR4-TRIF-caspase-8 axis in human macrophages. FASEB J. 2020;34(1):1819–1832. doi:10.1096/fj.201901239R
18. Manferdini C, Saleh Y, Dolzani P, et al. Impact of isolation procedures on the development of a preclinical synovial fibroblasts/macrophages in an in vitro model of osteoarthritis. Biology. 2020;9(12):459. doi:10.3390/biology9120459
19. Xu L, Chen Y, Nagashimada M, et al. CC chemokine ligand 3 deficiency ameliorates diet-induced steatohepatitis by regulating liver macrophage recruitment and M1/M2 status in mice. Metabolism. 2021;125:154914. doi:10.1016/j.metabol.2021.154914
20. Berges AJ, Ospino R, Lina IA, et al. Myeloid phenotypes in tracheostomy-associated granulation tissue. Laryngoscope. 2023;133(9):2346–2356. doi:10.1002/lary.30557
21. Xu L, Li F, Jiang M, et al. Immunosuppression by inflammation-stimulated amplification of myeloid-derived suppressor cells and changes in expression of immune checkpoint HHLA2 in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2023;18:139–153. doi:10.2147/COPD.S394327
22. Tong J, Flavell RA, Li HB. RNA m(6)A modification and its function in diseases. Front Med. 2018;12(4):481–489. doi:10.1007/s11684-018-0654-8
23. Li X, Kang J, Lv H, et al. CircPrkcsh, a circular RNA, contributes to the polarization of microglia towards the M1 phenotype induced by spinal cord injury and acts via the JNK/p38 MAPK pathway. FASEB J. 2021;35(12):e22014. doi:10.1096/fj.202100993R
24. Guan R, Wang J, Li Z, et al. Sodium Tanshinone IIA sulfonate decreases cigarette smoke-induced inflammation and oxidative stress via blocking the activation of MAPK/HIF-1α signaling pathway. Front Pharmacol. 2018;9:263. doi:10.3389/fphar.2018.00263
25. Huang Y, Gao J, Meng XM, et al. Involvement of mitogen-activated protein kinase activation in cyclooxygenase-2 and transforming growth factor-β production in alveolar macrophage from chronic bronchitis rats. Immunopharmacol Immunotoxicol. 2011;33(4):645–651. doi:10.3109/08923973.2011.557383
26. Arora S, Dev K, Agarwal B, Das P, Syed MA. Macrophages: their role, activation and polarization in pulmonary diseases. Immunobiology. 2018;223(4–5):383–396. doi:10.1016/j.imbio.2017.11.001
27. Wang C, Zhou J, Wang J, et al. Progress in the mechanism and targeted drug therapy for COPD. Signal Transduct Target Ther. 2020;5(1):248. doi:10.1038/s41392-020-00345-x
28. Zhang F, Ran Y, Tahir M, Li Z, Wang J, Chen X. Regulation of N6-methyladenosine (m6A) RNA methylation in microglia-mediated inflammation and ischemic stroke. Front Cell Neurosci. 2022;16:955222. doi:10.3389/fncel.2022.955222
29. Zhao M, Hou J, Zheng S, et al. Peucedanum praeruptorum Dunn polysaccharides regulate macrophage inflammatory response through TLR2/TLR4-mediated MAPK and NF-κB pathways. Biomed Pharmacother. 2022;152:113258. doi:10.1016/j.biopha.2022.113258
30. Cai L, Li D, Feng Z, Gu X, Xu Q, Li Q. YTHDF2 regulates macrophage polarization through NF-κB and MAPK signaling pathway inhibition or p53 degradation. Dis Markers. 2022;2022:3153362. doi:10.1155/2022/3153362
31. Jiang X, Liu B, Nie Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6(1):74. doi:10.1038/s41392-020-00450-x
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.