Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Tetracycline-grafted PLGA nanoparticles as bone-targeting drug delivery system
Authors Wang H, Liu J, Tao S, Chai G, Wang J, Hu F, Yuan H ![]()
Received 18 May 2015
Accepted for publication 13 August 2015
Published 8 September 2015 Volume 2015:10(1) Pages 5671—5685
DOI https://doi.org/10.2147/IJN.S88798
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Lei Yang
Hua Wang,1 Jun Liu,2 Shan Tao,2 Guihong Chai,2 Jianwei Wang,3 Fu-Qiang Hu,2 Hong Yuan2
1Center of Analysis and Measurement, 2College of Pharmaceutical Sciences, Zhejiang University, Hangzhou, 3The Second Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou, People’s Republic of China
Purpose: Nanoparticles (NPs) that target bone tissue were developed using poly(lactic-co-glycolic acid) (PLGA) copolymers and tetracycline (TC)-based bone-targeting moieties. These NPs are expected to enable the transport of drugs, such as simvastatin (SIM), for the treatment of osteoporosis.
Methods: The molecular structures of TC–PLGA were validated by 1H-NMR, and the SIM-loaded NPs were prepared using the solvent emulsification method. The surface properties, cytotoxicity, cellular uptake, cell mineralization, bone targeting potential, and animal pharmacodynamics of the TC–PLGA NPs were evaluated and compared to those of PLGA NPs.
Results: It was confirmed that the average particle size of the NPs was approximately 220 nm. In phosphate-buffered saline (PBS, pH 7.4), the SIM-loaded NPs exhibited a cumulative release of up to 80% within 72 hours. An in vitro cell evaluation indicated that the NPs had an excellent cellular uptake capacity and showed great biocompatibility with MC3T3-E1 cells, thereby reducing the cytotoxic effects of SIM. The cell mineralization assay showed that the SIM-loaded NPs induced osteogenic differentiation and mineralized nodule formation in MC3T3-E1 cells, thereby achieving the same effect as SIM. Preliminary findings from in vitro and in vivo bone affinity assays indicated that the TC–PLGA NPs may display increased bone-targeting efficiency compared to PLGA NPs lacking a TC moiety. The use of SIM-loaded TC–PLGA NPs in treating osteoporosis was tested through animal pharmacodynamics analyses performed in ovariectomized rats, and the results suggested that the SIM-loaded TC–PLGA NPs can improve the curative effects of SIM on the recovery of bone mineral density compared to either SIM-loaded PLGA NPs or SIM alone.
Conclusion: Bone-targeting NPs, which were based on the conjugation of TC to PLGA copolymers, have the ability to target bone. These NPs may be developed as a delivery system for hydrophobic drugs, and they are expected to improve the curative effects of drugs, reduce the administered drug doses, and reduce side effects in other organs.
Keywords: poly(lactic-co-glycolic acid), simvastatin, tetracycline, osteoporosis, bone targeting, nanoparticles
Introduction
Recent studies have indicated that statins have the potential ability to promote new bone formation in vitro and in animals.1 These exciting results point to a new generation of treatment options for osteoporosis, because current therapies for the treatment of osteoporosis are mostly anti-osteoclast agents, including estrogen and related compounds, bisphosphonates, calcitonin, vitamin D analogs; currently, limited methods are available to induce osteogenesis. It is known that more than 95% of gastrointestinally absorbed statins will be decomposed in the liver, resulting in a much lower concentration in the bone;2 this decomposition may be the reason why most clinical studies using routine dosage of statins result in inconsistent effects on the bone.3,4 Positive results were obtained in the laboratory in animals orally administered with extremely high dosages of statins,5 and it has also been found that exceedingly high doses of systemically applied statins will lead to significant, adverse, and systemic side effects.6 These results have motivated us to find a suitable deliver system for statins to reduce their side effects and to target statins’ delivery to the bone.
As a new type of drug delivery system, nanotechnology-based delivery systems have shown a broad variety of useful properties, such as the ability to target organs or tissues, improve drugs’ biocompatibility, reduce drugs’ toxic effects, and to allow controlled drug release or sustained drug release.7–10 Poly(lactic-co-glycolic acid) (PLGA) biopolymer is a hydrophobic synthetic polymer with good biodegradability and biocompatibility. It produces a biocompatible byproduct that can be eliminated through normal metabolic pathways and has been widely used in clinical treatments. Specific drugs that have very low bio-availabilities or cannot survive the environments of the gastrointestinal tract in vivo can be administered using PLGA nanoparticles (NPs) as carriers; their encapsulation into the PLGA NP delivery system not only enables their protection but also improves the bio-availability of these drugs.11–13 Some drugs with high cytotoxicity at low concentrations can be delivered with PLGA NPs, and a long-term sustained drug release can be achieved by their encapsulation into PLGA NPs.14 PLGA NPs are also a potential drug delivery device for drugs used in treating osteoporosis, and a chemical modification that targets these NPs directly to bone will be more beneficial to osteoporosis patients.
Tetracycline (TC) is a broad-spectrum antibiotic produced by the Streptomyces genus of the Actinobacteria phylum that is indicated for use against many bacterial infections. However, in clinical applications, many previous studies have shown that TC can be taken up by bone through a reaction that appears to occur directly between TC and hydroxyapatite Ca10(PO4)6(OH)2, the major constituent of bone tissue.15 Thus, TC can be a potent chemical compound for bone targeting.
In the present study, TC–PLGA was synthesized by an esterification reaction between the hydroxyl of TC and the carboxyl of PLGA. The TC-grafted PLGA NPs (TC–PLGA NPs) were prepared using the solvent emulsification method. We hoped that the TC would act as a bone targeting system that would lead the NPs directly to the bone. We chose simvastatin (SIM) as the model drug to induce the enhancement of osteogenesis. We expected the SIM-loaded TC-grafted PLGA NPs (SIM/TC–PLGA NPs) to increase the amount of SIM that reached the bone, thereby exerting a better curative effect on osteoporosis.
Materials and methods
Materials
PLGA (50:50 [the ratio of lactic acid and glycolic acid]; MW, 2,000 Da; MW =15,000 Da) was purchased from Jinan Daigang Biomaterial Co, Ltd (Jinan, Shandong, People’s Republic of China). 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide (EDC), hydroxybenzotriazole (HOBt), TC hydrochloride, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT), fluorescein isothiocyanate (FITC), β-Glycerophosphate, L-ascorbic acid, and alizarin red S were obtained from Sigma-Aldrich Co (St Louis, MO, USA). The bicinchoninic acid (BCA) protein assay kit was purchased from Beyotime Institute of Biotechnology (Haimen, Jiangsu, People’s Republic of China). Alpha-minimum essential medium (αMEM), fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific, Waltham, MA, USA. Hydroxyapatite powder (HAp), methylene chloride, N,N-Dimethylformamide (DMF), and polyvinyl alcohol (PVA) were purchased from Chemical Reagent Co, Ltd. (Shanghai, People’s Republic of China). 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl indotricarbocyanine Iodide (DiR) lipophilicity fluorescence dye was obtained from Molecular Probes (Eugene, OR, USA). Hydrogenated soya phosphatidyl choline (S100) was purchased from Lipoid GmbH (Ludwigshafen, Germany). SIM was purchased from Zhejiang Hisun Pharmaceutical Co, Ltd (Taizhou, People’s Republic of China). Otcadecylamine (ODA) was purchased from Sigma-Aldrich Co.
All mice were housed under a 12-hour light/dark cycle and had access to food and water ad libitum. All experimental protocols and animal handling procedures were performed in accordance with the US National Institutes of Health (NIH) guidelines for the care and use of laboratory animals and all experiments in the current study were approved by The Committee for Animal Experiments at Zhejiang University, Hangzhou, People’s Republic of China.
Synthesis and characterization of TC–PLGA2000 copolymer
A simple esterification reaction was used to synthesize the TC–PLGA2000.16,17 Briefly, 500 mg of PLGA (MW, 2,000), 156 mg of TC hydrochloride, 72 mg of EDC, and 50 mg of HOBt were dissolved in 20 mL of DMF. This solution was heated and stirred for 24 hours at 60°C. Then, the mixture was dialyzed against deionized water using a dialysis membrane (MW cut-off, 14 kDa; Spectrum Laboratories, Laguna Hills, CA, USA) for 48 hours. After centrifugation at 4,000 rpm for 10 minutes and after washing three times with deionized water, the final TC–PLGA2000 products were obtained. The 1H-NMR spectra of the products were obtained using a nuclear magnetic resonance spectrometer (BioSpin AC-80; Bruker Optik GmbH, Ettlingen, Germany). TC–PLGA2000 was dissolved in deuterated dimethyl sulfoxide (DMSO) at 50 mg/mL. PLGA2000 and TC were compared at a concentration of 20 mg/mL.
Preparation of fluorescein
A reaction between the amino group of ODA and the isothiocyanate group of FITC was used to synthesize the ODA–FITC. Briefly, 20 mg of ODA and 28 mg of FITC were dissolved in 6 mL of ethanol. The solution was heated and stirred for 24 hours at 50°C, protected from light. After the reaction was finished, the mixture was mixed with 50 mL of distilled water to precipitate the ODA–FITC. The precipitate was filtrated through a 0.45 mm filter (EMD Millipore, Billerica, MA, USA) and was washed three times with distilled water. The final product was lyophilized and stored in the dark for further use.
Preparation of PLGA NPs and TC–PLGA NPs
Over all, 5 mg of SIM and 10 mg of hydrogenated soya phosphatidyl choline (S100) were dissolved in 1.0 mL of methylene chloride and conjugated by stirring for 30 minutes. The solution was then evaporated under vacuum at room temperature. After all of the methylene chloride was evaporated, the final SIM–S100 drug compound was obtained.
PLGA NPs and TC–PLGA NPs were prepared via the solvent emulsification method.18,19 Briefly, the SIM–S100 drug compound (15 mg; only for SIM-loaded PLGA NPs and TC–PLGA NPs); ODA–FITC (5 mg; only for the ODA–FITC-labeled PLGA NPs and the TC–PLGA NPs); DiR (1.0 mg; only for the DiR-labeled PLGA NPs and the TC–PLGA NPs), 80 mg PLGA (MW =15,000); and 20 mg TC–PLGA2000 (only for the TC–PLGA NPs), or the PLGA2000 (only for the PLGA NPs) were dissolved in 2 mL methylene chloride. The mixture was quickly dispersed into 10 mL of PVA solution (2%; w/v) under high shear and dispersing at 20,000 rpm for 10 minutes, followed by homogenization via a probe sonicator. The emulsion was then evaporated under vacuum until almost all of the methylene chloride was evaporated. The PLGA NPs and TC–PLGA NPs were isolated by centrifugation at 20,000 rpm for 20 minutes and were washed with deionized water several times. The final PLGA NPs or TC–PLGA NPs were redispersed in deionized water.
High-performance liquid chromatography assay for SIM
High-performance liquid chromatography (HPLC) analysis of SIM was performed using a modular liquid chromatograph system (Agilent Technologies, Santa Clara, CA, USA) with a C18 analytical column (Dikma Technologies, Lake Forest, CA, USA) column (250 mm ×4.6 mm; 5 μm). The mobile phase was a mixture of acetonitrile and water containing 25 mM phosphoric acid (65:35; v:v), eluted at a flow rate of 1.0 mL/min and a sample injection volume of 100 μL. The UV detector wave length was set at 239 nm.20
Characterization of PLGA NPs and TC–PLGA NPs
Determination of particle size
The particle sizes of the PLGA NP and TC–PLGA NP suspensions (1.0 mg/mL) were determined by laser scattering technique using a Zetasizer analyzer (3000HS; Malvern Instruments Ltd).
Transmission electron microscopy observations
The morphological characteristics of the PLGA NPs and TC–PLGA NPs were examined using transmission electron microscopy (TEM; JEM-1230; JEOL, Tokyo, Japan). The samples were put on copper grids and were stained with 2% (w/v) phosphotungstic acid.
Determination of drug entrapment efficiency
The SIM-loaded PLGA NPs and SIM-loaded TC–PLGA NPs were destroyed by DMSO. The SIM content was measured by HPLC, and the SIM content in DMSO (Cs; mg/mL) was determined. The drug loading (DL%) of SIM and the entrapment efficiency (EE%) were calculated using Equations 1 and 2, respectively:
|
|
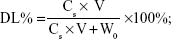
and
|
|
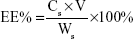
where Ws represents the weight of SIM (mg), W0 represents the weight of the synthetic polymer (mg) added into the system, and V denotes the volume of the DMSO (mL).
Drug release kinetics in vitro
The dialysis bag diffusion technique was used to study in vitro drug release from PLGA NPs and TC–PLGA NPs.21 The SIM-loaded PLGA NPs or TC–PLGA NP suspension equivalent to 50 μg SIM were added until they filled the dialysis bag (MW cut-off, 14 KDa; Spectrum Laboratories, Laguna Hills, CA, USA) and were placed in 15 mL of PBS (pH 7.4), were stirred at 60 r/min, and were maintained at a temperature of 37°C. At predetermined time intervals (ie, 1, 2, 4, 6, 9, 12, 24, 36, 48, and 72 hours), the dissolution medium was completely removed for analysis and was replaced with an equal volume of fresh dissolution medium. HPLC was used to measure the drug concentration.
Cell culture and evaluation
MC3T3-E1 cell culture
MC3T3-E1 cells, a clonal pre-osteoblastic cell line derived from newborn mouse calvaria, which were obtained from Keduo Biochemistry Co, Ltd (Hangzhou, People’s Republic of China), were cultured in αMEM containing 10% (v/v) FBS. When the cells were seeded for the osteogenic differentiation experiment, the basic medium was supplemented with 10 mmol/L β-glycerophosphate and 50 μg/L L-ascorbic acid. The medium was changed every 2 days.22
Cytotoxicity of PLGA NPs and TC–PLGA NPs
To determine the concentration of SIM that did not induce cytotoxicity, 200 μL of MC3T3-E1 cells were plated at a density of 2×104 cells/mL in a 96-well plate and were cultured with SIM-loaded PLGA NPs and TC–PLGA NPs for 48 hours. The concentrations of SIM loaded into the NPs ranged from 10−8 mol/L to 10−5 mol/L. After 48 hours of treatment, cell proliferation was quantified using the MTT assay. Briefly, 20 μL of MTT solution (5 mg/mL) was added, followed by incubation for another 4 hours. After MTT-containing medium was removed and formazan was solubilized in 200 μL DMSO, The optical density values of solution were measured at a wavelength of 570 nm, and cell viability was calculated by the following equation:23
|
|
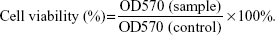
Cellular uptake investigation
For the cellular uptake study, MC3T3-E1 cells were plated in a 12-well plate at a density of 5×104 cells/well and were then treated with fresh αMEM containing ODA–FITC-loaded PLGA NPs and TC–PLGA NPs (100 μg/mL) for 24 hours at 37°C. At pre-determined intervals (ie, 2, 4, 7, 10 and 24 hours), the cells were washed three times with PBS and were collected. Then, the collected cells were subjected to repeated freezing and thawing with DMSO. After they were centrifuged at 10,000 rpm for 5 minutes, the fluorescence of supernatants was detected by fluorescence spectrophotometer and a Micro BCA protein assay kit was used to determine the intracellular protein contents. The cellular uptake percentage of the fluorescent loaded NPs was calculated by the following equation:
|
|
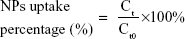
where Ct presents the intracellular ODA–FITC concentration at times t, and Ct0 denotes the initial ODA–FITC concentration at times t0.
Alizarin red S assay
To confirm the mineralization in differentiated cells, MC3T3-E1 cells were plated in a 12-well plate at a density of 5×104 cells/well and were treated with osteogenic differentiation medium containing SIM-loaded PLGA NPs and TC–PLGA NPs (10−8 mol/mL or 10−7 mol/mL of SIM equivalent) for 14 days. The osteogenic differentiation medium was refreshed every other day. After 14 days, 1.5% alizarin red S was prepared in distilled water, and the pH was adjusted to 4.0–4.2 using 0.5% ammonium hydroxide. Differentiated MC3T3-E1 cells were fixed with 70% ethanol (1 hour), were washed, and were stained with alizarin red S for 10–15 minutes. After the removal of the unreacted dye, the mineralized nodules were identified as red spots. Then, the alizarin red S staining was released from the cell matrix by incubating the samples in 10% cetylpyridinium chloride for 15 minutes. The alizarin red S concentration was determined by measuring the absorbance at 562 nm.24,25
Bone mineral binding ability in vitro
The adhesion between the bone-like substrates and the bone-targeting NPs was demonstrated in vitro. The binding capacity of NPs to HAp was evaluated using a method similar to that described in some previous reports.26,27 In brief, NPs loaded with the hydrophobic fluorescent dye ODA–FITC were used at a dilution of 4 mL (0.8 mg/mL of NPs equivalent). Their initial fluorescence emission intensities were measured (Ex [excitation wavelength] =516 nm, Em [emission wavelength] =532 nm, slit [width of slit] =5 nm, U [voltage of the photomultiplier] =700 V) using a fluorescence spectrometer (F-2500; Hitachi Ltd, Tokyo, Japan). Then, 25 mg of HAp with Ca10(PO4)6(OH)2 was added to 4 mL each of the ODA–FITC-loaded NPs. These suspensions were shaken under mechanical stirring at 60 rpm for 3 hours to allow for the adsorption of the ODA–FITC-loaded NPs onto the HAp. The amount of the ODA–FITC-labeled NPs adsorbed was determined by measuring the difference between the fluorescence emission intensity of the initial stock solution and that of the supernatant after adsorption, according to the following equation:
|
|
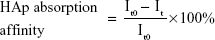
where It0 and It are the fluorescence emission intensities of the initial NP stock solution and that of the supernatant after 3 hours adsorption, respectively.
In vivo evaluation of the bone-targeting NPs in mice
The in vivo bone-targeting capacities of PLGA NPs and TC–PLGA NPs labeled with DiR, a lipophilic fluorescence dye, were evaluated in US Institute of Cancer Research (ICR) mice. The ICR mice were divided into two groups, one of which was administered with the PLGA NPs (the control) and the other with the bone-targeting TC–PLGA NPs. For each experiment, three animals per group were used, and 0.2 mL of the NPs (4 mg/mL) was injected into the tail veins of each mouse. After the injection, all the animals had free access to food and water. After 48 hours, the animals were sacrificed, and the major organs (heart, liver, spleen, lung, kidney, femur, tibia, and fibula) were collected. The fluorescent signals in these organs from three mice in each group were detected using the Maestro EX in vivo imaging system (Cambridge Research & Instrumentation, Inc, Woburn, MA, USA). The distribution of the fluorescent intensities as a percentage of the NPs was calculated from the following equation:
|
|
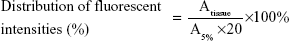
where Atissue represents the fluorescent intensities of the major tissues and A5% represents the fluorescent intensities of DiR-loaded NPs in suspension at the same injected dose at a concentration of 5%.
Animal pharmacodynamics to evaluate NP bone targeting
Twenty female Sprague Dawley rats (200–250 g each) were used. The animals were randomly assigned to five groups of four animals per treatment group as follows: group 1 control sham-operated rats (SHAM); group 2, ovariectomized rats (OVX); group 3, ovariectomized rats that were intravenously injected with SIM at a dose of 0.5 mg/kg/2 days (OVX + SIM); group 4, ovariectomized rats that were intravenously injected with SIM-loaded PLGA NPs at a dose of 0.5 mg/kg/2 days (OVX + SIM-loaded NPs); and group 5, ovariectomized rats that were intravenously injected with SIM-loaded TC–PLGA NPs at a dose of 0.5 mg/kg/2 days (OVX + SIM-loaded TC–NPs). All rats except those in group 1 were bilaterally ovariectomized under anesthesia using pentobarbital sodium (40 mg/kg),28 whereas the rats of group 1 were subjected to a sham operation under the same conditions of anesthesia and abdominal invasion. The injection of SIM-loaded NPs started 1 month after the bilateral ovariectomy or sham operation and continued for 2 months. After 2 months of injection, all animals were sacrificed. The left femurs of the animals were isolated and the bone mineral density (BMD) was measured using a dual-energy X-ray absorptiometer (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA).29,30 The repair of BMD as a percentage of the total bone was calculated from the following equation:
|
|
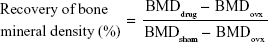
where BMDdrug, BMDovx and BMDsham represent the BMD of bone belonging to the rats of the administration, OVX, and SHAM groups, respectively.
Statistical analysis
The data were expressed as the mean ± standard deviation (SD) unless noted otherwise. The statistical analyses of the data were performed using Student’s t-test. Differences characterized by P<0.05 were considered statistically significant.
Results and discussion
Synthesis and characterization of TC–PLGA2000 copolymers
The TC–PLGA2000 was successfully synthesized by the direct conjugation of the carboxyl group of PLGA2000 with the hydroxyl group of TC via an esterification reaction, as described in Figure 1. To characterize the TC–PLGA2000, the product was evaluated by 1H-NMR spectroscopy. As shown in Figure 2A, the 1H-NMR spectra revealed peaks at 7.0–8.0 ppm that corresponded to the hydrogens of the benzene ring of TC. In Figure 2B, the peak at 4.0–6.0 ppm represents the methylene and methylidyne hydrogens of the PLGA copolymers. In Figure 2C, we observed all of the above-mentioned characteristics, as well as a peak at 13 ppm corresponding to the carboxyl of PLGA, which indicated that the carboxyl group of PLGA2000 had conjugated with the hydroxyl group of TC. These results all indicated that the synthesis of the TC–PLGA2000 was successful.
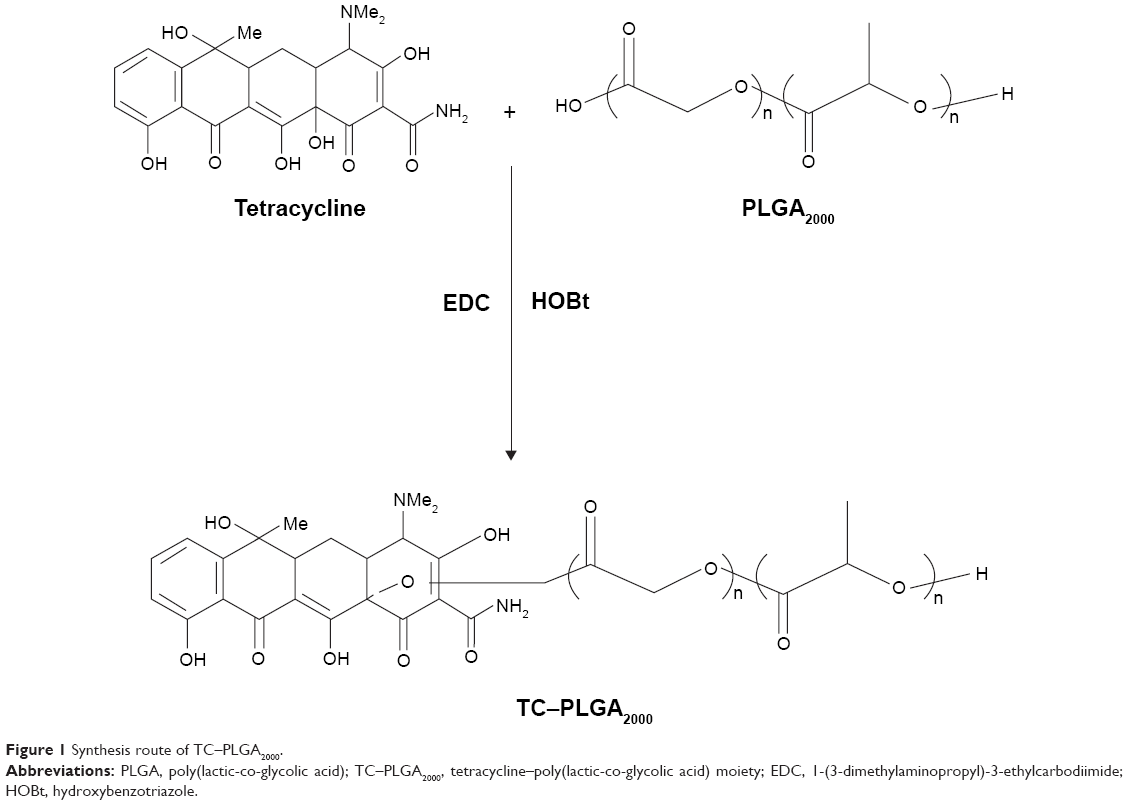 | Figure 1 Synthesis route of TC–PLGA2000. |
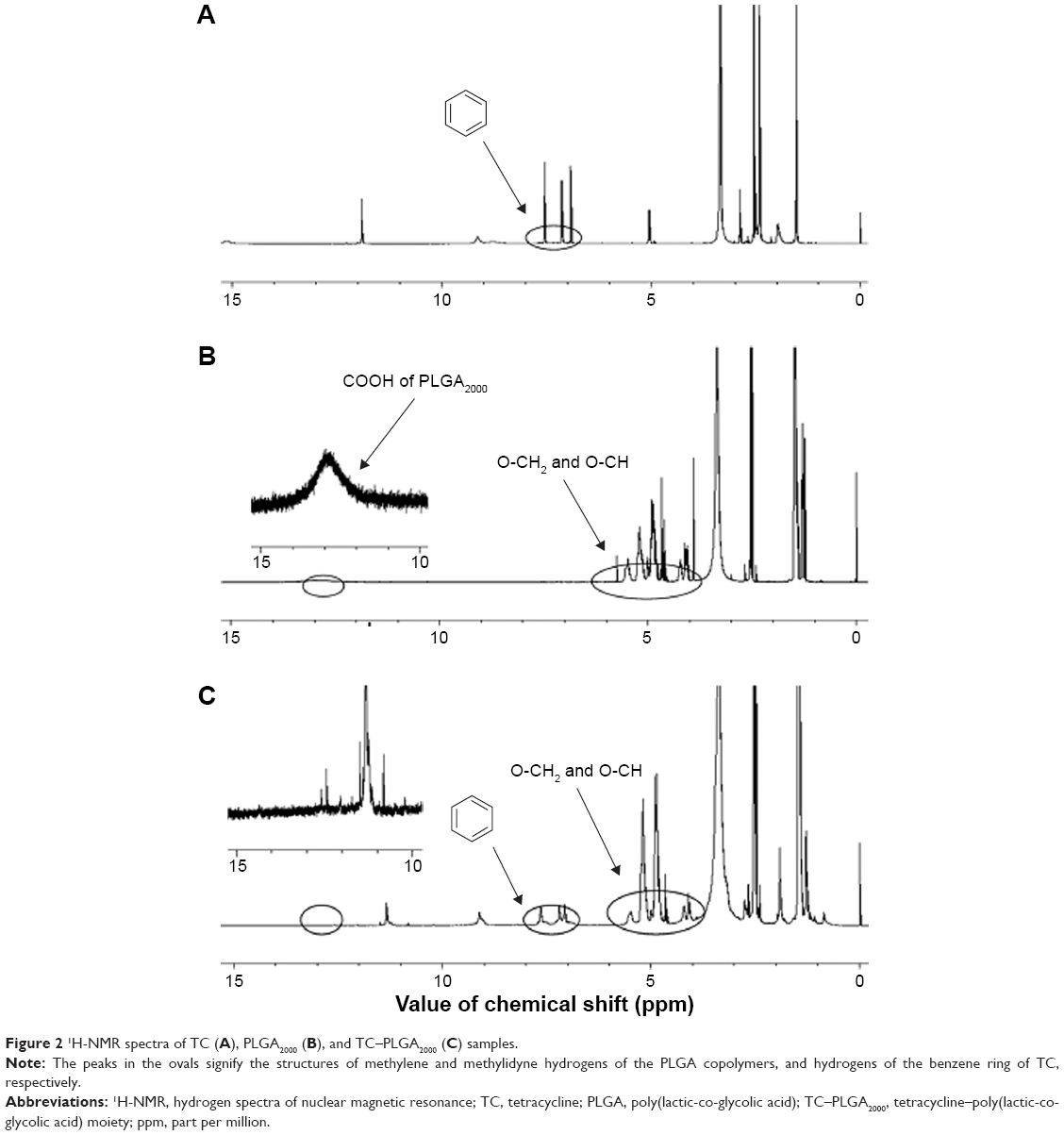 | Figure 2 1H-NMR spectra of TC (A), PLGA2000 (B), and TC–PLGA2000 (C) samples. |
Properties of blank and SIM-loaded PLGA NPs and TC–PLGA NPs
As shown in Table 1, the sizes of the blank PLGA NPs, SIM-loaded PLGA NPs and TC–PLGA NPs were approximately 220 nm. The TC graft had no significant effect on the particle size (P>0.05). The polydispersity index (PI) of these particles was approximately 0.3. These results demonstrated the narrow size distribution of the NPs.
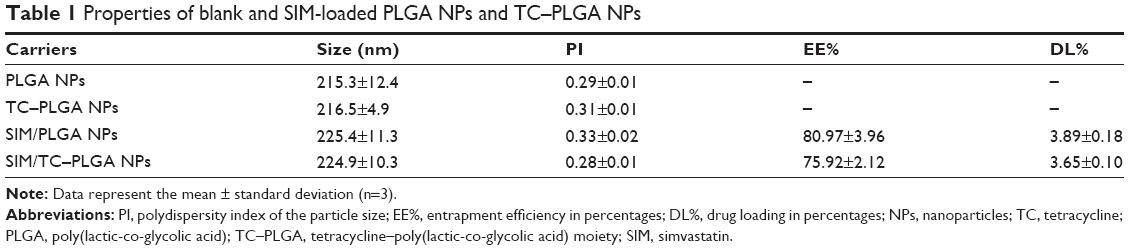 | Table 1 Properties of blank and SIM-loaded PLGA NPs and TC–PLGA NPs |
The NP size was further confirmed by using TEM. As shown in Figure 3, the blank and SIM-loaded NPs were all spherical or spherical-like particles, and were uniform in size and shape, as shown in TEM image (Figure 3). There were no obvious changes in particle size after TC graft.
 | Figure 3 TEM images of blank NPs and SIM-loaded PLGA or TC–PLGA NPs: PLGA NPs (A); TC–PLGA NPs (B); SIM-loaded PLGA NPs (C); and SIM-loaded TC–PLGA NPs (D). |
The DL% and EE% are shown in Table 1. The EE% of the SIM-loaded PLGA NPs was >75%. There were no obvious changes in EE% after TC graft (P>0.05).
The in vitro drug release behaviors of SIM-loaded NPs are shown in Figure 4. The SIM-loaded PLGA NPs exhibited a cumulative drug release >80% during a 72-hour time span. There were no significant differences in drug release profiles between the normal PLGA NP group and the TC–PLGA NP group (P>0.05). Hence, the TC graft could be considered to have no effect on drug release.
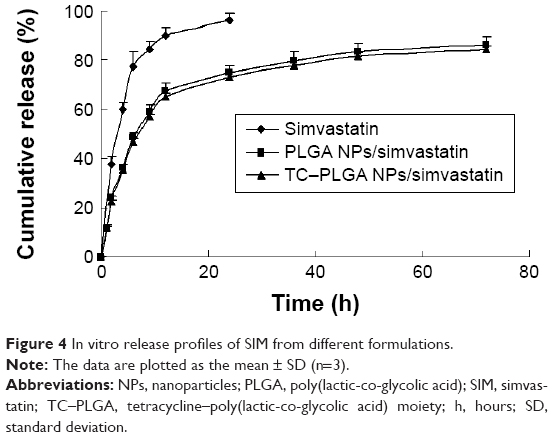 | Figure 4 In vitro release profiles of SIM from different formulations. |
Cell culture and evaluation
Cytotoxicity of NPs
Figure 5A shows the results of an MTT assay used to assess the cytotoxic effects of the PLGA NPs and TC–PLGA NPs on MC3T3-E1 cells. The results in Figure 5A demonstrate that the PLGA NPs and TC–PLGA NPs had no cytotoxicity, and that cell viability was nearly 100% at PLGA and TC–PLGA NP concentrations <100 μg/mL.
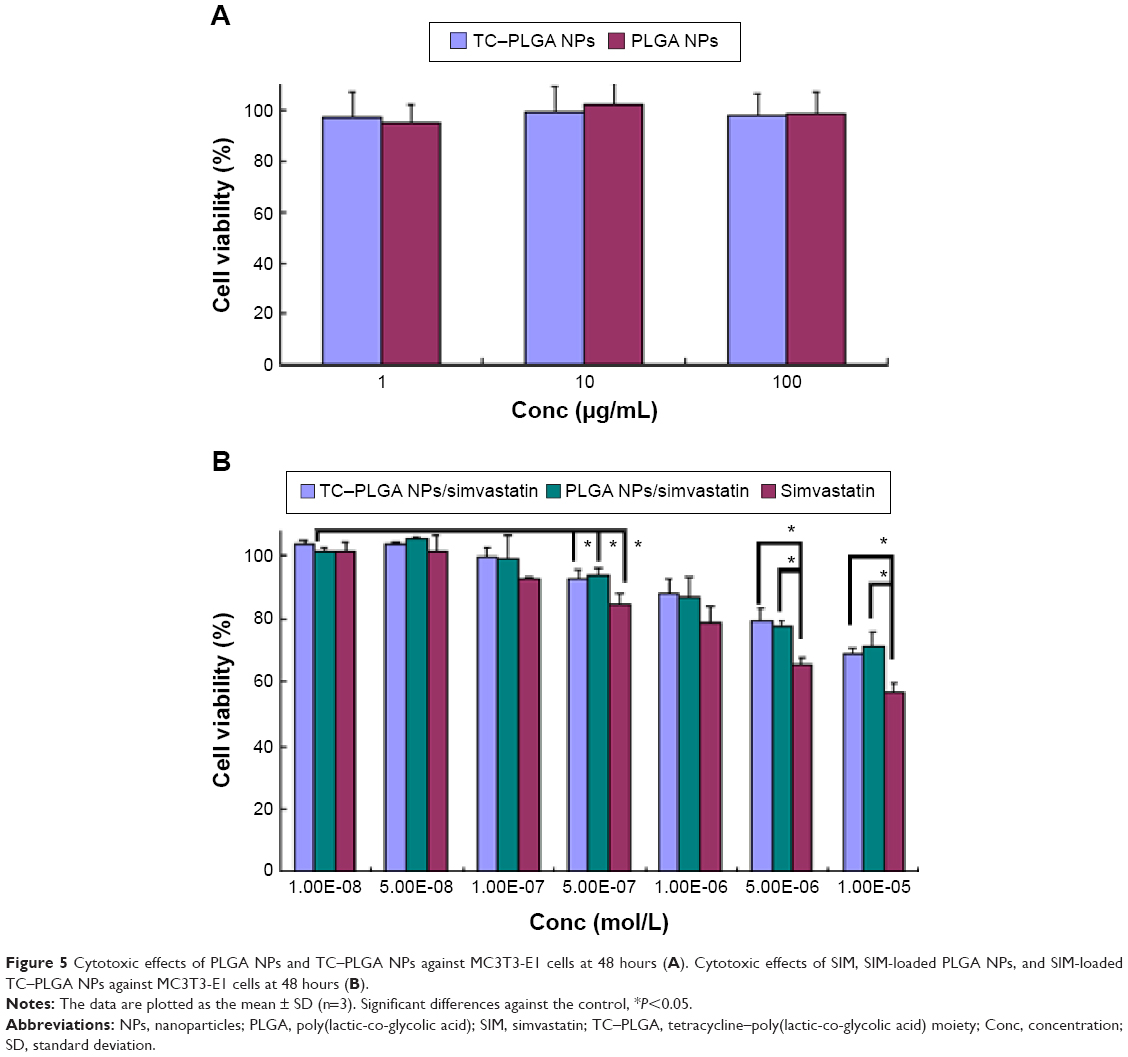 | Figure 5 Cytotoxic effects of PLGA NPs and TC–PLGA NPs against MC3T3-E1 cells at 48 hours (A). Cytotoxic effects of SIM, SIM-loaded PLGA NPs, and SIM-loaded TC–PLGA NPs against MC3T3-E1 cells at 48 hours (B). |
To determine the optimum concentration of SIM-loaded NPs for osteogenic differentiation, we cultured the MC3T3-E1 cells with varying concentrations of SIM-loaded NPs. As seen in Figure 5B, SIM-loaded NPs could reduce the cytotoxic effects and improve the biocompatibility of SIM, especially when the concentration of SIM was >5×10−6 mol/L. Meanwhile, the cytotoxic effects of the SIM-loaded PLGA NPs and the SIM-loaded TC–PLGA NPs were not significantly different, indicating that the TC graft had a negligible effect on cell viability.
When the concentrations of SIM were >5×10−7 mol/L, the cells treated with either SIM or SIM-loaded NPs displayed significant cytotoxic effects, resulting in a decrease in cell viability compared to the untreated control cells (P<0.05). These results suggested that the concentration of SIM should be kept below 10−7 mol/L in future experiments testing osteogenic differentiation.
Cell uptake investigation
Cellular uptake behaviors of PLGA NPs and TC–PLGA NPs were evaluated using MC3T3-E1 cells as model cells. ODA–FITC, which was water-insoluble and had almost no release in culture medium, was used as a label to quantify the internalization of the NPs into the cells. As shown in Figure 6, the uptake of NPs by MC3T3-E1 cells increased with passing time. After 24 hours, the uptake percentage was approximately 8.91% for the ODA–FITC-loaded PLGA NPs but was just 6.67% for the ODA–FITC-loaded TC–PLGA NPs, which also showed a weak decline over time. This may have been because the TC graft on the surface of the PLGA NPs may have improved the hydrophilicity of the PLGA NPs, leading to a decrease in the ability of the NPs to be internalized.
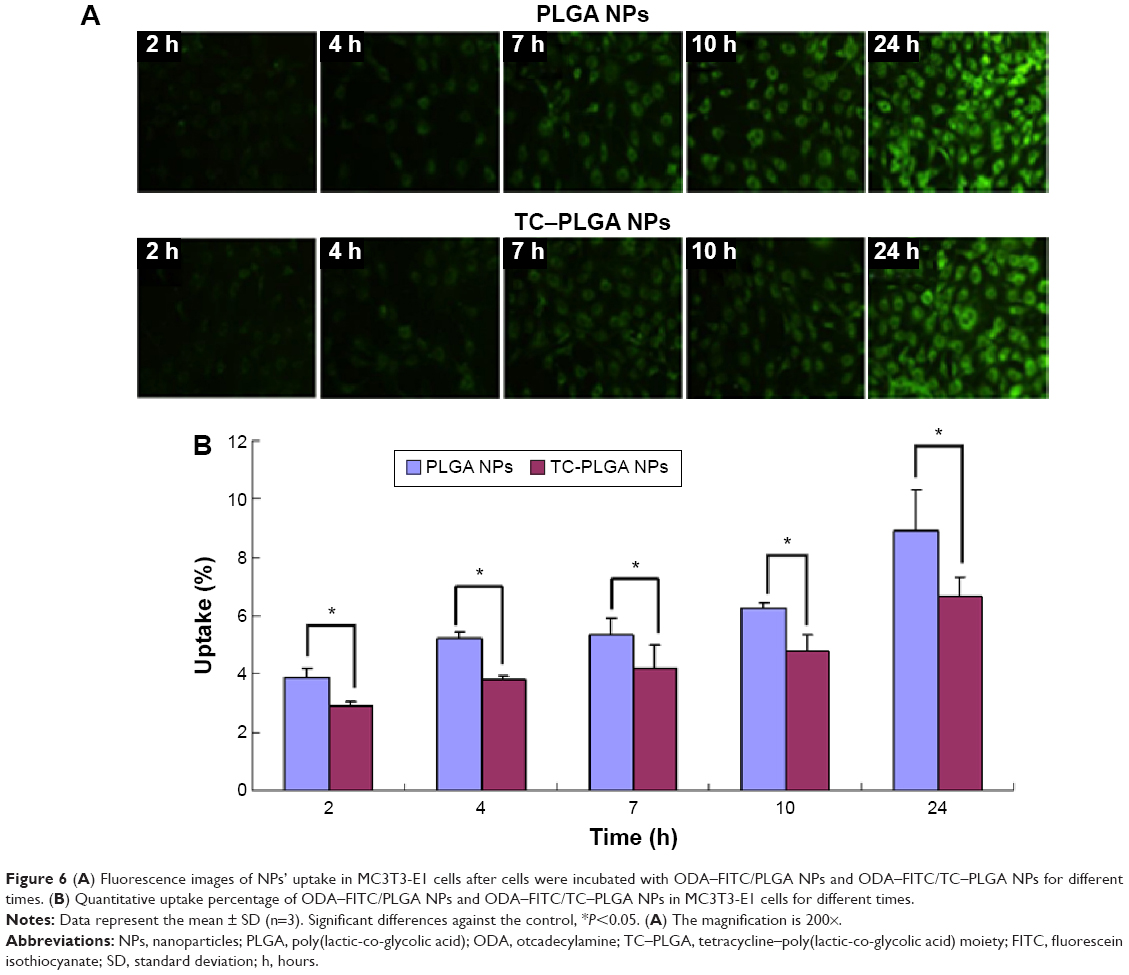 | Figure 6 (A) Fluorescence images of NPs’ uptake in MC3T3-E1 cells after cells were incubated with ODA–FITC/PLGA NPs and ODA–FITC/TC–PLGA NPs for different times. (B) Quantitative uptake percentage of ODA–FITC/PLGA NPs and ODA–FITC/TC–PLGA NPs in MC3T3-E1 cells for different times. |
Effects of SIM-loaded NPs on mineralized nodule formation
We tested the effects of SIM-loaded NPs on osteoblast differentiation, as evidenced by mineralization. MC3T3-E1 cells were cultured in osteogenic differentiation medium for 14 days with SIM-loaded PLGA NPs and SIM-loaded TC–PLGA NPs. The medium used in our in vitro experiments to induce osteogenic differentiation contained 50 μg/mL ascorbic acid and 10 mM β-glycerophosphate. This is a widely employed standard medium in osteogenic differentiation studies24,25 and has been reported to possess osteogenic activities.31 Due to its necessary involvement in the cell culture studies, we also examined the osteogenic effects of this medium, and we used differentiation medium treatment as a background in order to assess the actual osteogenic activities of the SIM-loaded NPs.
As shown in Figure 7, treatments with free SIM and SIM-loaded NPs all yielded an increase in cell mineralization over that of the differentiation medium-treated cells. The increase in the activity was significant both at SIM concentrations of 10−8 mol/L and 10−7 mol/L, though the SIM concentration of 10−7 mol/L had a better effect on cell mineralization. The effects of SIM, SIM-loaded PLGA NPs, and SIM-loaded TC–PLGA NPs on the formation of mineralized nodules were not significantly different at the same concentrations, indicating that the interference of the PLGA NPs and TC–PLGA NPs with osteogenic differentiation was negligible.
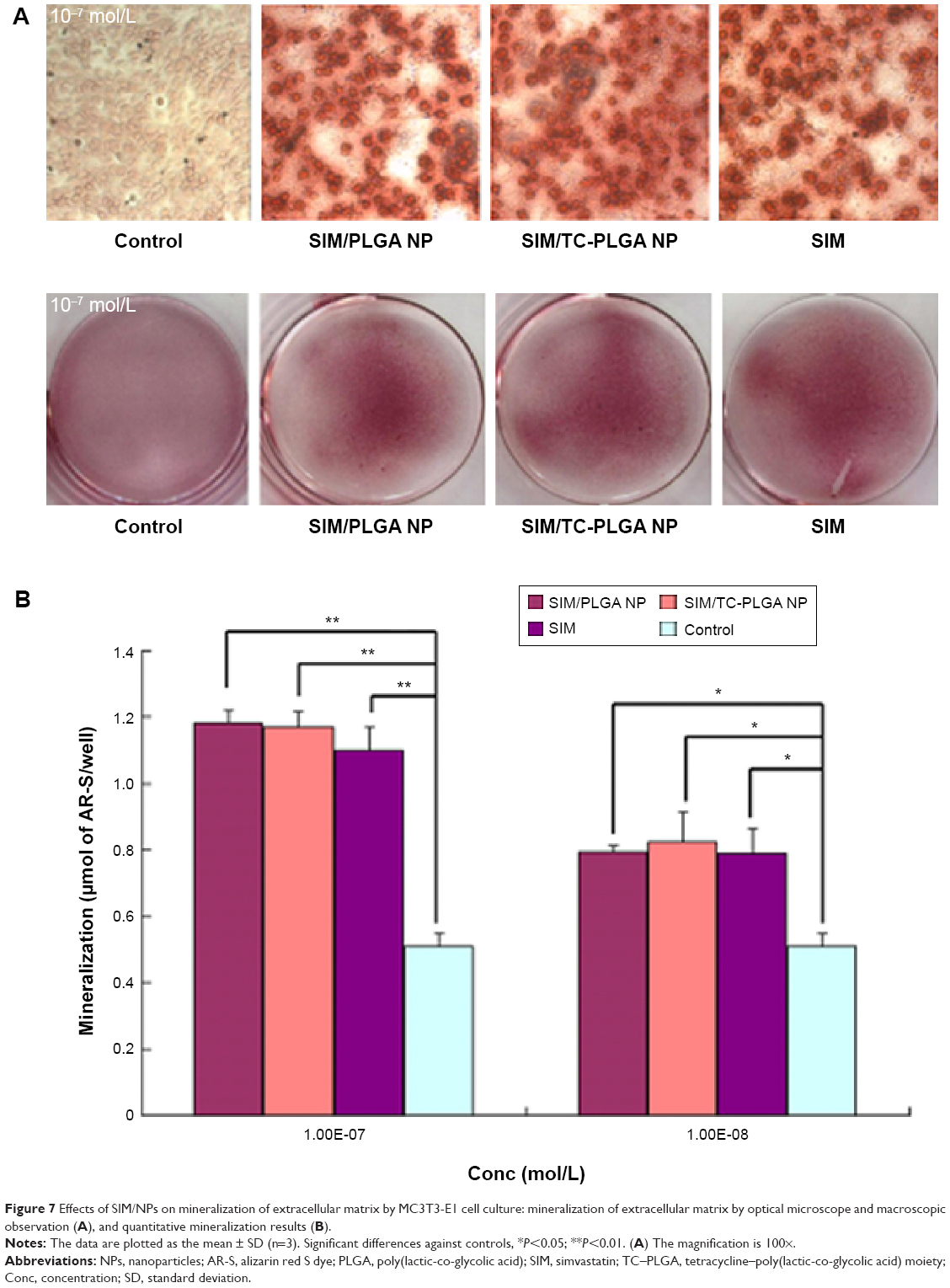 | Figure 7 Effects of SIM/NPs on mineralization of extracellular matrix by MC3T3-E1 cell culture: mineralization of extracellular matrix by optical microscope and macroscopic observation (A), and quantitative mineralization results (B). |
Bone mineral binding ability
Apatite is the main component of bone in its natural state. In vertebrate bones, this inorganic material is known as hydroxyapatite Ca10(PO4)6(OH)2, or carbonated hydroxyapatite. A HAp binding assay was performed in vitro (Figure 8A), and the affinity of the bone-targeting NPs for HAp are shown in Figure 8B. The data presented in Figure 8B clearly demonstrate that the proportion of the TC–PLGA NPs bound to HAp (87.94%) was greater than the proportion of bound PLGA NPs (18.59%). These results indicated that after grafting, TC could still maintain its affinity for HAp and that the ability for bone targeting of PLGA NPs was improved.
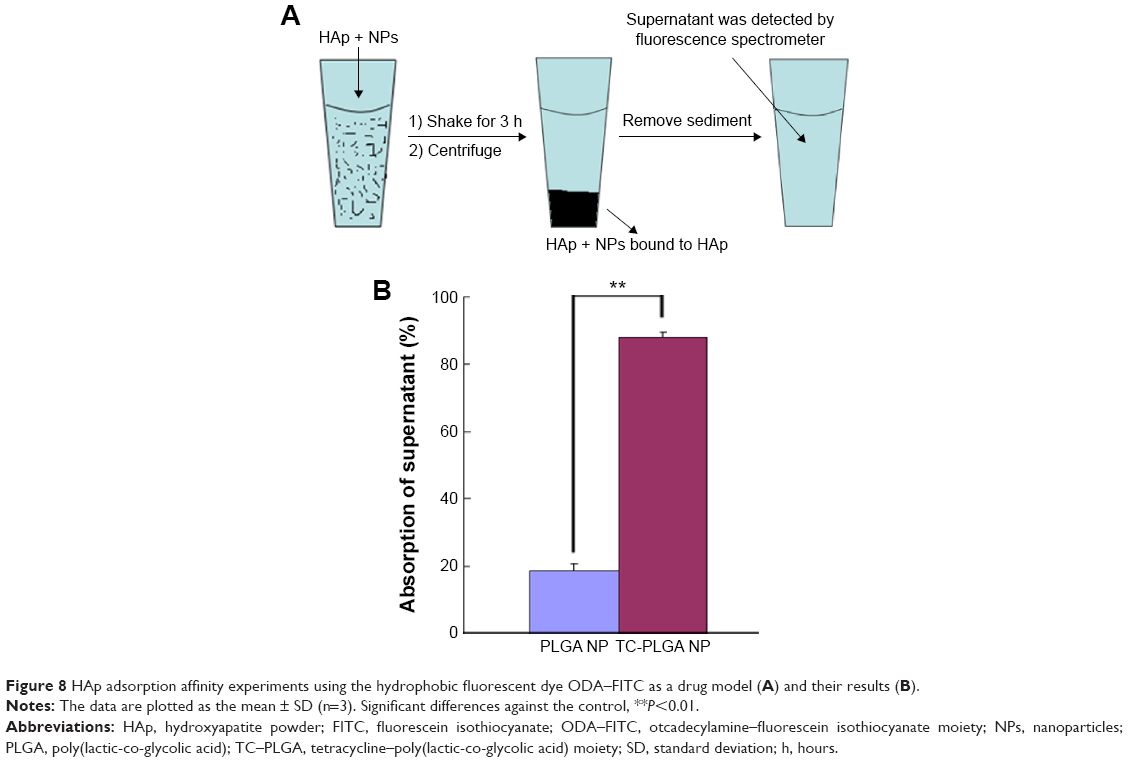 | Figure 8 HAp adsorption affinity experiments using the hydrophobic fluorescent dye ODA–FITC as a drug model (A) and their results (B). |
In vivo distribution assay
An in vivo biodistribution assay in ICR mice was performed to further examine the bone-targeting ability of TC–PLGA NPs. Figure 9B shows the accumulation of the NPs in the skeleton (represented by the femur, tibia, and fibula). The fluorescent intensities were evaluated through both NP groups (DiR-loaded PLGA NPs and DiR-loaded TC–PLGA NPs), which reached the skeleton 48 hours after intravenous administration via the tail vein in mice. After 48 hours, the fluorescent intensities of DiR in the bone due to the TC–PLGA NPs were clearly higher than those due to the PLGA NP control. These data were then quantified from the fluorescent intensities, and the results are shown in Figure 9D. The tissue distributions of the fluorescent intensities clearly indicated that 80.5% more DiR-loaded TC–PLGA NPs were present in the femur, tibia, and fibula than in the DiR-loaded PLGA NPs. Therefore, the TC–PLGA copolymer has a tendency to target and to accumulate in bone.
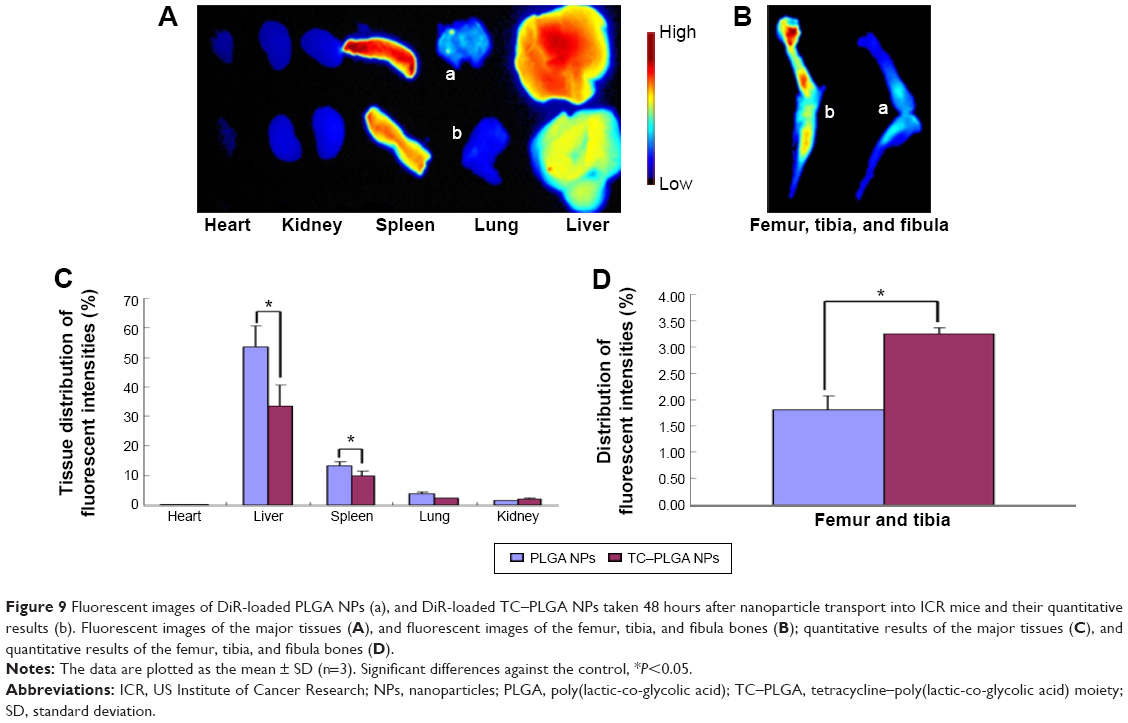 | Figure 9 Fluorescent images of DiR-loaded PLGA NPs (a), and DiR-loaded TC–PLGA NPs taken 48 hours after nanoparticle transport into ICR mice and their quantitative results (b). Fluorescent images of the major tissues (A), and fluorescent images of the femur, tibia, and fibula bones (B); quantitative results of the major tissues (C), and quantitative results of the femur, tibia, and fibula bones (D). |
Additional in vivo tests evaluating the whole-body distribution of NPs are needed to indirectly determine the efficacy of these targeted nano-polymeric delivery systems. As shown in Figure 9A, the fluorescence signals were detected mainly in the liver and spleen, and were barely detectable in the lungs, kidneys, and heart of the mice from the PLGA NP control group. Meanwhile, the fluorescence signals observed in the spleen and liver of the mice from the TC–PLGA NP group were significantly reduced. The fluorescence signals were also barely detectable in the heart, lungs, and kidneys of the rats from the TC–PLGA NP treatment group. The reason for the accumulation of the NPs in the liver and spleen was that the macrophage cells in these organs play a role in uptake and phagocytosis.32
The whole-body distribution data were also quantified from the fluorescence intensities, and the results are shown in Figure 9C. It is worth mentioning that compared to the DiR-loaded PLGA NPs, 37.6% and 24.0% fewer DiR-loaded TC–PLGA NPs were present in the liver and spleen, respectively. The results showed that TC–PLGA NPs could reduce the trapping of NPs in the liver and spleen, reducing side effects in other organs caused by drugs. This can be taken as indirect evidence that TC–PLGA NPs have a tendency to target and accumulate in bone.
Animal pharmacodynamics evaluation of bone-targeting NPs
Twelve weeks after ovariectomy operation, four animals died, one in the SHAM group and three in ovariectomized rats. As shown in Figure 10A, the femur BMD was distinctly different between the two groups (P<0.01), with values of 0.185±0.003 g/cm2 in the SHAM group and 0.149±0.004 g/cm2 in the OVX group, indicating the success of the osteoporotic model established in ovariectomized rats.
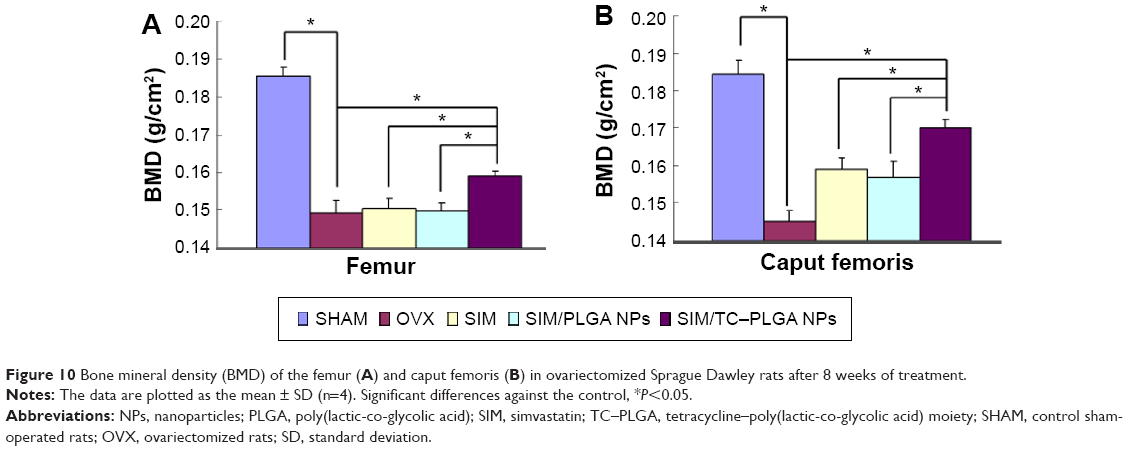 | Figure 10 Bone mineral density (BMD) of the femur (A) and caput femoris (B) in ovariectomized Sprague Dawley rats after 8 weeks of treatment. |
Eight weeks after treatment, all of the animals were sacrificed. As shown in Figure 10A, the femur BMD between groups 2 (OVX), 3 (OVX + SIM), and 4 (OVX + SIM-loaded NPs) showed no significant differences (P>0.05). However, compared to group 2 (OVX), the femur BMD in group 5 (OVX + SIM-loaded TC–NPs) was increased by 6.71%. The recovery of BMD in the femur was 4.14% in group 3 (OVX + SIM), 2.07% in group 4 (OVX + SIM-loaded NPs), and 17.59% in group 5 (OVX + SIM-loaded TC–NPs).
The caput femoris BMD was also measured, as shown in Figure 10B. A statistically significant increase in the caput femoris BMD was observed. The caput femoris BMD was increased by 9.47% in group 3 (OVX + SIM), 7.92% in group 4 (OVX + SIM-loaded NPs), and 17.04% in group 5 (OVX + SIM-loaded TC–NPs) when compared to the group 2 (OVX) group. The recovery of the caput femoris BMD was 35.26% in group 3 (OVX + SIM), 29.49% in group 4 (OVX + SIM-loaded NPs), and 63.46% in group 5 (OVX + SIM-loaded TC–NPs). The better recovery of the caput femoris from osteoporosis may be because cancellous bone structures such as the caput femoris are more sensitive than cortical bone to the inducement of osteoblast differentiation.
Overall, the results demonstrated that the TC–PLGA NPs have a good ability to target bone and to achieve high bone tissue drug accumulation, improving the curative effects of SIM on osteoporosis. These NPs may act as a delivery system for hydrophobic drugs and are expected to reduce the required drug dose administered or to reduce the side effects of drugs in other organs, thereby improving the curative effects of SIM.
Conclusion
Two types of NPs (PLGA NPs and TC–PLGA NPs) were designed and prepared, and then their bone-targeting ability and curative effects on osteoporosis were assessed. 1H-NMR was used to confirm that the final TC–PLGA2000 product was successfully synthesized. The average particle size was approximately 220 nm for the two types of copolymer NPs, and the SIM-loaded NPs exhibited a cumulative release of up to 80% within 72 hours in vitro. The in vitro cell evaluation indicated that the NPs had excellent cellular uptake ability and great biocompatibility with MC3T3-E1 cells, which would reduce the cytotoxic effects of SIM. The cell mineralization assay showed that the SIM-loaded NPs induced osteogenic differentiation and mineralized nodule formation in MC3T3-E1 cells, thus achieving the same effect as SIM. In addition, the TC moiety of the TC–PLGA NPs had enhanced bone-binding capacity when binding to apatite mineral surfaces, due to its affinity for HAp.
Biophotonic imaging technology was also used to examine the organ distribution of the DiR-labeled areas or of the fluorescent dye delivered by the PLGA and TC–PLGA NPs in ICR mice. The intensity of the intraosseous fluorescence signal was stronger in the bone after injection with the TC–PLGA NPs than after treatment with the PLGA NPs. The animal pharmacodynamics of treatment of osteoporosis in ovariectomized rats suggested that the SIM-loaded TC–PLGA NPs could improve the curative effects of SIM on the recovery of DMD compared to either the SIM-loaded PLGA NPs or free SIM. The bone-targeting NPs, based on the conjugation of TC to PLGA copolymers, had the ability to target bone, and their use for the treatment of osteoporosis with SIM was successfully achieved. These NPs may act as a delivery system for hydrophobic drugs and are expected to reduce the drug dose required for administration or to reduce the side effects in other organs, thereby improving the curative effects of SIM.
Disclosure
The authors report no conflicts of interest in this work.
References
Mundy G, Garrett R, Harris S, et al. Stimulation of bone formation in vitro and in rodents by statins. Science. 1999;286(5446):1946–1949. | ||
Blum CB. Comparison of properties of four inhibitors of 3-hdroxy-3methyglutary-coenzyme A reductase. Am J Cardiol. 1994;73:3D–11D. | ||
Montagnani A, Gonnelli S, Cepollaro C, et al. Effect of simvastatin treatment on bone mineral density and bone turnover in hypercholesterolemic postmenopausal women: a 1-year longitudinal study. Bone. 2003;32:427–433. | ||
Tikiz C, Tikiz H, Taneli F, Gumuser G, Tuzun C. Effects of simvastatin on bone mineral density and remodeling parameters in postmenopausal osteopenic subjects: 1-year follow-up study. Clin Rheumatol. 2005;24:447–452. | ||
Skoglund B, Forslund C, Aspenberg P. Simvastatin improves fracture healing in mice. J Bone Miner Res. 2002;17:2004–2008. | ||
Guyton JR. Benefit versus risk in statin treatment. Am J Cardiol. 2006;97:95C–97C. | ||
Shekunov BY, Chattopadhyay P, Seitzinger J, Huff R. Nanoparticles of poorly water-soluble drugs prepared by supercritical fluid extraction of emulsions. Pharm Res. 2006;23(1):196–204. | ||
Kwon GS, Kataoka K. Block copolymer micelles as long-circulating drug vehicles. Adv Drug Deliv Rev. 1995;16(2):295–309. | ||
Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliv Rev. 2001;47(1):113–131. | ||
Hans ML, Lowman AM. Biodegradable nanoparticles for drug delivery and targeting. Curr Opin Solid State Mater Sci. 2002;6(4):319–327. | ||
De Jong WH, Borm PJ. Drug delivery and nanoparticles: applications and hazards. Int J Nanomedicine. 2008;3(2):133. | ||
Wang G, Uludag H. Recent developments in nanoparticle-based drug delivery and targeting systems with emphasis on protein-based nanoparticles. Expert Opin Drug Deliv. 2008;5:499–515. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Ho ML, Fu YC, Wang GJ, et al. Controlled release carrier of BSA made by W/O/W emulsion method containing PLGA and hydroxyapatite. J Control Release. 2008;128(2):142–148. | ||
Perrin RA. Binding of tetracycline to bone. Nature. 1965;208(11):787–788. | ||
Mazaleyrat JP, Boutboul A, Lebars Y, Gaucher A, Wakselman M. Practical resolution of an atropoisomeric [alpha],[alpha]-disubstituted glycine with l-phenylalanine cyclohexylamide as chiral auxiliary. Tetrahedron Asymmetry. 1998;9(15):2701–2713. | ||
Gaucher A, Zuliani Y, Cabaret D, Wakselman M, Mazaleyrat JP. Synthesis and resolution of β 2, 2-HBin, the first enantiomerically stable β-amino acid with chirality only due to axial dissymmetry. Tetrahedron Asymmetry. 2001;12(18):2571–2580. | ||
Nath SD, Son S, Sadiasa A, Min YK, Lee BT. Preparation and characterization of PLGA microspheres by the electrospraying method for delivering simvastatin for bone regeneration. Int J Pharm. 2013;443(1–2):87–94. | ||
Ho MH, Chiang CP, Liu YF, et al. Highly efficient release of lovastatin from poly(lactic-co-glycolic acid) nanoparticles enhances bone repair in rats. J Orthop Res. 2011;29(10):1504–1510. | ||
Tiwari R, Pathak K. Nanostructured lipid carrier versus solid lipid nanoparticles of simvastatin: comparative analysis of characteristics, pharmacokinetics and tissue uptake. Int J Pharm. 2011;415(1):232–243. | ||
Wang CZ, Fu YC, Jian SC, et al. Synthesis and characterization of cationic polymeric nanoparticles as simvastatin carriers for enhancing the osteogenesis of bone marrow mesenchymal stem cells. J Colloid Interface Sci. 2014;432:190–199. | ||
Yang F, Zhao S, Zhang F, He FM, Yang GL. Simvastatin-loaded porous implant surfaces stimulate preosteoblasts differentiation: an in vitro study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;111(5):551–556. | ||
Park JB, Zhang H, Lin CY, et al. Simvastatin maintains osteoblastic viability while promoting differentiation by partially regulating the expressions of estrogen receptors α. J Surg Res. 2012;174(2):278–283. | ||
Gregory CA, Gunn WG, Peister A, Prockop DJ. An Alizarin red-based assay of mineralization by adherent cells in culture: comparison with cetylpyridinium chloride extraction. Anal Biochem. 2004;329(1):77–84. | ||
Maeda T, Matsunuma A, Kawane T, Horiuchi N. Simvastatin promotes osteoblast differentiation and mineralization in MC3T3-E1 cells. Biochem Biophys Res Commun. 2001;280(3):874–877. | ||
Ho ML, Fu YC, Wang GJ, et al. Controlled release carrier of BSA made by W/O/W emulsion method containing PLGA and hydroxyapatite. J Control Release. 2008;128:142–148. | ||
Fu YC, Fu TF, Wang HJ, et al. Aspartic acid-based modified PLGA–PEG nanoparticles for bone targeting: In vitro and in vivo evaluation. Acta Biomater. 2014;10(11):4583–4596. | ||
Waynforth HB. Experimental and Surgical Technique in the Rat. Academic Press, London: Academic Press; 1980. | ||
Wang JW, Xu SW, Yang DS, Lv RK. Locally applied simvastatin promotes fracture healing in ovariectomized rat. Osteoporos Int. 2007;18(12):1641–1650. | ||
Ito T, Takemasa M, Makino K, Otsuka M. Preparation of calcium phosphate nanocapsules including simvastatin/deoxycholic acid assembly, and their therapeutic effect in osteoporosis model mice. J Pharm Pharmacol. 2013;65(4):494–502. | ||
Maeda T, Matsunuma A, Kurahashi I, Yanagawa T, Yoshida H, Horiuchi N. Induction of osteoblast differentiation indices by statins in MC3T3-E1 cells. J Cell Biochem. 2004;92(3):458–471. | ||
Wong L, Bendayan R, Rauth AM, Li YQ, Wu XY. Chemotherapy with anticancer drugs encapsulated in solid lipid nanoparticles. Adv Drug Deliv Rev. 2007;59(6):491–504. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.