")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Tenofovir alafenamide in the treatment of chronic hepatitis B: design, development, and place in therapy
Authors Ogawa E, Furusyo N, Nguyen MH
Received 11 July 2017
Accepted for publication 18 October 2017
Published 6 November 2017 Volume 2017:11 Pages 3197—3204
DOI https://doi.org/10.2147/DDDT.S126742
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Eiichi Ogawa,1 Norihiro Furusyo,1 Mindie H Nguyen2
1Department of General Internal Medicine, Kyushu University Hospital, Fukuoka, Japan; 2Division of Gastroenterology and Hepatology, Department of Medicine, Stanford University Medical Center, Palo Alto, CA, USA
Abstract: Tenofovir alafenamide (TAF), a novel prodrug of tenofovir (TFV), has been approved for the treatment of chronic hepatitis B virus (HBV) infection. TAF has been shown to be a potent inhibitor of HBV replication at a low dose, with high intracellular concentration and more than 90% lower systemic TFV concentration than tenofovir disoproxil fumarate (TDF). In two randomized, double-blind, multinational, Phase 3, non-inferiority trials for hepatitis B e antigen (HBeAg)-positive and -negative patients (primary analysis: 48 weeks), TAF 25 mg orally once-daily was not inferior to TDF 300 mg in achieving an HBV DNA level <29 IU/mL at week 48. No amino-acid substitutions associated with viral breakthrough were detected by deep sequencing, and no resistance to TAF was found through week 96. In addition, no difference in the frequency of HBeAg or hepatitis B surface antigen loss was observed. However, TAF was associated with a significantly higher ALT normalization rate than was TDF, based on the American Association for the Study of Liver Diseases criteria (male: ALT ≤30 U/L and female: ALT ≤19 U/L). An analysis of renal safety showed that patients treated with TAF had a significantly lower decrease in the estimated glomerular filtration rate level than did patients treated with TDF. Similarly, the declines of hip and spine bone mineral density were significantly less in the TAF group. These trends of efficacy and renal/bone safety continued through week 96. Longer term follow-up and real-world data will be required to determine if the differences in viral/biochemical response and renal/bone safety seen with TAF in comparison with TDF are clinically relevant.
Keywords: hepatitis B virus, treatment, tenofovir alafenamide, tenofovir disoproxil fumarate, adverse effects
Introduction
Chronic hepatitis B virus (HBV) infection is one of the leading causes of cirrhosis, liver decompensation, and hepatocellular carcinoma (HCC). An estimated 257 million people are positive for hepatitis B surface antigen (HBsAg) globally.1 The availability of universal vaccination and effective antiviral agents for several decades in high endemic countries has contributed to a slow decrease in the estimated global prevalence of HBsAg to 3.6%–3.7%.2,3 However, the number of HBsAg positive people has increased (223 million in 1990 to 240 million in 2005) due to an increase in world population, especially in low and middle income countries in high endemic regions; over 5% in sub-Saharan Africa.2 Due to interaction among various host, environmental, and viral factors, chronic HBV infection can range from chronic infection with active viral replication but relatively normal biochemical profiles to chronic hepatitis with elevated ALT.4,5 Serial monitoring of ALT, HBV DNA level, and hepatitis B e antigen (HBeAg) sero-status is essential for characterization of the phase of infection.6
There are currently two classes of treatment options for chronic HBV infection: pegylated interferon and nucleos(t)ide analog. Treatment with pegylated interferon involves immune system control of HBV infection, and thus is limited to patients who can better respond to interferon, such as patients with HBV genotype A/B, wild type pre-core and basal core promotor sequences, low HBV DNA, and higher ALT levels at baseline or those who are younger.7–9 Interferon-based therapies are also contraindicated in the presence of hepatic decompensation and should be used with caution in patients with cirrhosis. Nucleos(t)ide analogs inhibit HBV replication and are generally well tolerated; however, lamivudine, telbivudine, and adefovir are no longer recommended because of the high risk of resistance.4,6 Alternatives to these drugs, entecavir and tenofovir disoproxil fumarate (TDF), are recommended by most management guidelines as the first-line oral agents for chronic HBV infection and can be used for patients with hepatic decompensation or those who have had a liver transplant.4,6,10 Long-term nucleos(t)ide analog treatment has been shown to be effective in the suppression of HBV replication to levels below the detection limits of PCR assays, in histologic improvement,11 and in reducing the incidence of HCC,12 although the loss or seroconversion of HBsAg is very rare.13 In this context, long-term treatment is required in almost all cases. As such, long-term safety of therapy including renal and bone dysfunction with Fanconi-like syndrome from adefovir or TDF use can be of concern, albeit low.14–16
Tenofovir alafenamide (TAF) was approved recently by the United States Food and Drug Administration; the European Medicines Agency; and the Health, Labor, and Welfare Ministry in Japan. Both TAF and TDF are phosphonoamidate prodrugs of tenofovir (TFV) that share the same intracellular active metabolite, TFV diphosphate (TFV-DP), which is effective against both HBV and HIV-1 infection.17,18 TAF has greater plasma stability, which allows for more efficient uptake by hepatocytes at lower plasma concentrations than TDF, thus the circulating concentration of TFV is 90% lower after administration of a 25 mg dose of TAF than after a 300 mg dose of TDF.19 This difference likely contributes to the better safety profile of TAF compared with TDF, especially for renal and bone dysfunction. This review focuses on two Phase 3 trials of the clinical use of TAF for patients with chronic HBV infection.20,21
Pharmacokinetics
The structure of TFV and its prodrugs, TAF and TDF, are presented in Figure 1.22 After intestinal absorption to plasma, TAF enters hepatocytes by passive diffusion facilitated by organic anion-transporting polypeptides 1B1 and 1B3, then it is metabolized by carboxylesterase-1 to form TFV, which is phosphorylated to the active metabolite TFV-DP. TAF is more stable in plasma than TDF: the in vitro plasma T1/2 of TAF was 30–90 minutes compared to 0.4 minutes for TDF:19,23 which allows for efficient uptake by hepatocytes. TFV released in the body is eliminated renally by the combined action of active tubular secretion in the proximal tubule and passive glomerular filtration; therefore, lowering the TFV equivalents administered reduces kidney exposure. Effective therapy is thus achieved at approximately 90% lower systemic exposure to TFV, translating to statistically significant improvement in safety parameters associated with bone mineral density (BMD) and markers of renal function on short-term follow-up.
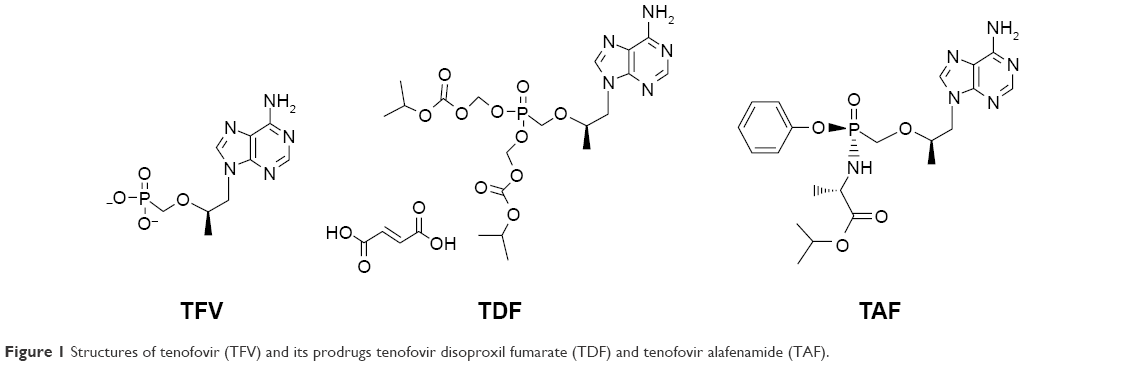 | Figure 1 Structures of tenofovir (TFV) and its prodrugs tenofovir disoproxil fumarate (TDF) and tenofovir alafenamide (TAF). |
In vitro incubation of primary human hepatocytes with TAF results in a high intracellular concentration of TFV-DP, approximately 5- and 120-fold higher than the concentration observed when incubated with TDF and TFV, respectively.24 Moreover, intracellular TFV-DP levels persistently increased over a 24-hour period. TAF is efficiently taken up and activated by hepatocytes by a multistep process, resulting in persistent intracellular levels of TFV-DP, a potent inhibitor of HBV replication.
According to the TFV pharmacokinetics of patients with severe hepatic impairment (Child-Pugh C) treated with TAF 25 mg, TFV exposure was only modestly lower compared to healthy controls with normal hepatic function.25 For patients with severe renal impairment (estimated glomerular filtration rate [eGFR] 15–29 mL/min), TFV exposure was significantly higher (26.4 vs 9.50 ng/mL) than for matched healthy patients with normal renal function (eGFR ≥90 mL/min).26 However, the observed TFV exposure in treatment with TAF 25 mg for patients with severe renal impairment in this same study, was still lower than plasma TFV levels historically seen in pharmacokinetics studies with TDF as part of various antiretroviral regimens for HIV-infected patients with normal renal function. A study of the effect of food on TAF pharmacokinetics showed that TAF exposure was decreased under fasted compared to fed conditions.27 However, no significant trend in exposure-response/safety was found within the range of TAF exposure under both fasted and fed conditions.19–21
Phase 1 trial
A Phase 1 trial was done for non-cirrhotic, treatment-naïve adults with chronic HBV infection who were randomized to TDF 300 mg or to different doses of TAF (8, 25, 40, or 120 mg) for 4 weeks. Across the TAF groups, the mean reduction of serum HBV DNA level at week 4 (−2.81, −2.55, −2.19, and −2.76 log10IU/mL for the 8, 25, 40, and 120 mg groups, respectively) was similar to that of the TDF group (−2.68 log10IU/mL).19 Moreover, the HBV kinetics during treatment were similar among the groups. The TAF pharmacokinetics were linear and proportional to the dose range of 8–120 mg, but none of the participants experienced grade 3/4 adverse events. When TAF was given at doses of 25 mg or lower, TFV exposure was significantly reduced, with over 92% relative to TDF. From these efficacy and safety data, TAF 25 mg was selected for use in Phase 3 trials.
Design of Phase 3 trials
There are two identically designed Phase 3 non-inferiority trials that are ongoing, randomized, double-blind, multinational, and have continued since September 2013 for treatment-naïve and -experienced adult (≥18 years) patients with HBeAg-positive20 or -negative21 chronic hepatitis B (CHB). For both the HBeAg-positive and -negative groups, the principal inclusion criteria are a plasma HBV DNA level ≥20,000 IU/mL, ALT ≥60 U/L for male or ≥38 U/L for female, and no more than ten times the upper limit of normal and estimated creatinine (Cr) clearance ≥50 mL/min (by Cockcroft-Gault method). The principal exclusion criteria included evidence of decompensation, the presence of HCC, coinfection with hepatitis C virus, hepatitis D virus, HIV, and specified hematological and liver function abnormalities. In both trials, patients received TAF 25 mg or TDF 300 mg (randomly assigned 2:1, respectively) orally once a day for 96 weeks, after which all patients began open-label treatment with TAF 25 mg until week 144. A longer follow-up (384 weeks: 8 years) is ongoing to evaluate the incidence of bone and renal events over the long term. The primary efficacy endpoint was the proportion of patients who had an HBV DNA level <29 IU/mL at week 48. The major results for efficacy are shown in Table 1. The secondary safety endpoints at week 48 included percentage change in hip and spine BMD and renal parameter change from baseline. The major results for safety are shown in Table 2.
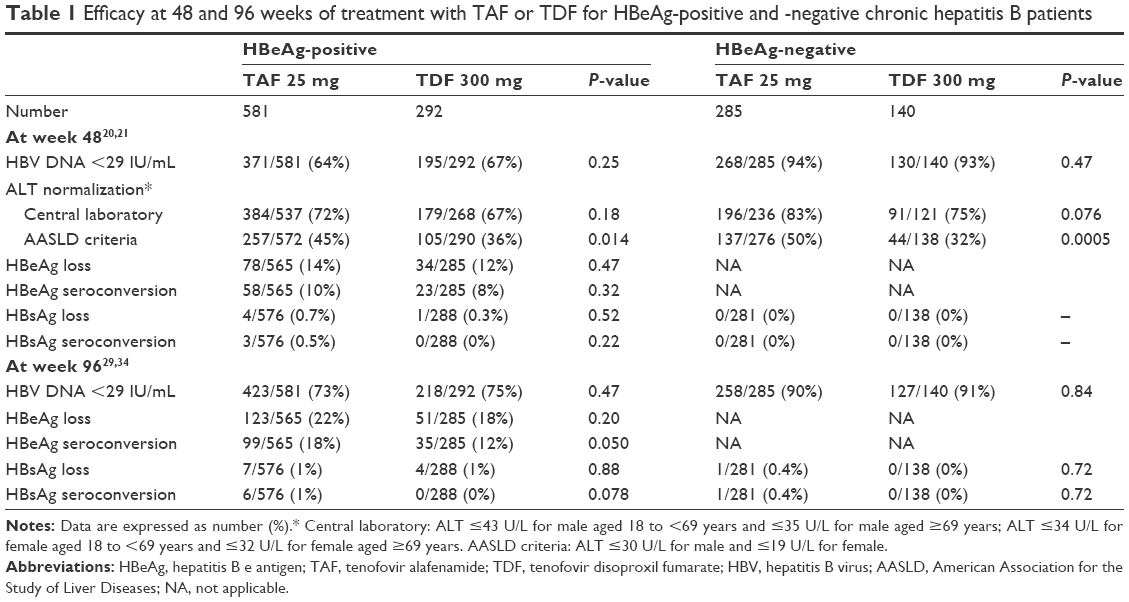 | Table 1 Efficacy at 48 and 96 weeks of treatment with TAF or TDF for HBeAg-positive and -negative chronic hepatitis B patients |
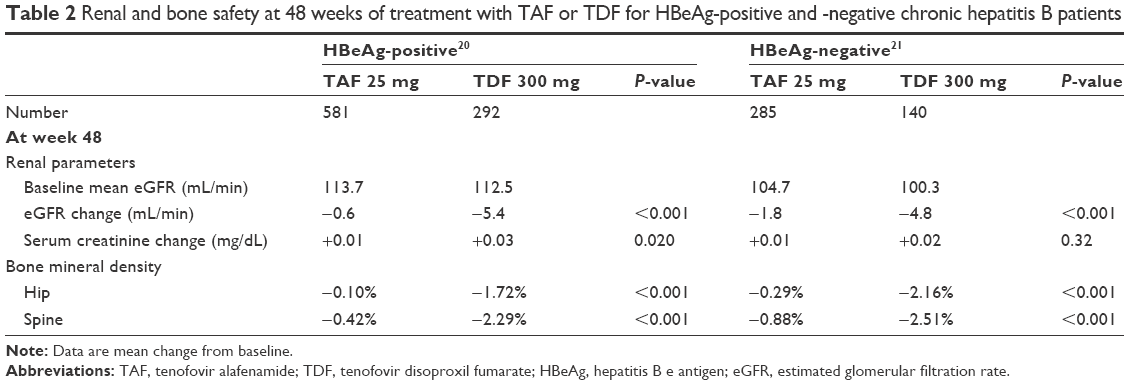 | Table 2 Renal and bone safety at 48 weeks of treatment with TAF or TDF for HBeAg-positive and -negative chronic hepatitis B patients |
Phase 3 trial for HBeAg-positive patients
In total, 873 patients were randomly assigned to receive either TAF 25 mg (n=581) or TDF 300 mg (n=292). Most of the patients were Asian (TAF 83% and TDF 79%), the most common HBV genotype was genotype C (both groups 52%), and approximately a quarter of the patients had been previously treated with other nucleos(t)ide analog(s). The baseline mean HBV DNA level was 7.6 log10IU/mL for both groups.20
The primary efficacy endpoint, an HBV DNA level <29 IU/mL at week 48, was achieved by 371 (64%) patients in the TAF group and 195 (67%) in the TDF group (difference in proportions: −3.6% [95% CI: −9.8 to 2.6], P=0.25), which suggests non-inferiority of TAF 25 mg antiviral efficacy over 48 weeks of treatment in comparison with TDF 300 mg. Moreover, there were no significant differences in the rates of patients achieving an HBV DNA level <29 IU/mL at week 48 in analyses of subgroups that included age (<50 vs ≥50 years), sex, race (Asian vs non-Asian), baseline HBV DNA level (<8 vs ≥8 log10IU/mL), prior treatment status (naïve vs experienced), region (East Asia, Europe, North America, and other), treatment adherence (<95 vs ≥95%), HBV genotype, baseline ALT level (by central laboratory normal range), or baseline FibroTest score (<0.75 vs ≥0.75). There was also no significant difference in the rate of HBV DNA suppression (<29 IU/mL) between the TAF and TDF groups at weeks 72 (TAF 72% vs TDF 72%)28 and 96 (TAF 73% vs TDF 75%).29
In regard to biochemical response, the primary 48-week analysis indicated that treatment with TAF was associated with a significantly higher ALT normalization rate than was TDF (TAF 45% vs TDF 36%) (difference: 8.7% [95% CI: 1.8 to 15.6], P=0.014) based on the American Association for the Study of Liver Diseases (AASLD) criteria (male: ALT ≤30 U/L and female: ALT ≤19 U/L). This trend continued to week 96, at which time the rates of ALT normalization based on AASLD criteria were 52% and 42% in the TAF and TDF groups, respectively (P=0.0003).29 Moreover, the FibroTest score showed a significantly greater decrease in the TAF than in the TDF groups (TAF −0.07 vs TDF −0.04) (difference: −0.03 [95% CI: −0.04 to −0.01], P=0.007) at week 48.
The rates of HBeAg loss after 48 weeks of treatment were similar between the TAF and TDF groups (TAF 14% vs TDF 12%). The factors associated with HBeAg loss were older age, higher baseline ALT, and lower baseline HBV DNA level.30 Patients with HBeAg loss were likely to achieve earlier HBV DNA suppression. A total of 10% of the TAF and 8% of the TDF group also experienced seroconversion to anti-HBe at week 48, but only four (0.7%) TAF and one (0.3%) TDF treated patient experienced HBsAg loss.
Phase 3 trial for HBeAg-negative patients
In total, 426 patients were randomly assigned to receive either TAF 25 mg (n=285) or TDF 300 mg (n=141). Most of the patients were Asian (72% in both groups), the most common HBV genotype was genotype C (TAF 40% and TDF 34%), and approximately 20% of the patients had previously been treated with other nucleos(t)ide analog(s). The baseline mean HBV DNA levels were 5.7 and 5.8 log10IU/mL for the TAF and TDF groups, respectively.21
The primary efficacy endpoint, an HBV DNA level <29 IU/mL at week 48, was achieved by 268 (94%) patients in the TAF group and 130 (93%) in the TDF group (difference in proportions: 1.8% [95% CI: −3.6 to 7.2], P=0.47), which suggests that the antiviral efficacy of TAF 25 mg was not inferior to that of TDF 300 mg. Moreover, there were no significant differences in the rate of patients achieving an HBV DNA level <29 IU/mL at week 48 in analyses of subgroups that included age (<50 vs ≥50 years), sex, race (Asian vs non-Asian), baseline HBV DNA level (<7 vs ≥7 log10IU/mL), prior treatment status (naïve vs experienced), region (East Asia, Europe, North America, and other), treatment adherence (<95 vs ≥95%), HBV genotype, or baseline ALT level (by central laboratory normal range). There was no significant difference in the rate of HBV DNA suppression (<29 IU/mL) between the TAF and TDF groups at weeks 72 (TAF 93% vs TDF 92%)28 and 96 (TAF 90% vs TDF 91%).29
As also seen in the Phase 3 trial for HBeAg-positive patients, the primary 48-week analysis indicated that treatment with TAF was associated with a significantly higher ALT normalization rate than was TDF (TAF 50% vs TDF 32%) (difference: 17.9% [95% CI: 8.0 to 27.7], P=0.0005) based on the AASLD criteria for the upper normal limit of ALT level. This trend continued to week 96, at which time the rates of ALT normalization based on the AASLD criteria were 50% and 40% for the TAF and TDF groups, respectively (P=0.035).29 None of the patients in either group had HBsAg loss at week 48, and the mean change in quantitative HBsAg from baseline to week 48 was minimal in both groups (TAF −0.09 vs TDF −0.06 log10IU/mL).
Pooled analyses of Phase 3 trials
Several pooled analyses from the two Phase 3 trials have been conducted. In an evaluation of rapid biochemical and viral responses, the percentage of patients who achieved both ALT normalization (AASLD criteria) (TAF 9% vs TDF 5%, P=0.01) and HBV DNA suppression (<29 IU/mL) (TAF 24% vs TDF 22%) at week 12 of treatment was higher in the TAF group than in the TDF group.31 ALT normalization and HBV DNA suppression at week 12 were associated with lower baseline HBV DNA, HBeAg negativity, and male sex.31 In evaluation of predictive factors associated with a normalized ALT level at week 48, patients with features of metabolic syndrome, such as high body mass index, hypertension, and hyperlipidemia, were less likely to achieve ALT normalization with TAF or TDF treatment.32
In regards to virologic response, an HBV DNA level ≥2,000 IU/mL after 48 weeks of treatment was considered an indication of viral persistence. In total, 50 patients from both Phase 3 trials (TAF: n=35 [4%], TDF: n=15 [4%]) had viral persistence at week 48. Baseline HBV DNA ≥8 log10IU/mL, treatment adherence, and HBV genotype D were strongly associated with having viral persistence with an HBV DNA level ≥2,000 IU/mL at week 48, but treatment assignment (TAF or TDF) was not.28 Of the 38 patients who had viral breakthrough (HBV DNA ≥69 IU/mL or ≥1 log10 increase from nadir), 17 (45%) were found to be non-adherent to the study medication.33 No amino-acid substitutions associated with viral breakthrough were detected by deep sequencing, and no resistance to TAF was found through week 96.28,33,34
In pooled analyses of the HBsAg kinetics of TAF and TDF for 48 weeks, HBsAg change was similar for the TAF and TDF treatment regimens across HBV genotypes.35 HBeAg-positive patients with HBV genotype A or B had greater HBsAg declines, which increased with treatment duration. In multivariable logistic regression analysis, HBV genotype B was independently associated with an HBsAg >0.5−log10 reduction (OR 5.92, 95% CI 3.44–10.10, P<0.0001).35 In addition, higher HBV DNA, higher ALT level, and male sex were independently associated with an HBsAg >0.5−log10 reduction. Although the reasons of the advantage of male sex in ALT normalization and HBsAg reduction have not been fully elucidated, possible reasons include differences in the inclusion and normalization criteria for ALT level between males and females.
Safety profile
Both TAF and TDF treatment were reported to be well tolerated. Most adverse events were of mild to moderate severity, with only 1% of the patients discontinuing treatment due to an adverse event.20,21 The most common adverse events were nasopharyngitis (TAF 10% vs TDF 7%), upper respiratory tract infection (TAF 10% vs TDF 7%), and headache (TAF 9% vs TDF 8%) through week 48. Thirty-six patients (4%) who received TAF and 21 (5%) who received TDF experienced serious adverse events, none of which was considered to be related to the study drug. The incidence of grade 3/4 abnormalities was similar between the TAF and TDF groups (TAF 31% vs TDF 29%). The most common grade 3/4 abnormality was elevation of the ALT level (TAF 8% vs TDF 9%), especially for HBeAg-positive patients. Of these, three (<1%) patients treated with TAF and four (1%) with TDF experienced an ALT flare during the early treatment period, within 1–3 months, but all resolved without sequelae.
Renal safety
None of the patients experienced serious renal-related adverse effects or proximal renal tubulopathy, including Fanconi syndrome, in either the TAF or TDF treatment groups.20,21 At week 48, patients treated with TAF had a significantly lower decrease in median eGFR level than did those treated with TDF in both HBeAg-positive (TAF −0.6 vs TDF −5.4 mL/min, P<0.0001)20 and HBeAg-negative (TAF −1.8 vs TDF −4.8 mL/min, P=0.004)21 studies. Analysis of factors associated with an eGFR decline of ≥25% from baseline extracted TDF treatment, higher baseline eGFR level, and FibroTest score >0.75.36 Moreover, chronic kidney disease (CKD) stage decline was lower with TAF than with TDF treatment, especially for female patients and those with comorbid conditions, such as hypertension, diabetes mellitus, and cardiovascular diseases.36 However, the study cohorts only included patients with stage 1 (eGFR ≥90 mL/min) and stage 2 (eGFR 60–89 mL/min) CKD at baseline; and of these, only eight of the 825 (0.96%) evaluable TAF patients and six of the 414 (1.4%) evaluable TDF patients developed stage 3 disease (eGFR <60 mL/min) at week 48 (P=0.45). In pooled analyses with longitudinal eGFR levels to week 96, the eGFR decline of the TAF patients continued to be lower than the decline seen in the TDF group (at week 96: TAF −1.2 vs TDF −4.8 mL/min, P<0.001).37
TAF treatment resulted in smaller changes in renal markers of glomerular function at week 48 than were seen with TDF treatment, including the urine protein to Cr ratio (median change: TAF 6.0% vs TDF 16.5%, P=0.010) and the urine albumin to Cr ratio (median change: TAF 6.9% vs TDF 12.2%, P=0.073).38 For renal markers of tubular function, patients treated with TAF had significantly smaller changes in the urine RBP/Cr ratio (median change: TAF −0.3% vs TDF 25.1%, P<0.001) and the β2M/Cr ratio (median change: TAF −3.5% vs TDF 37.9%, P<0.001).38 Similar results were observed at week 96 for RBP/Cr (median change: TAF 21.8% vs TDF 49.1%, P<0.001) and β2M/Cr (median change: TAF 8.4% vs TDF 48.9%, P<0.001).39 These results suggest reduced impact on renal function with TAF treatment compared with TDF treatment.
In the first 24 weeks of the open-label extension phase (from weeks 96 to 120), significant improvement in the eGFR level (median change: 1.8%, P=0.018) and renal tubular marker (β2M/Cr median change: −21.4%, P=0.016) were found for patients who switched from TDF to TAF at week 96.39
Bone safety
Seven patients (TAF 6/866 [0.7%] and TDF 1/432 [0.2%]) experienced a bone fracture; however, six were associated with injury and the other was considered to be an old spinal compression fracture.20,21 No treatment-related fracture events occurred after 48 weeks of treatment. The declines in hip and spine BMD at week 48 were significantly less in the TAF group (HBeAg-positive: hip −0.10% and spine −0.42%, HBeAg-negative: hip −0.29% and spine −0.88%) than in the TDF group (HBeAg-positive: hip −1.72% and spine −2.29%, HBeAg-negative: hip −2.16% and spine −2.51%) (all P<0.0001).20,21 In a pooled analysis, similar results for BMD change were observed at week 96 in the TAF and TDF groups (hip: TAF −0.33% vs TDF −2.52%, P<0.001 and spine: TAF −0.75% vs TDF −2.59%, P<0.001).40 According to an analysis with multiple risk factors for osteoporosis that included female sex, age ≥50 years, Asian race, and eGFR <90 mL/min, the percentage of patients on TAF with >3% hip BMD decline remained 8%–10% at week 48, irrespective of the number of risk factors.41 In contrast, the percentage of patients on TDF with >3% hip BMD decline increased to 58% (19/33) for patients with all four risk factors. At week 96, the percentage of patients on TAF and TDF with >3% hip BMD decline increased to 34% (16/47) and 75% (24/32), respectively, for patients with all four risk factors.40
In the first 24 weeks of the open-label extension phase (from weeks 96 to 120), significant improvement in hip (−2.7% to −2.1%, P<0.001) and spine BMD (−3.1% to −1.6%, P<0.001) was observed at week 120 for patients who switched from TDF to TAF at week 96.39
The observed reduction of BMD decline was supported by biomarkers of bone metabolism including markers of bone formation (procollagen type 1 N-terminal propeptide, bone-specific alkaline phosphatase, osteocalcin), resorption (C-type collagen sequence), and regulation (parathyroid hormone).42 Although parathyroid hormone had poor correlation with biomarkers of bone formation, greater BMD decline was generally associated with increasing changes in bone biomarker levels.
Place in therapy and conclusion
According to primary 48-week analyses of Phase 3 trials,20,21 the efficacy of TAF was not inferior to that of TDF for both HBeAg-positive and -negative patients in regards to virologic outcomes. However, the rate of ALT normalization by the more stringent AASLD criteria was significantly higher for TAF than for TDF. Parameters for renal and bone safety markers are also more favorable with TAF compared to TDF, though these differences are modest, at least in the short-term (up to 96 weeks). Long-term safety is an important consideration in the therapeutic management of patients with CHB because treatment is often life-long. Recent studies have reported that CHB patients are aging with higher rates of comorbidities including CKD and osteoporosis.43,44 However, while safer renal and bone profiles would be of benefit for patients with CHB, determining if TAF is sufficiently safe or the observed differences in renal and bone markers between TAF and TDF treated patients from Phase 3 trials are clinically relevant would require further studies with long-term follow-up and patients from real-world settings. For the time being, for patients with existing renal or bone diseases or high risk for such disorders, TAF would be preferable to TDF given the more favorable renal and bone safety profiles observed in registration trials.
Disclosure
Norihiro Furusyo: grant/research support: MSD K.K., Gilead Sciences K.K., Daiichi Sankyo Co., Ltd., and Bristol-Myers Squibb. Mindie H Nguyen: grant/research support: Bristol-Myers Squibb, Gilead Sciences, Janssen Pharmaceutical; Advisory board/consultant: Dynavax Laboratories, Gilead Sciences, Intercept Pharmaceutical; Anylam Pharmaceutical; Roche Laboratories; and Novartis Pharmaceuticals. The authors have no other conflicts of interest to report.
References
World Health Organization [homepage on the Internet]. Medical Centre, Fact sheets-Hepatitis B. WHO. Available from: http://www.who.int/mediacentre/factsheets/fs204/en. Accessed October 19, 2017. | ||
Ott JJ, Stevens GA, Groeger J, Wiersma ST. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine. 2012;30(12):2212–2219. | ||
Schweitzer A, Horn J, Mikolajczyk RT, Krause G, Ott JJ. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet. 2015;386(10003):1546–1555. | ||
European Association for the Study of the Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J Hepatol. 2017;67(2):370–398. | ||
Gish RG, Given BD, Lai CL, Locarnini SA, Lau JY, Lewis DL, Schluep T. Chronic hepatitis B: Virology, natural history, current management and a glimpse at future opportunities. Antiviral Res. 2015;121:47–58. | ||
Terrault NA, Bzowej NH, Chang KM, Hwang JP, Jonas MM, Murad MH; American Association for the Study of Liver Diseases. AASLD guidelines for treatment of chronic hepatitis B. Hepatology. 2016;63(1):261–283. | ||
Janssen HL, van Zonneveld M, Senturk H, et al. Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: a randomised trial. Lancet. 2005;365(9454):123–129. | ||
Lau GK, Piratvisuth T, Luo KX, et al. Peginterferon Alfa-2a, lamivudine, and the combination for HBeAg-positive chronic hepatitis B. N Engl J Med. 2005;352(26):2682–2695. | ||
Buster EH, Hansen BE, Lau GK, Piratvisuth T, Zeuzem S, Steyerberg EW, Janssen HL. Factors that predict response of patients with hepatitis B e antigen-positive chronic hepatitis B to peginterferon-alfa. Gastroenterology. 2009;137(6):2002–2009. | ||
Sarin SK, Kumar M, Lau GK, et al. Asian-Pacific clinical practice guidelines on the management of hepatitis B: a 2015 update. Hepatol Int. 2016;10(1):1–98. | ||
Woo G, Tomlinson G, Nishikawa Y, et al. Tenofovir and entecavir are the most effective antiviral agents for chronic hepatitis B: a systematic review and Bayesian meta-analyses. Gastroenterology. 2010;139(4):1218–1229. | ||
Wu CY, Lin JT, Ho HJ, et al. Association of nucleos(t)ide analogue therapy with reduced risk of hepatocellular carcinoma in patients with chronic hepatitis B: a nationwide cohort study. Gastroenterology. 2014;147(1):143–151. | ||
Wong RJ, Nguyen MT, Trinh HN, et al. Hepatitis B surface antigen loss and sustained viral suppression in Asian chronic hepatitis B Patients: A community-based real world study. J Viral Hepat. Epub 2017 Jun 5. | ||
Lampertico P, Chan HL, Janssen HL, Strasser SI, Schindler R, Berg T. Review article: long-term safety of nucleoside and nucleotide analogues in HBV-monoinfected patients. Aliment Pharmacol Ther. 2016;44(1):16–34. | ||
Maggi P, Montinaro V, Leone A, et al. Bone and kidney toxicity induced by nucleotide analogues in patients affected by HBV-related chronic hepatitis: a longitudinal study. J Antimicrob Chemother. 2014;70(4):1150–1154. | ||
Shimizu M, Furusyo N, Ikezaki H, et al. Predictors of kidney tubular dysfunction induced by adefovir treatment for chronic hepatitis B. World J Gastroenterol. 2015;21(7):2116–2123. | ||
Sax PE, Wohl D, Yin MT, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate, coformulated with elvitegravir, cobicistat, and emtricitabine, for initial treatment of HIV-1 infection: two randomised, double-blind, phase 3, non-inferiority trials. Lancet. 2015;385(9987):2606–2615. | ||
Mills A, Arribas JR, Andrade-Villanueva J, et al. Switching from tenofovir disoproxil fumarate to tenofovir alafenamide in antiretroviral regimens for virologically suppressed adults with HIV-1 infection: a randomised, active-controlled, multicentre, open-label, Phase 3, non-inferiority study. Lancet Infect Dis. 2016;16(1):43–52. | ||
Agarwal K, Fung SK, Nguyen TT, et al. Twenty-eight day safety, antiviral activity, and pharmacokinetics of tenofovir alafenamide for treatment of chronic hepatitis B infection. J Hepatol. 2015;62(3):533–540. | ||
Chan HL, Fung S, Seto WK, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of HBeAg-positive chronic hepatitis B virus infection: a randomised, double-blind, Phase 3, non-inferiority trial. Lancet Gastroenterol Hepatol. 2016;1(3):185–195. | ||
Buti M, Gane E, Seto WK, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of patients with HBeAg-negative chronic hepatitis B virus infection: a randomised, double-blind, Phase 3, non-inferiority trial. Lancet Gastroenterol Hepatol. 2016;1(3):196–206. | ||
Ray AS, Fordyce MW, Hitchcock MJ. Tenofovir alafenamide: A novel prodrug of tenofovir for the treatment of Human Immunodeficiency Virus. Antiviral Res. 2016;125:63–70. | ||
Lee WA, He GX, Eisenberg E, et al. Selective intracellular activation of a novel prodrug of the human immunodeficiency virus reverse transcriptase inhibitor tenofovir leads to preferential distribution and accumulation in lymphatic tissue. Antimicrob Agents Chemother. 2005;49(5):1898–1906. | ||
Murakami E, Wang T, Park Y, Hao J, Lepist EI, Babusis D, Ray AS. Implications of efficient hepatic delivery by tenofovir alafenamide (GS-7340) for hepatitis B virus therapy. Antimicrob Agents Chemother. 2015;59(6):3563–3569. | ||
Custodio JM, Ma G, Cuvin J, Ting L, Flaherty JF, Zack JZ. Pharmacokinetics and safety of tenofovir alafenamide in subjects with severe hepatic impairment [abstract no. FRI-127/poster]. J Hepatol. 2016;64(1 Suppl):S594–S595. | ||
Custodio JM, Fordyce M, Garner W, et al. Pharmacokinetics and safety of tenofovir alafenamide in HIV-uninfected subjects with severe renal impairment. Antimicrob Agents Chemother. 2016;60(9):5135–5140. | ||
Custodio JM, Ma G, Sajwani K, Ling KH, Flaherty JF, Kearney BP. Lack of clinically relevant effect of food on the pharmacokinetics of tenofovir alafenamide [abstract no. P_46/poster]. Reviews in Antiviral Therapy & Infectious Diseases. 2016;6:52–53. | ||
Gane EJ, Saito M, Bae SH, et al. Characterization of host, viral, and treatment-related factors associated with antiviral efficacy of tenofovir alafenamide and tenofovir disoproxil fumarate [abstract no. 1879/poster]. Hepatology. 2016;64(1 Suppl):929A–930A. | ||
Agarwal K, Fung S, Seto WK, et al. A Phase 3 study comparing tenofovir alafenamide (TAF) to tenofovir disoproxil fumarate (TDF) in patients with HBeAg-positive, chronic hepatitis B (CHB): efficacy and safety results at week 96 [abstract no. FRI-153/poster]. J Hepatol. 2017;66(1 Suppl):S478. | ||
Pan CQ, Li MK, Lee KS, et al. Predictors of HBeAg loss in HBeAg-positive patients with chronic hepatitis B during treatment with TAF or TDF [abstract no. 1882/poster]. Hepatology. 2016;64(1 Suppl):931A. | ||
Brunetto MR, Ahn SH, Chang TT, et al. Correlation of early biochemical and virologic responses during oral antiviral therapy for chronic hepatitis B [abstract no. 1857/poster]. Hepatology. 2016;64(1 Suppl):917A. | ||
Fung S, Tatsuhashi H, Tak WY, et al. Features of the metabolic syndrome are associated with lack of serum ALT normalization during therapy for chronic hepatitis B [abstract no. 1852/poster]. Hepatology. 2016;64(1 Suppl):914A–915A. | ||
Chan HL, Fung S, Cathcart AL, et al. No resistance to tenofovir alafenamide detected through 48 weeks of treatment in patients with chronic hepatitis B [abstract no. 1843/poster]. Hepatology. 2016;64(1 Suppl):909A. | ||
Brunetto M, Lim YS, Gane E, et al. A Phase 3 study comparing tenofovir alafenamide to tenofovir disoproxil fumarate in patients with HBeAg-negative, chronic hepatitis B: efficacy and safety results at week 96 [abstract no. PS-042/oral]. J Hepatol. 2017;66(1 Suppl):S25–S26. | ||
Marcellin P, Seto W-K, Hu CT, et al. Genotype-specific differences in magnitude of HBsAg reduction during tenofovir disoproxil fumarate or tenofovir alafenamide therapy in CHB patients [abstract no. 1858/poster]. Hepatology. 2016;64(1 Suppl):918A. | ||
Agarwal K, Furusyo N, Byun KS, et al. Improved renal laboratory parameters in CHB patients treated with TAF compared with TDF [abstract no. 1844/poster]. Hepatology. 2016;64(1 Suppl):910A. | ||
Chuang WL, Agarwal K, Hwang JS, et al. Continued improvement in renal laboratory parameters in CHB patients treated with tenofovir alafenamide (TAF) compared with tenofovir disoproxil fumarate (TDF) over 96 weeks [abstract no. SAT-171/poster]. J Hepatol. 2017;66(1 Suppl):S695. | ||
Lim YS, Ide T, Strasser SI, et al. Minimal changes in urine markers of tubular dysfunction in CHB patients receiving tenofovir alafenamide compared with tenofovir disoproxil fumarate [abstract no. 1901/poster]. Hepatology. 2016;64(1 Suppl):940A. | ||
Chan HL, Fung S, Seto WK, et al. Improved bone and renal safety of switching from tenofovir disoproxil fumarate to tenofovir alafenamide: preliminary results from 2 Phase 3 studies in HBeAg-positive and HBeAg-negative patients with chronic hepatitis B [abstract no. PS-041/oral]. J Hepatol. 2017;66(1 Suppl):S25. | ||
Fung S, Chuang WL, Nishiguchi S, et al. Smaller decreases in bone mineral density in chronic hepatitis B patients receiving tenofovir alafenamide compared with tenofovir disoproxil fumarate over 96 weeks [abstract no. SAT-162/poster]. J Hepatol. 2017;66(1 Suppl):S691–S692. | ||
Seto WK, Asahina Y, Peng CY, et al. Reduced changes in bone mineral density in chronic HBV (CHB) patients receiving tenofovir alafenamide (TAF) compared with tenofovir disoproxil fumarate (TDF) [abstract no. 67/oral]. Hepatology. 2016;64(1 Suppl):35A. | ||
Chuang WL, Seto WK, Inokuma T, et al. Comparison of markers of bone turnover demonstrates less changes in CHB patients receiving tenofovir alafenamide compared with tenofovir disoproxil fumarate [abstract no. 1856/poster]. Hepatology. 2016;64(1 Suppl):916A–917A. | ||
Nguyen MH, Lim J, Liou I, et al. Higher and rising prevalence and incidence of renal impairment and chronic kidney disease in chronic hepatitis B patients compared to matched non-chronic hepatitis B controls in the United States: results of a real-world analysis [abstract no. SAT-132/poster]. J Hepatol. 2017;66(1 Suppl):S677–S678. | ||
Gordon SC, Lim J, Liou I, et al. Prevalence of osteoporosis and bone fracture was 2-fold higher in chronic hepatitis B compared to non-chronic hepatitis B patients and continued to increase in the last decade: results of a United States population-based cohort study [abstract no. PS-108/oral]. J Hepatol. 2017;66(1 Suppl):S61. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.