Back to Journals » Infection and Drug Resistance » Volume 16
TBX5 Variants are Associated with Susceptibility to and the Incidence of Liver Cirrhosis and Hepatocellular Carcinoma in the Chinese Population: A Multicenter and Follow-Up Study
Authors Yao J, Mao X, Sun Q, Wu B, Yu W, Huang Y, Luo S, Zeng J, Lin J
Received 2 March 2023
Accepted for publication 19 April 2023
Published 2 May 2023 Volume 2023:16 Pages 2653—2665
DOI https://doi.org/10.2147/IDR.S410151
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
JinJian Yao,1,2,* Xiaochun Mao,3,* Qigang Sun,4,* Biao Wu,5 Weiling Yu,6 Yanjing Huang,7 Shuai Luo,1 Jia Zeng,1 Jusheng Lin2
1Department of Emergency, Hainan General Hospital, Haikou, Hainan, People’s Republic of China; 2Institute of Liver and Gastrointestinal Diseases, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, People’s Republic of China; 3Department of Ophthalmology, Xiangyang Central Hospital Affiliated to Hubei University of Arts and Science, Xiangyang, Hubei, People’s Republic of China; 4Department of Hepatobiliary and Pancreatic Surgery, Hainan General Hospital, Haikou, Hainan, People’s Republic of China; 5Infectious Disease, Hainan General Hospital, Haikou, Hainan, People’s Republic of China; 6Oncology Department, Haikou City People’s Hospital, Haikou, Hainan, People’s Republic of China; 7Oncology Department of Hainan General Hospital, Haikou, Hainan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: JinJian Yao; Jusheng Lin, Email [email protected]; [email protected]
Purpose: Liver cirrhosis (LC) and hepatocellular carcinoma (HCC) are progressions affected by genetic predispositions, and persistent hepatitis B virus infection also demonstrates genetic susceptibility. All HBV-related outcomes have been compared in parallel to identify risk polymorphism in HBV progression.
Methods: The multiple-stage association study filtered and validated the risk SNPs for HBV progression and explored their association with persistent infection, with a total of 8906 subjects in China from three sites. Cox proportional hazards models and Kaplan–Meier Log rank tests were used to determine the time to the progressive event in relation to the risk SNPs.
Results: Rs3825214 in TBX5 replicated a specific association with LC and HCC in 4 progression cohorts and was not related to persistent infection, naivety to HBV infection and natural clearance in 3 persistent cohorts. In combined samples, rs3825214 was associated with an increased risk of LC (P< 0.001; OR = 1.98) and HCC (P< 0.001; OR = 1.68). The results of bioinformatics analysis indicated that rs3825214 genotypes change RNA structure and intron excision ratio. In the follow-up of 571 hospital-based persistent HBV infection patients, ninety-three (16.29%) developed LC, and seventy-four (12.96%) progressed to HCC at a median follow-up of 5.1 years. Rs3825214 was associated with HCC and LC events in Cox proportional hazards models (P< 0.001).
Conclusion: We identified and confirmed that genetic variants in TBX5 are significantly associated with susceptibility to and the incidence of LC and HCC.
Keywords: TBX5, polymorphism, liver cirrhosis, hepatocellular carcinoma, follow-up, hepatitis B virus
Introduction
Chronic hepatitis B virus (HBV) infection occurs worldwide but is endemic in East Asia and Africa, and the progression of chronic HBV infection includes liver cirrhosis (LC) and hepatocellular carcinoma (HCC), which account for 500000 to 1.2 million deaths per year worldwide.1 Despite the identical cause, the progression of chronic infection and outcome of primary infection are highly variable. More than 350 million people have chronic HBV infection, but only a subset of these individuals, 50% of male carriers and 14% of female carriers,2,3 will eventually die due to progression to LC and HCC.
Progression is a multifactorial and interindividual-variation disorder involving treatment for HBV infection, coinfection with other hepatotropic viruses4 and human immunodeficiency virus (HIV); sex; age;5 alcohol; smoking; and human genetic variants. The association of human genetic variants with progression and persistent infection has been recently emphasized in Asia,6–10 thereby broadening our understanding of genetic predisposition. The familial clustering of hepatocellular carcinoma in HBV endemic areas also consolidated the genetic role in disease progression.11 LC is a precancerous lesion, and some cases of HCC are accompanied by concurrent LC, indicating that there might be some common genetic susceptibility and suggesting that the internal genetic relationship might be an informative and unnoticed part of previous association studies. Therefore, it is of great interest to identify clear susceptibility loci by conducting a parallel comparison among progressive subgroups and primary infection.
A joint analysis strategy was used, including a DNA pooling screen in the genome-wide analysis (GWAS) and subsequent independent replications, to explore genetic susceptibility to LC and HCC, to examine their relation to outcomes of primary infection, explore significance of risk single nucleotide polymorphism (SNP) and to further assess the effect of risk loci on the incidence of progression in follow-up analysis.
Methods
Genetic Association Design and Subject Enrollment
More than 906,600 SNPs in the screen phase stage were initially genotyped in DNA pools, including the pool of HCC (case), the pool of LC (case) and the pool of asymptomatic chronic HBV carriers (AsC) (control), to identify the progressive genetic loci. Associations detected in this pool-based GWAS phase were validated in four independent cohorts using individual genotyping, and the combined samples from four independent cohorts were used to evaluate the genotypic effects on progression.12 Additionally, the risk loci from progressive cohorts were assessed for susceptibility to consequences of primary HBV infection in three persistent infection cohorts, consisting of subjects with persistent infection (case), subjects who were naivety to HBV infection (control) and subjects who have spontaneously recovered (SR) (control) in each cohort.
The subgroup of persistent HBV infection in Hubei Province was followed-up to weigh the contribution of risk genotypes on the incidence of progressive outcomes from enrollment to December 31, 2017.
All unrelated Han Chinese subjects were recruited at four tertiary hospitals in Hubei Province, central China; three tertiary hospitals in Guangdong; and three tertiary hospitals in Hainan, southern China. Replication 1 progressive cohort, replication 2 progressive cohort and persistent infection cohort 1 were from Hubei Province; replication 3 progressive cohort and persistent infection cohort 2 comprised subjects from Guangdong; and replication 4 progressive cohort and persistent infection cohort 3 comprised subjects from Hainan.
Participants were enrolled according to the uniform predesigned protocol (Supplemental Table S1) in these centers. To maintain homogeneity in progression, the natural history and diagnosis were confirmed in detail by reviewing medical records and obtaining the medical history of all subjects. The patients with persistent infection, LC, HCC and AsC had no serologic evidence for coinfection with hepatitis C virus (HCV), hepatitis E virus (HEV), hepatitis D virus (HDV), cytomegalovirus (CMV), HIV, or medications associated with liver injuries.
The study was approved by the Ethics Committee of Hainan General Hospital and was performed in accordance with the Helsinki Declaration II. Written informed consent was obtained from patients and their next of kin for patients.
DNA Isolation and Progressive DNA Pooling Construction
Genomic DNA was isolated from whole blood collected in EDTA tubes using a QuickGene DNA whole blood kit S (DB-S) with QuickGene-Mini80 equipment (Fujifilm, Tokyo, Japan). The yield and purity of DNA was qualified by ultraviolet light spectroscopy and divided into three aliquots at −70°C for DNA analysis.
Eighty-eight patients in the LC group, 90 patients in the HCC group, and 66 patients in the AsC group from replication were used to create DNA pools, and the detailed process of DNA pooling has been released in GEO (GSE26034). In accordance with the protocol of DNA pooling,13 the DNA concentration of all samples was quantified four times using a Quant-iT PicoGreen dsDNA assay kit (Invitrogen, Carlsbad, CA) and tested by real-time PCR. Each individual contributed a total of 120 ng DNA to create the DNA pooling and de novo a total of three times.12,13 A total of 12 DNA pools were established, and once created, each pool was diluted to 50 ng/µL for GWAS.14
DNA-Pool Analysis and Significant SNP Selection
The DNA pools were genotyped according to the Genome-Wide Human SNP 6.0 guide and workflow (Affymetrix, Santa Clara, CA). Sex chromosome markers were excluded in the analysis stage. The silhouette method was employed to find frequency differences between pools by calculating the ratio of allele signal intensities PMA/(PMA+PMB).15 A silhouette value greater than 0.6 was used as a threshold for further replication, thereby minimizing the possibility of false-positives at the apparent cast of false-negatives. SNPs with MAF below 0.05 were excluded because of the limited power to detect a significant association and the decrease in the accuracy of DNA pooling with rear variants. Two statistical strategies were used to select the top risk variants. For the first strategy, the SNP was significant in single-point (single method) of all three progressive outcomes with a Silhouette value >0.6 as the cutoff. This strategy was proposed by Kaufman and Rousseeuw, and they posited that a Silhouette value greater than 0.5 indicated an acceptable association. For the second strategy, the SNP was significant in window-points (sliding-window method) within the range of 7.5 kb,10 kb,12.5 kb and 15 kb throughout whole genome.
Replicated Individual Genotyping
All subjects were individually genotyped according to the manufacturer’s instructions using the TaqMan 7900HT sequence Detection System (Applied Biosystems, Foster City, Calif.). Each assay was carried out using 10 ng DNA in a 5-µL reaction consisting of TaqMan universal polymerase chain reaction master mix (Applied Biosystems, Foster City, Calif.), forward and reverse primers, and 6-carboxyfluorescein (FAM) and 4,7,2’-trichloro-7’-phenyl-6-carboxyfluorescein (VIC)-labeled probes designed by Applied Biosystems (ABI Assays-on-demand). Allelic discrimination was measured automatically using Sequence Detection Systems 2.1 software (automatic confidence level 95%). Approximately 8% of all genotypes were repeated in independent polymerase chain reactions to check for consistency and to ensure intraplate and interplate genotyping quality control. No genotyping discrepancies were detected between the repeated samples (Figure 1).
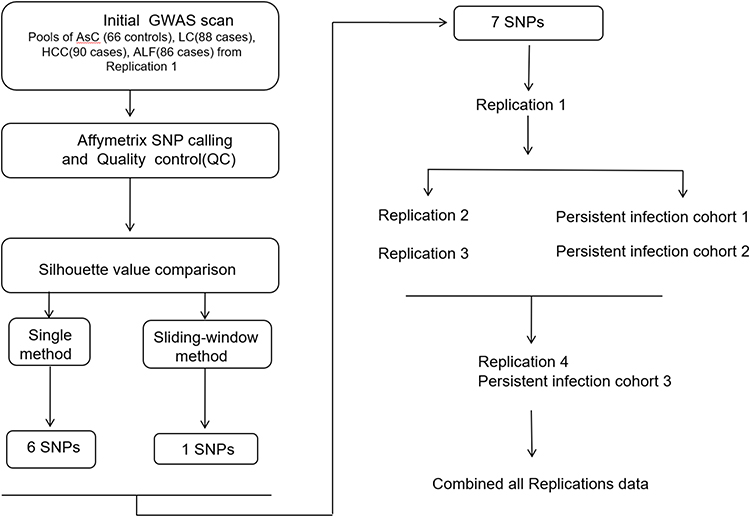 |
Figure 1 The workflow of association analysis. |
Genotypic Function Exploration by Web-Based Bioinformatics
We used RNAfold (http://rna.tbi.univie.ac.at//cgi-bin/RNAWebSuite/RNAfold.cgi) to predict the biological effect of the significant SNPs on RNA structure. RNAfold is a classic database to predict RNA structure and energy change in RNA formation according to RNA sequence. Free energy represents the energy required to change the secondary structure from the current RNA structure. Additionally, we further explored the effects of SNPs on gene expression by investigating a public database of the GTEx portal (http://www.gtexportal.org/home/).
Follow-Up Outcomes
The follow-up time was calculated from the date of enrollment to either the date of patients with LC or HCC occurrence or December 30, 2017, for those without LC or HCC occurrence. Progressive events were ascertained at an average of three months by phone, out-of-hospital and hospitalized medical records. Patients will be diagnosed with HCC if they have LC and HCC at the same time. Family history defined the direct relatives who had HBV-related complications. LC was diagnosed by liver ultrasonography and abdominal computerized tomography features suggestive of cirrhosis based on a quantitative scoring system derived from the appearance of the liver margins, portal vein calibre and spleen diameter, supplemented with the cirrhotic complications (portal hypertension or bleeding in the esophageal varices or gastric varices, swelling in the legs and abdomen and splenomegaly). HCC was diagnosed based on pathologic confirmation or a positive lesion detected by imaging techniques (liver ultrasonography and abdominal computerized tomography) and a-fetoprotein levels exceeding 400 mg/l.
Statistical Analysis
The silhouette value was used to estimate genotypic cluster differences between cases and controls by the R package (www.r-project.org). This process was performed in a blinded manner, so the analyzer did not know the case/control status of each subgroup. The replication analysis was calculated using STATA software (version 10.0/SE, Stata, College Station, Tex.). Hardy-Weinberg equilibrium (HWE) assumptions were independently tested in each persistent infection cohort, chi-square analysis was employed to examine the differences in allele frequencies and genotype, the Cochran-Armitage test was used for genotypic trend analysis, and three genetic models fit the genetically susceptible type.
Unconditional logistic analysis was employed to estimate the odds ratio (OR) and 95% CI. In the combined analysis, the ORs and 95% CIs were adjusted by sex, age and cohort.
In follow-up analysis, the effects of risk genotype(s) were assessed by Kaplan‒Meier analyses and Cox regression analysis. In multivariate analysis, all available baseline covariates with P < 0.20 in unadjusted analysis were considered potential independent predictors. By using a backward stepwise approach with factors sequentially eliminated according to the results of the likelihood ratio test, we constructed Cox proportional hazards models to examine the association between the risk SNPs and progressive events.
Results
Patient Characteristics
All subjects were self-reported Han Chinese ancestry. A total of 8906 participants were recruited from June 2007 to June 2020, comprising 1610 HCC patients, 1430 LC patients, 1540 AsC patients, 1406 persistent infection patients, 1406 naïve to HBV infection participants and 1514 natural clearance participants. The subjects naïve to HBV infection and natural clearance in each replication showed no significant deviation from HWE. The characteristics of the participants in each cohort are shown in Table 1.
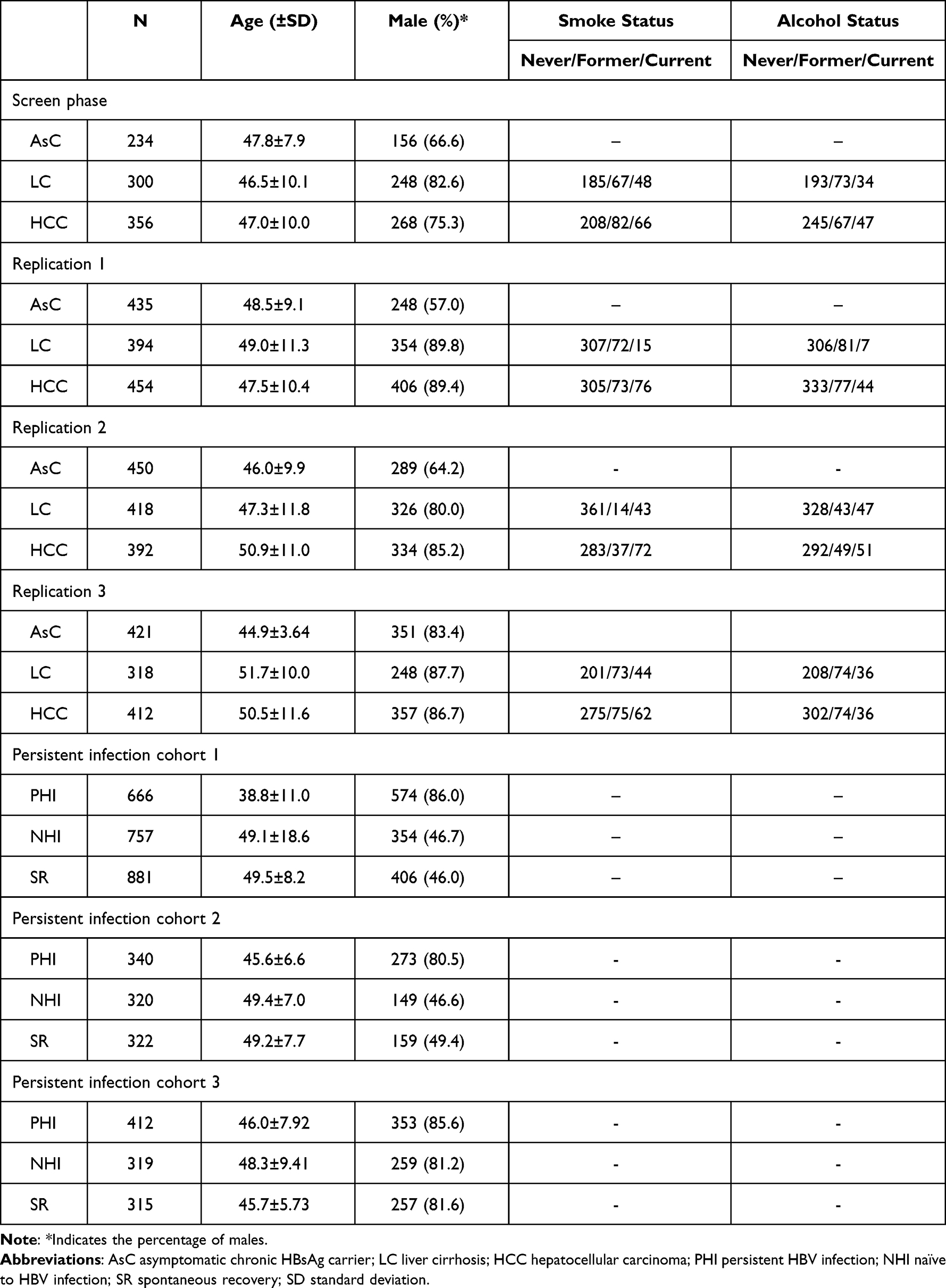 |
Table 1 Characteristics of Participants in Each Cohort |
Pooled GWAS Data for Screening Risk SNPs for Replication
After quality control and silhouette score ranking, 1848 SNPs with average silhouette widths were more than 0.7 in the single-point method (data not shown). According to predesigned SNP selection strategies, 7 SNPs (Table 2) met the selection criteria and were prioritized for further replications: rs11119081, rs2638121, rs7021235, rs3825214, rs6457617 and rs71699 from the single method, and rs2996466 from the sliding-window method.
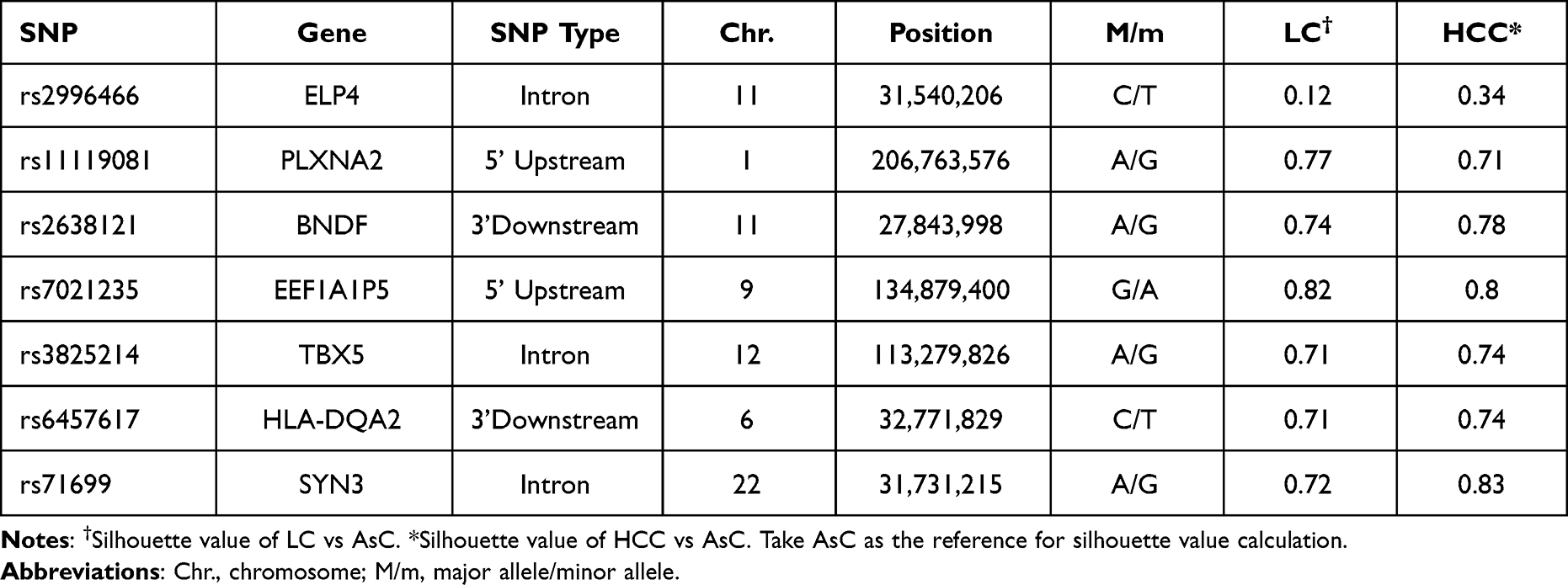 |
Table 2 The Selected SNPs for Replication Genotyping |
Replication 1 Panel for Progression
To test the potential association of 7 SNPs, 1009 subjects consisting of 234 AsC patients, 300 LC patients and 300 HCC patients were individually genotyped in the screening stage. We compared the frequency of both allele and genotype between AsC versus LC and AsC versus HCC. The Pallele and Pgenotype values for each SNP are shown in Table 3. Of these SNPs, rs3825214 within TBX5 (T-box transcription factor 5) was strongly associated with LC (Pdominant=0.004) and HCC (Pdominant= 0.0009). The remaining SNPs showed no significant associations with either LC or HCC (Table 3).
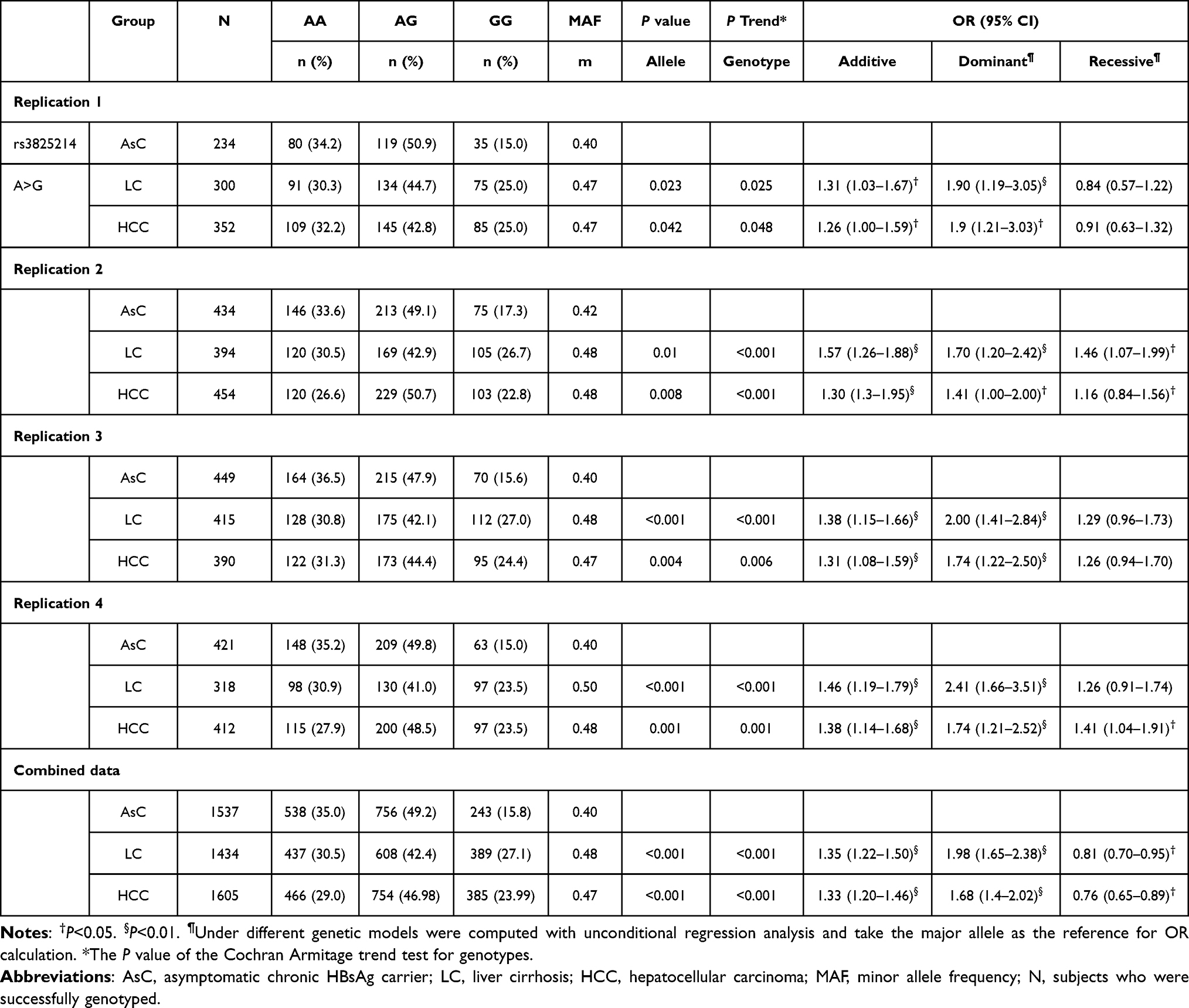 |
Table 3 Association of rs3825214 in Each Progressive Cohort |
Replication 2 Panel for Progression
To confirm the significance in additional Hubei cohorts, 1383 subjects consisting of 435 patients with AsC, 394 patients with LC and 454 patients with HCC were further assessed. The Pallele and Pgenotype values for rs3825214 are shown in Table 3. Again, the association of rs3825214 with LC was observed (Pdominant=0.0027), and the association of rs3825214 with HCC was discovered. (Pdominant=0.0089). However, the remaining four SNPs were not nominally significant.
Replication 3 Panel for Progression
To validate the results, 1381 subjects from Guangdong, including 450 patients with AsC, 418 patients with LC and 392 patients with HCC, were evaluated. The Pallele and Pgenotype values for rs3825214 are presented in Table 3. Consistently, rs3825214 of TBX5 showed an association in both LC (Pdominant<0.001) and HCC (Pdominant=0.0014).
Replication 4 Panel for Progression
To further validate the results, 1151 subjects from Hainan, including 421 patients with AsC, 318 patients with LC and 412 patients with HCC, were genotyped. The Pallele and Pgenotype values for rs3825214 are shown in Table 3. Consistently, rs3825214 of TBX5 showed evidence of association in both LC (Pdominant<0.001) and HCC (Pdominant=0.0014).
Combined Data Analysis for Progression
For all the subjects of Han Chinese ancestry, rs3825214 within TBX5 was in the same direction among the cohorts, and the genotype frequencies of the risk loci in persistent infection cohorts conformed to HWE expectations. Therefore, we combined the samples across all progressive cohorts. In the combined analysis, rs3825214GG was associated with an increased risk of LC (versus AA/AG P < 0.001; OR = 1.98; 95% CI: 1.65–2.38) and HCC (P < 0.001; OR, 1.68; 95% CI: 1.4–2.02).
Association of rs3825214 with Persistent HBV Infection
The susceptible relationship of rs3825214 with persistent HBV infection, natural clearance and naivety to HBV infection was assessed in three independent panels: persistent infection cohort 1 comprised 666 individuals with persistent HBV infection, 757 individuals who were naïve to HBV infection, and 881 individuals with natural clearance; persistent infection cohort 2 comprised individuals 340 with persistent HBV infection, 320 individuals who were naivety to HBV infection, and 321 individuals with natural clearance; and persistent infection cohort 3 included 409 individuals with persistent HBV infection, 332 individuals who were naivety to HBV infection, and 315 individuals with natural clearance.
The genotype and allele of rs3825214 presented no significant association in the two-two comparison among persistent HBV infection, natural clearance and naïve to HBV infection (Table 4).
 |
Table 4 Association of rs3825214 with Persistent HBV Infection, Natural Clearance and Naïve to HBV Infection |
Function Exploration and Expression Alternation
We used RNAfold and the GTEx portal to predict the function of rs3825214 (G>A). In the RNAfold analysis, the centroid secondary and minimum free energy (minimum free energy, MFE) structures of rs3825214 are shown in Figure 2. The genotype of rs3825214 alters the minimum free energy, free energy of the thermodynamic ensemble, frequency of the MFE structure in the ensemble and ensemble diversity, minimum free energy (G: −41.60 kcal/mol vs A: −39.50 kcal/mol), free energy of the thermodynamic ensemble (G: −46.13 kcal/mol vs A: −44.78 kcal/mol), frequency of the MFE structure in the ensemble (G: 0.06% vs A: 0.02%), and ensemble diversity (G: 44.91 vs A: 55.83) (Figure 2).
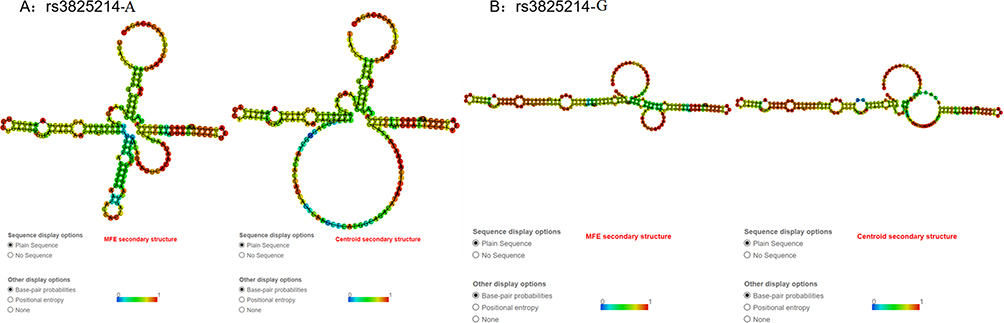 |
Figure 2 The RNAfold algorithm predicts the genotypic impact of rs3825214 on centroid secondary and minimum free energy. (A) MFE and centroid secondary structure of rs3825214-A. (B) MFE and centroid secondary structure of rs3825214-G. |
In expression analysis, we found that rs3825214 contributed to expression quantitative trait loci (eQTL) based on the public database GTEx Portal. The rs3825214 genotype significantly altered the intron excision ratio of TBX5 in the heart (P=1.924e-8) (Figure 3).
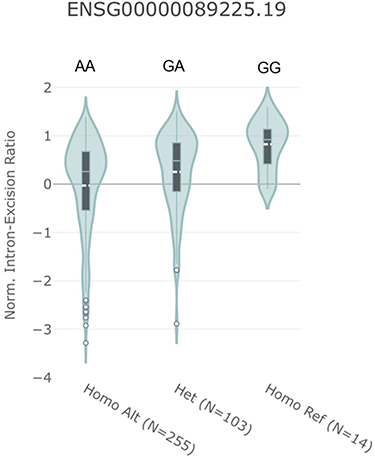 |
Figure 3 Violin plots showing the association between normalized intron-excision ratio and rs3825214 genotype. The GG allele was significantly correlated with increased intron excision in the heart. Data were generated using the Single‐Tissue eQTL function in the GTEx Data Portal. |
Multivariate Analysis of Follow-Up Outcomes
A total of 652 persistent infection patients at baseline were included for follow-up in persistent infection cohort 1. Twenty-one (3.2%) patients lost their phone connection and were without hospitalized register records, 31 (4.7%) declined to offer disease information, and 29 (4.4%) patients refused follow-up. To preclude the potential bias created by loss to follow-up, the 81 subjects were compared, and there was no significant difference in genotype distribution of rs3825214 (MM:Mm:mm 25:44:12/190:279:106, P=0.58), sex (male 62 versus 496, P=0.51), or age (40.2 versus 38.6, P=0.21) compared with subjects who did complete follow-up examinations.
A total of 571 patients were included in the follow-up analysis. During a median of 5.1 years (range 0.098–7.31 years) of follow-up with a total of 2876.2 person-years, 93 (16.29%) patients had LC, and 74 (12.96%) had HCC.
In univariate Cox analysis, the risk factors independently predicting LC included age (P<0.001, OR = 1.04; 95% CI: 1.02–1.06), alcohol never (versus former, P=0.002, OR = 2.3; 95% CI: 1.34–3.92; versus current, P=0.009, OR = 1.97; 95% CI: 1.19–3.27), INR (P<0.001, OR = 3.13; 95% CI: 1.92, 5.11), HBeAg persistence (P<0.001, OR = 2.98; 95% CI: 1.94–4.59), HBV positive (P<0.001, OR = 2.21; 95% CI: 1.44–3.37), and rs3825214GG (versus AG+AA, P<0.001, OR = 3.03; 95% CI: 1.95–4.70) (Table 5).
 |
Table 5 Univariate Cox Analysis of Baseline Characteristics of Follow-Up Subjects |
The predictive risk factors for HCC included age (P<0.001, OR = 1.07; 95% CI: 1.05, 1.09), HBeAg persistence (P<0.001, OR = 3.01; 95% CI: 1.84, 4.91), HBV positivity (P<0.001, OR = 2.14; 95% CI: 1.48, 3.93), and rs3825214GG (versus AG+AA, P<0.001, OR = 4.32; 95% CI: 2.68, 6.89) (Table 6).
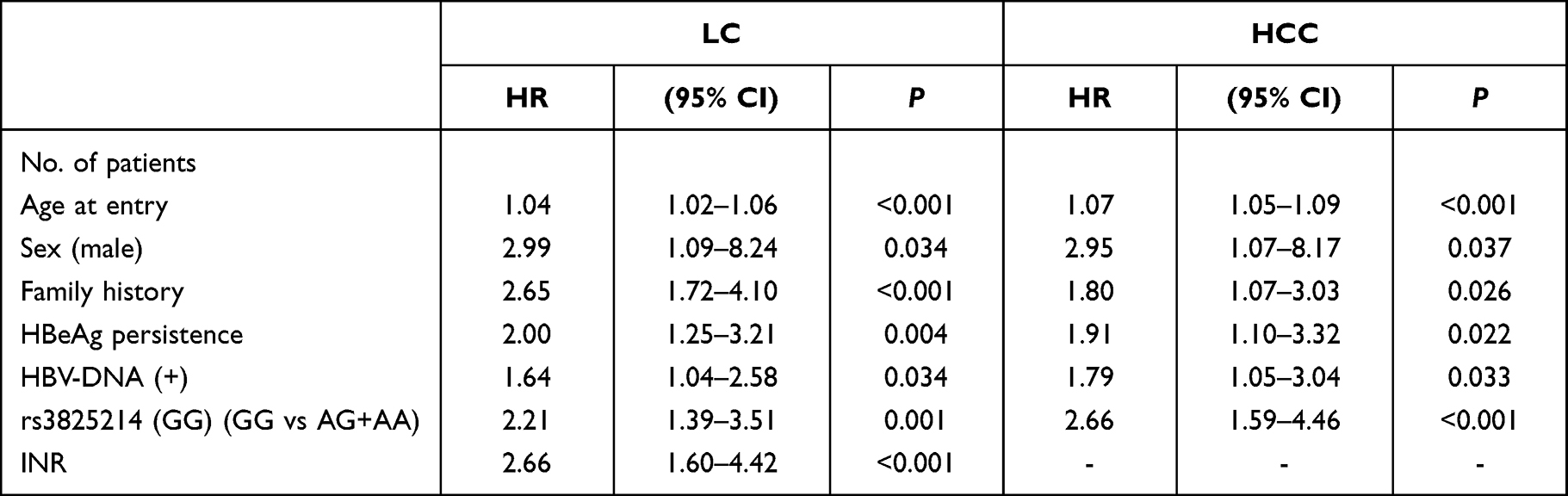 |
Table 6 HBV-Related LC and HCC in Cox Proportional Hazard Regression Models |
After accounting for covariates, we also found that rs3825214GG remained associated with the incidence of LC and HCC in Kaplan‒Meier analyses (Figure 4).
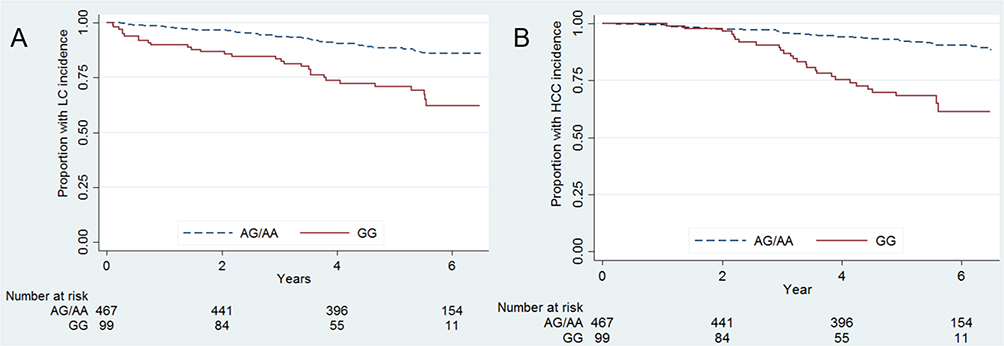 |
Figure 4 Kaplan‒Meier graphs of time to progressive events, (A) rs3825214 GG (21/85, 24.7%) vs AG/AA (25/444, 5.6%) with log-rank P value <0.0001 in HCC incidence; (B) rs3825214 GG (24/82, 29.3%) vs AG/AA (46/423, 10.9%) with log-rank P value <0.0001 in LC incidence. |
Discussion
By investigating a large number of HBV-related subjects in the Chinese Han population from multiple centers, rs3825214 within TBX5 was specifically associated with LC and HCC. With at least 5 years of follow-up of persistent HBV infection patients, we observed that the susceptibility variants increased the incidence of LC and HCC, which highlights the genotype-phenotype link between risk genotypes and progressive events.
These results underscore several important principles. First, consistent with previous analysis, susceptibility exploration on a genome-wide scale has the potential to detect novel risk loci and provide independent information beyond candidate genes for progression, shedding light on the pathophysiology/function of HBV-related progression. Second, the associations are detectable in both LC and HCC, considering etiologic homogeneity, suggesting that the progressive phenotypes might share the pathological pathway, which indicates the transferability of preventive and therapeutic options for progression. Third, the genotype(s) modified the risk of LC and HCC provide incremental clues about genetic risk discrimination for chronic HBV-infected individuals. Fourth, the risk polymorphism was related to the phenotype and outcomes in long-term observation.
The rs3825214 is located in the intron region of TBX5 at 12q24.1, harboring a 5-kb linkage disequilibrium region at the 3’-end region, indicating that the 5-kb haplotype block including the promoter region as a whole passes genetic information into the next generation (Supplementary Figure 1). The LD structure and biologic rationale are supportive clues for TBX5 genetic variant involvement in disease progression. TBX5 encodes a transcription factor involved in multiple organ differentiation at the embryogenesis stage, and mutations in TBX5 cause Holt-Oram syndrome in humans, characterized by congenital cardiac defects and forelimb malformations. The genetic variants in TBX5 have also been correlated with blood pressure16 and cardiac arrhythmias17 in GWAS for adults. Additionally, genotype rs3825214 was related to lone atrial fibrillation in the Chinese Han population,18 indicating that genetic variations in TBX5 are likely related to organ function defects. The TBX5 rs3825214 genotype from GTEx data regulated alternative splicing to affect heart development (Figure 3 and Supplementary Figure 1). Additionally, RNAfold analysis showed that the TBX5 rs3825215 genotype affected RNA formation energy and stability, so functionally, the TBX5 rs3825215 genotype influenced epigenetics. Our results in combination with existing evidence suggest that genetic variants in TBX5 could result in deficient liver tissue development and enhance vulnerability to HBV-related liver immune injuries and/or the inability to repair damaged cells; thus, enduring injuries are likely attributed to LC, which also increases the risk of HCC. Interestingly, the current study reported that hypermethylation of the TBX5 promoter region was associated with HCC in the Japanese population,19 posing a potential link between genetic variations and methylation status, an alternative explanation for the influence of genetic variations on HCC development.
Our previous studies demonstrated the two susceptible directions to either persistent infection20 or progression.21 In interest of specificity, the association of the risk loci with persistent infection, natural clearance and naïve to HBV infection was extensively evaluated in a well-defined population collected from the same region of a progressive cohort. Given that HBV vaccination was available in mainland China in 1980, an age above 40 years22 and no vaccination history are preconditions for natural exposure to HBV. Based on preconditions, community-based subject enrollment, including vertical and horizontal exposure, is representative of the general population in HBV endemic regions. This exploration confirmed the specific association; rs3825214-GG demonstrated no progressive characteristics in the general population once the rs3825214-GG individual had chronic HBV infection that increased the risk of developing LC or/and HCC.
The risk loci have been related to the clinical severity and HCC development in Asian chronic HBV infection patients,23 while the relationship of the risk genotypes to HBV-related progressive events has not previously been defined. In our hospital-based follow-up, 167 of 575 patients developed progressive outcomes overall. The exploration of this is significant, as the findings not only confirm that rs3825214 is associated with LC and HCC but also suggests that risk genotypes from other association studies might have an effect on prognosis when no evidence supports biological differences. Accumulating genetic data improve the prediction accuracy of progressive outcomes.
Several limitations of our study need to be mentioned, since GWAS based on the DNA pool was applied as the first step to obtain major genetic predisposition in overview and lost its susceptibility in detail. There are potential biases in statistical methods, and different references for imputation and prioritizing strategies of SNP selection might yield a different association. The mechanisms underlying the association of TBX5 with LC and HCC need to be systematically elucidated. Moreover, in the functional annotation, the condition should keep in mind that the risk genotypes that we tested may not actually be causal but rather correlate the causal genotypes by LD linkage relations; thus, further studies are first needed to fine-map the risk region of TBX5 and clarify the influence of risk genotype(s) or haplotype(s) on the gene regulation of expression and function processes, which in turn provide mechanistic plausibility. The follow-up study is limited to hospital persistent infection patients, which might generate bias.
To the best of our knowledge, our report has extended our understanding of the genetic architecture underlying LC and HCC. Accumulating genetic information in an unbiased manner indicated that susceptibility might be higher; thus, it remains to be determined whether the new high-resolution genotype technology can systematically and specifically reveal the progressive genetic profile. In aggregate, it is important to gain a better understanding of the genetic profile that mediates prognostic differences in addition to clinical risk factors.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Hainan General Hospital. Informed Consent Statement: Informed consent was obtained from all subjects involved in the study.
Acknowledgments
We thank our colleagues for their valuable help in collecting clinical data.
Funding
This research was funded by the Province Natural Science Foundation of Hainan, grant number: 818MS131 and National Basic Research Program of China (973 Program, No. 2007CB512903).
Disclosure
The authors declare no conflict of interest.
References
1. Ganem D, Prince AM. Hepatitis B virus infection--natural history and clinical consequences. N Engl J Med. 2004;350:1118–1129. doi:10.1056/NEJMra031087
2. Lai CL, Yuen MF. Chronic hepatitis B--new goals, new treatment. N Engl J Med. 2008;359:2488–2491. doi:10.1056/NEJMe0808185
3. Beasley RP, Hwang LY, Lin CC, Chien CS. Hepatocellular carcinoma and hepatitis B virus. A prospective study of 22 707 men in Taiwan. Lancet. 1981;2:1129–1133. doi:10.1016/S0140-6736(81)90585-7
4. Liao B, Zhang F, Lin S, et al. Epidemiological, clinical and histological characteristics of HBV/HDV co-infection: a retrospective cross-sectional study in Guangdong, China. PLoS One. 2014;9:e115888. doi:10.1371/journal.pone.0115888
5. Szpakowski JL, Tucker LY. Causes of death in patients with hepatitis B: a natural history cohort study in the United States. Hepatology. 2013;58:21–30. doi:10.1002/hep.26110
6. Hu L, Zhai X, Liu J, et al. Genetic variants in human leukocyte antigen/DP-DQ influence both hepatitis B virus clearance and hepatocellular carcinoma development. Hepatology. 2012;55:1426–1431. doi:10.1002/hep.24799
7. Hu Z, Liu Y, Zhai X, et al. New loci associated with chronic hepatitis B virus infection in Han Chinese. Nat Genet. 2013;45:1499–1503. doi:10.1038/ng.2809
8. Kamatani Y, Wattanapokayakit S, Ochi H, et al. A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nat Genet. 2009;41:591–595. doi:10.1038/ng.348
9. Zhang H, Zhai Y, Hu Z, et al. Genome-wide association study identifies 1p36.22 as a new susceptibility locus for hepatocellular carcinoma in chronic hepatitis B virus carriers. Nat Genet. 2010;42:755–758. doi:10.1038/ng.638
10. Zhu ZZ, Di JZ, Gu WY, et al. Association of genetic polymorphisms in STAT1 gene with increased risk of hepatocellular carcinoma. Oncology. 2010;78:382–388. doi:10.1159/000320521
11. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi:10.1016/S0140-6736(03)14964-1
12. Skol AD, Scott LJ, Abecasis GR, Boehnke M. Joint analysis is more efficient than replication-based analysis for two-stage genome-wide association studies. Nat Genet. 2006;38:209–213. doi:10.1038/ng1706
13. Sham P, Bader JS, Craig I, O’Donovan M, Owen M, Pooling: DNA. a tool for large-scale association studies. Nat Rev Genet. 2002;3:862–871. doi:10.1038/nrg930
14. Papassotiropoulos A, Stephan DA, Huentelman MJ, et al. Common Kibra alleles are associated with human memory performance. Science. 2006;314:475–478. doi:10.1126/science.1129837
15. Pearson JV, Huentelman MJ, Halperin RF, et al. Identification of the genetic basis for complex disorders by use of pooling-based genomewide single-nucleotide-polymorphism association studies. Am J Hum Genet. 2007;80:126–139. doi:10.1086/510686
16. Levy D, Ehret GB, Rice K, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41:677–687. doi:10.1038/ng.384
17. Holm H, Gudbjartsson DF, Arnar DO, et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet. 2010;42:117–122. doi:10.1038/ng.511
18. Zang X, Zhang S, Xia Y, et al. SNP rs3825214 in TBX5 is associated with lone atrial fibrillation in Chinese Han population. PLoS One. 2013;8:e64966 . doi:10.1371/journal.pone.0064966
19. Deng YB, Nagae G, Midorikawa Y, et al. Identification of genes preferentially methylated in hepatitis C virus-related hepatocellular carcinoma. Cancer Sci. 2010;101:1501–1510 . doi:10.1111/j.1349-7006.2010.01549.x
20. Li J, Yang D, He Y, et al. Associations of HLA-DP variants with hepatitis B virus infection in southern and Northern Han Chinese populations: a multicenter case-control study. PLoS One. 2011;6(8):e24221 . doi:10.1371/journal.pone.0024221
21. Liu L, Li J, Yao J, et al. A genome-wide association study with DNA pooling identifies the variant rs11866328 in the GRIN2A gene that affects disease progression of chronic HBV infection. Viral Immunol. 2011;24(5):397–402. doi:10.1089/vim.2011.0027
22. Lara Cristina da Cunha G, Brunini S, Guimarãe RA, et al. Epidemiology of hepatitis B virus infection in people living in poverty in the central-west region of Brazil. BMC Public Health. 2019;19(1):443. doi:10.1186/s12889-019-6828-8
23. Jung SW, Park NH, Shin JW, et al. Polymorphisms of DNA repair genes in Korean hepatocellular carcinoma patients with chronic hepatitis B: possible implications on survival. J Hepatol. 2012;57:621–627. doi:10.1016/j.jhep.2012.04.039
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Impact of Metabolic Dysfunction Associated Fatty Liver Disease on the Prognosis of Patients with Hepatitis B Virus-Related Hepatocellular Carcinoma Based on Propensity Score Matching Analysis
Xue J, Wang QX, Xiao HM, Shi MJ, Xie YB, Li S, Lin M, Chi XL
Cancer Management and Research 2022, 14:2193-2202
Published Date: 14 July 2022
Indocyanine Green Retention Test as a Predictor of Postoperative Complications in Patients with Hepatitis B Virus-Related Hepatocellular Carcinoma
Mai RY, Bai T, Luo XL, Wu GB
Therapeutics and Clinical Risk Management 2022, 18:761-772
Published Date: 2 August 2022
The Age, Gamma-Glutamyl Transpeptidase and Platelet Index: A Novel Noninvasive Model for Predicting Hepatocellular Carcinoma in Patients with Hepatitis B Virus-Related Liver Cirrhosis
Liu K, Huang Z, Yang S, Lin L, Zheng S, Zhang X, Xue Y, Xie W
Journal of Hepatocellular Carcinoma 2022, 9:1057-1063
Published Date: 8 October 2022
Prognosis of Primary Liver Cancer Based on LI-RADS Classification with Extracellular Agent-Enhanced MRI
Li Y, Ni X, Liu X, Yang C, Wang Y, Lu X, Zhou C
Journal of Hepatocellular Carcinoma 2023, 10:399-411
Published Date: 9 March 2023
Contribution of Aflatoxin B1 Exposure to Liver Cirrhosis in Eastern Ethiopia: A Case-Control Study
Mekuria A, Xia L, Ahmed TA, Bishaw S, Teklemariam Z, Nedi T, Abula T, Engidawork E, Gong YY
International Journal of General Medicine 2023, 16:3543-3553
Published Date: 16 August 2023