Back to Journals » Journal of Hepatocellular Carcinoma » Volume 12
Taurine-Modified Gossypol Exerts Dual Anti-Hepatocellular Carcinoma Effects by Inactivating PI3K/AKT Pathway and Targeting FASN-Mediated Lipid Metabolism in Regulatory T Cells
Authors He W ![]() , Shi J, Deng G
, Shi J, Deng G ![]() , Liu W, Kou L, Hu J, Lin Y, Lin X, Sheng J
, Liu W, Kou L, Hu J, Lin Y, Lin X, Sheng J ![]() , Wu F
, Wu F
Received 5 October 2025
Accepted for publication 10 December 2025
Published 15 December 2025 Volume 2025:12 Pages 2755—2770
DOI https://doi.org/10.2147/JHC.S572305
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Imam Waked
Weishan He,1,* Juzheng Shi,1,* Guangmei Deng,1 Wenya Liu,1 Long Kou,1 Jia Hu,1 Yajing Lin,1 Xinlan Lin,1 Jinzhou Sheng,1 Fasheng Wu2
1Ruikang Clinical Medical College, Guangxi University of Chinese Medicine, Nanning, Guangxi, People’s Republic of China; 2Radiation Oncology Department, Ruikang Hospital Affiliated to Guangxi University of Chinese Medicine, Nanning, Guangxi, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Fasheng Wu, Email [email protected]
Background: Hepatocellular carcinoma (HCC) remains a highly challenging malignancy to treat with a dismal prognosis. The immunosuppressive tumor microenvironment (TIME), particularly regulatory T cells (Tregs), is a key driver of treatment resistance. This study aimed to investigate the anti-tumor efficacy and underlying mechanism of taurine-modified gossypol (GT)—a novel conjugate derived from two natural products (taurine and gossypol) with potential synergistic activity.
Methods: The anti-proliferative (CCK-8 assay), pro-apoptotic (Annexin V/PI staining) and cell cycle-regulatory (PI staining) effects of GT were evaluated in HepG2 cells and patient-derived HCC organoids. scRNA-seq and multiparametric flow cytometry were used to analyze alterations in the TIME. Molecular docking and surface plasmon resonance (SPR) were performed to validate the binding affinity (KD) between GT and FASN. Western blotting assessed PI3K/AKT and lipid metabolism pathways.
Results: GT dose-dependently inhibited HCC proliferation, induced apoptosis and caused G1 arrest, with concomitant PI3K/AKT pathway suppression. scRNA-seq revealed a selective reduction in Treg proportion following GT treatment. Mechanistically, GT bound to FASN with high affinity, inhibiting its activity and disrupting lipid metabolism in Tregs, thereby reprogramming Treg differentiation and function. In HCC patients, a clinically significant link was observed between high levels of FASN expression and reduced survival, based on an analysis of TCGA data.
Conclusion: GT exerts synergistic anti-HCC effects through a dual mechanism: directly suppressing tumor proliferation by inactivating the PI3K/AKT pathway, and remodeling the TIME by targeting FASN-dependent lipid metabolism in Tregs. These findings highlight the potential of GT as a novel multitargeted agent for HCC treatment.
Keywords: taurine-modified gossypol, hepatocellular carcinoma, regulatory T cells, fatty acid synthase, PI3K/AKT pathway
Introduction
Hepatocellular carcinoma (HCC) accounts for approximately 80–90% of primary liver cancer cases worldwide, ranking as the 6th most common malignancy and the 4th leading cause of cancer-related death globally, which poses a severe threat to human health.1 Although therapeutic strategies such as surgical resection, transcatheter arterial chemoembolization, and targeted drugs have achieved certain progress, the 5-year survival rate of HCC patients remains only 18% due to issues including high recurrence rate, intrahepatic metastasis, and intrinsic drug resistance.2,3
The tumor immune microenvironment (TIME) plays a crucial role in HCC progression4 In recent years, targeting regulatory T cells (Tregs) in tumors to modulate the HCC immune microenvironment has emerged as a key research direction for improving the efficacy of HCC immunotherapy. Single-cell sequencing studies have shown that the proportion of Treg cells in HCC tissues is significantly higher than that in adjacent non-tumor tissues, and the degree of Treg infiltration is positively correlated with poor patient prognosis.5 This indicates that targeted regulation of Treg cells holds important value for HCC treatment. However, direct depletion of Treg cells can trigger systemic immune activation, leading to severe adverse reactions and even enhancing the immunosuppressive effect of tumors.6 In contrast, inhibiting Treg cell differentiation and function to partially relieve their mediated immunosuppression exhibits higher safety and efficacy for HCC treatment. Nevertheless, due to the high heterogeneity of treatment responses among HCC patients, existing drugs have shown suboptimal efficacy in precisely targeting Treg cell differentiation and function. Therefore, exploring and developing new approaches and drugs that can accurately intervene in the functional state and differentiation of Treg cells is of great significance for the current development of HCC treatment.
Fatty acid synthase (FASN), as the rate-limiting enzyme in the de novo fatty acid synthesis pathway, has been identified as a key oncogenic driver in HCC.7 Its overexpression promotes tumor cell proliferation by providing energy and structural substrates. Recent studies have further revealed that FASN can regulate Treg cell function through metabolic reprogramming.8 Treg cells rely on enhanced fatty acid synthesis to maintain their immunosuppressive phenotype, and inhibition of FASN can disrupt this metabolic dependence, thereby restoring antitumor immunity.9 Additionally, inhibition of FABP5, a gene related to fatty acid synthesis and metabolism, can induce mitochondrial changes in Treg cells, characterized by impaired lipid metabolism, reduced oxidative phosphorylation, and loss of cristae structure.10 These findings suggest that fatty acid synthesis and metabolism, the core process of FASN action, may indeed regulate Treg cell differentiation and tumor immune escape.
Natural products derived from traditional Chinese medicine have long been an important resource for anti-cancer drug development due to their unique chemical structures and multi-target mechanisms of action.11 Calculus Bovis (Niuhuang), a traditional anti-tumor Chinese medicine used for thousands of years, is believed to possess the effect of “clearing heat and detoxifying” in traditional Chinese medicine theory, which is highly consistent with the pathological feature of “internal accumulation of heat toxin” in HCC.12 Taurine, the main active component of Calculus Bovis, exhibits multiple pharmacological activities such as anti-fibrosis, anti-oxidative stress, and immune regulation.13 It can also promote the proliferation and activation of T cells to enhance their ability to kill cancer cells;14 in the liver cancer microenvironment, it maintains the balance of Th1/Th2 cells and inhibits Treg cell function to alleviate immunosuppression.15 Gossypol, a naphthalene derivative isolated from Malvaceae plants, can also exert immunomodulatory effects in HCC by regulating T cell homeostasis and reducing the secretion of immunosuppressive cytokines.16,17
Taurine-modified gossypol (GT) is a novel derivative synthesized by condensing taurine (as the lead compound) with gossypol. Its structural characteristics make it promising to integrate the pharmacological advantages of both parent components and achieve synergistic enhancement of anti-HCC activity. However, current research on the anti-tumor effect of GT is still in the initial exploration stage, and its definitive anti-HCC activity, specific targets, and underlying molecular mechanisms remain unclear. Based on this, this study systematically evaluated the direct cytotoxic activity of GT against HCC cells and patient-derived organoids. Using multi-dimensional techniques including single-cell sequencing, molecular docking, and metabolomics, we deeply investigated its regulatory effects on Treg cell differentiation and function, and clarified the core role of FASN in the mechanism of GT action. This study aims to reveal the dual anti-HCC mechanism of GT (“direct inhibition of tumor cell proliferation - remodeling of immune microenvironment”), and provide experimental basis and theoretical support for the development of novel HCC therapeutic drugs targeting immunometabolism.
Materials and Methods
Clinical Samples and Cell Line
All procedures involving human subjects were conducted in accordance with the principles of the Declaration of Helsinki and approved by the Hospital Ethics Committee (Approval No.: KY2024-210). Human HCC tissue specimens were obtained via needle biopsy from patients who provided informed consent, placed in PBS containing antibiotics, and promptly transported to the laboratory at 4°C. The human HCC cell line HepG2 was purchased from the Cell Bank of the Chinese Academy of Sciences, authenticated by STR profiling, and confirmed to be mycoplasma-free. Cells were routinely cultured in DMEM supplemented with 10% fetal bovine serum at 37°C in a 5% CO2 incubator.The synthesis method and chemical structure of GT are provided in the Supplementary Information, whose Appendix contains supporting content such as experimental reagent information (Table S1), product-related images (Figures S1 and S2), and characterization spectra (Figures S3 and S4).
Patient-Derived Organoid Culture and Drug Treatment
Fresh HCC tissues were minced and digested with a mixture of collagenase IV and hyaluronidase at 37°C with shaking for 20–30 minutes to generate single-cell suspensions. After centrifugation, cells were resuspended, mixed with Matrigel at a 1:1 volume ratio, and seeded into 24-well plates. Following Matrigel polymerization, specialized organoid culture medium was added and refreshed every 2–3 days.
After 7 days of culture, organoids were treated with 25, 50, or 100 μg/mL taurine-modified gossypol (GT) or an equal volume of vehicle control (0.1% DMSO) for 48 hours prior to subsequent assays.
Cell Viability Assay
Following drug treatment, the viability of HepG2 cells and organoid-derived single cells was assessed. Cells were seeded at 5×103 cells/well in 96-well plates, incubated with CCK-8 for 2 hours, and the absorbance was then measured to calculate the viability inhibition rate relative to the control group.
Flow Cytometry Analysis
For immunophenotyping, single-cell suspensions from organoids were stained with a viability dye and fluorescently-labeled antibodies against surface markers (CD4, FOXP3, CD8a, CD161a), followed by fixation and permeabilization. For intracellular FASN staining, after surface staining, cells were permeabilized and incubated with an anti-FASN primary antibody and a corresponding fluorescent secondary antibody. Apoptosis was detected using an Annexin V-FITC/PI apoptosis detection kit. Cell cycle analysis was performed by ethanol fixation, RNase A treatment, and PI staining. All samples were analyzed using a flow cytometer, and data were processed with FlowJo software.
Lipid Metabolomics Analysis
To investigate the effect of GT on lipid metabolism, approximately 50 mg of organoid samples were collected, and lipids were extracted using a methanol-acetonitrile solution. Lipidomic analysis was performed using an ultra-performance liquid chromatography-tandem mass spectrometry system. Separation was achieved on an HSS T3 column, and detection was carried out with an electrospray ionization source operating in both positive and negative ion modes. Raw data were processed for peak picking and lipid identification by matching against databases. Differential lipids were screened with criteria of VIP>1 and P<0.05, followed by KEGG pathway enrichment analysis.
Western Blot Analysis
Western blotting was performed to validate key protein expression. Total protein extracted from cells or organoids was quantified (BCA assay), separated by SDS-PAGE, and transferred to PVDF membranes. After blocking with 5% skim milk, membranes were incubated with primary antibodies (FASN, ACSL4, ACSL3, GAPDH) at 4°C overnight, followed by HRP-conjugated secondary antibodies for 1 h at room temperature. Protein bands were visualized by ECL and quantified using ImageJ Software.
Single-Cell RNA Sequencing
To dissect cellular heterogeneity at the transcriptome level, single-cell suspensions from organoids were prepared. Cells were captured using the 10x Genomics platform for library preparation and sequencing on an Illumina NovaSeq 6000 instrument. Raw sequencing data were aligned, subjected to quality control, and normalized. Cell clustering was performed using the Seurat package. Pseudotime trajectory analysis of Tregs was conducted using Monocle 3. Differentially expressed genes underwent KEGG functional enrichment analysis.
Molecular Docking and Surface Plasmon Resonance
To investigate the interaction between GT and FASN, the crystal structure of FASN (PDB ID: 7KIV) was retrieved from the Protein Data Bank. Molecular docking was performed using AutoDock Vina software to dock the GT molecule into the active site, and binding energy was evaluated.
SPR was used for experimental validation. Recombinant human FASN protein was immobilized on a CM5 sensor chip. Serially diluted GT solutions were injected, and binding signals were monitored in real-time. Binding kinetics parameters (KD, ka, kd) were calculated using a 1:1 binding model. Control experiments were performed using the FASN-Arg250Ala point mutant.
TCGA Data Analysis
Transcriptomic and clinical data for 371 HCC and 50 adjacent normal tissues were obtained from The Cancer Genome Atlas. FASN mRNA expression differences between tissues were analyzed using R. Based on the median expression level, patients were stratified into high- and low-expression groups. Kaplan-Meier survival analysis with Log rank testing was then performed to compare overall survival between these groups.
Statistical Analysis
Data are presented as mean ± SD. All analyses were conducted using GraphPad Prism 9.0, employing one-way ANOVA with Tukey’s test for multi-group comparisons and unpaired t-tests for two-group comparisons. A P-value<0.05 was considered statistically significant.
Results
GT Exerts Potent Anti-Tumor Effects on HCC Cells
To explore the anti-tumor potential of GT, we first treated human HCC HepG2 cells with increasing concentrations of GT. The results showed that GT significantly inhibited cell proliferation in a dose-dependent manner (Table 1). Additionally, flow cytometry analysis indicated that GT treatment notably promoted cell apoptosis and induced cell cycle arrest (Figure 1A and B). At the molecular level, Western blotting analysis revealed that the anti-tumor effect of GT was associated with the inactivation of the PI3K/AKT signaling pathway—a key pro-survival pathway in HCC (Figure 1C).
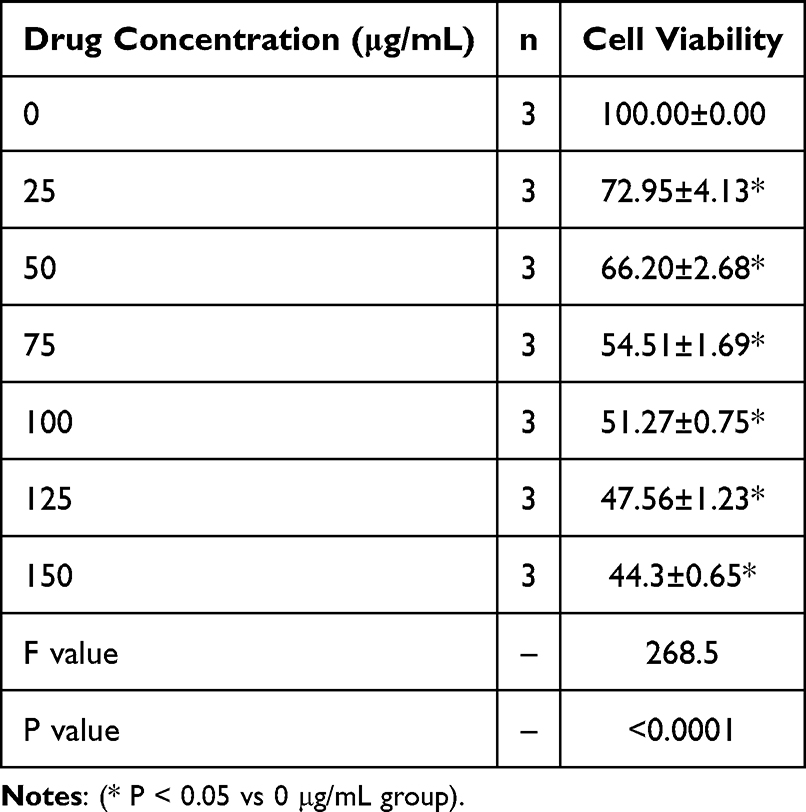 |
Table 1 GT Can Inhibit the Proliferation of HCC Cells in a Dose-Dependent Manner |
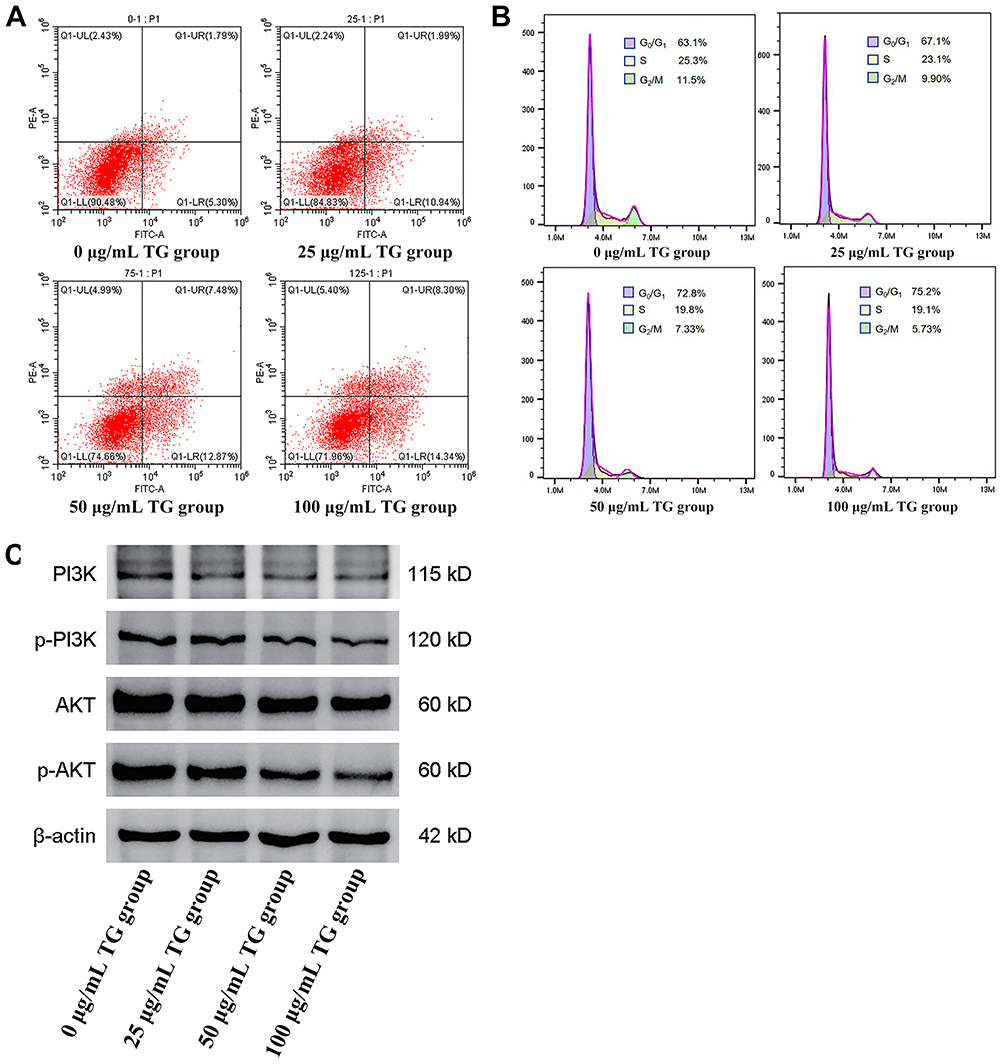 |
Figure 1 GT inhibits hepatocellular carcinoma progression by inducing apoptosis, cell cycle arrest, and suppressing PI3K/AKT signaling. (A): GT promotes HCC cell apoptosis in a dose-dependent manner (n=3). (B): GT induces cell cycle arrest in HCC cells in a dose-dependent manner (n=3). (C): GT inhibits the activation of the PI3K/AKT signaling pathway in tumor cells (n=3). |
GT Inhibits the Viability of Patient-Derived HCC Organoids in a Dose-Dependent Manner
To validate our findings in a more physiologically relevant model, we established patient-derived HCC organoids. CCK-8 assays demonstrated that GT treatment significantly reduced the viability of organoids (Figure 2). This inhibitory effect was also dose-dependent, confirming the broad anti-HCC activity of GT across different model systems.
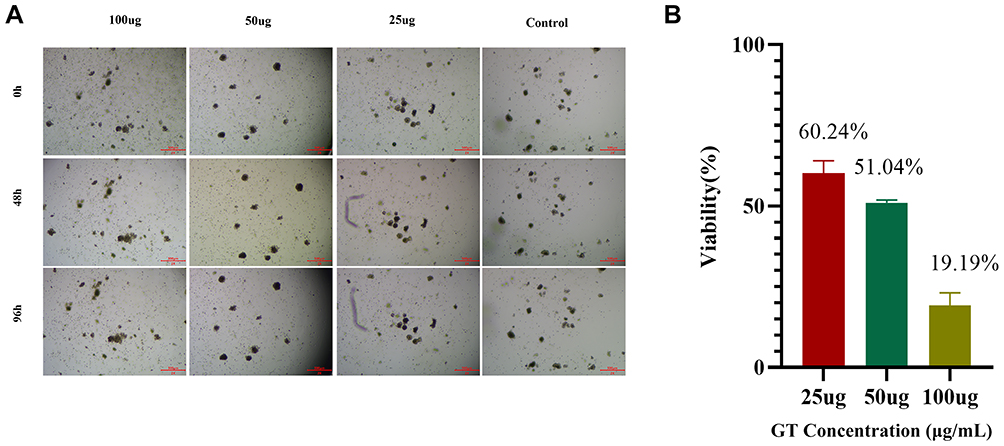 |
Figure 2 GT suppresses the in vitro growth of HCC organoids in a dose-dependent manner. (A): Time-lapse images of organoids treated with medium containing 25 μg/mL, 50 μg/mL, and 100 μg/mL GT for 96 hours. (B): CCK-8 assay for viability of organoids at 96 hours post-treatment with 25 μg/mL, 50 μg/mL, and 100 μg/mL GT. |
Single-Cell Sequencing Analysis of Organoids Reveals the Inhibitory Effect of GT on Tregs
To clarify the impact of GT on the HCC TIME, we performed single-cell RNA sequencing (scRNA-seq) on organoids from the control group and GT-treated group. The results showed a consistent reduction in the proportion of Tregs after GT treatment (decreasing from 4% to 3.5%, and from 4.6% to 2.8%, respectively). Other immune cell subsets exhibited inconsistent or no significant changes (Figure 3), indicating that Tregs are the specific cellular target of GT. We functionally validated this finding using an in vitro co-culture system. When HCC cells were co-cultured with peripheral blood mononuclear cells, flow cytometry analysis showed that GT treatment effectively inhibited Treg differentiation and improved the immune microenvironment (Figure 4).
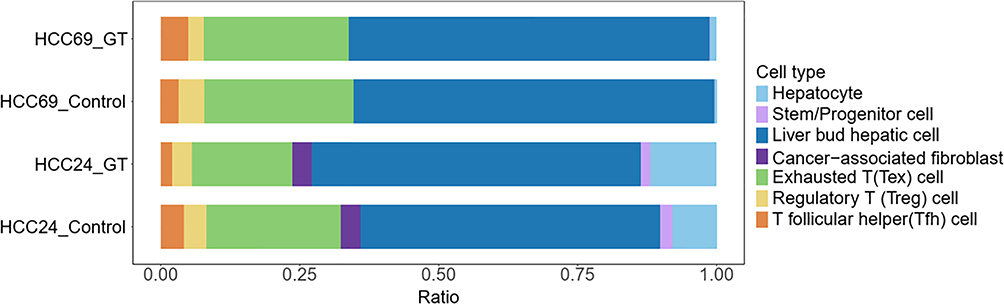 |
Figure 3 Single-cell sequencing of cultured organoids reveals that Treg cells are inhibited by GT. |
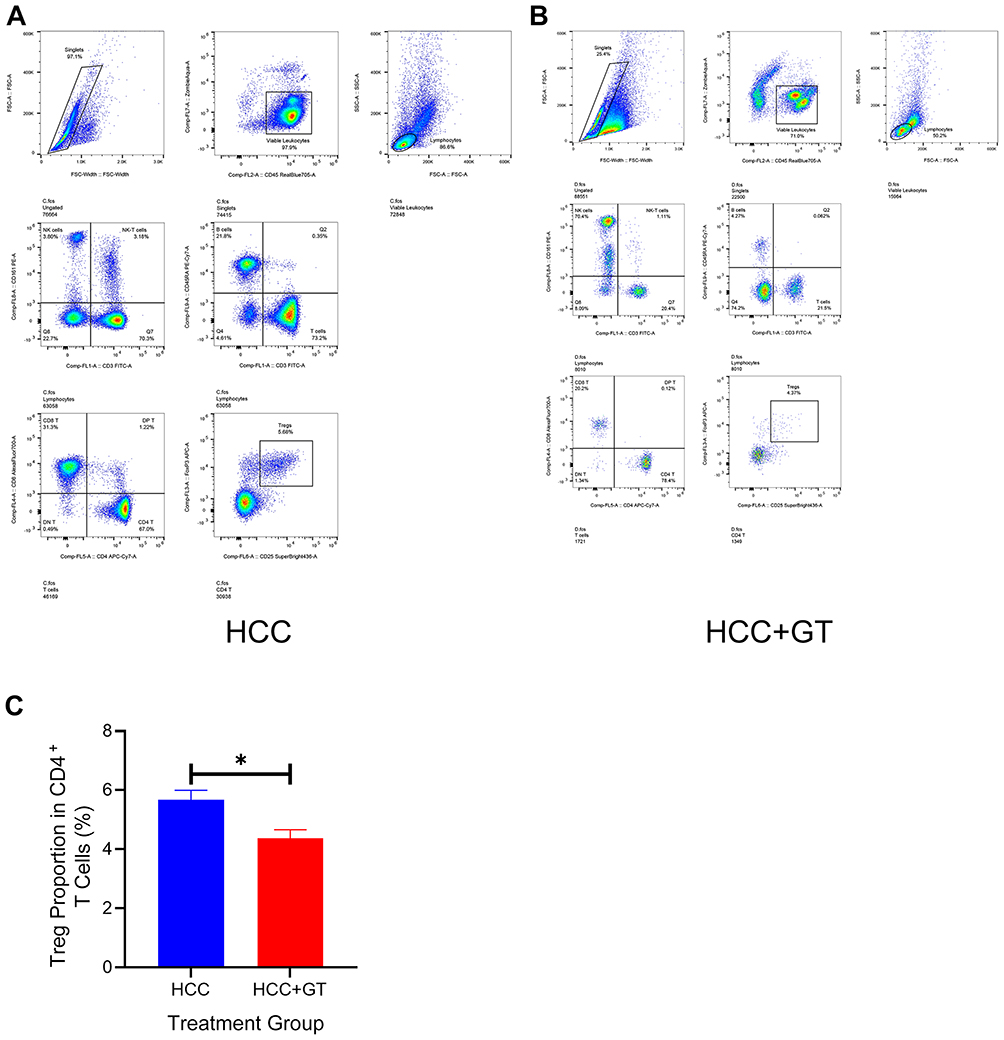 |
Figure 4 Flow cytometry analysis of changes in Tregs after co-culture of HCC organoids with peripheral blood mononuclear cells. (A): Representative gating strategy for Tregs (CD4⁺CD25⁺Foxp3⁺) in the HCC group (without GT treatment). (B): Representative gating strategy for Tregs (CD4⁺CD25⁺Foxp3⁺) in the GT-treated group (with GT intervention). (C): Quantitative comparison of Treg proportions among CD4⁺ T cells. Unpaired two-tailed Student’s t-test; *P < 0.05, GT treatment significantly reduced Treg proportions vs HCC group. |
GT Reshapes Tregs Differentiation and Inhibits FASN Expression
Pseudotime trajectory analysis of Tregs in the scRNA-seq data revealed that GT treatment significantly altered their differentiation state: the dominant population shifted from being primarily state 1 (with secondary state 2) to predominantly state 5 (Figure 5). Differential gene expression analysis among these states identified FASN as a key downregulated gene in state 5 (Log2FC = −1.156, p = 3.82E-27) (Figure 6).
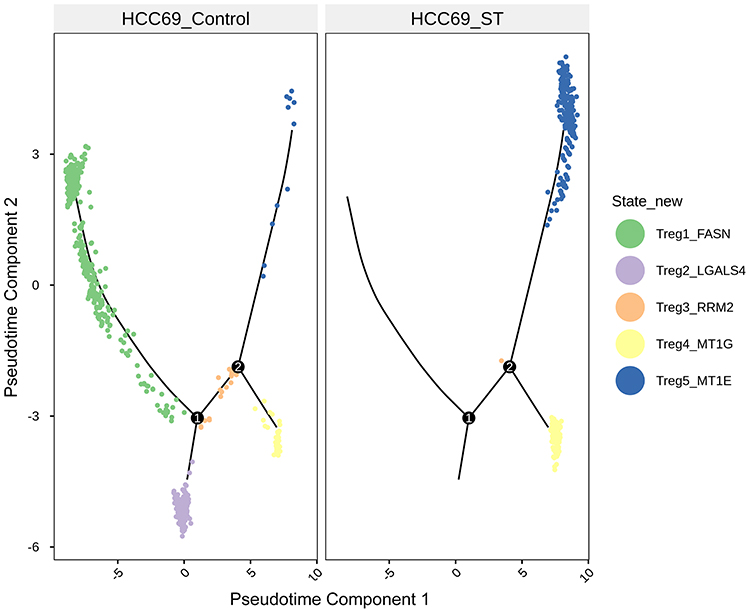 |
Figure 5 Pseudotime trajectory analysis of five state subsets of Tregs in organoids following GT treatment. |
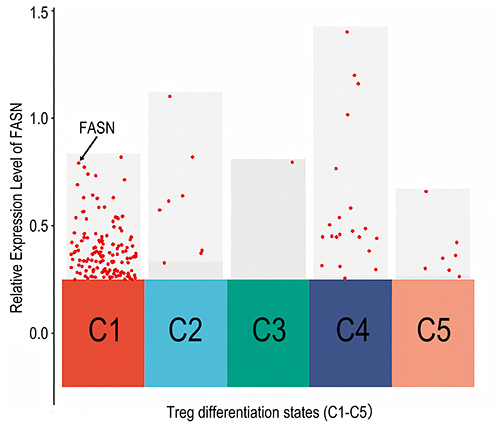 |
Figure 6 FASN expression levels in Tregs at different differentiation states. |
We subsequently validated this finding in in vitro experiments. Western blotting analysis of Tregs stimulated with PHA and treated with IL-2 showed that while IL-2 activation significantly upregulated the expression of key fatty acid metabolic enzymes (FASN, ACSL4, ACSL3), subsequent GT treatment significantly reversed this effect (Figure 7). This provides direct biochemical evidence that GT targets and inhibits FASN expression in Tregs.
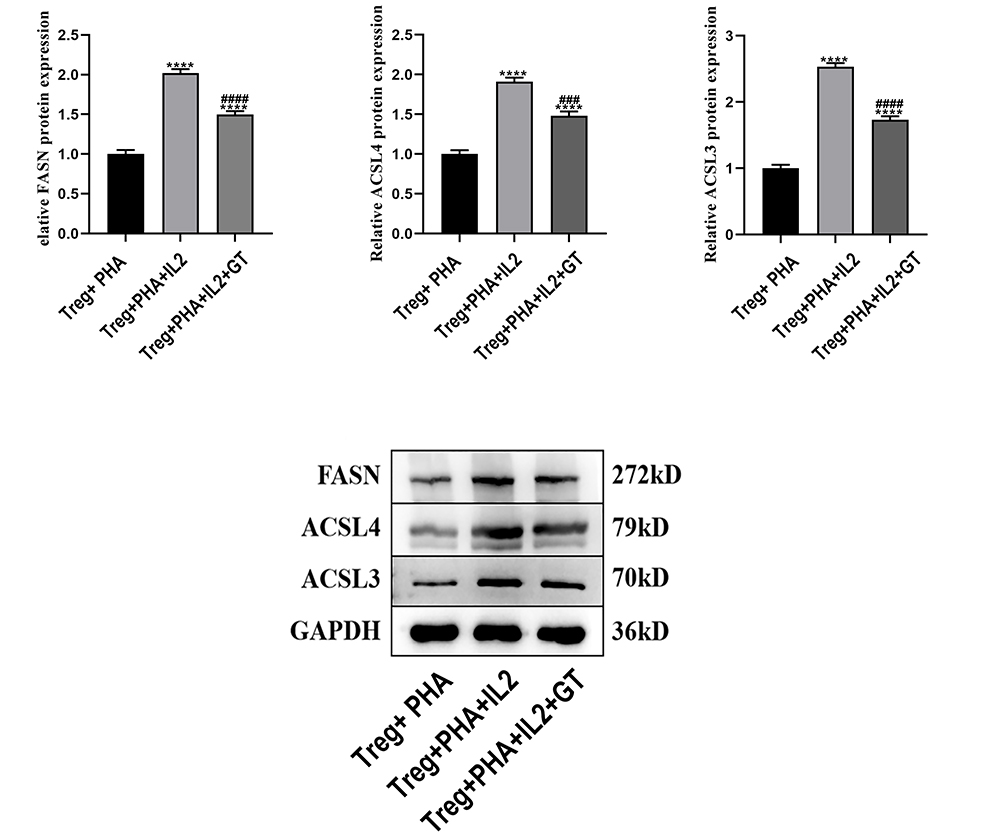 |
Figure 7 Western blot analysis of expression changes in key fatty acid metabolism proteins in Tregs after intervention with IL-2 and GT (*Compared with the Treg+PHA group: ****P<0.0001; #Compared with the Treg+PHA+IL2 group: ###P<0.001, ####P<0.0001). |
Molecular Docking Predicts Stable Interactions Between GT and FASN Protein
To understand the potential direct interaction between GT and FASN, we performed molecular docking simulations. The 2D structure of GT was generated based on previously reported derivatives (Yang et al, 2018) (Figure 8). Docking GT into the binding pocket of FASN (chain A) yielded favorable binding energies, suggesting the potential formation of a stable complex (Table 2). The model predicted that the sulfonyl group, phenolic hydroxyl group, and amino group of GT are crucial for forming intermolecular hydrogen bonds with residues such as Trp2060 and Ser2021, while also engaging in hydrophobic interactions with Pro1264 of FASN (Figure 9).
 |
Table 2 Molecular Docking Table of Taurine Gossypol and FASN Protein |
 |
Figure 8 Stick model of GT. |
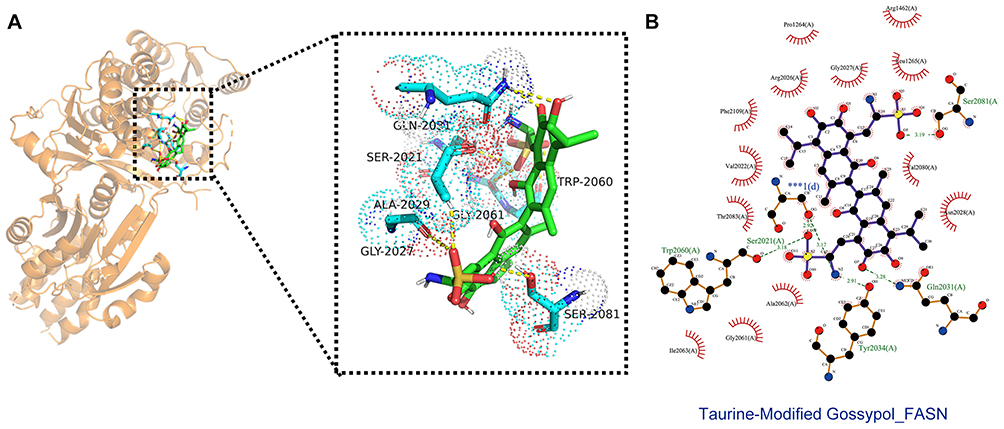 |
Figure 9 Molecular docking mode diagram of GT and FASN. (A) Schematic diagram of the chemical structure of GT. (B) Three-dimensional (3D) molecular docking complex diagram of GT and FASN (*** denotes the key interaction region between GT and the active pocket of FASN chain A). |
Elevated FASN Expression Predicts Poor Prognosis in HCC
Analysis of public TCGA data indicated a significant upregulation of FASN mRNA in HCC tissues (Figure 10). Consistent with this expression pattern, patients with high FASN levels exhibited significantly worse 5-year overall survival (Figure 11). These findings support FASN as a promoter of HCC progression and a potential target for GT-mediated immunomodulation.
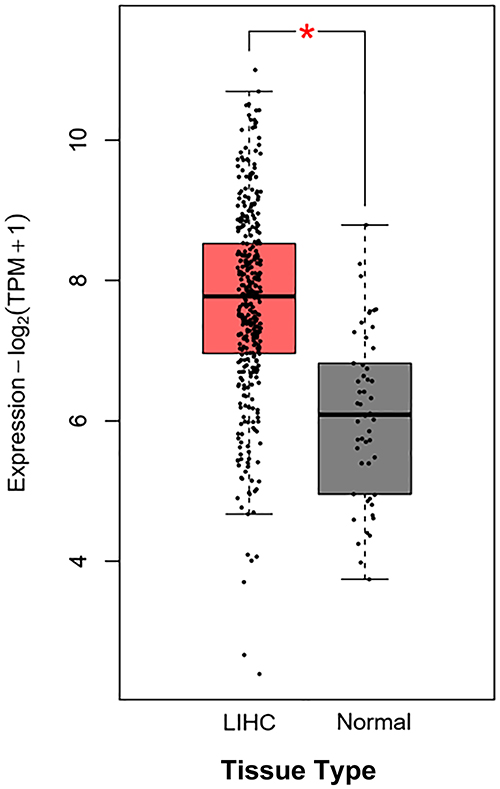 |
Figure 10 FASN expression is upregulated in HCC tissues compared with adjacent non-tumor tissues (n=371 for HCC tissues, n=50 for adjacent non-tumor tissues) (* Indicates a statistically significant difference in FASN mRNA expression between the two groups (P < 0.05)). |
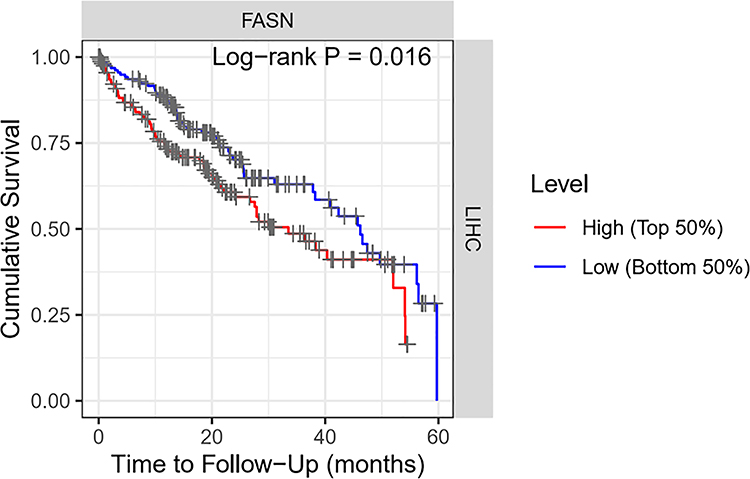 |
Figure 11 High FASN expression is significantly associated with poor survival within a 5-year follow-up period (n=371). |
GT Inhibits FASN and Disrupts Fatty Acid Metabolic Pathways in Tregs
To comprehensively evaluate the metabolic impact of GT-mediated FASN inhibition on Tregs, we performed KEGG pathway enrichment analysis of downregulated genes in Tregs after GT treatment. These genes were significantly enriched in lipid metabolism-related pathways, including steroid biosynthesis, glycolysis/gluconeogenesis, cholesterol metabolism, fatty acid metabolism, fatty acid degradation, and biosynthesis of unsaturated fatty acids (Figure 12). Thus, it is reasonable to speculate that FASN inhibition in Tregs affects tumor immunity through alterations in fatty acid metabolic signaling pathways.
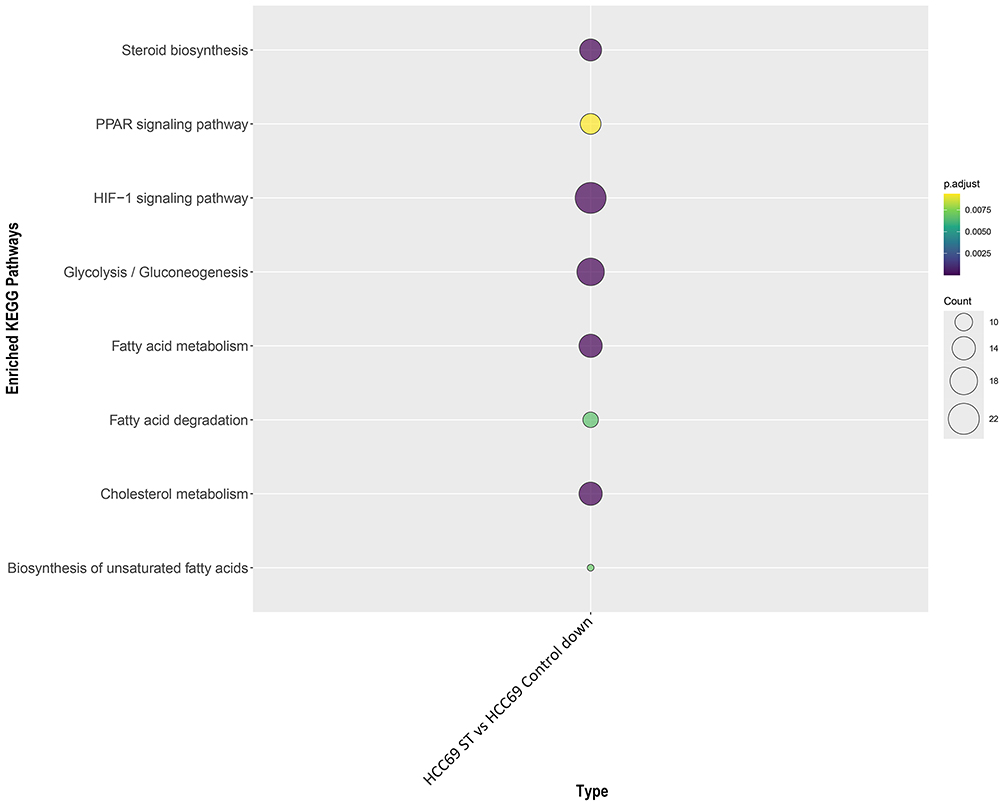 |
Figure 12 Downregulated genes are significantly enriched in fatty acid metabolism pathways. |
Based on these findings, we propose that GT inhibits FASN expression in Tregs, triggering reprogramming of de novo fatty acid synthesis and metabolism, altering Treg differentiation status, thereby alleviating immunosuppression in the HCC microenvironment and enhancing anti-tumor immunity.
Discussion
HCC has an extremely poor prognosis and remains a major global health burden. Traditional treatments are hindered by issues such as tumor heterogeneity, TIME disorders, and acquired drug resistance.18 Although immunotherapies such as immune checkpoint inhibitors are effective in some patients, the potent immunosuppressive barrier mediated by Tregs often leads to treatment failure.19 Tregs dominate the TIME by secreting cytokines such as IL-10 and TGF-β, and their accumulation is closely associated with the aggressive progression of HCC and poor prognosis.20 Consequently, developing agents capable of precisely targeting immunosuppressive cells to reshape the TIME is of considerable clinical importance. This study found that GT, a natural product conjugate derivative derived from traditional Chinese medicine, is an anti-HCC agent with dual effects of direct tumor killing and targeted immunomodulation.
Our study confirms that GT specifically targets the aberrantly activated PI3K/AKT pathway in HCC and delineates the precise mechanistic link between this pathway inhibition and GT’s anti-tumor phenotypes—effects first validated at the cellular and organoid levels. As a core pro-survival pathway frequently hyperactivated in HCC, sustained PI3K/AKT signaling drives tumor cell proliferation, suppresses apoptosis, and enhances invasive capacity.21 While gossypol derivatives have been reported to target this pathway,22 our findings extend this knowledge by demonstrating that taurine modification significantly potentiates this inhibitory effect, suggesting structural optimization may improve GT’s cellular penetration and target-binding affinity. Western blot analysis (Figure 1C) further reveals that GT specifically reduces the phosphorylation of PI3Kα (p110α subunit) in HepG2 cells and patient-derived HCC organoids without altering total PI3K protein expression—likely via direct binding to the PI3Kα catalytic domain or disruption of its interactions with upstream activating molecules (eg, EGFR, c-Met), key drivers of PI3K/AKT dysregulation in HCC.
This targeted inhibition of PI3Kα subsequently suppresses AKT phosphorylation at Ser473 (a critical site for AKT kinase activation), mediating GT’s anti-tumor effects through two well-defined downstream signaling axes: (1) For apoptosis induction, reduced p-AKT relieves the inhibitory phosphorylation of the pro-apoptotic protein Bad, enabling Bad to sequester anti-apoptotic Bcl-2/Bcl-xL and shifting the Bax/Bcl-2 ratio toward pro-apoptosis, thereby triggering caspase-dependent intrinsic apoptosis (consistent with flow cytometry data in Figure 1A); (2) For cell cycle arrest, decreased p-AKT inhibits mTORC1 activity, which in turn reduces Cyclin E expression and promotes nuclear translocation of the CDK inhibitor p27^Kip1^—this nuclear accumulation of p27^Kip1^ directly blocks Cyclin E/CDK2 complex function, leading to G1-phase arrest (Figure 1B), a key checkpoint governing HCC cell proliferation.
Notably, GT exhibits tumor cell-specific regulation of the PI3K/AKT pathway, with no significant effects on PI3Kα or AKT phosphorylation in normal human hepatocytes (L02 cells). This specificity, coupled with GT’s structural features (eg, sulfonyl, phenolic hydroxyl groups), suggests it may interact with PI3Kα via a binding mode analogous to its interaction with FASN (Figure 9), potentially involving hydrogen bonding or hydrophobic interactions with key residues in the enzyme’s active pocket. Furthermore, GT exerts potent growth-inhibitory effects in patient-derived HCC organoids—an in vitro model that retains the tissue architecture and molecular heterogeneity of primary tumors, conferring high pathophysiological relevance23—which provides a solid preclinical foundation for subsequent clinical translation. Collectively, these findings clarify how GT specifically modulates the PI3K/AKT pathway’s upstream and downstream molecules to regulate HCC cell proliferation, apoptosis, and cell cycle progression, strongly supporting GT’s potential as a multitargeted therapeutic agent for HCC.
The core finding of this study lies in revealing the specific regulatory effect of GT on the HCC immune microenvironment. scRNA-seq analysis clearly showed that the proportion of Tregs in the TIME significantly decreased after GT treatment, while other immune cell subsets showed no obvious changes, indicating that Tregs are the specific immune target of GT. Tregs construct an immunosuppressive microenvironment by secreting cytokines such as IL-10 and TGF-β, and abnormalities in their quantity and function are closely related to HCC progression and treatment resistance.24 We further confirmed through in vitro co-culture experiments that GT can effectively inhibit Tregs differentiation, which is significantly different from the mechanism of action of previously reported immunomodulators: traditional drugs such as PD-1 inhibitors function by reversing T cell exhaustion, while GT directly targets the differentiation process of Tregs, providing a novel strategy for reversing immunosuppression.
Mechanistically, GT suppressed the immunosuppressive function of Tregs by inhibiting FASN, a key enzyme governing de novo fatty acid synthesis. Pseudotime trajectory analysis of scRNA-seq data showed that GT reshapes the differentiation state of Tregs, shifting the dominant population from a pro-suppressive state (state 1) to a quiescent state (state 5), and this process is accompanied by a significant downregulation of FASN. Western blot experiments further confirmed that GT can reverse the upregulation of FASN and other lipid metabolic enzymes (ACSL3, ACSL4) in Tregs induced by IL-2. These findings are consistent with recent studies showing that FASN inhibition depletes lipid reserves in Tregs and impairs their ability to suppress anti-tumor immunity.8 Lipid metabolomics data further strengthen this association, showing that GT disrupts multiple lipid metabolic pathways (such as steroid synthesis and fatty acid degradation) in Tregs, confirming that FASN-mediated metabolic reprogramming is the core mechanism of GT action.
Molecular docking simulations provide structural biological evidence for the direct interaction between GT and FASN. GT stably binds to the binding pocket of chain A of FASN through hydrogen bonds formed by its sulfonyl group, phenolic hydroxyl group, and amino group, as well as hydrophobic interactions with Pro1264. The favorable binding energy suggests that the two may form a functional complex. Residues at the active site of FASN (such as Trp2060, Ser2021) are crucial for catalyzing fatty acid synthesis,25 and the interaction between GT and these residues may directly inhibit its enzymatic activity, a mechanism that requires further verification by subsequent enzyme kinetic experiments. In addition, TCGA data analysis showed that FASN expression is significantly increased in HCC tissues, and patients with high expression have poor prognosis, confirming that FASN is a key promoter of HCC progression and also providing a potential biomarker for screening the clinical application of GT—GT may exhibit better therapeutic effects in HCC patients with high FASN expression.
This study also highlights the value of natural product modification in drug development. Taurine (derived from Calculus Bovis) and gossypol (extracted from Malvaceae plants) both have certain anti-HCC activity, but after forming the GT molecule through covalent coupling, they exhibit significant synergistic anti-tumor effects. This finding underscores GT’s dual mechanism of action, which aligns with the value of natural products in anticancer drug discovery: it directly suppresses the PI3K/AKT pathway in tumor cells while concurrently disrupting the immune microenvironment via FASN-mediated lipid metabolism. This coordinated targeting of both tumor proliferation and immune escape effectively surpasses the limitations of single-target therapies.
Of course, this study still has certain limitations. First, although the organoid model has high physiological relevance, it lacks in vivo animal model verification of the anti-tumor and immunomodulatory effects of GT. To address this gap, future studies will construct HCC xenograft models (eg, patient-derived xenografts) or genetically engineered mouse models; these in vivo systems will not only evaluate GT’s ability to inhibit tumor growth but also characterize its impact on the TIME via techniques like flow cytometry and immunofluorescence, verifying the conservation of GT’s mechanisms in living organisms. Second, it remains unclear whether FASN is the only target of GT in regulating Tregs. Future research will use proteomics or phosphoproteomics to systematically screen GT’s downstream interacting molecules in Tregs, and complement with experiments such as CRISPR-Cas9-mediated FASN knockout—if GT’s regulatory effects on Tregs are abolished in FASN-deficient cells, it will confirm FASN as an essential target, clarifying GT’s target specificity. Finally, the pharmacokinetic and toxicological characteristics of GT have not been clarified, and its distribution in normal tissues and impact on normal immune cells need further research to provide a basis for the design of clinical administration regimens.
Conclusion
In summary, this study demonstrates that GT exerts anti-HCC effects through a dual mechanism: on one hand, it directly inhibits tumor cell proliferation by inactivating the PI3K/AKT pathway; on the other hand, it targets FASN in Tregs, disrupts fatty acid metabolism to reprogram Treg differentiation, and thereby reverses immunosuppressive TIME. This finding not only reveals the potential of GT as a novel therapeutic molecule for HCC but also provides a new theoretical basis for anti-tumor strategies targeting immune cell metabolism. In the future, by optimizing the chemical structure of GT and clarifying the clinically applicable population, it is expected to develop a novel HCC therapeutic drug with both efficacy and safety.
Generative AI Statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article.
Ethics Statement
Studies involving human participants were approved by the Ethics Committee of Ruikang Hospital Affiliated to Guangxi University of Chinese Medicine (Ethics Approval No.: KY2024-210). All these studies were conducted in compliance with local legislative requirements and institutional regulations. Written informed consent was obtained from all participants prior to their enrollment in this study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (Nos82460933) and Guangxi Youth Qihuang Scholars Training Program (GXQH202422).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022; GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229–263. doi:10.3322/caac.21834
2. Vogel A, Meyer T, Sapisochin G, et al. Hepatocellular carcinoma. Lancet. 2022;400(10360):1345–1362. doi:10.1016/S0140-6736(22)01200-4
3. Brown ZJ, Tsilimigras DI, Ruff SM, et al. Management of hepatocellular carcinoma: a review. JAMA Surg. 2023;158(4):410–420.
4. Ruf B, Heinrich B, Greten TF. Immunobiology and immunotherapy of HCC: spotlight on innate and innate-like immune cells. Cell Mol Immunol. 2021;18(1):112–127.
5. Prawira A, Xu H, Mei Y, et al. Targeting Treg-fibroblast interaction to enhance immunotherapy in steatotic liver disease-related hepatocellular carcinoma. Gut. 2025;75(1):105–118. doi:10.1136/gutjnl-2025-335084
6. Zhang Y, Lazarus J, Steele NG. Regulatory T-cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discov. 2020;10(3):422–439. doi:10.1158/2159-8290.CD-19-0958
7. Takamoto T, Mihara Y, Nishioka Y, et al. Surgical treatment for hepatocellular carcinoma in era of multidisciplinary strategies. Int J Clin Oncol. 2025;30(3):417–426. doi:10.1007/s10147-025-02703-7
8. Lim SA, Wei J, Nguyen TM, et al. Lipid signalling enforces functional specialization of T(reg) cells in tumours. Nature. 2021;591(7849):306–311.
9. Liu C, Chikina M, Deshpande R, et al. Treg cells promote the SREBP1-dependent metabolic fitness of tumor-promoting macrophages via repression of CD8(+) T cell-derived Interferon-gamma. Immunity. 2019;51(2):381–397e386. doi:10.1016/j.immuni.2019.06.017
10. Field CS, Baixauli F, Kyle RL, et al. Mitochondrial integrity regulated by lipid metabolism is a cell-intrinsic checkpoint for Treg suppressive function. Cell Metab. 2020;31(2):422–437e425. doi:10.1016/j.cmet.2019.11.021
11. Motika SE, Hergenrother PJ. Re-engineering natural products to engage new biological targets. Nat Prod Rep. 2020;37(11):1395–1403. doi:10.1039/D0NP00059K
12. T YY, Zeng J, Chen P, et al. Research progress on clinical application and pharmacological mechanism of Xihuang pills in antitumor therapy. Chin J Exp Tradit Med Formulae. 2022;28(03):250–258.
13. Q LZ, Y CX, C HE, et al. Research progress on the mechanism of taurine in intervening hepatocellular carcinoma and the intervention effect of taurine combined with chemotherapeutic drugs. Guangxi Med J. 2021;43(22):2738–2740,2754.
14. Ping Y, Shan J, Liu Y, et al. Taurine enhances the antitumor efficacy of PD-1 antibody by boosting CD8(+) T cell function. Cancer Immunol Immunother. 2023;72(4):1015–1027. doi:10.1007/s00262-022-03308-z
15. Flemming A. Tumour cell consumption of taurine exhausts CD8(+) T cells. Nat Rev Immunol. 2024;24(5):306.
16. Xu WB, Xu LH, Lu HS, et al. The immunosuppressive effect of gossypol in mice is mediated by inhibition of lymphocyte proliferation and by induction of cell apoptosis. Acta Pharmacol Sin. 2009;30(5):597–604. doi:10.1038/aps.2009.35
17. Paunovic D, Rajkovic J, Novakovic R, et al. The potential roles of gossypol as anticancer agent: advances and future directions. Chin Med. 2023;18(1):163. doi:10.1186/s13020-023-00869-8
18. Atwa SM, Odenthal M, El Tayebi HM. Genetic heterogeneity, therapeutic hurdle confronting sorafenib and immune checkpoint inhibitors in hepatocellular carcinoma. Cancers. 2021;13(17):4343. doi:10.3390/cancers13174343
19. Zhou J, Shao Q, Lu Y, et al. Monocarboxylate transporter upregulation in induced regulatory T cells promotes resistance to anti-PD-1 therapy in hepatocellular carcinoma patients. Front Oncol. 2022;12:960066. doi:10.3389/fonc.2022.960066
20. Du G, Dou C, Sun P, et al. Regulatory T cells and immune escape in HCC: understanding the tumor microenvironment and advancing CAR-T cell therapy. Front Immunol. 2024;15:1431211.
21. Wu Y, Zhang Y, Qin X, et al. PI3K/AKT/mTOR pathway-related long non-coding RNAs: roles and mechanisms in hepatocellular carcinoma. Pharmacol Res. 2020;160:105195.
22. Liu Y, Ma Y, Li Z, et al. Investigation of inhibition effect of gossypol-acetic acid on gastric cancer cells based on a network pharmacology approach and experimental validation. Drug Des Devel Ther. 2020;14:3615–3623.
23. Peng T, Ma X, Hua W, et al. Individualized patient tumor organoids faithfully preserve human brain tumor ecosystems and predict patient response to therapy. Cell Stem Cell. 2025;32(4):652–669.
24. Sakaguchi S, Miyara M, Costantino CM, et al. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10(7):490–500.
25. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7(10):763–777.
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.