")
Back to Journals » International Journal of Nanomedicine » Volume 15
Targeting Tumorigenicity of Breast Cancer Stem Cells Using SAHA/Wnt-b Catenin Antagonist Loaded Onto Protein Corona of Gold Nanoparticles
Authors Shamsian A, Sepand MR , Javaheri Kachousangi M, Dara T, Ostad SN , Atyabi F, Ghahremani MH
Received 14 October 2019
Accepted for publication 11 May 2020
Published 11 June 2020 Volume 2020:15 Pages 4063—4078
DOI https://doi.org/10.2147/IJN.S234636
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Azam Shamsian,1,2 Mohammad Reza Sepand,3 Marziye Javaheri Kachousangi,1,2 Tahereh Dara,4 Seyed Nasser Ostad,3 Fatemeh Atyabi,1,2 Mohammad Hossein Ghahremani1,3
1Nanotechnology Research Center, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran; 2Department of Pharmaceutical Nanotechnology, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran; 3Department of Toxicology and Pharmacology, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran; 4Department of Pharmaceutics, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran
Correspondence: Mohammad Hossein Ghahremani
Department of Pharmacology and Toxicology, Faculty of Pharmacy, Tehran University of Medical Sciences, New Building, Room 2-318, Tehran 1417614411, Iran
Tel/Fax +982166959102
Email [email protected]
Background: Among various theories for the origin of cancer, the “stemness phenotype model” suggests a dynamic feature for tumor cells in which non-cancer stem cells (non-CSCs) can inter-convert to CSCs. Differentiation with histone-deacetylase inhibitor, vorinostat (SAHA), can induce stem cells to differentiate as well as enforces non-CSCs to reprogram to CSCs. To avoid this undesirable effect, one can block the Wnt-βcatenin pathway. Thus, a dual delivery system of SAHA and a Wnt-βcatenin blocker will be beneficial in the induction of differentiation of CSCs. Protein corona (PC) formation in nanoparticle has a biologic milieu, and despite all problematic properties, it can be employed as a medium for dual loading of the drugs.
Materials and Methods: We prepared sphere gold nanoparticles (GNPs) with human plasma protein corona loaded with SAHA as differentiating agent and PKF118-310 (PKF) as a Wnt-βcatenin antagonist. The MCF7 breast cancer stem cells were treated with NPs and the viability and differentiation were evaluated by Western blotting and sphere formation assay.
Results: We found that both drugs loaded onto corona-capped GNPs had significant cytotoxicity in comparison to bare GNP-corona. Data demonstrated an increase in stem cell population and upregulation of mesenchymal marker, Snail by SAHA-loaded GNPs treatment; however, the combination of PKF loaded GNPs along with SAHA-loaded GNPs resulted in a reduction of stem cell populations and Snail marker. We have shown that in MCF7 and its CSCs simultaneous treatment with SAHA and PKF118-310 induced differentiation and inhibition of Snail induction.
Conclusion: Our study reveals the PC-coated GNPs as a biocompatible career for both hydrophilic (PKF) and hydrophobic (SAHA) agents which can decrease breast cancer stem cell populations along with reduced stemness state regression.
Keywords: co-delivery, HDAC inhibitor, Wnt, cancer stem cell, protein corona
Introduction
Breast cancer accounts for about a quarter of the newly diagnosed cancers and was the leading cause of cancer-related mortality among women in the world in 2018.1 In spite of the advances in the treatment of breast cancer in recent years, disease recurrence over time is the most important barrier to a perfect treatment.1 Over recent years, several hypotheses have been proposed about the formation, proliferation, and development of tumors and the stem cell hypothesis has challenged others like the clonal or stochastic model.2 The stochastic model suggests that cell populations with various phenotypes in the normal tissue undergo genetic or epigenetic alterations and can develop a tumor with different heterogeneous cell populations in which all the tumor cells have the same proliferative ability.3 Otherwise, the acquisition of aggressive and metastatic phenotypes and extraordinary proliferation and placement in this model are not justified. In the classic cancer stem cell model, a minority of cells in the tumor called cancer stem cells promote a tumor with the hierarchy of cells that transforms into differentiated cells. These cells provide heterogeneity in the tumor and each hierarchy of the cells has a degree of plasticity.3 The term “stemness” defines the long-term self-renewal and differentiation capability into a variety of cell lines. These characteristics are considered as dynamic properties in the “stemness phenotype model”, in which, unlike the CSC theory, all of the tumor cells, including the CSCs and other differentiated cancer cells, are in a dynamic balance. Therefore, most of the cells in a tumor have the potential to reinitiate tumorigenesis, since conversion between different phenotypes is possible depending on the tumor-mediating microenvironment, because following the destruction of the CSCs by chemotherapy, other more differentiated tumor cells can undergo reprogramming, mainly by epigenetic changes, and reproduce a constant population of CSCs in order to survive the tumor against chemotherapy.4 Thus, to avoid re-initiation, it is logical to treat all the cells in the tumor simultaneously, whether stem cells or non-stem cell, contrary to the classic theory of CSC that recommends targeting only CSCs for tumor eradication.
Investigations have proposed that histone deacetylase (HDAC) 1, 2, 3 and 6, as epigenetic eraser enzymes, are upregulated in more progressive and hormone receptor-negative breast cancers.5–7 In addition, HDAC 1 and 7 are strongly implicated in maintaining pluripotency in breast cancer stem-like cells in comparison to non-CSCs.8
In this regard, HDAC inhibitors (HDACI) have been implicated in reversing the epithelial-to-mesenchymal transition (EMT) reprogramming, reducing the CSC population, downregulating miRNAs involved in the CSC phenotype, CSC proliferation and survival in breast cancer.9 Vorinostat or suberoylanilide hydroxamic acid (SAHA), a pan-histone deacetylase inhibitor (HDACI) approved by the FDA for cutaneous T cell lymphoma in 2006, induces G1 and G2-M cycle arrest along with morphological differentiation followed by apoptosis in breast cancer cell lines.10 On the other hand, Debeb et al found that the epithelial-mesenchymal equilibrium induced by HDACIs in CSCs and non-CSCs did not necessarily shift to a differentiation status. In other words, while HDACIs differentiate CSCs, they can induce reprogramming of residual non-CSCs through activating alternative pathways like the WNT/β-Catenin signaling pathway.11 A Wnt/β-catenin blocker can be used to avoid this effect. Among the blockers of the Wnt/β-catenin signaling pathway, PKF118-310 (PKF) inhibits the binding of β-catenin to TCF/LEF transcription factors complex at the terminal phase of the signaling pathway.12 Previous research into the role of TCF/LEF antagonists in mice breast cancer cells showed that PKF is able to specifically hamper the ability of secondary tumor formation in breast cancer stem-like cells rather than normal mammary progenitor cells.13 Therefore, to induce differentiation and avoid reprogramming of non-stem cells, an HDACI, Vorinostat, and a Wnt inhibitor, PKF, were used in our study.
Protein corona (PC) is a protein enclosure that forms around nanoparticles following exposure of NP to the biological fluids. PC is constantly exchanged with surrounding free proteins due to its continuous dynamism. A soft corona is formed within a few seconds to a few minutes with looser bindings, while proteins with tighter interactions form hard corona over a longer time. The shielding of the NP surface with PC totally changes the behavior and fate of NPs.14,15 PC can be used as a suitable medium for loading drugs with hydrophilic or hydrophobic moiety.16 Gold nanoparticle (GNP) as a conventional nanocarrier has some unique features such as biocompatibility, relative stability, optical properties and application in photothermal therapy17 and has been used as a scaffold to form PC for drug delivery. The objective of the present study was to examine the effect of a co-delivery system composed of an HDACI (SAHA) and Wnt/β-catenin antagonist (PKF) on the PC of GNPs in the MCF7 breast cancer stem cells.
Materials and Methods
Materials
Suberoylanilide hydroxamic acid (SAHA) or Vorinostat (Cayman, Germany), Lysotracker Red DND-99 (Invitrogen, Germany), B27 supplement 1:50 (Invitrogen, UK), Anti-E-cadherin (Cell signaling, Danvers, MA, USA), Anti-Snail (Cell signaling, Danvers, MA, USA), Anti-β-actin (Santa Cruz Biotech, CA, USA) primary antibodies and Tetrachloroauric acid (HAuCl4‚3H2O)(Acros organics, USA) were purchased. Sodium borohydride (NaBH4), L-ascorbic acid, Cetyltrimethylammonium bromide (CTAB), Sodium thiosulfate, Silver nitrate (AgNO3), RPMI-Medium (Biosera, France), Fetal bovine serum (FBS)(Gibco, USA), Penicillin/streptomycin (Biowest, France). TWEEN20, Dimethyl sulfoxide (DMSO), and formaldehyde were all purchased from Merck (Merck, Germany). PKF118-310, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), 4′,6-diamidino-2-phenylindole (DAPI), Fluorescein isothiocyanate (FITC), and Human Epidermal Growth Factor were purchased from Sigma Aldrich. Human plasma was obtained from the Blood Transfusion Organization (IRAN) and then aliquoted and stored at −80°C. SAHA was dissolved in deionized water (pH 12, 5 mg/mL) and PKF was dissolved in deionized water (5mg/mL) as stock solutions and were used for further experiments.
Preparation of Gold Nanoparticle
The seed-mediated growth method with some modifications was used for the synthesis of gold nanoparticles (GNPs).18 Au seeds were prepared by pouring an aqueous solution of HAuCl4·3H2O (250 µL, 0.01 M) into CTAB solution (10 ccs, 0.1 M) in a glass vial. While the mixture was stirred vigorously, an aqueous solution of NaBH4 (sodium borohydride) (600 µL, 0.01 M in ice) was added to the above solution. The color of the solution changed to a brown-yellow color due to the conversion of Au3+ to Au0 within about 10 mins. The solution could be used for the next step of the synthesis within about 5 hrs. For gold nanosphere synthesis, CTAB solution (40 mL, 0.1 M) was mixed with HAuCl4·3H2O solution (800 µL, 0.01 M) to form a growth solution under vigorous stirring. Then, ascorbic acid solution (250 µL, 0.1 M) was added until the solution was colorless. After about 2 mins, seed solution (2 mL) was added and the color of the mixture changed to dark purple within 5 mins. The UV/visible spectra of synthesized gold nanosphere solution were recorded from 200 to 800 nm by CE7500 (CECIL, UK).
Preparation of Corona-Coated Gold Nanoparticle and Loading Drugs
To coat GNPs with protein corona, 10% human plasma solution in PBS was prepared and equivalent volumes of 40 μg GNPs from synthesized solution were aliquoted in some vials. The temperature of all solutions was adjusted to 37°C in an incubator prior to mixing. Then, 400 μg PKF and SAHA from their stock solutions were separately added to 900 μL of 10% plasma solution and placed in an incubator (New Brunswick Scientific, NJ) at 37°C for 30 mins. Afterward, GNPs were added to plasma-drug solutions and stirred gently at 37°C for 3 hrs. Finally, the mixtures were centrifuged twice at 13,500 RPM for 30 mins. Bare corona-coated GNPs (Bare GNP-corona) were prepared as control as stated above, except for adding the drugs; instead, the same volume of vehicles was added. All supernatants were kept for further measurements. A brief schematic image of GNP-corona-drug preparation is illustrated in Figure 1C.
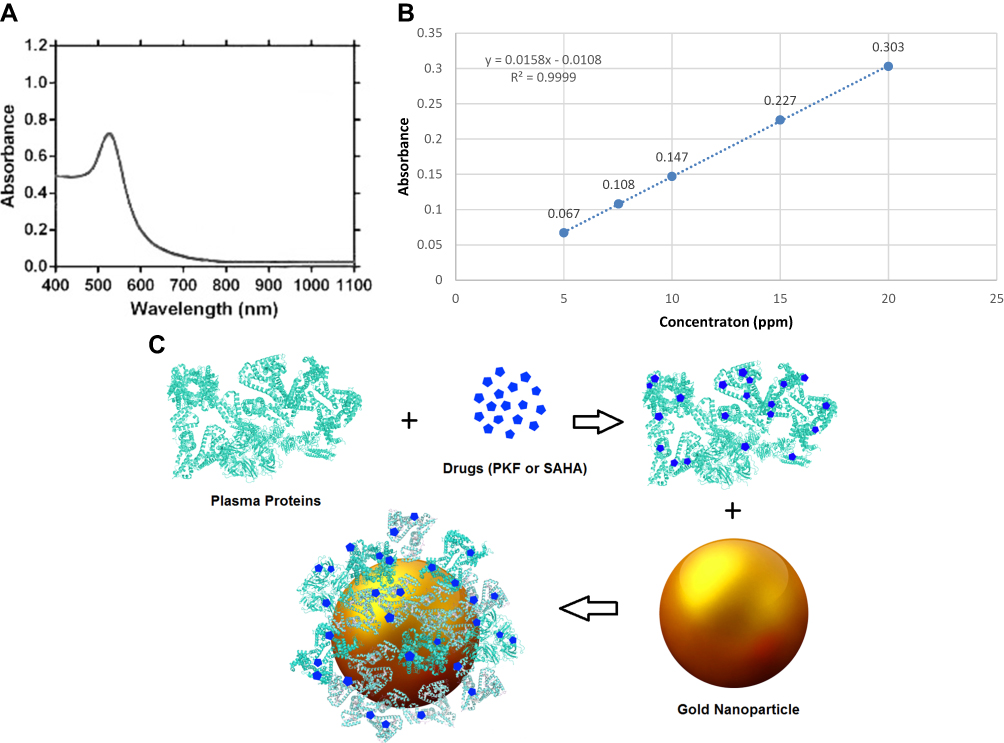 |
Figure 1 (A) UV-visible absorbance of gold nanospheres; (B) Calibration curve of Au element concentration (ppm) measured by atomic absorption spectroscopy. The calculated equation of linear regression is above graph; (C) Schematic illustration of step by step physically binding of small molecules (PKF118-310 or SAHA) onto plasma proteins and formation of protein corona-drugs on the surface of gold nanospheres. Abbreviation: SAHA, suberoylanilide hydroxamic acid. |
Characterization of GNPs, Bare GNP-Corona, and GNP-Corona-Drug
Concentration of Synthesized GNP Solution
The weight content of GNPs in the unit volume of synthesized solution was determined by using atomic absorption spectrometer (model 180–80, Hitachi). In order to prepare a sample for measurement, GNP colloidal solution was dissolved in a 1:1 (v/v) ratio of nitric acid: hydrochloric acid. A calibration curve with known variable concentrations of HAuCl4 as a source of gold atoms was plotted. Finally, the concentration of the synthesized GNP solution was obtained by placing the sample absorbance in the calibration curve equation.
Size and Zeta Potential
The colloidal dispersions of GNPs, GNP-corona, and GNP-corona-drugs were sufficiently diluted with deionized water prior to measurement. The mean particle diameter, polydispersity index (PDI), and ζ-potential were analyzed by the dynamic light scattering (DLS) technique using the Zetasizer (Malvern Instruments, Malvern, Germany) at a fixed scattering angle of 90º and a temperature of 25ºC. Data were provided by the Nano DTS software.
Drug Loading Efficiency and Entrapment Efficiency
All supernatants obtained from two centrifugation rounds of drug-loaded corona-coated GNPs and bare corona-coated GNPs were kept. The control supernatants were used as blank groups. UV/vis absorbance was determined in clear supernatants by UV–vis spectrophotometer at 239 nm for SAHA and 395 nm for PKF. The absorbance of the unloaded drugs in the supernatant was calculated by subtracting the UV absorbance (at λmax of drugs) of the supernatant of bare corona-coated GNPs as the blank group from the absorbance of the supernatant of drug-loaded GNPs. The unloaded drug content was calculated by replacing the unloaded drug absorbance in the equation of calibration curves for each drug. Loaded drug content was quantified by subtracting the amount of corona-unbound drug in the supernatant from the total amount of drug used for loading. The drug entrapment efficiency was calculated as the mount of loaded drug divided by total drug*100. The experiments were repeated 5 times and the mean ± SD was recorded.
Protein Corona Pattern Characterization
Prepared bare GNP-corona and drug-loaded GNP-corona (GNP-corona-drug) were boiled for 15 mins and an identical mount of samples was loaded into wells of sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) 10% to separate the protein content of the corona of GNPs. The separated protein bands on the gel were detected by silver nitrate staining. Briefly, the gel was incubated and shaken in the fixation solution (25 mL methanol, 6 mL acetic acid, 25 µL formaldehyde 37%, 19 mL deionized water) for 2 hrs at room temperature. After washing with ethanol 96% (18 mL) and deionized (DI) water (32 mL) for 20 mins, the gel was incubated in sodium thiosulfate 0.03% for 2 mins. The gel was washed in DI water three times and stained by silver nitrate 0.2% containing 38 µL formaldehyde 37% for 20 mins. The gel was then shaken in 50 mL detection solution (12% v/w sodium carbonate, sodium thiosulfate 0.03%, formaldehyde 37%, 50 µL) until the bands became visible. The detection reaction was stopped using the same fixation solution without formaldehyde and photos were taken from detected protein bands.
In vitro Release Pattern
The in vitro release pattern of PKF and SAHA from gold nanoparticles was evaluated by resuspending the pellets of freshly prepared drug-loaded nanoparticles and bare corona-coated GNPs as the blank sample in 1 mL phosphate-buffered saline (pH 7.4). Bare GNP-corona as the blank group and GNP-corona drug dispersions were kept in a shaker incubator at 37°C and 80 RPM. The samples were centrifuged at 13,500 RPM for 10 mins at predetermined time intervals (6, 24, 48, and 72 hr) and were resuspended in 1 mL PBS. The supernatants were removed and maintained for UV/Visible absorption spectral measurements of drugs using a CE7500 (CECIL, UK) according to the method described above. The same procedure was performed by a medium of 1% TWEEN20 in PBS. The percentage of the cumulative release of drugs was plotted versus time.
Cell Culture
The human breast cancer stem cells isolated from MCF-7 cell lines (CSCs) were gratefully supplied by Dr. S. N. Ostad19 and cultured in RPMI-Medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37°C in a humidified 5% CO2 atmosphere. When needed, half of the culture medium was removed and replaced with fresh medium.
Cellular Uptake Studies
Initially, FITC fluorescent dye was loaded on CTAB-coated GNPs by mixing GNPs with 100μL of 1 mg/mL FITC solution in PBS and stirring overnight at 4°C in the dark. Then, the mixture was centrifuged at 13,500 RPM for 15 mins and the unloaded FITC was removed. FITC-labeled GNPs were exposed to 10% plasma solution to form a corona.
The cellular uptake of FITC-labeled GNP-corona was assessed by confocal laser scanning microscopy. The cells were cultured at a density of 2×105 on glass slides in a 6-well culture plate and treated with FITC-loaded GNP-corona (2μg/mL) for 5 hrs in the dark. For tracking nanoparticles in lysosomes, the cells were washed with PBS and incubated with 50 nM LysoTracker Red® (Thermofisher, USA) for 10 mins. After washing with PBS, the cells were fixed with 4% paraformaldehyde for 4 mins. The cell nuclei were also stained with DAPI for 5 mins. Glass slides were then removed and observed under a confocal microscope (CLSM; Nikon Eclipse Ti, Japan) using the sequential scanning mode.
The uptake was also tested by flow cytometry. For flow cytometric study, the seeded cells into 6-well plates were trypsinized, centrifuged and resuspended in PBS. In each treatment, 10,000 cells were evaluated by BD FACSCalibur (BD Biosciences, San Jose, CA, USA) and the data were analyzed using the Flowjo software.
Cell Viability Measurement
MCF-7 cells and CSCs were seeded in 96-well tissue culture plates at a density of 4000 cells/well. The cell lines were routinely cultured in RPMI-1640 supplemented with 10% FBS and 1% penicillin/streptomycin and incubated at 37°C in a 5% CO2 humidified incubator. After 24 hrs, the medium of each well was removed and replaced by various concentrations of PKF, SAHA, GNPs, GNP-corona, GNP-corona-PKF, or GNP-corona-SAHA. In predetermined times (72, 96, and 120 hrs), MTT solution (20 µL, 5 mg/mL in PBS) was added after removing the medium and the cells were incubated at 37°C for 3 hrs. The purple formazan crystals were dissolved in dimethyl sulfoxide (DMSO, 100 µL) in each well and the optical density was measured at 540 and 690 nm using the UV/vis microplate spectrophotometer (Anthos 2020, UK). All experiments were performed in octuplets, and the cell viability was calculated as percent control.
Sphere-Forming Assay
A sphere-forming assay was performed according to a previously reported protocol with some modifications.20 Briefly, the CSCs (2×105 cells per well) were seeded in 10% FBS-supplemented Dulbecco’s modified Eagle’s medium (DMEM, Biowest, France) and incubated at 37°C in a humidified atmosphere containing 5% CO2 for 24 hrs. Cell treatment was carried out for a period of 6 days with an appropriate concentration of bare and drug-loaded nanoparticles. The cells were then collected and using a needle-syringe, a single cell suspension in cold PBS was prepared. Then, 1×104 cells/well were plated in non-adherent 1% agarose-coated 6-well culture plates and incubated in serum-free DMEM-F12, supplemented with B27 (1:50, Invitrogen), 20 ng/mL hEGF (Sigma, USA) and 1% penicillin/streptomycin. The medium was changed every 48 hrs and the formation of 3D spheres was evaluated after 7 days. Spheres larger than 50 μm were counted manually and sphere-forming efficiency was calculated as percent control.
Western Blotting Analysis of Protein Expression
CSCs were seeded and treated based on the procedure employed for the sphere-forming assay. The cells were lysed by lysis buffer (Tris buffer 62.5 mM pH 6.8, sodium dodecyl sulfate 2%, glycerol 1%, dithiothreitol 50 mM and bromophenol blue) and whole-cell lysates were stored at −80°C. For Western blotting, cell lysates were resolved in 10% SDS-PAGE. The gel as then transferred onto polyvinylidene difluoride membrane (Roche, Mannheim, Germany) and blocked with 5% non‐fat dry skimmed milk in TBST for 2 hrs at room temperature. Then, the membranes were incubated with anti-E-cadherin (1:1000, Cell signaling, Danvers, MA, USA), anti Snail (1:500, Cell signaling, Danvers, MA, USA) or anti-β-actin (1:1000; Santa Cruz Biotech, CA, USA) primary antibodies at 4°C overnight. After primary antibody incubation, the membranes were washed with TBST and incubated with horseradish peroxidase‐conjugated secondary antibody (1:5000; BioRad, Hercules, CA, USA) at room temperature for 1 hr. The protein bands were detected using the BM chemiluminescence detection system (Roche) and the images were captured using the Fusion FX gel-doc system (VilberLourmat, France). The protein bands were digitized by the Image J software and reported as a ratio to the corresponding β‐actin band intensity.
Statistical Analysis
Sigmaplot and GraphPad Prism software were used for processing experimental data and plotting graphs, respectively. The data were analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s comparison for the samples of more than two groups. P-value <0.05 were considered significant. All graphical error bars represent the standard deviation.
Results and Discussion
Characterization of GNPs, GNP-Corona, and GNP-Corona-Drugs
GNPs were prepared using the seed-mediated growth method and UV/Visible spectroscopy was done to prove the existence of gold spherical nanoparticles. The UV spectra of GNPs exhibited only one absorbance peak at 520–530 nm (Figure 1A). The absence of additional peaks between 520 and 700 nm, which could show the presence of triangular or cubic nanoparticles, indicated that only spherical nanoparticles were present in the prepared solution.
Atomic absorption spectroscopy was done to assess the concentration of the synthesized solution. A calibration curve was plotted for Au element at various concentrations (Figure 1B) (R2=0.999) and the concentration of the solution was measured at 76.5 μg/mL based on the synthesized GNP dispersion and the absorbance.
Table 1 presents the average particle size, polydispersity index (PDI) and Zeta potential of gold nanoparticles at various corona concentrations with and without drugs. The average size of gold nanoparticles was ~90 nm; however, the size of the nanoparticles increased at higher serum concentrations. Moreover, drug loading on corona nanoparticles increased the average particle size compared to the bare GNP-corona. The zeta potential for bare nanoparticles was +37 mV; however, the addition of corona reduced the zeta potential. The results were in agreement with previous studies.21
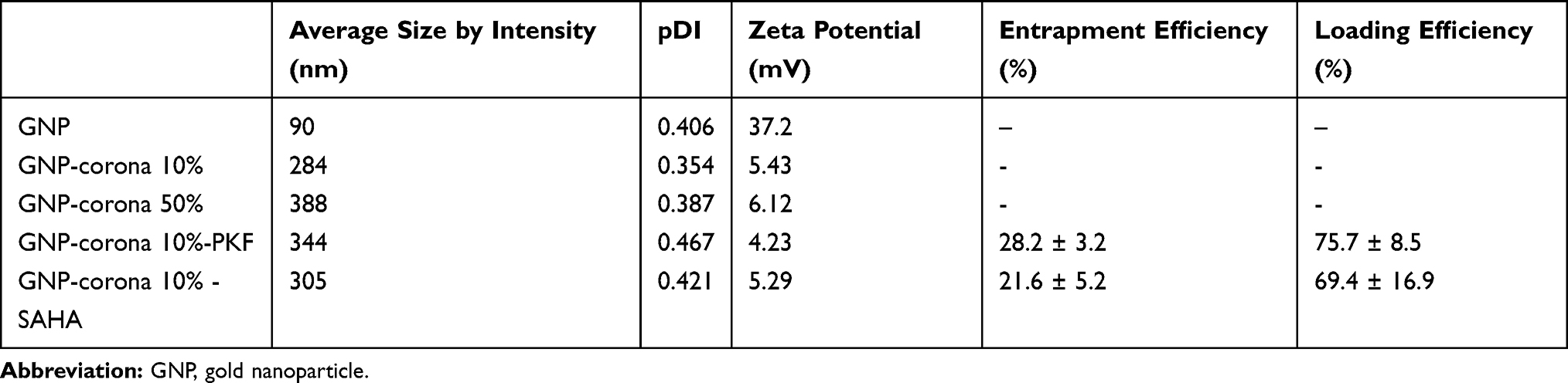 |
Table 1 Physicochemical Characterization of GNPs, Bare GNP-Corona and Drug-Loaded GNPs and Efficiency Percent of Drug Loadings Onto Corona-Coated GNPs |
The pattern of protein corona was analyzed by SDS-PAGE (Figure 2B). The protein pattern and contents in drug-loaded GNPs differed from bare GNPs. Given that the same serum concentration and content were introduced to the same gold nanoparticles, it could be suggested that the presence of any external substance in the serum contents particularly affects the protein-protein and protein-nano surface interaction. Thus, the nanoparticles behave differently toward the serum protein mixture with other agents related to pure serum proteins to adsorb stable proteins in order to form a hard corona.
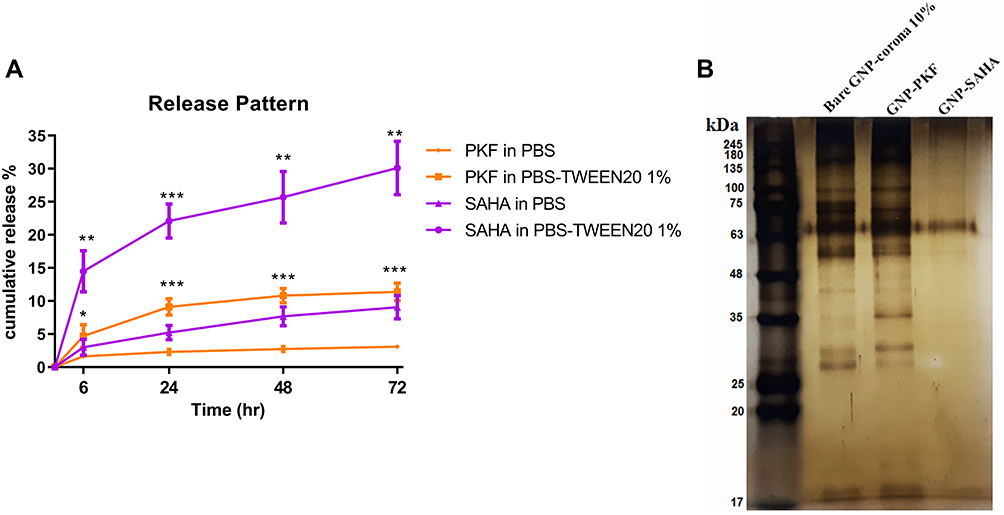 |
Figure 2 (A) Cumulative release pattern of PKF and SAHA from corona-capped GNPs versus time in PBS and PBS containing 1% Tween 20 (PBS-Tween20 1%) at 37°C. Error bars represent mean ± standard error among the three independent experiments. *P< 0.05, **P<0.01, ***P<0.001 significant differences between PKF/SAHA in PBS-TWEEN20 1% and corresponding PKF/SAHA in PBS; (B) Protein corona pattern of Bare GNP-corona 10%, GNP-corona 10%-PKF and GNP-corona 10%-SAHA using SDS-PAGE 10%. Protein size marker is indicated by molecular weights (kDa). Abbreviations: PBS, phosphate buffer Saline; GNP, gold nanoparticle; PKF, PKF118-310. |
Drug Loading on Corona-Coated GNPs
PKF and SAHA were successfully loaded on the corona of GNPs. The weight ratio of total drugs loaded on GNPs was about 10.5:1. The entrapment efficiency of SAHA and PKF was 21.6% and 28.2%, respectively (Table 1). Co-delivery of two or more drugs with various and even opposite chemical properties like hydrophilic and hydrophobic drugs has always been challenging. SAHA is a hydrophobic drug with log P=+1.5 and PKF is a hydrophilic drug (log P=−1.5). In other words, SAHA tends to enter the n-octanol 1000 times more than PKF, which represents the dramatic difference in their chemical traits.
Considering the surface chemistry and net charge of GNPs as the core of PC formation, the shell of GNPs has been chosen to be CTAB-capped since positively charged gold nanorods are more prone to be internalized into cells. Besides, the positively charged GNPs can adsorb more serum protein than negatively charged ones.22,23 Thus, we used the GNPs to deliver drugs by serum protein corona coating. Despite the reported CTAB-induced cell membrane and mitochondrial cytotoxicity,23 no significant toxicity was observed for corona-coated CTAB-capped GNPs used in this study (Figure 3).
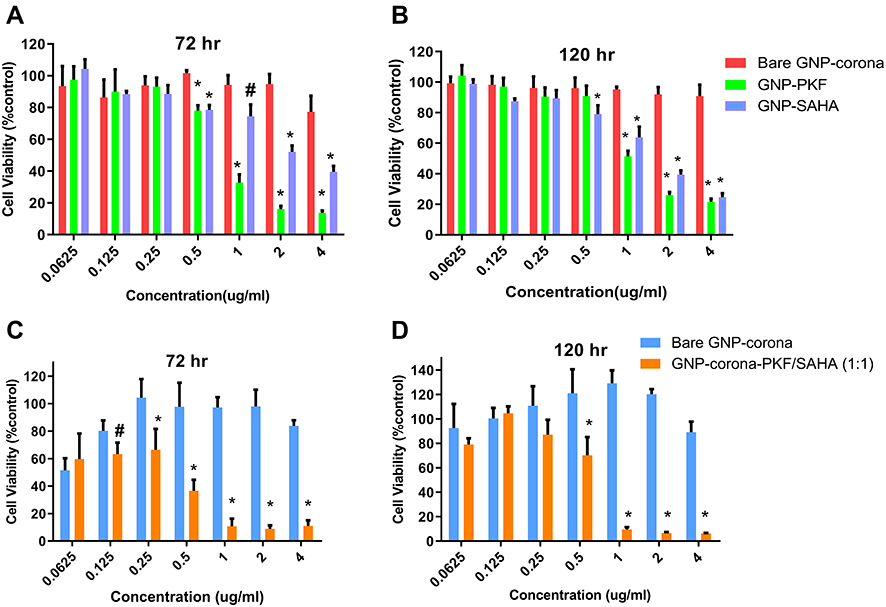 |
Figure 3 The Cell viability evaluation by MTT assay of (A) Bare-GNP-corona, GNP-corona-PKF and GNP-corona-SAHA against CSCs at 72 h and (B) at 120 h, (C) Bare GNP-corona and combination of GNP-corona-PKF and GNP-corona-SAHA (1:1 w/w) against CSCs at 72 h and (D) at 120 h. Data were presented as mean ± standard deviation of four separated experiments. *Shows P< 0.01 and #P< 0.05 significant differences between treatments and corresponding bare GNP-corona concentration. Abbreviations: MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide; CSCs, cancer stem cells; PKF, PKF118-310; SAHA, suberoylanilide hydroxamic acid; GNP, gold nanoparticle. |
The human serum contains different types of proteins with various functional groups on amino acid residues that can interact physically with any other small molecule through hydrogen bonds, van der Waals forces, hydrophobic forces, or electrostatic attraction. Most of the drugs, either hydrophilic or hydrophobic, entering into the bloodstream can bind to the serum proteins regardless of their chemical features. In this regard, SAHA is a high protein-bound drug (~71%)24 but PKF clinical protein binding has not been reported.
On the other hand, protein corona has an extremely unpredictable and complex function due to changes in the biocompatibility and cell uptake of NPs, nullifying specific targeting agents25 and causing uncertainty about the cellular fate.26 According to these problems, researchers sought to eliminate or minimize PC effects at nano-bio interfaces. However, considering the inevitability of the PC formation and its useful properties, the approach is to consider tailoring the desired profile of PC adopted in favor of specific therapeutic purposes.27,28 Some of the benefits that arise from PC formation include colloidal stability maintenance,28,29 biocompatibility enhancement and lack of recognition by the immune system,30 more control over the cellular uptake and the cellular fate of NPs. Therefore, PC could be exploited as a biocompatible drug depot beside the therapeutic applications of NP itself.31 In a preliminary study in this field in 2012, Kah et al29 confirmed the possibility of DNA oligonucleotides and Doxorubicin loading onto PC around gold nanorods. By the same rationale, Shahabi et al16 succeeded in an attempt to prepare a co-delivery system by loading hydrophilic doxorubicin and hydrophobic meloxicam onto fetal calf serum protein corona. Therefore, the findings and previous reports inspired us to exploit protein corona as a sponge-like drug depot for the drugs in this study. In this regard, the PC around gold nanorods was used for loading a hydrophobic photosensitizer chlorin e6 solely for photodynamic/thermal therapy31 and in combination with Doxorubicin for additional simultaneous chemotherapy.32
In the previous studies, the colloidal stability of the nanoparticles was in a good condition after coating by PC.28,31 One study showed that the aggregation index of CTAB-capped gold nanorods increased in ionic media of PBS and culture medium whereas the aggregation of corona-coated gold nanorods decreased in both media,29 although excess amounts of proteins in the solution may induce more aggregation of corona-coated nanoparticles.28
Once the nanoparticles (NPs) enter the body, PC around pristine NPs inevitably alters with time depending on multifactorial physiologic conditions. However, it seems that PC preserves the history of its former profiles.33 Therefore, it might be better to design a PC for nanoparticles before administration for various reasons. In this way, it is more likely to predict the behavior of the body with NPs and vice versa. For instance, dysopsonin-enriched PC could increase the circulation time of NPs in the bloodstream or functionalize NPs with natural non-immunogenic proteins in the plasma for targeted drug delivery.33 Moreover, pristine NPs can cause hemolysis, endothelial cell damage and thrombosis within the first minutes before coating by plasma proteins.33,34 NPs are allowed to pass through endothelial cells to reach the site of mature tumors by the EPR (enhanced permeation retention) effect. Inorganic NPs like GNP not only diffuse across endothelial cells via EPR passive targeting but also they are capable of being delivered to immature tumors by NanoEL (Nanomaterial-induced endothelial leakiness) active targeting. However, NanoEL for nanoparticle transition through blood vessels is associated with adverse effects such as metastasis and worsens pathologic conditions that depend on increased vascular permeability. It can be assumed that the surface optimization of NPs with tailored-PC could minimize these undesirable effects.34
In vitro Release Pattern
After loading the drugs onto PC, the rate and pattern of release for each drug were accumulatively plotted versus time in two media of PBS (pH 7.4) and 1% tween 20 in PBS at 37°C (Figure 2A). To determine the released drugs, UV/visible spectroscopy of the supernatant was performed for each drug against blank. The release percentage of PKF and SAHA from corona-coated GNPs in PBS was below 10%, approximately 3% for PKF and 9% for SAHA, after 72 hrs. This mild sustained mode of release is consistent with previous studies indicating that protein corona significantly attenuates the burst effect in the release profile of drugs loaded on PC,35 NP surface and protein conjugated nanocarriers.36 Adding 1% Tween 20 as a surfactant to PBS can simulate an in vitro culture medium because some components in the culture medium act like surfactants. According to the data, the release rate increased by 3.7 and 3.3 times for PKF and SAHA in PBS-Tween 1% after 72 hrs, respectively. These findings appear to be well supported by a study that found that Tween 20 enabled desorption of human plasma proteins, especially albumin, from polymeric surfaces.37 Human serum albumin is one of the most abundant proteins in all protein coronas of different samples, as what we observed in protein corona patterns (Figure 2B).
Cellular Uptake of Corona-Coated GNPs
The cellular uptake of GNPs in MCF-7 cells and CSCs was evaluated by FITC bound GNPs using confocal microscopy and flow cytometry after 6-hrs incubation at 37°C (Figure 4A and B). The cells were also treated with DAPI and Lysotracker Red® to stain the nucleus and lysosomes. The images highlighted that the FITC-labeled GNPs in green were internalized efficiently into cytoplasmic areas of the cells. Furthermore, the Lysotracker Red confirmed that the nanoparticles reached the lysosomes through the endocytosis mechanism (Figure 4A).
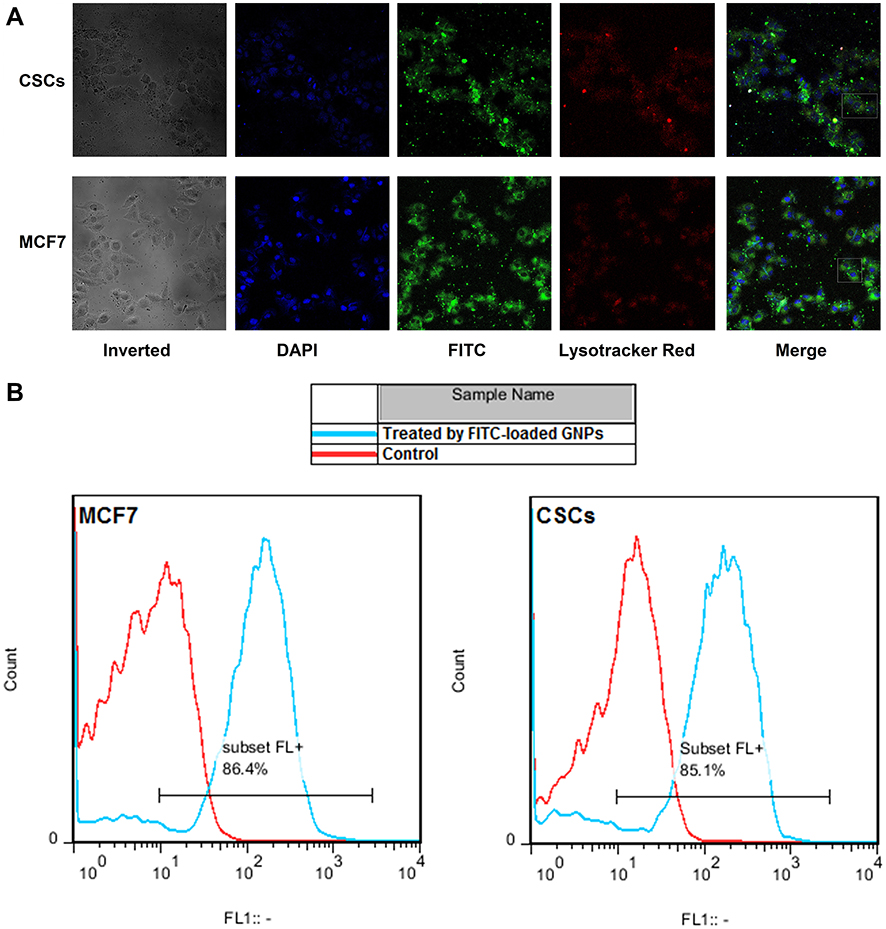 |
Figure 4 CSCs and MCF-7 cells were incubated for 6 hrs with FITC-loaded GNP-corona and following 8-mins incubation with LysoTracker Red; (A) Confocal microscopy images of CSCs and MCF-7 cells. From the left side, inverted indicates a bright field photo of cells, Nucleus stained by DAPI in blue, FITC-labeled GNPs in green, Lysosomes stained with Lysotracker Red® in red and Merge indicates overlayed three previous fluorescent images from left to right. Scale bars: 100μm; The CSCs and MCF7 cells in white rectangles are shown in larger size below the image; (B) Flow cytometry of treated CSCs and MCF-7 cells with FITC-loaded GNP-corona for 6 hrs versus untreated cells as control. Ten thousand cells were analyzed in each experiment. Abbreviations: CSCs, cancer stem cells; FITC, fluorescein isothiocyanat; DAPI, 4′,6-diamidino-2-phenylindole. |
It has been shown that the effect of protein corona on cell uptake of GNPs depends on the NP size, cell type38 and surface charge.22 Moreover, cellular uptake analysis by flow cytometry showed an uptake rate of 86.4% for MCF-7 cells and 85.1% for CSCs (Figure 4B) and confocal images emphasized a significant internalization of GNPs in the first 5 hrs. Although some studies found that protein corona commonly reduced cell uptake of GNPs due to the inhibition of corona from adhesion of GNPs to the cell membrane,22,38 significant uptake was found in MCF-7 and CSCs in the present study. It is worthwhile to point out that the protein corona inhibition is more severe for larger GNPs.38 Furthermore, positively charge GNPs have the greatest amount of cell uptake, even after reduction by corona coating.22 Thus, in the present study, the inhibitory effect of corona on cellular uptake of GNPs was not significant since the GNPs had a small size and positive charge.
The Cytotoxicity Effects of Drug-Loaded GNPs
The cytotoxic effects of drugs and NP loaded drugs were investigated in MCF-7 and CSCs at 24, 48, 72, 96 and 120 hrs after the treatments using the MTT assay. PKF induced cytotoxicity in MCF-7 cells following 72–120 hrs treatment has been shown (Figure 5A). In MCF7 cells, PKF showed dose-dependent cytotoxicity after 72 hrs, which increased in longer incubations. However, in CSCs, cytotoxicity started later after 96 hours of treatment (Figure 5B). Besides, unlike MCF-7 cells, the effect of PKF on CSCs did not increase in longer treatments (120 hrs) or at doses higher than 0.74 uM (Figure 5A and B). The results suggested resistance to the toxicity of PKF in CSCs compared to MCF7 cells. Similar to PFK, SAHA induced cytotoxicity in a dose-dependent manner in MCF7 cells and CSCs (Figure 5C, D). The effect of SAHA was stronger than PKF at 72 hrs and the CSCs were more resistant to the cytotoxicity of SAHA compared to MCF7 cells (Figure 5C, D). The calculated IC50s indicated resistance to SAHA cytotoxicity in CSCs compared to MCF7 cell (Table 2). The drugs were also tested for shorter times; however, there were no considerable toxic effects in 24 and 48 hrs except at high doses of 10 and 20 μM (Data not shown). When GNP loaded drugs were tested in CSCs, the cytotoxicity of PKF or SAHA-loaded GNPs increased at 72 hrs (Figure 3A). The IC50 of PKF and SAHA in CSCs at 72 hrs reduced to 0.81 and 2.46 uM, respectively (Table 3). Thus, GNP-PKF and GNP-SAHA could decrease the viability of CSCs in shorter incubation times, perhaps suggesting more efficient drug entry into CSCs. According to the IC50 data (Table 2), it could be concluded that the delayed cytotoxicity effect of SAHA in CSCs might be due to the main effect of SAHA on the induction of differentiation in tumor cells. Therefore, more time may be required for the expression of differentiation markers resulting in a differentiated phenotype and cell death. However, it is not clear whether the cytotoxic effects of SAHA were due to direct apoptotic effects or the induction of differentiation followed by cell cycle arrest and apoptosis, which requires further experiments.
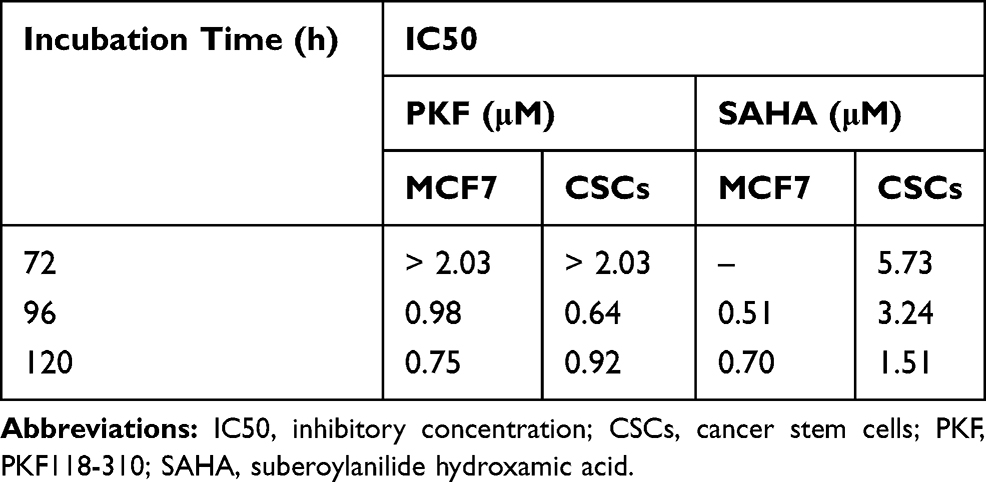 |
Table 2 IC50 (μM) Values of PKF and SAHA Against MCF-7 and CSC-Derived MCF-7 (CSCs) in Enriched CSCs at Various Incubation Times (IC50s Not Shown are Related to Experiments Have Not Been Performed) |
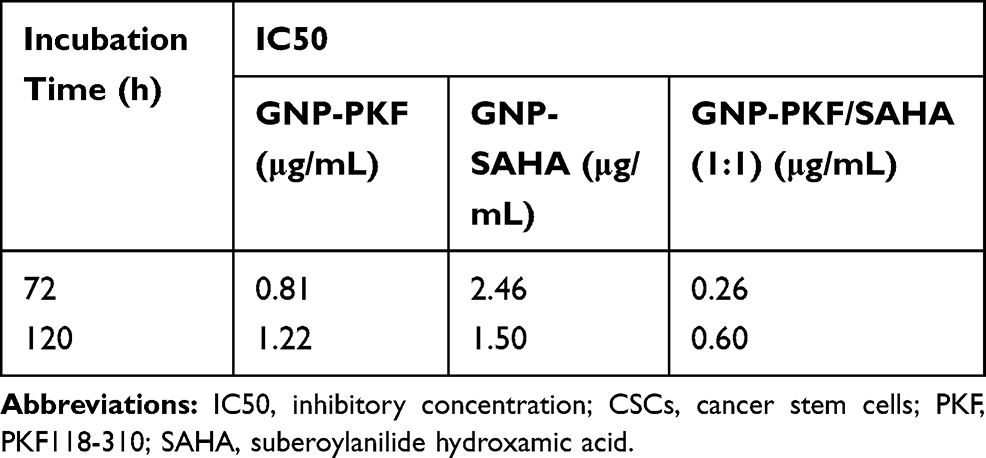 |
Table 3 IC50 (μg/Ml) Values of GNP-Corona-PKF, GNP-Corona-SAHA and Their Combination with the Ratio of 1:1 w/w in Enriched CSCs at 72 and 120 hrs |
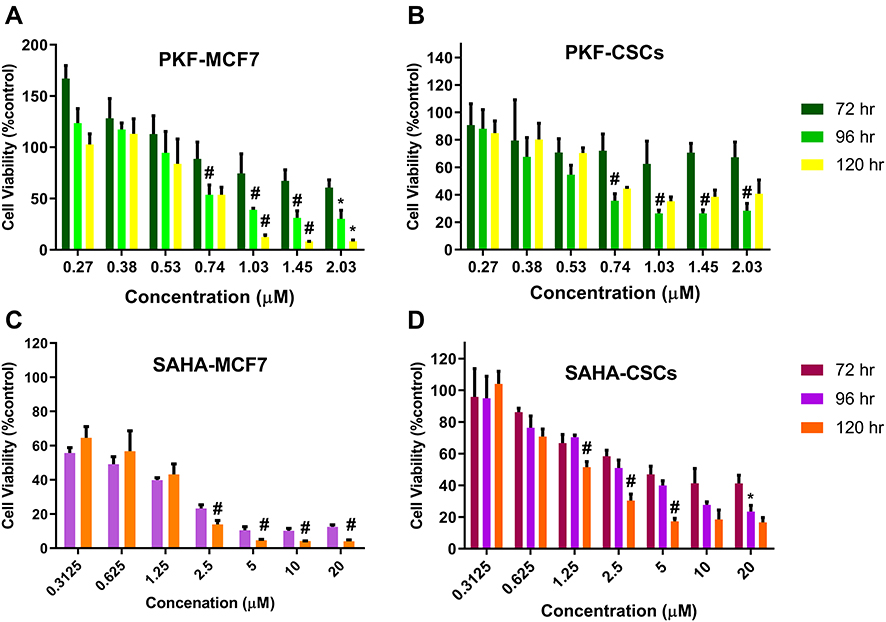 |
Figure 5 The Cell viability evaluation by MTT assay of (A) PKF in 72, 96, and 120 hrs against MCF-7 and (B) CSCs, (C) SAHA in 72, 96 and 120 hrs against MCF-7 and (D) against CSCs. Data were presented as mean ± standard deviation of four separated experiments. *Indicates P< 0.05 and #P< 0.01 significant differences between corresponding 96 vs 72 hrs (on 96 h bars) and 120 vs 96 hrs (on 120 hrs bars). Abbreviations: MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide; CSCs, cancer stem cells; PKF, PKF118-310; SAHA, suberoylanilide hydroxamic acid; GNP, gold nanoparticle. |
The IC50 values showed that CSCs were more resistant to both SAHA and PKF compared to MCF-7 cells (Table 2). As shown in Figure 3A and B, GNP-corona-PKF had statistically different cytotoxicity at concentrations of 0.5, 1, 2 and 4 μg/mL compared to bare GNP-corona at 72 hrs. Conversely, the delayed effect of GNP-corona-SAHA supports this idea that it requires more time for cytotoxicity. According to Figure 3B, the cytotoxicity of drug-loaded GNP-corona at 72 hrs was significantly different at 1ug/mL compared to bare GNP-corona. The cytotoxicity at 120 hrs indicated a significant increase at lower doses for drug-loaded nanoparticles (Figure 3B).
Meanwhile, treatment with a combination of SAHA and PKF loaded GNPs (w/w ratio 1:1) was associated with a stronger toxicity in CSCs in comparison to single treatment of either drug (Figure 3C and D). In this regard, at doses of 0.25–2 μg/mL of GNP-corona-PKF/SAHA with a ratio of 1:1, the toxic effect to CSCs was stronger at 72 and 120 hrs. These results suggest a synergistic effect for this combination (Figure 3C and D), for instance, the combination showed significant toxicity at 0.25 μg/mL at 72 hrs (Figure 3A and B) that was not present at 0.125 ug/mL of each GNP-drug alone (Figure 3A and B). Moreover, the IC50 values showed markedly enhanced toxicity of the GNP-PKF/SAHA combination compared to each GNP-drug (Table 3). Taken together, it seems that blocking the Wnt pathway along with inhibition of HDAC resulted in better proliferation control in CSCs.
Effect of Drug-Loaded GNPs on Gene Expression and Sphere Formation
Western blot immunoassay showed altered mesenchymal and epithelial protein markers in CSCs following treatment with GNP-corona-drug. The CSCs were treated with bare-GNP and drug-loaded GNPs for 6 days and the cells were lysed and stored at −80°C. Since the 1:1 ratio of GNP-SAHA: PKF had strong toxicity in CSCs (Figure 3C and D), lower doses of GNP-PKF were used to evaluate the protein expression in longer periods and sphere formation. Thus, the concentrations of GNP-SAHA:PKF were as 4:1, 2:1 and 1:1 ratios as indicated (Figure 6). The results of Western blot analysis indicated that the expression of Snail as a mesenchymal protein marker increased dramatically when the cells were exposed to GNP-SAHA (1.2 μg/mL) for 6 days. Addition of GNP-PKF (0.33 μg/mL) in the presence of a fixed concentration of GNP-SAHA (1.2 μg/mL) suppressed Snail expression and the reduction continued with increasing the dose of GNP-PKF (Figure 6C). E-cadherin as an epithelial protein marker was slightly augmented by increasing the GNP-PKF concentration compared to the control group.
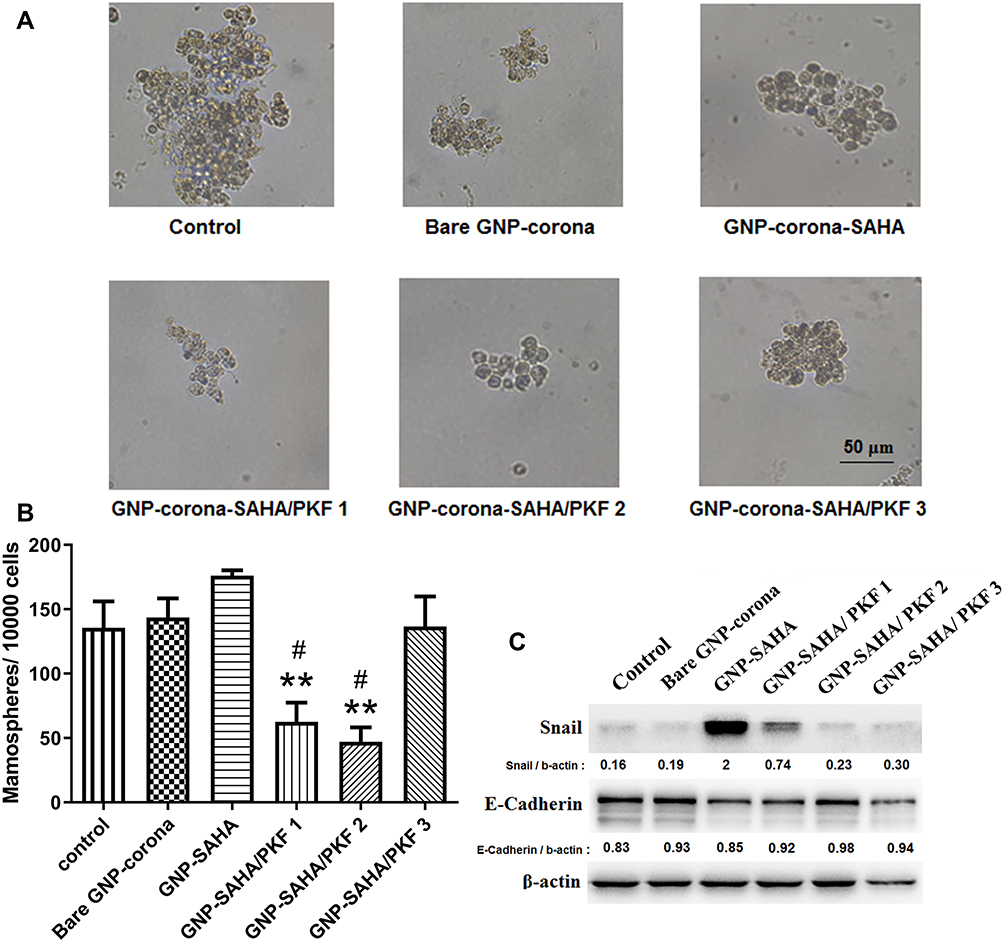 |
Figure 6 (A) Images of 3D spheres were formed in sphere-forming assay in each sample; (B) Number of sphere formation after 7 days per 10,000 treated cells was seeded at low-attachment culture plates in DMEM/F12 medium supplemented with B27 and hEGF. Error bars are indicated as mean ± standard deviation of two independent experiments. (**P<0.01 versus GNP-corona-SAHA, #P<0.05 versus bare GNP-corona and control); (C) Expression level of Snail and E-cadherin by Western blot analysis of lysed samples. Numbers were indicated as the ratio of each sample intensity to corresponding b-actin intensity analyzed by ImageJ software. Concentrations of GNP-corona-SAHA and GNP-corona-PKF (μg/mL) in each sample in Western blot and sphere-forming assay, respectively: Control 0:0, Bare GNP-corona 0:0, GNP-SAHA 1.2:0, GNP-SAHA/PKF1 1.2:0.3, GNP-SAHA/PKF2 1.2:0.6, GNP-SAHA/PKF3 1.2:1.2. Abbreviations: hEGF, human epidermal growth factor; PKF, PKF118-310; SAHA, suberoylanilide hydroxamic acid; GNP, gold nanoparticle. |
The stem cell phenotype in the cells can be evaluated by sphere-forming capability. In other words, sphere formation shows the stemness phenotype in the stem cell population. Although there are reports of the capability of GNPs solely to induce osteoblastic differentiation of mesenchymal stem cells,39 no significant difference was observed in the number of sphere formation (Figure 6B) or expression of EMT markers (Figure 6C) between the GNP-corona-treated group and the control group. It is possible that the corona coating influences the effects of naked GNPs on cell differentiation status.
Images of spheres formed in each sample are presented in Figure 6A. The results of sphere formation (Figure 6B) showed that GNP-SAHA induced ~30% more sphere formation compared to bare GNP although the difference was not statistically significant. However, GNP-PKF significantly reduced sphere formation compared to the bare GNP-corona group and the GNP-SAHA group. These results were in accordance with the results of the gene expression level by Western blot analysis. Taken together, the findings indicated that SAHA induced EMT marker and sphere formation in enriched CSC populations.
As expected, increased stem cell population following GNP-corona-SAHA treatment was consistent with a previous study which stated that SAHA could induce acquisition of EMT phenotype and up-regulation of mesenchymal-related genes like vimentin and N-cadherin in prostate cancer cell lines due to hyperactivity of HDACs in hyper-acetylation of mesenchymal genes promotors.40 It is noteworthy that SAHA has been approved by the FDA for T-cell lymphoma due to its promising therapeutic responses in hematologic neoplasms; however, contradictory results have raised doubts about its clinical application in solid tumors. Therefore, SAHA, as a single agent, fails to fulfill the Response Evaluation Criteria In Solid Tumors (RECIST),41 which is why SAHA is part of a combination therapy for breast cancer in various clinical trials in most cases.40–42 Several studies found that SAHA could promote differentiation, revert CSC characteristics,43 weaken invasiveness,44 heighten drug susceptibility,43,45 induce autophagy and facilitate apoptosis.46 However, a common aspect has been neglected in almost all of these studies, which is that they only examined the effects of SAHA in enriched stem-like cell subpopulations irrespective of probable conversion of other non-CSC populations to CSCs. In fact, SAHA is a double-edged sword for the cells in solid tumors with varying degrees of stemness. This idea was satisfactorily corroborated by Kuo et al who found the growth inhibition of human lung tumor xenografts in mice by SAHA treatment, while the remaining tumor CSCs exhibited more aggressiveness, stemness, and expression of malignant genes.47
The reason for the positive/negative concurrent effect of SAHA may be associated with hyper-activation of the Wnt/β-Catenin pathway, a possible mechanism by which HDACIs including SAHA induce apoptosis in colorectal carcinoma48 and lymphoblastic leukemia.49 On the contrary, another study found that decreased levels of Wnt transcription factor TCF7L2 induced apoptosis in colorectal carcinoma cells.50 While the intracellular β-Catenin level is strictly under control in order to maintain cell survival and proliferation, its up/down-regulation results in apoptosis depending on the cell content and condition.49
It seems that hyper-activation of the Wnt/β-Catenin pathway mediates SAHA-induced apoptosis in breast cancer. However, on the other hand, it also contributes to the undesirable reprogramming of non-CSCs to stem-like phenotype;11 a report that encouraged us to assess co-treatment with SAHA and PKF118-310, a Wnt antagonist. A remarkable finding of the present study was that adding GNP-corona-PKF to GNP-corona-SAHA at a low concentration of 0.25 μg/mL reduced the stem cell population to about 54% compared to the control group. A combination of GNP-SAHA/PKF blocked the sphere formation at low concentrations, too. In this respect, low concentrations of GNP-PKF had significant effects on the sphere compared to higher concentrations of GNP-PKF.
A review of the literature showed a similar effort to target triple-negative breast cancer by a triple therapy with an HDACI (valproic acid), a Wnt signaling antagonist (BC21), and tamoxifen.51 HDACI and Wnt antagonist upregulate estrogen receptors and improve the efficacy of tamoxifen; with this in mind, the authors found that HDACI and tamoxifen could only inhibit CSCs, indicating the importance of Wnt antagonist as an inhibitor of non-CSC to CSC conversion in this triple therapy.51 Besides, HDCI (Panobinostat)/Wnt antagonist (BC2059) dual therapy of primary AML progenitor cells causes efficient apoptosis.52 In brief, these reports are in good agreement with our results. Similarly, in this study, CSC increase induced by HDACI (SAHA) was inhibited using a Wnt antagonist (PKF) and the cells were maintained in a differentiated form.
A relatively low drug loading and release rate were the main limitations of this research. Cytotoxicity of CTAB-capped GNPs prevented the use of large weights for drug loading. It is thus recommended that more biocompatible capping agents should be used in the synthesis procedure.
PC could be engineered on a basis of what profile of drug release is expected. For a higher drug release rate, hard corona proteins should differ from those in the environment, whereas similar types of proteins in the hard corona and environment cause the exchange at the level of only soft corona, indicating a more sustained and steady release profile.28
Conclusion
Although serious attention has been paid to specific targeting of cancer stem cells in recent years, smart cancer cells change their phenotypes when exposed to drugs and tumor microenvironment changes to ensure the tumor survival. Thus, the specific destruction of cancer stem cells is not solely an ideal solution for complete tumor eradication.
In summary, in this study, CSCs and non-CSCs were targeted in order to reduce the number of breast cancer stem cell populations using a combination of Vorinostat (SAHA), as a breast cancer stem cell differentiator, and PKF118-310, as a Wnt/β-Catenin pathway blocker. In fact, the cells were directed towards differentiation while the dedifferentiation path was blocked. These results indicate a step towards differentiation with reduced stemness. A combination therapy was accomplished with proper internalization of drugs into cells using a not much sophisticated nanoscale drug delivery system. The present findings might help to solve the simultaneous co-delivery of hydrophilic and hydrophobic drugs by gold nanoparticle systems with protein corona coating. It is hoped that the results of this study will help to design optimized drug delivery systems based on PC loading and cast light on more efficient treatment methods of CSCs annihilation considering the shortcuts that cancer cells use to bypass drug mechanisms.
Acknowledgments
This research was made possible by grant No. 92-03-33-24377 from Tehran University of Medical Sciences. The Authors would also like to appreciate Dr. Faeze Vakhshite and Dr. Shima Ebrahimi for their valuable contributions.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Rahman M, Deleyrolle L, Vedam-Mai V, Azari H, Abd-El-Barr M, Reynolds BA. The cancer stem cell hypothesis: failures and pitfalls. Neurosurgery. 2011;68(2):531–545. doi:10.1227/NEU.0b013e3181ff9eb5
3. Garvalov BK, Acker T. Cancer stem cells: a new framework for the design of tumor therapies. J Mol Med. 2011;89(2):95–107. doi:10.1007/s00109-010-0685-3
4. Cruz M, Sidén Å, Calaf G, Delwar Z, Yakisich J. The stemness phenotype model. ISRN Oncol. 2012;2012:1–10. doi:10.5402/2012/392647
5. Kawai H, Li H, Avraham S, Jiang S, Avraham HK. Overexpression of histone deacetylase HDAC1 modulates breast cancer progression by negative regulation of estrogen receptor α. Int J Cancer. 2003;107(3):353–358. doi:10.1002/ijc.11403
6. Müller BM, Jana L, Kasajima A, et al. Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer-overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer. 2013;13(1):215. doi:10.1186/1471-2407-13-215
7. Saji S, Kawakami M, Hayashi S-I, et al. Significance of HDAC6 regulation via estrogen signaling for cell motility and prognosis in estrogen receptor-positive breast cancer. Oncogene. 2005;24(28):4531–4539. doi:10.1038/sj.onc.1208646
8. Witt A, Lee C, Lee T, et al. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene. 2017;36(12):1707. doi:10.1038/onc.2016.337
9. Dvorakova M, Vanek T. Histone deacetylase inhibitors for the treatment of cancer stem cells. MedChemComm. 2016;7(12):2217–2231. doi:10.1039/C6MD00297H
10. Munster PN, Troso-Sandoval T, Rosen N, Rifkind R, Marks PA, Richon VM. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces differentiation of human breast cancer cells. Cancer Res. 2001;61(23):8492–8497.
11. Debeb BG, Lacerda L, Xu W, et al. Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through WNT/β‐catenin signaling. Stem Cells. 2012;30(11):2366–2377. doi:10.1002/stem.1219
12. Lepourcelet M, Chen Y-NP, France DS, et al. Small-molecule antagonists of the oncogenic Tcf/β-catenin protein complex. Cancer Cell. 2004;5(1):91–102. doi:10.1016/S1535-6108(03)00334-9
13. Hallett RM, Kondratyev MK, Giacomelli AO, et al. Small molecule antagonists of the Wnt/beta-catenin signaling pathway target breast tumor-initiating cells in a Her2/Neu mouse model of breast cancer. PLoS One. 2012;7(3):e33976. doi:10.1371/journal.pone.0033976
14. Lacerda SHDP, Park JJ, Meuse C, et al. Interaction of gold nanoparticles with common human blood proteins. ACS Nano. 2009;4(1):365–379. doi:10.1021/nn9011187
15. Walczyk D, Bombelli FB, Monopoli MP, Lynch I, Dawson KA. What the cell “sees” in bionanoscience. J Am Chem Soc. 2010;132(16):5761–5768. doi:10.1021/ja910675v
16. Shahabi S, Treccani L, Dringen R, Rezwan K. Utilizing the protein corona around silica nanoparticles for dual drug loading and release. Nanoscale. 2015;7(39):16251–16265. doi:10.1039/C5NR04726A
17. Singh P, Pandit S, Mokkapati V, Garg A, Ravikumar V, Mijakovic I. Gold nanoparticles in diagnostics and therapeutics for human cancer. Int J Mol Sci. 2018;19(7):1979. doi:10.3390/ijms19071979
18. Mirsadeghi S, Dinarvand R, Ghahremani MH, et al. Protein corona composition of gold nanoparticles/nanorods affects amyloid beta fibrillation process. Nanoscale. 2015;7(11):5004–5013. doi:10.1039/C4NR06009A
19. Goodarzi N, Ghahremani MH, Amini M, et al. CD 44‐targeted docetaxel conjugate for cancer cells and cancer stem‐like cells: a novel hyaluronic acid‐based drug delivery system. Chem Biol Drug Des. 2014;83(6):741–752. doi:10.1111/cbdd.12288
20. Shaw FL, Harrison H, Spence K, et al. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J Mammary Gland Biol Neoplasia. 2012;17(2):111–117. doi:10.1007/s10911-012-9255-3
21. Piella J, Bastús NG, Puntes V. Size-dependent protein–nanoparticle interactions in citrate-stabilized gold nanoparticles: the emergence of the protein corona. Bioconjug Chem. 2016;28(1):88–97. doi:10.1021/acs.bioconjchem.6b00575
22. Li Y, Monteiro-Riviere NA. Mechanisms of cell uptake, inflammatory potential and protein corona effects with gold nanoparticles. Nanomedicine. 2016;11(24):3185–3203. doi:10.2217/nnm-2016-0303
23. Qiu Y, Liu Y, Wang L, et al. Surface chemistry and aspect ratio mediated cellular uptake of au nanorods. Biomaterials. 2010;31(30):7606–7619. doi:10.1016/j.biomaterials.2010.06.051
24. Available from: https://www.drugbank.ca/drugs/DB02546. https://www.drugbank.ca/drugs/DB02546.
25. Jain P, Pawar R, Pandey R, et al. In-vitro in-vivo correlation (IVIVC) in nanomedicine: is protein corona the missing link? Biotechnol Adv. 2017;35(7):889–904. doi:10.1016/j.biotechadv.2017.08.003
26. Corbo C, Molinaro R, Parodi A, Toledano Furman NE, Salvatore F, Tasciotti E. The impact of nanoparticle protein corona on cytotoxicity, immunotoxicity and target drug delivery. Nanomedicine. 2016;11(1):81–100. doi:10.2217/nnm.15.188
27. Hamad-Schifferli K. Exploiting the novel properties of protein coronas: emerging applications in nanomedicine. Nanomedicine. 2015;10(10):1663–1674. doi:10.2217/nnm.15.6
28. Cifuentes-Rius A, de Puig H, Kah JCY, Borros S, Hamad-Schifferli K. Optimizing the properties of the protein corona surrounding nanoparticles for tuning payload release. ACS Nano. 2013;7(11):10066–10074. doi:10.1021/nn404166q
29. Kah JCY, Chen J, Zubieta A, Hamad-Schifferli K. Exploiting the protein corona around gold nanorods for loading and triggered release. ACS Nano. 2012;6(8):6730–6740. doi:10.1021/nn301389c
30. Simon J, Müller LK, Kokkinopoulou M, et al. Exploiting the biomolecular corona: pre-coating of nanoparticles enables controlled cellular interactions. Nanoscale. 2018;10(22):10731–10739. doi:10.1039/C8NR03331E
31. Yeo ELL, Joshua U, Cheah J, et al. Exploiting the protein corona around gold nanorods for low-dose combined photothermal and photodynamic therapy. J Mater Chem B. 2017;5(2):254–268. doi:10.1039/C6TB02743A
32. Yeo ELL, Cheah -JU-J, Lim BY, Thong PSP, Soo KC, Kah JCY. Protein corona around gold nanorods as a drug carrier for multimodal cancer therapy. ACS Biomater Sci Eng. 2017;3(6):1039–1050. doi:10.1021/acsbiomaterials.7b00231
33. Setyawati MI, Tay CY, Docter D, Stauber RH, Leong DT. Understanding and exploiting nanoparticles’ intimacy with the blood vessel and blood. Chem Soc Rev. 2015;44(22):8174–8199. doi:10.1039/C5CS00499C
34. Tee JK, Yip LX, Tan ES, et al. Nanoparticles’ interactions with vasculature in diseases. Chem Soc Rev. 2019;48(21):5381–5407. doi:10.1039/C9CS00309F
35. Chakraborty D, Tripathi S, Ethiraj K, Chandrasekaran N, Mukherjee A. Human serum albumin corona on functionalized gold nanorods modulates doxorubicin loading and release. New J Chem. 2018;42(20):16555–16563. doi:10.1039/C8NJ03673J
36. Behzadi S, Serpooshan V, Sakhtianchi R, et al. Protein corona change the drug release profile of nanocarriers: the “overlooked” factor at the nanobio interface. Colloids Surf B Biointerfaces. 2014;123:143–149. doi:10.1016/j.colsurfb.2014.09.009
37. Feng M, Berdugo Morales A, Poot A, Beugeling T, Bantjes A. Effects of tween 20 on the desorption of proteins from polymer surfaces. J Biomater Sci Polym Ed. 1996;7(5):415–424. doi:10.1163/156856295X00427
38. Cheng X, Tian X, Wu A, et al. Protein corona influences cellular uptake of gold nanoparticles by phagocytic and nonphagocytic cells in a size-dependent manner. ACS Appl Mater Interfaces. 2015;7(37):20568–20575. doi:10.1021/acsami.5b04290
39. Yi C, Liu D, Fong -C-C, Zhang J, Yang M. Gold nanoparticles promote osteogenic differentiation of mesenchymal stem cells through p38 MAPK pathway. ACS Nano. 2010;4(11):6439–6448. doi:10.1021/nn101373r
40. Kong D, Ahmad A, Bao B, Li Y, Banerjee S, Sarkar FH. Histone deacetylase inhibitors induce epithelial-to-mesenchymal transition in prostate cancer cells. PLoS One. 2012;7(9):e45045. doi:10.1371/journal.pone.0045045
41. Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst. 2000;92(3):205–216. doi:10.1093/jnci/92.3.205
42. Available from: https://www.clinicaltrials.gov/ct2/results?cond=breast+cancer&term=vorinostat&cntry=&state=&city=&dist=.
43. Li R, Huang J, Ma M, et al. Two-stage induced differentiation of OCT4+/Nanog+ stem-like cells in lung adenocarcinoma. Oncotarget. 2016;7(42):68360.
44. Feng J, Zhang S, Wu K, et al. Combined effects of suberoylanilide hydroxamic acid and cisplatin on radiation sensitivity and cancer cell invasion in non–small cell lung cancer. Mol Cancer Ther. 2016;15(5):842–853. doi:10.1158/1535-7163.MCT-15-0445
45. Lu S, Labhasetwar V. Drug-resistant breast cancer cell line displays cancer stem cell phenotype and responds sensitively to epigenetic drug SAHA. Drug Deliv Transl Res. 2013;3(2):183–194. doi:10.1007/s13346-012-0113-z
46. Chiao M-T, Cheng W-Y, Yang Y-C, Shen -C-C, Ko J-L. Suberoylanilide hydroxamic acid (SAHA) causes tumor growth slowdown and triggers autophagy in glioblastoma stem cells. Autophagy. 2013;9(10):1509–1526. doi:10.4161/auto.25664
47. Kuo W-Y, Wu C-Y, Hwu L, et al. Enhancement of tumor initiation and expression of KCNMA1, MORF4L2 and ASPM genes in the adenocarcinoma of lung xenograft after vorinostat treatment. Oncotarget. 2015;6(11):8663. doi:10.18632/oncotarget.3536
48. Bordonaro M, Lazarova DL, Sartorelli AC. The activation of beta-catenin by Wnt signaling mediates the effects of histone deacetylase inhibitors. Exp Cell Res. 2007;313(8):1652–1666. doi:10.1016/j.yexcr.2007.02.008
49. Shao N, Zou J, Li J, et al. Hyper-activation of WNT/β-catenin signaling pathway mediates anti-tumor effects of histone deacetylase inhibitors in acute T lymphoblastic leukemia. Leuk Lymphoma. 2012;53(9):1769–1778. doi:10.3109/10428194.2012.663085
50. Götze S, Coersmeyer M, Müller O, Sievers S. Histone deacetylase inhibitors induce attenuation of Wnt signaling and TCF7L2 depletion in colorectal carcinoma cells. Int J Oncol. 2014;45(4):1715–1723. doi:10.3892/ijo.2014.2550
51. Sulaiman A, Sulaiman B, Khouri L, et al. Both bulk and cancer stem cell subpopulations in triple‐negative breast cancer are susceptible to Wnt, HDAC, and ERα coinhibition. FEBS Lett. 2016;590(24):4606–4616. doi:10.1002/1873-3468.12496
52. Fiskus W, Sharma S, Saha S, et al. Pre-clinical efficacy of combined therapy with novel β-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia. 2015;29(6):1267. doi:10.1038/leu.2014.340
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.