Back to Journals » OncoTargets and Therapy » Volume 15
Targeting TP53-Mutated Acute Myeloid Leukemia: Research and Clinical Developments
Authors Granowicz EM, Jonas BA ![]()
Received 31 January 2022
Accepted for publication 7 April 2022
Published 21 April 2022 Volume 2022:15 Pages 423—436
DOI https://doi.org/10.2147/OTT.S265637
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr William C. Cho
Eric M Granowicz, Brian A Jonas
Department of Internal Medicine, Division of Hematology/Oncology, University of California Davis Comprehensive Cancer Center, Sacramento, CA, USA
Correspondence: Brian A Jonas, Department of Internal Medicine, Division of Hematology/Oncology, University of California Davis Comprehensive Cancer Center, 4501 X Street, Suite #3016, Sacramento, CA, 95817, USA, Tel +1 916-734-3772, Fax +1 916-734-7946, Email [email protected]
Abstract: TP53 is a key tumor suppressor gene that plays an important role in regulating apoptosis, senescence, and DNA damage repair in response to cellular stress. Although somewhat rare, TP53-mutated AML has been identified as an important molecular subgroup with a prognosis that is arguably the worst of any. Survival beyond one year is rare after induction chemotherapy with or without consolidative allogeneic stem cell transplant. Although response rates have been improved with hypomethylating agents, outcomes remain particularly poor due to short response duration. Improvements in our understanding of AML genetics and biology have led to a surge in novel treatment options, though the clinical applicability of these agents in TP53-mutated disease remains largely unknown. This review will focus on the epidemiology, molecular characteristics, and clinical significance of TP53 mutations in AML as well as emerging treatment options that are currently being studied.
Keywords: acute myeloid leukemia, TP53 mutation, venetoclax, eprenetapopt, magrolimab
Introduction
Acute myeloid leukemia (AML) is a heterogeneous aggressive malignancy arising from clonal expansion of neoplastic hematopoietic precursor cells, which can occur de novo, after exposure to cytotoxic treatments, or after transformation from an antecedent hematologic disease. It is the most common acute leukemia in adults with an annual age-adjusted incidence rate of 4.3/100,000 rising to 15–20/100,000 above the age of 60 years.1 Recent advances in next-generation sequencing (NGS) have revealed a stepwise process of mutational events, epigenetic dysregulation, and acquirement of copy number aberrations as main drivers of AML pathogenesis.2–5 Leukemogenic mutations can often be detected in a small percentage of hematopoietic stem cell clones without any evidence of cytopenias or malignancy, a phenomenon referred to as clonal hematopoiesis of indeterminate potential (CHIP). Though the overall rate of progression from CHIP to AML is only 0.5–1.0% per year, mutations in the TP53 gene have been associated with higher rates of AML than most other implicated genes.6 In AML, certain genetic aberrations are associated with inferior outcomes, with those that affect the TP53 gene being among the absolute worst. Somatically acquired TP53 mutations constitute early events in the transformation of hematopoietic stem cells into pre-leukemia stem cells (preLSCs) and subsequently AML, and contribute substantially to its therapeutic resistance.7 This review will focus on the role of TP53 mutations in the prognosis and treatment of AML, with an emphasis on emerging therapies with the potential to improve outcomes in this challenging molecular subgroup.
Biology
Mutations in TP53 are seen in nearly half of all tumors, making it the most consistently mutated gene in human malignancies.8,9 Immunohistochemistry and real-time reverse transcription PCR (rt-PCR) can be used to detect TP53 mutations, though NGS is preferred given its higher sensitivity.10 Situated on chromosome 17p13.1, the TP53 gene encodes a 393 amino acid phosphoprotein, p53, a transcription factor with critical tumor suppressor functions.11,12 It contains five hallmark domains including the DNA-binding, oligomerization, proline-rich SH3, C-terminal regulatory, and N-terminal transactivation domains, the latter of which interacts with the negative regulator MDM2.13,14 The TP53 gene is known as the “guardian of the genome” given its importance in regulating cellular proliferation/differentiation associated with aberrant oncogene expression.15 When provoked by cellular stressors such as DNA damage, hypoxia, and oncogene activation, it regulates a multitude of transcriptional targets involved in DNA repair, cell-cycle arrest, anti-angiogenesis, senescence, and induction of apoptosis.8,11,16,17
Thus, inactivation of TP53 through gene mutation or deletion favors the action of oncogenes, ultimately promoting uncontrolled proliferation of neoplastic cells.8,11,18,19 The role of p53 in mediating apoptosis is especially important in the setting of cytotoxic chemotherapy, where it has been associated with an inherent resistance to DNA damaging agents traditionally used to treat AML and other malignancies.20–22
Epidemiology
TP53 mutations are found in about 5–15% of AML cases.23,24 In 75% of these cases, it is the only mutated gene identified on NGS.25 TP53 alterations are associated with older age in AML, occurring in up to 25% of cases in elderly individuals.24 Though TP53 mutations occur across all morphological subtypes of AML, they are more frequently seen in acute erythroblastic leukemia, therapy-related AML (t-AML), and in AML that has progressed from an underlying myeloproliferative neoplasm, where it can be found in up to a third of cases.26–28 The mutation can be germline, in which case there is a tendency to develop other solid tumor malignancies, a condition referred to as Li-Fraumeni syndrome.29,30 Sporadic mutations are also seen, in which case carcinogen exposure is often implicated.31,32 The higher frequency of TP53 mutations in t-AML appears to be related to expansion of pre-existing chemotherapy-resistant hematopoietic stem cell clones carrying age-related TP53 mutations rather than direct induction of TP53 mutation by cytotoxic chemotherapy.33
TP53 Mutation Characteristics
Greater than 80% of oncogenic TP53 mutations have been reported as missense mutations, with “hot spots” noted in various arginine residues (R175H, R248Q, R273C, R282). Other mutation types have been reported, including insertions, deletions, nonsense, and frameshift mutations.34,35 In most cases, the mutation occurs in the DNA-binding domain, with about a quarter of the remaining mutations located widely throughout the other domains.36 These mutations typically lead to loss of function of p53ʹs tumor suppressive capabilities, although some mutations (in codons 175, 248, 173, 282) can lead to gain of function, often through binding of mutated p53 to other tumor suppressor proteins such as p63 and p73.14,37–39 TP53 can also be deleted through loss of the short arm of chromosome 17 (band 17p13.1).40 In addition to somatic mutations and cytogenetic aberrancies involving the TP53 gene, p53 dysregulation can occur through overexpression of its canonical negative regulators (Mdm2, Mdm4, p14ARF).41 Co-occurring mutations involving other commonly mutated AML genes (eg, DNMT3A, FLT3, IDH1, IDH2, NPM1, TET2) is seen in only a minority of cases, while chromothripsis, complex karyotype, and recurrent karyotypic structural aberrations, especially involving chromosomes 5, 7, and 17, are seen more frequently.2,42,43
Prognosis
Risk stratification in AML plays an important role in both prognostication and determining the best overall treatment approach. Various risk stratification models exist, with the most widely utilized in clinical practice being the European LeukemiaNet (ELN) system. This system was largely based on cytogenetic analysis prior to 2017.44
In recent years, improvements in rt-PCR and genomic sequencing have allowed for more routine access to molecular analysis and subsequent adjustments to the ELN risk stratification. In 2012, a molecular analysis of 841 AML patients defined five distinct prognostic subgroups that outperformed cytogenetic analysis alone. TP53-mutated disease represented the “very unfavorable group” associated with a three-year OS of 0%.45 In a 2016 study regarding 1540 patients enrolled in three trials investigating treatment with intensive chemotherapy, driver mutations involving 111 cancer-associated genes along with cytogenetic findings were combined to identify 11 molecular classes with distinct clinical outcomes. In addition to the eight recurrent cytogenetic abnormalities that had already been described in AML, three additional classes emerged: AML with TP53 mutations, chromosomal aneuploidies, or both (13%), AML with mutations of chromatin and RNA-splicing regulators (18%), and AML with IDH2R172 mutations (1%). The TP53-mutated class was associated with a particularly dismal prognosis, especially when a complex karyotype was also present, with each having an independent and additive effect on prognosis.23 A 2015 analysis of the genetic ontogeny of AML used sequencing of 82 myeloid malignancy target genes to identify three clinicopathologically distinct subgroups in a cohort of patients enrolled in the ACCEDE trial with secondary-AML (s-AML) or t-AML. These subgroups included secondary-type, TP53 de novo/pan-AML-type, and TP53-mutated, the latter of which was associated with reduced median OS (4.0 vs 8.5 months) in s-AML and increased chemoresistance in t-AML.46 The ELN subsequently updated its risk stratification in 2017 to include TP53 mutation as an independent adverse-risk indicator, associated with an estimated five-year overall survival (OS) of 20% and 6% in those treated with intensive chemotherapy aged <60 and ≥60, respectively.47
The variant allele frequency (VAF) for TP53 mutations appears to play a role in prognosis as well. Higher TP53 VAFs were associated with a significant increase in the relative hazards on OS in elderly patients treated with low-intensity therapies.48 In patients treated with low-dose cytarabine, a VAF >40% was independently associated with higher rates of relapse and worse RFS and OS, which was not true for patients that were treated with a hypomethylating agent (HMA).49 Outcomes with high-intensity therapies appear to depend less on TP53 VAFs, with similar OS, EFS, and CR rates even with VAF <20%.50 This may be the result of expanding subclones that are selected for during treatment and suggests that subclonal TP53 mutations should be considered in prognostication.
The relationship between prognosis and the TP53 mutation allelic state is still largely unknown at this time in AML. In the MDS setting, multi-hit TP53-mutant disease is an independent predictor of OS and transformation to AML, while monoallelic TP53-mutant disease may have a prognosis that is similar to wild-type.51 Further analysis will be required to assess for a similar possibility in the AML setting.
The functional subtype of TP53 mutation may also have an impact on prognosis. In an analysis of 9833 DNA sequence variants in human p53-null cells, certain variants were associated with greater expansion in in vitro cultures, which was used to generate a relative fitness score (RFS).52 This score was applied to 83 TP53-mutated patients intensively treated with German-Austrian AML study group protocols, which demonstrated better median OS (12.9 vs 5.5 months) in patients with low-risk scores when compared to those with high-risk scores.53 This indicates that functional characterization of TP53 mutants may play an important role in refining the prognostic significance of TP53 mutations.
Treatments
Intensive Chemotherapy
High-intensity chemotherapy, typically with a combination of cytarabine and an anthracycline, has long been considered the standard treatment approach in fit patients. However, the most recent NCCN guidelines recommend considering alternative induction strategies in TP53-mutated disease.54 The discouraging outcomes after treatment with aggressive chemotherapy is reflected by the inclusion of TP53 mutations in the adverse-risk category in the ELN risk-stratification system. This scoring system was developed by analyzing outcomes from patients enrolled in Phase 3 clinical trials involving treatment with cytarabine plus doxorubicin or mitoxantrone-based induction.47 Other studies have revealed response rates ranging from 20% to 42% with a median OS of 4–9 months after treatment with induction chemotherapy.23,24,26,42,55,56 TP53 mutation has also been identified as a predictor of inferior response to CPX-351, a newer liposomal form of cytarabine and daunorubicin that is approved for treatment of t-AML and AML with MRC.57 Median OS was similar for TP53-mutant patients treated with CPX-351 vs 7+3 induction.58 Despite these observations, intensive cytotoxic therapy does still offer an advantage over no treatment, supported by an improved median OS (8 vs 1.7 months).55 Although high-intensity treatments may be appropriate in the right clinical situation, newer therapies that do not require activation of TP53 in response to DNA damage are likely the future to overcoming these poor outcomes.
Hypomethylating Agents
Epigenetic modifications resulting from DNA hypermethylation events at CpG islands have been associated with transcriptional silencing of genes involved in cell cycle regulation and other important growth regulators that suppress leukemogenesis.59,60 The HMAs, decitabine (DEC) and azacitidine (AZA), are cytosine analogs that inhibit DNA methyltransferases, ultimately leading to reversal of DNA hypermethylation patterns and renewed transcription of previously silenced tumor suppressor genes.60,61 These agents have shown surprising activity in TP53-mutated disease and ELN adverse-risk in general, with higher response rates than those historically seen with high-intensity chemotherapy.
The first study to report such results evaluated a 10-day course of DEC in 113 patients with AML or MDS, revealing a complete response with (CR) or without hematologic recovery (CRi) in 100% of the 21 patients with TP53-mutated disease in comparison to 41% of the 46 patients with TP53 wild-type disease, and no difference in OS [Table 1].62 A Phase 2 trial compared 5-day and 10-day DEC dosing schedules in elderly AML patients, which demonstrated similar response rates and OS for each dosing schedule. A subgroup analysis of the TP53-mutated cases revealed response rates of 29% (2/7) and 47% (8/17) in the 5- and 10-day dosing schedules, respectively, which were not significantly different from response rates seen in the other subgroups (diploid cytogenetics, adverse-risk cytogenetics, de novo AML, and t-AML). Median OS was also similar between TP53-mutated and wild-type patients in the 5-day arm (5.5 vs 4.9 months), with a trend towards worse median OS in the 10-day arm (4.7 vs 8.3 months) that was not statistically significant.63 Like DEC, AZA has also demonstrated similar response rates in TP53-mutated disease, although this has not translated into improved survival outcomes.64,65,66 Despite the higher response rates with HMA therapy, these do not appear to be durable, with the longest remissions occurring in rare patients achieve TP53 mutation clearance.63,67
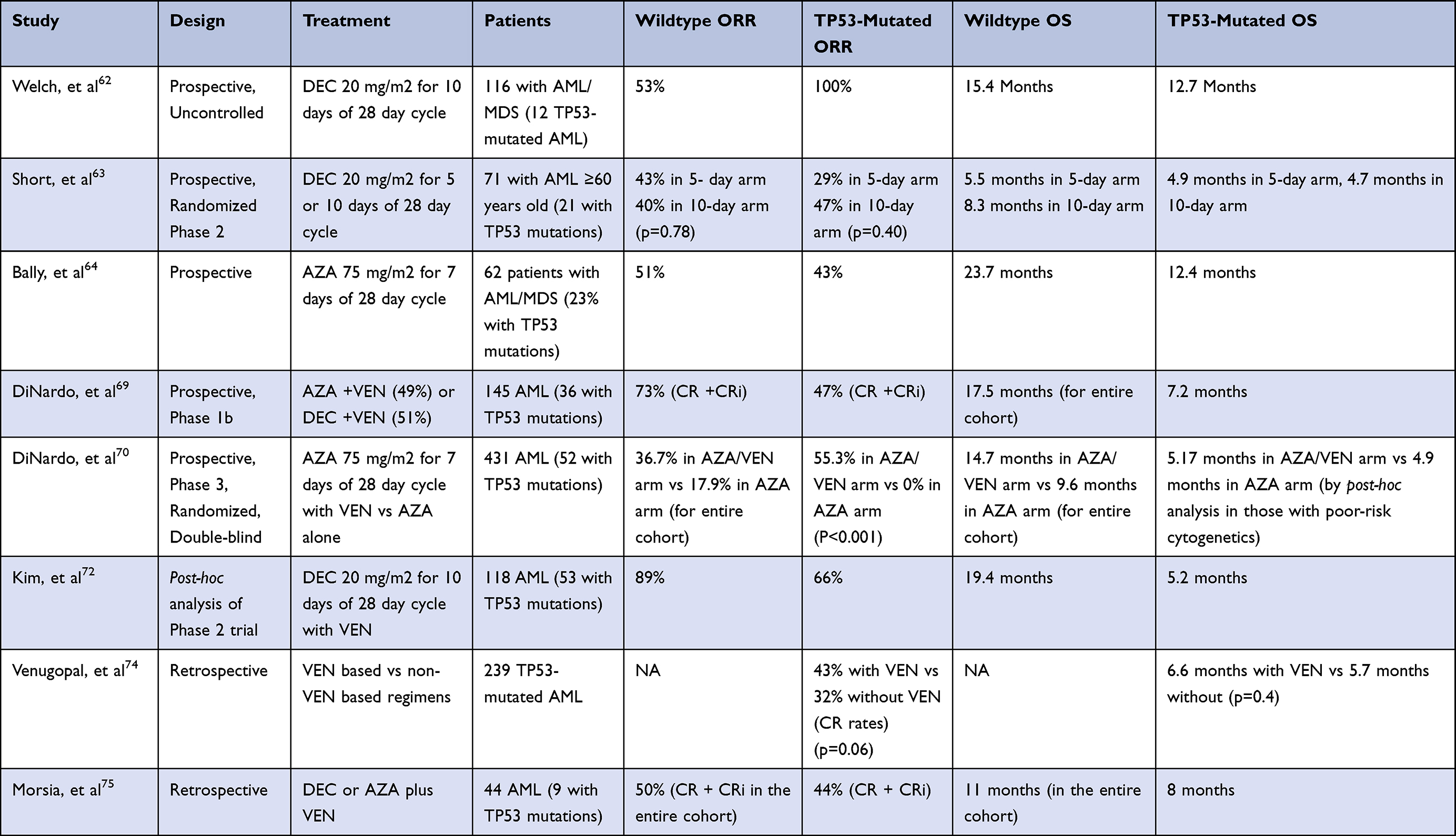 |
Table 1 Approved Drugs with Activity in TP53-Mutated AML |
The mechanism accounting for the increased sensitivity of TP53-mutated disease to HMAs remains largely unknown at this time. One investigation into the gene-regulatory effects of HMAs in AML cells with monosomal karyotype (including deletion of 17p) revealed that hemizygous tumor suppressor genes are more sensitive to decitabine-mediated induction, resulting in derepression and restoration to expression levels seen in diploid cells.68 This could partially explain the activity of HMAs when p53 aberration is due to del 17p or in TP53-mutated disease that is associated with a monosomal karyotype. Further studies will be needed to elucidate the mechanism of HMA activity in TP53-mutated disease without any karyotypic abnormalities.
Venetoclax
The BCL-2 inhibitor, venetoclax (VEN), is approved for newly diagnosed and relapsed/refractory AML in combination with a HMA, a regimen which has become an important tool in treating elderly patients and those who are not candidates for high-intensity therapy.69,70 Initial trials involving DEC plus VEN found TP53 status to be a statistically significant predictor of response in a post-hoc exploratory analysis. The CR/CRi rate of 47%, median duration of response (DoR) of 5.6 months, and median OS of 7.2 months appeared favorable when compared to historical controls.69 A subsequent retrospective analysis involving 32 patients with TP53 mutations echoed these results, demonstrating a CR/CRi rate of 67% and 38% in the frontline and relapsed/refractory setting, respectively, with responses seen in those that underwent previous hematopoietic stem cell transplantation. Baseline VAF was comparable among responders vs nonresponders and similar outcomes were seen with 5- and 10-day schedules of DEC.71
More recent data have reported conflicting results. A post-hoc analysis of a phase 2 trial involving 10-day DEC plus VEN revealed significantly inferior rates of CR/CRi (57% vs 77%), MRD negativity by flow cytometry (29% vs 59%), median OS (5.2 vs 19.4 months), and median RFS (3.4 vs 18.9 months) in TP53-mutated disease when compared to wild-type. These patients were also compared to patients receiving 10-day DEC alone on a separate prospective clinical trial with similar baseline characteristics, which demonstrated higher ORR (66% vs 53%) and MRD negativity (29% vs 25%) with the addition of VEN, but no significant difference in OS or RFS.72 An analysis of TP53-mutated patients with poor-risk cytogenetics enrolled in the phase 3 trial that led to the approval of AZA plus VEN revealed higher CR rates (41% vs 17%) similar median DoR (6.54 vs 6.7 months), and similar median OS (5.17 vs 4.9 months) for the combination when compared to AZA alone.73 A retrospective study involving 238 patients treated with VEN and non-venetoclax-based regimens similarly showed higher response rates, but no difference in OS or RFS with venetoclax-based regimens, regardless of age or intensity of treatment.74 TP53 mutation was also found to be associated with worse OS in the relapsed/refractory setting in retrospective analyses.75–77 In vitro studies have supported inactivation of TP53 as a mediator of VEN resistance, as well as other genes that regulate the mitochondrial apoptotic network (BAX, PMAIP1, TFPD1).78,79 Although venetoclax-based combination therapies have high response rates and improved ability to attain disease control, the short-lived nature of these responses does not necessarily translate into improved survival outcomes [Table 1], highlighting the continued need for adequate consolidation strategies.
Magrolimab
CD47 is a macrophage checkpoint protein that is expressed on the surface of a variety of cells where it functions as a “don’t eat me” signal by interacting with signal regulatory protein α (SIRPα) on the surface of macrophages and inhibiting phagocytosis.80 CD47 expression is elevated on AML cells, where it aids in evasion of the immune system.81,82 Magrolimab is an anti-CD47 IgG4 monoclonal antibody that stimulates antibody-dependent cellular phagocytosis and T-cell mediated cytotoxicity through inhibition of the CD47/SIRPα signaling access.83
Preclinical studies have demonstrated the ability of AZA to upregulate the “eat me” signal, calreticulin, in mouse xenograft models, which has inspired the combination of magrolimab plus AZA that is currently being evaluated in a Phase 1b trial.84 The trial includes untreated, induction chemotherapy-ineligible or relapsed/refractory AML patients and intermediate to very-high risk untreated or relapsed/refractory MDS patients. Preliminary results have revealed an ORR of 63% (27/43) for the AML cohort, with elimination of LSCs (defined as CD34+/CD38-) in 71% of responding patients. The TP53-mutant cohort ORR, CR, and CRi rates were 69% (20/29), 45% (13/29), and 14% (4/29), respectively, with a median DoR of 7.6 months. The median OS for TP53 mutant and wild-type patients was 12.9 months and 18.9 months, respectively. The combination was well tolerated in both studies with a safety profile comparable to AZA monotherapy. Aged red blood cells express CD47 leaving them susceptible to on-target hemolytic anemia with magrolimab treatment. Despite this, worsening anemia was only reported in about one-third of patients, with >50% becoming red blood cell transfusion-independent at some point throughout the course of therapy.85
Given the results of these studies, phase 3 trials are currently ongoing comparing AZA plus magrolimab to AZA plus VEN or intensive chemotherapy. A phase 1/2 trial is also assessing the safety and efficacy of magrolimab plus AZA in combination with VEN in newly diagnosed and relapsed/refractory AML. Preliminary results have reported a CR/CRi rate of 94% (15/16) in newly diagnosed patients with complete cytogenetic response and MRD negativity by flow cytometry rates of 75% and 55%. The TP53-mutant cohort achieved a CR/CRi rate of 100% (7/7), with and MRD negative rate of 57% (4/7).86 Additional follow up will be needed to determine whether these responses are associated with improved survival outcomes [Table 2].
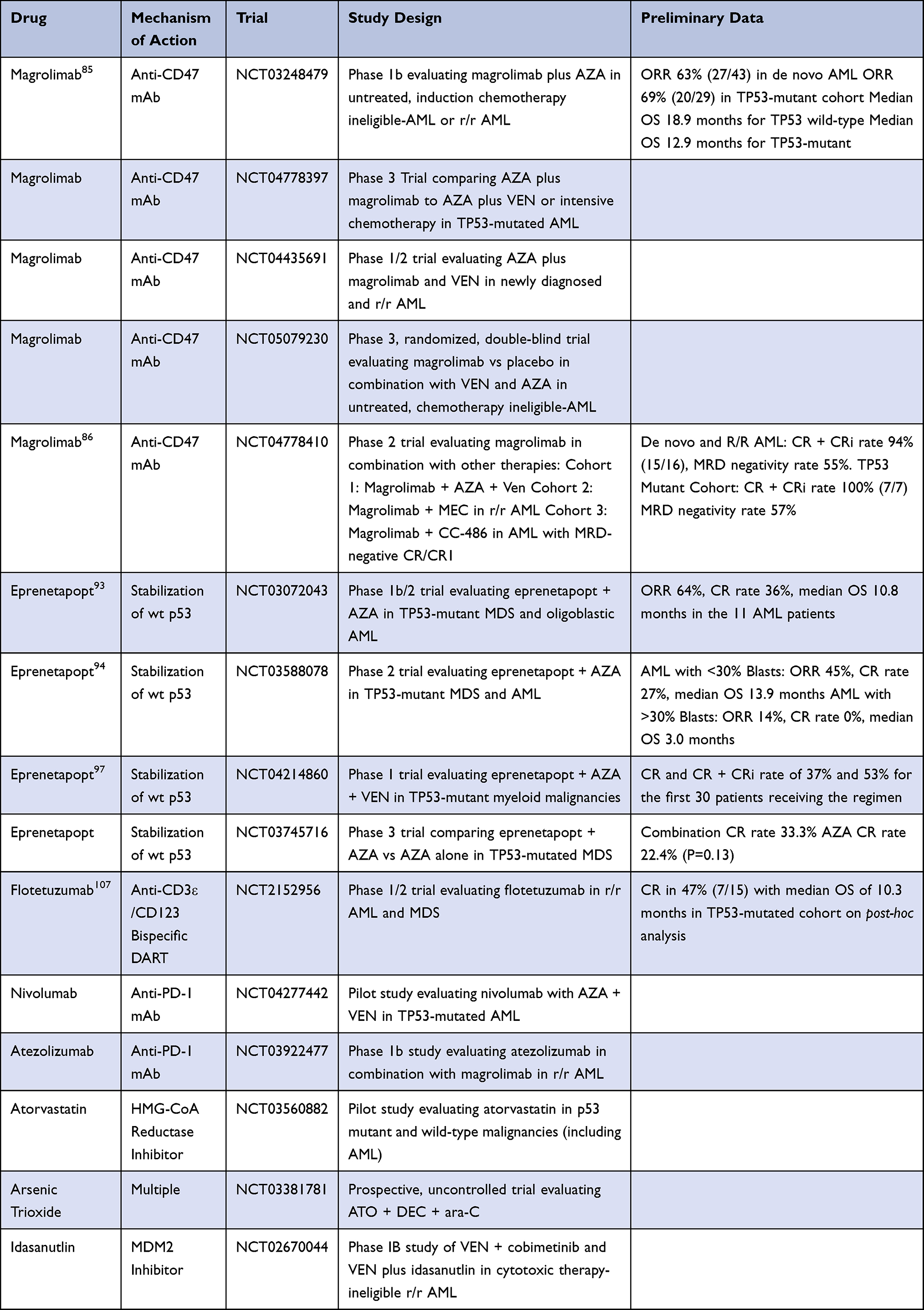 |
Table 2 Emerging Therapies for TP53-Mutated AML |
Eprenetapopt (APR-246)
Eprenetapopt (APR-246), a PRIMA-1 analog, is a first-in-class small molecule that targets TP53-mutant cancer cells through reactivation of p53 function.87,88 The active component is methylene quinuclidinone, a decomposition product of eprenetapopt that covalently binds cysteine residues in mutant p53, shifting the equilibrium in favor of the wild-type p53 conformation through thermodynamic stabilization.89 Increasing oxidative stress through depletion of glutathione and induction of ferroptosis also contributes to eprenetapopt’s mechanism of action.90,91
Two phase 1b/2 studies have been conducted evaluating the combination of eprenetapopt with AZA, which was shown to have synergistic cytotoxicity in TP53-mutated AML cell lines and in vivo models.92 The first of these trials involved patients with TP53-mutated intermediate to very high risk MDS or oligoblastic AML (20–30% blasts) [Table 2].93 The second study involved a similar patient population, though AML with any blast percentage was permitted and eprenetapopt/AZA maintenance could be administered for up to one year if allogeneic HCT was performed.94 An analysis of the 100 patients combined in these two trials revealed an ORR, CR rate, NGS negativity rate, MRD negativity rate, and median OS after allogeneic HCT of 69%, 43%, 40%, 6%, 11.8 months, and 16.1 months, respectively. In the AML population, ORR and CR rate were 64% and 36%, respectively. Isolated TP53 mutation was predictive of higher CR rate (52% vs 30%). Those with biallelic TP53 mutations or complex karyotype experienced higher CR rates than those without either of these features (49% vs 8%). Patients who responded and proceeded to allogeneic HCT had a median OS that was not reached vs 9.1 months in allogeneic HCT patients who did not respond.95
Ongoing trials are evaluating eprenetapopt in other clinical settings and combinations. A phase 2 study evaluating eprenetapopt with AZA as maintenance therapy for up to one year in TP53-mutated MDS and AML has released preliminary results for the 33 patients that have been enrolled, demonstrating a median RFS of 368 days and median OS of 586 days.96 A phase 1 trial involving eprenetapopt in combination with AZA and VEN has also published preliminary data for the first 30 patients, with a CR and CR/CRi rate of 37% and 53%, respectively.97 A phase 3 trial comparing AZA to AZA plus eprenetapopt in MDS patients failed to reach its primary endpoint according to a 2020 press release, though there was a statistically insignificant trend towards improved CR rate in the combination arm (33.3% vs 22.4%). The most common grade ≥3 adverse events reported with eprenetapopt in these trials were reversible neurologic phenomena (40%), febrile neutropenia (33–37%), diarrhea (50%), vomiting (39%), and hematologic toxicity that was similar to AZA monotherapy.94–97
Immunotherapy
Aside from magrolimab, several other immunotherapeutic approaches have been tried in AML, mostly with limited success. Checkpoint inhibition with PD-1, PD-L1, and CTLA-4 blockade has revolutionized treatment of various solid tumors. Increased expression of PD-L1 and PD-1 has been described on bone marrow AML blasts and infiltrating CD8+ T cells, with TP53-mutation, relapsed/refractory disease, and disease that has progressed after treatment with HMAs even more likely to express PD-L1.98 Previous exposure to HMAs has also been associated with upregulation of CTLA-4.99 Other biomarkers often associated with response to immunotherapies that have been identified in TP53-mutated AML include greater infiltration by CD8+ T-cells, resting memory NK/CD4+T cells, and higher tumor mutational burden.100
Trials have been completed involving checkpoint immune blockade either alone or in combination with other agents. Though activity was demonstrated with both AZA plus nivolumab in the relapsed/refractory setting and nivolumab plus cytarabine/idarubicin in the frontline, TP53 mutation was not predictive of response in the former and was associated with nonresponse in the latter.101,102 A phase 2 trial of pembrolizumab after high-dose cytarabine in the relapsed/refractory setting revealed a CR rate of 40% in the TP53-mutated population, although there were only 5 patients included in the study.103
Bispecific dual affinity retargeting antibodies (DARTs) have provided an additional immunotherapeutic approach in lymphoid neoplasms that has been more challenging in AML given its greater diversity of cell surface antigens. Flotetuzumab is a bispecific DART against CD3ε and CD123 which encourages the formation of an immunologic synapse between cytotoxic T cells and AML cells in an MHC-independent fashion.104 Given that CD123 expression is augmented in primary induction failure and early relapse AML, a phase 1/2 study was conducted involving this patient population where the CR/CRh/CRi rate was 30.0% and median OS was 10.2 months.105,106 A post-hoc analysis of bone marrow samples collected from patients on this trial demonstrated a CR in 47% (7/15) of TP53-mutated cases with a median OS of 10.3 months and two CRs that persisted for >6 months. Responders were also more likely to have higher tumor inflammation signature, FOXP3, CD8A, inflammatory cytokine, and PD1 gene expression scores at baseline.107 Novel combinations including immunotherapeutics are currently being assessed in clinical trials [Table 2].
Allogeneic Hematopoietic Stem Cell Transplantation
Allogeneic stem cell transplant is the most common consolidation strategy offered to young patients with adverse-risk AML, as it is generally the only reasonable chance for cure. Outcomes in the TP53-mutated population tend to be among the poorest. A retrospective analysis by the European Society for Blood and Marrow Transplantation of 139 patients with 17p abnormalities who underwent hematopoietic stem cell transplant (HCT) in CR1 revealed a 2-year OS and leukemia-free survival (LFS) of 28% and 24%, respectively.108 Another study of AML patients undergoing allogeneic HCT after achieving CR1 with intensive chemotherapy induction found TP53 mutation to be a member of the “adverse molecular-genetic profile” group associated with the worst outcomes, including a lower 2-year OS (24.9% vs 57.9%) and lower relapse-free survival (23.7% vs 57.9%) in comparison to patients not in this group.109 Despite these findings, some patients do appear to achieve long-term survival in the presence of certain clinical features. An analysis of 83 patients who underwent allogeneic HCT for TP53-mutated AML or MDS revealed three independent factors that predicted worse OS, including Karnofsky performance status ≤80%, HCT comorbidity index >4, and disease not in CR1/2 at the time of transplant. Patients with 0, 1, and ≥2 of these factors had one-year OS rates of 67%, 39%, and 17%, respectively.110 With the improved CR rates and lesser toxicity associated with many novel agents, allogeneic HCT will likely continue to remain an important tool in the curative approach to TP53-mutated AML.
Future Treatment Approaches
Several other strategies are currently being tested to overcome treatment resistance in TP53-mutated AML by direct targeting of the p53 protein in an effort to degrade its mutant form or restore its wild-type functions [Table 2]. Statins have demonstrated the ability to induce degradation of mutant p53 by reducing mevalonate-5-phosphate formation, which increases CHIP ubiquitin ligase-mediated degradation of mutant p53.111 Preclinical studies demonstrating the ability of these agents to synergize with doxorubicin and inhibit growth of TP53-mutated AML cells as well as a general vulnerability to mevalonate reduction in TP53-mutated tumors have inspired the opening of a phase 1 trial of atorvastatin in TP53-mutated malignancies.112–114 Arsenic trioxide has been shown to induce degradation of mutant p53 via a proteasomal pathway involving upregulation of Pirh2 E3 ligase, which is currently being studied in a clinical trial in combination with DEC and cytarabine in patients with TP53-mutated AML.115,116 Histone deacetylase inhibitors and heat shock protein 90 inhibitors have proven ability to degrade mutant p53, though this has not translated into clinical activity in early phase trials for AML and other myeloid neoplasms.117–121 MDM2 inhibitors have proven clinical activity in TP53 wild-type disease, though their use in TP53-mutant disease remains in question and may require the use of additional agents in combination.120 Other agents are aimed at restoring the wild-type function of mutant p53 or targeting the G2M point and other pathways that TP53-mutant cancers depend on. These agents have demonstrated promising preclinical data that has not yet been translated into clinical studies.121–123
Conclusion
The presence of a molecular aberration impairing the function of the TP53 gene has been identified as a poor prognostic indicator in AML, with dismal outcomes following standard induction chemotherapy and consolidation with allogeneic HCT. Higher VAFs have been associated with worse outcomes and will likely play a role in risk-stratifying patients with even finer precision than currently available models. The use of hypomethylating agents with or without VEN has produced higher response rates than induction chemotherapy, though median OS continues to be around a year with these treatments. Emerging treatments involve novel therapeutic mechanisms that have led to increased optimism about the ability to exploit TP53 mutations therapeutically. Efforts to improve immunotherapeutics have included targeting of new receptors and use of bispecific DARTs. Direct targeting of mutated p53 protein with eprenetapopt has led to some initial success. Although these therapies have demonstrated promising results in early phase clinical trials, larger randomized trials are still needed to confirm their benefit over approved treatments, and it may ultimately require combination of these therapies in order to achieve adequate responses that are also durable. Recent data have suggested that TP53-mutated AML and MDS with excess blasts (MDS-EB) should be one disease entity due to their similar biological behavior and survival outcomes.124 Given the relative rarity of TP53-mutated AML, this adaptation could help simplify future clinical trial design and eliminate existing drug approval barriers, allowing more efficient access to TP53 targeted therapy. As further treatments for AML and other myeloid disorders are developed, continued focus on outcomes in the TP53-mutated population will be required to identify additional agents with activity in this setting. TP53-mutated disease remains a challenging AML subgroup with a continued need for more effective treatment options.
Author Contributions
EMG and BAJ: All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
Brian A Jonas reports consultant/advisor for AbbVie, BMS, Celgene, Genentech/Roche, Gilead, GlycoMimetics, Jazz, Pfizer, Servier, Takeda, Tolero, and Treadwell; protocol steering committee for GlycoMimetics; data monitoring committee for Gilead; travel reimbursement from AbbVie; and research funding to his institution from 47, AbbVie, Accelerated Medical Diagnostics, Amgen, AROG, BMS, Celgene, Daiichi Sankyo, F. Hoffmann-La Roche, Forma, Genentech/Roche, Gilead, GlycoMimetics, Hanmi, Immune-Onc, Incyte, Jazz, Loxo Oncology, LP Therapeutics, Pfizer, Pharmacyclics, Sigma Tau, and Treadwell. The authors report no other conflicts of interest in this work.
References
1. Shallis RM, Wang R, Davidoff A, Ma X, Zeidan AM. Epidemiology of acute myeloid leukemia: recent progress and enduring challenges. Blood Rev. 2019;36:70–87. doi:10.1016/j.blre.2019.04.005
2. Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. doi:10.1056/NEJMoa1301689
3. Tyner JW, Tognon CE, Bottomly D, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562(7728):526–531. doi:10.1038/s41586-018-0623-z
4. Zjablovskaja P, Florian MC. Acute myeloid leukemia: aging and epigenetics. Cancers. 2019;12(1):103. doi:10.3390/cancers12010103
5. Maher M, Diesch J, Le Pannérer MM, Buschbeck M. Epigenetics in a spectrum of myeloid diseases and its exploitation for therapy. Cancers. 2021;13(7):1746. doi:10.3390/cancers13071746
6. Abelson S, Collord G, Ng SWK, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. 2018;559(7714):400–404.
7. Lal R, Lind K, Heitzer E, et al. Somatic TP53 mutations characterize preleukemic stem cells in acute myeloid leukemia. Blood. 2017;129(18):2587–2591. doi:10.1182/blood-2016-11-751008
8. Vousden KH, Lu X. Live or let die: the cell’s response to p53. Nat Rev Cancer. 2002;2(8):594–604. doi:10.1038/nrc864
9. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–310. doi:10.1038/35042675
10. Levine RL, Valk PJM. Next-generation sequencing in the diagnosis and minimal residual disease assessment of acute myeloid leukemia. Haematologica. 2019;104(5):868–871. doi:10.3324/haematol.2018.205955
11. Levine AJ. P53, the cellular gatekeeper for growth and division. Cell. 1997;88(3):323–331. doi:10.1016/S0092-8674(00)81871-1
12. Carr AM. Cell cycle. Piecing together the p53 puzzle. Science. 2000;287(5459):1765–1766. doi:10.1126/science.287.5459.1765
13. Bullock AN, Fersht AR. Rescuing the function of mutant p53. Nat Rev Cancer. 2001;1(1):68–76. doi:10.1038/35094077
14. Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4(10):793–805. doi:10.1038/nrc1455
15. Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Awakening guardian angels: drugging the p53 pathway. Nat Rev Cancer. 2009;9(12):862–873. doi:10.1038/nrc2763
16. Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24(17):2899–2908. doi:10.1038/sj.onc.1208615
17. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14(5):359–370. doi:10.1038/nrc3711
18. Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364(26):2496–2506. doi:10.1056/NEJMoa1013343
19. Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell. 2017;170(6):1062–1078. doi:10.1016/j.cell.2017.08.028
20. Lowe SW, Bodis S, McClatchey A, et al. p53 status and the efficacy of cancer therapy in vivo. Science. 1994;266(5186):807–810. doi:10.1126/science.7973635
21. Hientz K, Mohr A, Bhakta-Guha D, Efferth T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget. 2017;8(5):8921–8946. doi:10.18632/oncotarget.13475
22. Huang Y, Liu N, Liu J, et al. Mutant p53 drives cancer chemotherapy resistance due to loss of function on activating transcription of PUMA. Cell Cycle. 2019;18(24):3442–3455. doi:10.1080/15384101.2019.1688951
23. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–2221. doi:10.1056/NEJMoa1516192
24. Stengel A, Kern W, Haferlach T, Meggendorfer M, Fasan A, Haferlach C. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: an analysis of 3307 cases. Leukemia. 2017;31(3):705–711. doi:10.1038/leu.2016.263
25. Kuykendall A, Duployez N, Boissel N, Lancet JE, Welch JS. Acute myeloid leukemia: the good, the bad, and the ugly. Am Soc Clin Oncol Educ Book. 2018;38(38):555–573. doi:10.1200/EDBK_199519
26. Hou HA, Chou WC, Kuo YY, et al. TP53 mutations in de novo acute myeloid leukemia patients: longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015;5(7):e331. doi:10.1038/bcj.2015.59
27. Rose D, Haferlach T, Schnittger S, Perglerová K, Kern W, Haferlach C. Subtype-specific patterns of molecular mutations in acute myeloid leukemia. Leukemia. 2017;31(1):11–17. doi:10.1038/leu.2016.163
28. Cerquozzi S, Tefferi A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J. 2015;5(11):e366. doi:10.1038/bcj.2015.95
29. Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26(15):2157–2165. doi:10.1038/sj.onc.1210302
30. Bougeard G, Renaux-Petel M, Flaman JM, et al. Revisiting li-fraumeni syndrome from TP53 mutation carriers. J Clin Oncol. 2015;33(21):2345–2352. doi:10.1200/JCO.2014.59.5728
31. Hainaut P, Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res. 2000;77:81–137. doi:10.1016/s0065-230x(08)60785-x
32. Vom Brocke J, Schmeiser HH, Reinbold M, Hollstein M. MEF immortalization to investigate the ins and outs of mutagenesis. Carcinogenesis. 2006;27(11):2141–2147. doi:10.1093/carcin/bgl101
33. Wong TN, Ramsingh G, Young AL, et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature. 2015;518(7540):552–555. doi:10.1038/nature13968
34. Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat. 2002;19(6):607–614. doi:10.1002/humu.10081
35. Weisz L, Oren M, Rotter V. Transcription regulation by mutant p53. Oncogene. 2007;26(15):2202–2211. doi:10.1038/sj.onc.1210294
36. Baugh EH, Ke H, Levine AJ, Bonneau RA, Chan CS. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018;25(1):154–160. doi:10.1038/cdd.2017.180
37. Sabapathy K, Lane DP. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol. 2018;15(1):13–30. doi:10.1038/nrclinonc.2017.151
38. Yue X, Zhao Y, Xu Y, Zheng M, Feng Z, Hu W. Mutant p53 in cancer: accumulation, gain-of-function, and therapy. J Mol Biol. 2017;429(11):1595–1606. doi:10.1016/j.jmb.2017.03.030
39. Muller PA, Vousden KH. P53 mutations in cancer. Nat Cell Biol. 2013;15(1):2–8. doi:10.1038/ncb2641
40. Soenen V, Preudhomme C, Roumier C, Daudignon A, Lai JL, Fenaux P. 17p Deletion in acute myeloid leukemia and myelodysplastic syndrome. Analysis of breakpoints and deleted segments by fluorescence in situ. Blood. 1998;91(3):1008–1015. doi:10.1182/blood.V91.3.1008
41. Quintás-Cardama A, Hu C, Qutub A, et al. p53 pathway dysfunction is highly prevalent in acute myeloid leukemia independent of TP53 mutational status. Leukemia. 2017;31(6):1296–1305. doi:10.1038/leu.2016.350
42. Rücker FG, Schlenk RF, Bullinger L, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119(9):2114–2121. doi:10.1182/blood-2011-08-375758
43. Fontana MC, Marconi G, Feenstra JDM, et al. Chromothripsis in acute myeloid leukemia: biological features and impact on survival. Leukemia. 2018;32(7):1609–1620. doi:10.1038/s41375-018-0035-y
44. Döhner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453–474. doi:10.1182/blood-2009-07-235358
45. Grossmann V, Schnittger S, Kohlmann A, et al. A novel hierarchical prognostic model of AML solely based on molecular mutations. Blood. 2012;120(15):2963–2972. doi:10.1182/blood-2012-03-419622
46. Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367–1376. doi:10.1182/blood-2014-11-610543
47. Herold T, Rothenberg-Thurley M, Grunwald VV, et al. Validation and refinement of the revised 2017 European LeukemiaNet genetic risk stratification of acute myeloid leukemia. Leukemia. 2020;34(12):3161–3172. doi:10.1038/s41375-020-0806-0
48. Döhner H, Dolnik A, Tang L, et al. Cytogenetics and gene mutations influence survival in older patients with acute myeloid leukemia treated with azacitidine or conventional care. Leukemia. 2018;32(12):2546–2557. doi:10.1038/s41375-018-0257-z
49. Prochazka KT, Pregartner G, Rücker FG, et al. Clinical implications of subclonal TP53 mutations in acute myeloid leukemia. Haematologica. 2019;104(3):516–523. doi:10.3324/haematol.2018.205013
50. Short NJ, Montalban-Bravo G, Hwang H, et al. Prognostic and therapeutic impacts of mutant TP53 variant allelic frequency in newly diagnosed acute myeloid leukemia. Blood Adv. 2020;4(22):5681–5689. doi:10.1182/bloodadvances.2020003120
51. Bernard E, Nannya Y, Hasserjian RP, et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. 2020;26(10):1549–1556. doi:10.1038/s41591-020-1008-z
52. Kotler E, Shani O, Goldfeld G, et al. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol Cell. 2018;71(5):873. doi:10.1016/j.molcel.2018.08.013
53. Dutta S, Pregartner G, Rücker FG, et al. Functional Classification of TP53 Mutations in Acute Myeloid Leukemia. Cancers. 2020;12(3):637. doi:10.3390/cancers12030637
54. National Comprehensive Cancer Network. AML. Version 1.2022. Available from: https://www.nccn.org/professionals/physician_gls/pdf/aml.pdf.
55. Yanada M, Yamamoto Y, Iba S, et al. TP53 mutations in older adults with acute myeloid leukemia. Int J Hematol. 2016;103(4):429–435. doi:10.1007/s12185-016-1942-1
56. Metzeler KH, Herold T, Rothenberg-Thurley M, et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood. 2016;128(5):686–698. doi:10.1182/blood-2016-01-693879
57. Chiche E, Rahmé R, Bertoli S, et al. Real-life experience with CPX-351 and impact on the outcome of high-risk AML patients: a multicentric French cohort. Blood Adv. 2021;5(1):176–184. doi:10.1182/bloodadvances.2020003159
58. Lindsely RC, Gibson CJ, Murdock HM, et al. Genetic characteristics and outcomes by mutation status in a phase 3 study of CPX-351 versus 7+3 in older adults with newly diagnosed, high-risk/secondary acute myeloid leukemia. Blood. 2019;134(Supplement_1):15. doi:10.1182/blood-2019-124500
59. Subramaniam D, Thombre R, Dhar A, Anant S. DNA methyltransferases: a novel target for prevention and therapy. Front Oncol. 2014;1(4):80.
60. Plass C, Oakes C, Blum W, Marcucci G. Epigenetics in acute myeloid leukemia. Semin Oncol. 2008;35(4):378–387. doi:10.1053/j.seminoncol.2008.04.008
61. Fennell KA, Bell CC, Dawson MA. Epigenetic therapies in acute myeloid leukemia: where to from here? Blood. 2019;134(22):1891–1901. doi:10.1182/blood.2019003262
62. Welch JS, Petti AA, Miller CA, et al. TP53 and decitabine in acute myeloid leukemia and myelodysplastic syndromes. N Engl J Med. 2016;375(21):2023–2036. doi:10.1056/NEJMoa1605949
63. Short NJ, Kantarjian HM, Loghavi S, et al. Treatment with a 5-day versus a 10-day schedule of decitabine in older patients with newly diagnosed acute myeloid leukaemia: a randomised phase 2 trial. Lancet Haematol. 2019;6(1):e29–e37. doi:10.1016/S2352-3026(18)30182-0
64. Bally C, Adès L, Renneville A, et al. Prognostic value of TP53 gene mutations in myelodysplastic syndromes and acute myeloid leukemia treated with azacitidine. Leuk Res. 2014;38(7):751–755. doi:10.1016/j.leukres.2014.03.012
65. Müller-Thomas C, Rudelius M, Rondak IC, et al. Response to azacitidine is independent of p53 expression in higher-risk myelodysplastic syndromes and secondary acute myeloid leukemia. Haematologica. 2014;99(10):e179–e181. doi:10.3324/haematol.2014.104760
66. Ayala R, Rapado I, Onecha E, et al. The mutational landscape of acute myeloid leukaemia predicts responses and outcomes in elderly patients from the PETHEMA-FLUGAZA phase 3 clinical trial. Cancers. 2021;13(10):2458. doi:10.3390/cancers13102458
67. Uy GL, Duncavage EJ, Chang GS, et al. Dynamic changes in the clonal structure of MDS and AML in response to epigenetic therapy. Leukemia. 2017;31(4):872–881. doi:10.1038/leu.2016.282
68. Greve G, Schüler J, Grüning BA, et al. Decitabine Induces gene derepression on monosomic chromosomes: in vitro and in vivo effects in adverse-risk cytogenetics AML. Cancer Res. 2021;81(4):834–846. doi:10.1158/0008-5472.CAN-20-1430
69. DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17. doi:10.1182/blood-2018-08-868752
70. DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617–629. doi:10.1056/NEJMoa2012971
71. Aldoss I, Zhang J, Pillai R, et al. Venetoclax and hypomethylating agents in TP53-mutated acute myeloid leukaemia. Br J Haematol. 2019;187(2):e45–e48. doi:10.1111/bjh.16166
72. Kim K, Maiti A, Loghavi S, et al. Outcomes of TP53-mutant acute myeloid leukemia with decitabine and venetoclax. Cancer. 2021;127(20):3772–3781. doi:10.1002/cncr.33689
73. Pollyea DA, Pratz KW, Wei AH, et al. Outcomes in patients with poor-risk cytogenetics with or without TP53 mutations treated with venetoclax combined with hypomethylating agents. Blood. 2021;138:224. doi:10.1182/blood-2021-145639
74. Venugopal S, Shoukier M, Konopleva M, et al. Outcomes in patients with newly diagnosed TP53-mutated acute myeloid leukemia with or without venetoclax-based therapy. Cancer. 2021;127(19):3541–3551. doi:10.1002/cncr.33675
75. Morsia E, McCullough K, Joshi M, et al. Venetoclax and hypomethylating agents in acute myeloid leukemia: mayo clinic series on 86 patients. Am J Hematol. 2020;95(12):1511–1521. doi:10.1002/ajh.25978
76. Stahl M, Menghrajani K, Derkach A, et al. Clinical and molecular predictors of response and survival following venetoclax therapy in relapsed/refractory AML. Blood Adv. 2021;5(5):1552–1564. doi:10.1182/bloodadvances.2020003734
77. Wang YW, Tsai CH, Lin CC, et al. Cytogenetics and mutations could predict outcome in relapsed and refractory acute myeloid leukemia patients receiving BCL-2 inhibitor venetoclax. Ann Hematol. 2020;99(3):501–511. doi:10.1007/s00277-020-03911-z
78. DiNardo CD, Tiong IS, Quaglieri A, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood. 2020;135(11):791–803. doi:10.1182/blood.2019003988
79. Nechiporuk T, Kurtz SE, Nikolova O, et al. The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML cells. Cancer Discov. 2019;9(7):910–925. doi:10.1158/2159-8290.CD-19-0125
80. Swoboda DM, Sallman DA. The promise of macrophage directed checkpoint inhibitors in myeloid malignancies. Best Pract Res Clin Haematol. 2020;33(4):101221. doi:10.1016/j.beha.2020.101221
81. Chao MP, Jaiswal S, Weissman-Tsukamoto R, et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med. 2010;2(63):63ra94. doi:10.1126/scitranslmed.3001375
82. Jaiswal S, Jamieson CH, Pang WW, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271–285. doi:10.1016/j.cell.2009.05.046
83. Advani R, Flinn I, Popplewell L, et al. CD47 blockade by Hu5F9-G4 and rituximab in non-hodgkin’s lymphoma. N Engl J Med. 2018;379(18):1711–1721. doi:10.1056/NEJMoa1807315
84. Chao MP, Takimoto CH, Feng DD, et al. Therapeutic targeting of the macrophage immune checkpoint CD47 in myeloid malignancies. Front Oncol. 2020;9:1380. doi:10.3389/fonc.2019.01380
85. Sallman DA, Asch AS, Kambhampati S, et al. The first-in-class anti-CD47 antibody magrolimab combined with azacitidine is well-tolerated and effective in AML patients: phase 1B results. Blood. 2019;134:569. doi:10.1182/blood-2019-126271
86. Daver N, Konopleva M, Maiti A, et al. 371 Phase I/II study of azacitidine with venetoclax and magrolimab in patients with newly diagnosed older/unfit or high-risk acute myeloid leukemia (AML) and relapsed/refractory (R/R) AML. Blood. 2021;138:371. doi:10.1182/blood-2021-153638
87. Furukawa H, Makino T, Yamasaki M, et al. PRIMA-1 induces p53-mediated apoptosis by upregulating Noxa in esophageal squamous cell carcinoma with TP53 missense mutation. Cancer Sci. 2018;109(2):412–421. doi:10.1111/cas.13454
88. Rangel LP, Ferretti GDS, Costa CL, et al. p53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells. J Biol Chem. 2019;294(10):3670–3682. doi:10.1074/jbc.RA118.004671
89. Lambert JM, Gorzov P, Veprintsev DB, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009;15(5):376–388. doi:10.1016/j.ccr.2009.03.003
90. Haffo L, Lu J, Bykov VJN, et al. Inhibition of the glutaredoxin and thioredoxin systems and ribonucleotide reductase by mutant p53-targeting compound APR-246. Sci Rep. 2018;8(1):12671. doi:10.1038/s41598-018-31048-7
91. Birsen R, Larrue C, Decroocq J, et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica. 2021;107(2):403. doi:10.3324/haematol.2020.259531
92. Maslah N, Salomao N, Drevon L, et al. Synergistic effects of PRIMA-1(Met) (APR-246) and 5-azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia. Haematologica. 2020;105(6):1539–1551. doi:10.3324/haematol.2019.218453
93. Sallman DA, DeZern AE, Garcia-Manero G, et al. Eprenetapopt (APR-246) and azacitidine in TP53-mutant myelodysplastic syndromes. J Clin Oncol. 2021;39(14):1584–1594. doi:10.1200/JCO.20.02341
94. Cluzeau T, Sebert M, Rahmé R, et al. Eprenetapopt plus azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia: a Phase II study by the groupe francophone des myélodysplasies (GFM). J Clin Oncol. 2021;39(14):1575–1583.
95. Sallman DA, Komrokji RS, DeZern AE, et al. Long term follow-up and combined phase 2 results of eprenetapopt (APR-246) and azacitidine in patients with tp53 mutant myelodysplastic syndromes and oligoblastic acute myeloid leukemia. Blood. 2021;138:224. doi:10.1182/blood-2021-153286
96. Mishra A, Tamari R, DeZern A, et al. Allogeneic transplantation: long-term follow-up and disease recurrence phase ii trial of eprenetapopt (APR-246) in combination with azacitidine as maintenance therapy for TP53 mutated AML or MDS following allogeneic stem cell transplantation. Blood. 2021;138(Supplement 1):409. doi:10.1182/blood-2021-147962
97. Garcia-Manero G, Goldberg A, Winer E, et al. 3409 Phase I and expansion study of eprenetapopt (APR-246) in combination with venetoclax and azacitidine in TP53-mutant acute myeloid leukemia. Blood. 2021;138:3409. doi:10.1182/blood-2021-148940
98. Williams P, Basu S, Garcia-Manero G, et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer. 2019;125(9):1470–1481. doi:10.1002/cncr.31896
99. Yang H, Bueso-Ramos C, DiNardo C, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014;28(6):1280–1288. doi:10.1038/leu.2013.355
100. Wen XM, Xu ZJ, Jin Y, et al. Association analyses of TP53 mutation with prognosis, tumor mutational burden, and immunological features in acute myeloid leukemia. Front Immunol. 2021;12:717527. doi:10.3389/fimmu.2021.717527
101. Daver N, Garcia-Manero G, Basu S, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: a nonrandomized, open-label, phase II study. Cancer Discov. 2019;9(3):370–383. doi:10.1158/2159-8290.CD-18-0774
102. Ravandi F, Assi R, Daver N, et al. Idarubicin, cytarabine, and nivolumab in patients with newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome: a single-arm, phase 2 study. Lancet Haematol. 2019;6(9):e480–e488. doi:10.1016/S2352-3026(19)30114-0
103. Zeidner JF, Vincent BG, Ivanova A, et al. Phase II trial of pembrolizumab after high-dose cytarabine in relapsed/refractory acute myeloid leukemia. Blood Cancer Discov. 2021;2(6):616–629. doi:10.1158/2643-3230.BCD-21-0070
104. Johnson S, Burke S, Huang L, et al. Effector cell recruitment with novel Fv-based dual-affinity re-targeting protein leads to potent tumor cytolysis and in vivo B-cell depletion. J Mol Biol. 2010;399(3):436–449. doi:10.1016/j.jmb.2010.04.001
105. Kandeel EZ, El Sharkawy N, Hanafi M, Samra M, Kamel A. Tracing leukemia stem cells and their influence on clinical course of adult acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2020;20(6):383–393. doi:10.1016/j.clml.2019.11.018
106. Uy GL, Aldoss I, Foster MC, et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood. 2021;137(6):751–762. doi:10.1182/blood.2020007732
107. Vadakekolathu J, Lai C, Reeder S, et al. TP53 abnormalities correlate with immune infiltration and associate with response to flotetuzumab immunotherapy in AML. Blood Adv. 2020;4(20):5011–5024. doi:10.1182/bloodadvances.2020002512
108. Poiré X, Labopin M, Maertens J, et al. Allogeneic stem cell transplantation in adult patients with acute myeloid leukaemia and 17p abnormalities in first complete remission: a study from the acute leukemia working party of the European Society for Blood and Marrow Transplantation (EBMT). J Hematol Oncol. 2017;10(1):20. doi:10.1186/s13045-017-0393-3
109. Daher-Reyes G, Kim T, Novitzky-Basso I, et al. Prognostic impact of the adverse molecular-genetic profile on long-term outcomes following allogeneic hematopoietic stem cell transplantation in acute myeloid leukemia. Bone Marrow Transplant. 2021;56(8):1908–1918. doi:10.1038/s41409-021-01255-4
110. Ciurea SO, Chilkulwar A, Saliba RM, et al. Prognostic factors influencing survival after allogeneic transplantation for AML/MDS patients with TP53 mutations. Blood. 2018;131(26):2989–2992. doi:10.1182/blood-2018-02-832360
111. Parrales A, Ranjan A, Iyer SV, et al. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat Cell Biol. 2016;18(11):1233–1243. doi:10.1038/ncb3427
112. Martirosyan A, Clendening JW, Goard CA, Penn LZ. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: potential therapeutic relevance. BMC Cancer. 2010;10(1):103. doi:10.1186/1471-2407-10-103
113. Moon SH, Huang CH, Houlihan SL, et al. p53 represses the mevalonate pathway to mediate tumor suppression. Cell. 2019;176(3):564–580.e19. doi:10.1016/j.cell.2018.11.011
114. Burke LP, Kukoly CA. Statins induce lethal effects in acute myeloblastic leukemia [corrected] cells within 72 hours. Leuk Lymphoma. 2008;49(2):322–330. doi:10.1080/10428190701760011
115. Yan W, Zhang Y, Zhang J, Liu S, Cho SJ, Chen X. Mutant p53 protein is targeted by arsenic for degradation and plays a role in arsenic-mediated growth suppression. J Biol Chem. 2011;286(20):17478–17486. doi:10.1074/jbc.M111.231639
116. Yan W, Jung YS, Zhang Y, Chen X. Arsenic trioxide reactivates proteasome-dependent degradation of mutant p53 protein in cancer cells in part via enhanced expression of Pirh2 E3 ligase. PLoS One. 2014;9(8):e103497. doi:10.1371/journal.pone.0103497
117. Craddock CF, Houlton AE, Quek LS, et al. Outcome of azacitidine therapy in acute myeloid leukemia is not improved by concurrent vorinostat therapy but is predicted by a diagnostic molecular signature. Clin Cancer Res. 2017;23(21):6430–6440. doi:10.1158/1078-0432.CCR-17-1423
118. Prebet T, Sun Z, Ketterling RP, et al. Azacitidine with or without entinostat for the treatment of therapy-related myeloid neoplasm: further results of the E1905 North American Leukemia Intergroup study. Br J Haematol. 2016;172(3):384–391. doi:10.1111/bjh.13832
119. Lazenby M, Hills R, Burnett AK, Zabkiewicz J. The HSP90 inhibitor ganetespib: a potential effective agent for Acute Myeloid Leukemia in combination with cytarabine. Leuk Res. 2015;39(6):617–624. doi:10.1016/j.leukres.2015.03.016
120. Khurana A, Shafer DA. MDM2 antagonists as a novel treatment option for acute myeloid leukemia: perspectives on the therapeutic potential of idasanutlin (RG7388). Onco Targets Ther. 2019;12:2903–2910. doi:10.2147/OTT.S172315
121. Zhao D, Tahaney WM, Mazumdar A, Savage MI, Brown PH. Molecularly targeted therapies for p53-mutant cancers. Cell Mol Life Sci. 2017;74(22):4171–4187. doi:10.1007/s00018-017-2575-0
122. Winey M, Huneycutt BJ. Centrosomes and checkpoints: the MPS1 family of kinases. Oncogene. 2002;21(40):6161–6169. doi:10.1038/sj.onc.1205712
123. Toledo LI, Murga M, Zur R, et al. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. 2011;18(6):721–727. doi:10.1038/nsmb.2076
124. Grob T, Al Hinai AS, Sanders MA, et al. Molecular characterization of mutant TP53 acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood. 2022. 2021014472. doi:10.1182/blood.2021014472
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.