Back to Journals » Lung Cancer: Targets and Therapy » Volume 6
Targeting PD-1/PD-L1 in lung cancer: current perspectives
Authors González-Cao M, Karachaliou N, Viteri S, Morales Espinosa D, Teixidó C, Ruiz J, Molina-Vila MA, Santarpia M, Rosell R
Received 23 March 2015
Accepted for publication 23 April 2015
Published 31 July 2015 Volume 2015:6 Pages 55—70
DOI https://doi.org/10.2147/LCTT.S55176
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Pan-Chyr Yang
Video abstract presented by María Gonzalez-Cao and Cristina Teixidó
Views: 717
María González-Cao,1 Niki Karachaliou,1 Santiago Viteri,1 Daniela Morales-Espinosa,1 Cristina Teixidó,2 Jesús Sánchez Ruiz,3 Miquel Ángel Molina-Vila,2 Mariacarmela Santarpia,4 Rafael Rosell1,2,5,6
1Translational Cancer Research Unit, Instituto Oncológico Dr Rosell, Quirón Dexeus University Hospital, Barcelona, Spain; 2Pangaea Biotech SL, Barcelona, Spain; 3Centro Nacional de Investigación Oncología (CNIO), Madrid, Spain; 4Medical Oncology Unit, Human Pathology Department, University of Messina, Messina, Italy; 5Cancer Biology and Precision Medicine Program, Catalan Institute of Oncology, Germans Trias i Pujol Health Sciences Institute and Hospital, Campus Can Ruti, Badalona, Barcelona, Spain; 6Fundación Molecular Oncology Research, Barcelona, Spain
Abstract: Increased understanding of tumor immunology has led to the development of effective immunotherapy treatments. One of the most important advances in this field has been due to pharmacological design of antibodies against immune checkpoint inhibitors. Anti-PD-1/PD-L1 antibodies are currently in advanced phases of clinical development for several tumors, including lung cancer. Results from Phase I–III trials with anti-PD-1/PD-L1 antibodies in non-small-cell lung cancer have demonstrated response rates of around 20% (range, 16%–50%). More importantly, responses are long-lasting (median duration of response, 18 months) and fast (50% of responses are detected at time of first tumor evaluation) with very low grade 3–4 toxicity (less than 5%). Recently, the anti-PD-1 antibody pembrolizumab received US Food and Drug Administration (FDA) breakthrough therapy designation for treatment of non-small-cell lung cancer, supported by data from a Phase Ib trial. Another anti-PD-1 antibody, nivolumab, has also been approved for lung cancer based on survival advantage demonstrated in recently released data from a Phase III trial in squamous cell lung cancer.
Keywords: immunotherapy, immunoncology, cancer, checkpoint inhibitors
Introduction
Lung cancer is the most common cause of cancer death. Although improvements in overall survival (OS) and progression-free survival (PFS) have been achieved in recent years with the use of targeted drugs, 5-year survival for advanced disease remains very low.
Until recently, lung cancer was considered an immunoresistant disease, and immunotherapy treatment mainly focused on melanoma and renal cell carcinoma. During the last decade, understanding has grown with regard to the mechanisms by which the immune system recognizes tumors, as well as how cancer evades this recognition. Most of the promising new immunotherapies are antibodies directed against immune checkpoints.1
The most relevant immune checkpoint inhibitors are the cytotoxic T lymphocyte antigen-4 (CTLA-4), programmed death-1 (PD-1), lymphocyte activation gene 3 (LAG-3; also known as CD223), 2B4 (also known as CD244), B and T lymphocyte attenuator (BTLA; also known as CD272), T-cell membrane protein 3 (TIM-3; also known as HAVcr2), adenosine A2a receptor (A2aR), and the family of killer inhibitory receptors. Antibodies targeting these receptors, either alone or in combination with a second immune checkpoint blocker, have been shown to enhance antitumor immunity in animal models of cancer.
These immunotherapy treatments, such as the anti-CTLA-4 antibody ipilimumab and anti-PD-1 antibodies pembrolizumab and nivolumab, were first approved for advanced melanoma. Recently, results of clinical trials have also shown activity of anti-PD-1/PD-L1 antibodies in lung cancer,2,3 leading to US Food and Drug Administration (FDA) approval of pembrolizumab at the end of 2014 and nivolumab in March 2015.
Biological basis of cancer immunotherapy
It is well known that immunosuppression is a risk factor for cancer development. Mice lacking particular components of the innate or adaptive immune system are more susceptible to spontaneous and carcinogen-induced tumors as compared with wild-type mice. Also, immunosuppressed patients develop tumors more frequently than immunocompetent individuals.4 Solid tumors provoke an immunologic response that is normally insufficient for the elimination of cancer cells. Immunologic responses are induced by tumor-activated specific T lymphocytes CD8+ when the antigens are presented by antigen presenting cells (APCs) into peptides complexed with major histocompatibility complex class I (MHC-I), and the positive regulator CD28 on T-cells binds to CD80 (B-7 or B7-1) and CD86 (B7-2) on dendritic cells (Figure 1). The presence of cancer cells also increases release of soluble mediators such as interferon gamma (IFN-γ) and tumor necrosis factor-α (TNF-α) by CD4+ T helper lymphocytes.5–7
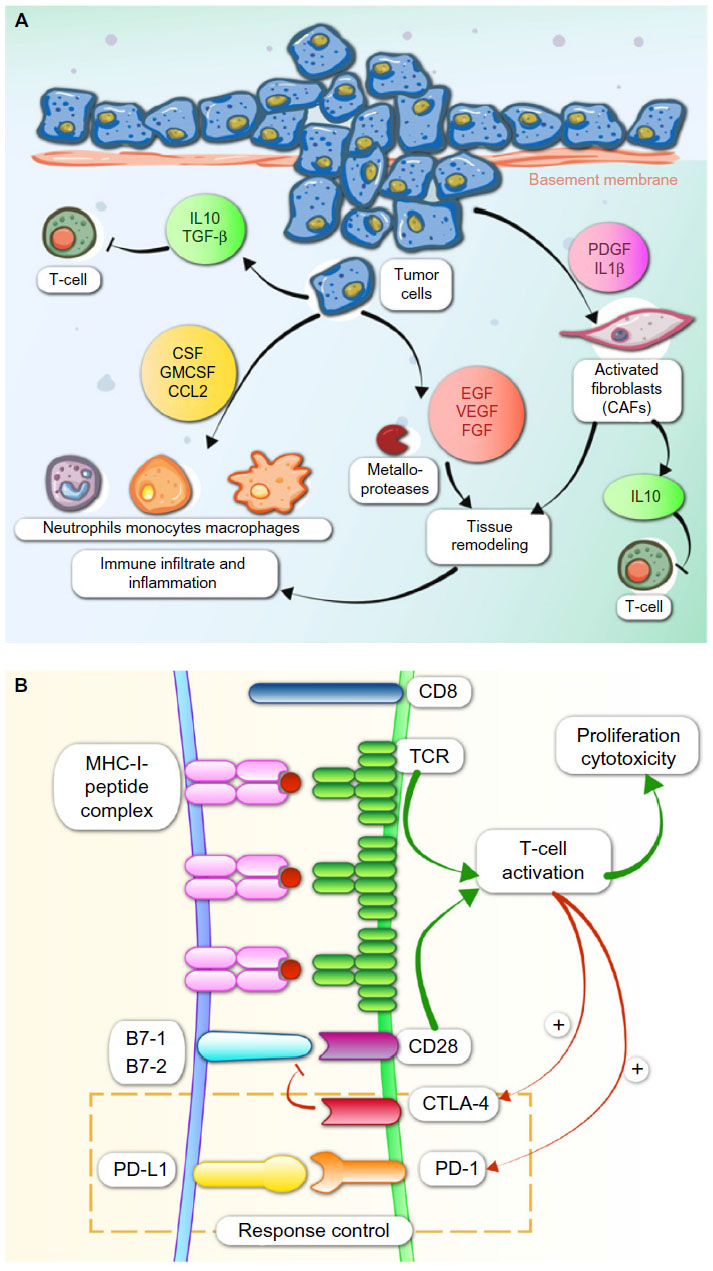 | Figure 1 Major immunological processes involved in cancer. |
Tumors have a peritumoral and intratumoral immune cell infiltrate consisting of macrophages, T-cells, B-cells, natural killer (NK) cells, neutrophils, dendritic cells, and eosinophils. These immunologic cells are enrolled due to the cytokine secretion by local inflammatory, stromal, and cancer cells. T-cells must migrate to the tumor and penetrate tumor stroma, which requires upregulation of specific receptors on the T-cells (Figure 1).8 To eradicate cancer, T-cells are required at high density in the tumor parenchyma. T helper 1 cells and cytotoxic T lymphocytes (CTLs) modulate the microenvironment to support an effective ongoing immune reaction. Number, location, and phenotype of the infiltrating T-cells affect prognosis.9,10 Strong infiltration of the tumor center and invasive tumor margin by CTLs confers good prognosis.11 Since CTL cells are often already present within cancers, therapeutics directed toward increasing their function or number may provide clinical benefit.12,13
Tumor-infiltrating lymphocytes (TILs) are a heterogeneous population. Recent studies have identified different subpopulations of TILs in samples from 24 melanoma patients. Expression of the checkpoint inhibitor molecules PD-1 (CD279), T-cell immunoglobulin and mucin protein 3 (TIM-3) and LAG-3 (CD223) on CD8+ TILs identified the autologous tumor reactive subpopulation. Only a fraction of the tumor reactive population expressed the costimulatory receptor 4-1BB (CD137). TCR-β deep sequencing revealed oligoclonal expansion of specific TCR-β clonotypes in CD8+PD-1+ compared with CD8+PD-1– TILs. PD-1 was the most overexpressed receptor in CD8+ TIL compared with TIM-3 (13%), 4-1BB (11%), and LAG-3 (6%). Antitumor activity of clones that coexpressed PD-1+/4-1BB+ or PD-1+/4-1BB– was similar but inferior to PD-1-negative clones.14
Inhibitory cells also exist within the tumor microenvironment, such as FOXP3+ regulatory T-cells (Tregs), cancer-associated fibroblasts, myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages. Suppressive Tregs are CD4+ T-cells that secrete cytokines such as transforming growth factor-β (TGF-β) and interleukin 10 (IL-10) to limit CTL function. MDSCs are a heterogeneous population of myeloid cells which share the ability to suppress T-cells and support generation of an immunosuppressive T-cell population and inhibit tumor lysis by NKs through production of arginase, expression of inducible nitric oxide synthase, and other mechanisms. Major MDSCs are tumor-associated macrophages and tumor-associated neutrophils that secrete platelet-derived growth factor (PDGF) and vascular endothelial growth factor (VEGF).
Tumor cells produce immunosuppressive factors such as IL-10 and TGF-β that produce recruitment of Tregs and MDSCs. In the case of TGF-β, also inhibition of NK activity is produced (Figure 1). Tumor cells induce the cytokine secretion by Tregs and macrophages and upregulation of Tregs by the expression of indoleamine 2,3-dioxygenase (IDO) by APCs. Tumor cells produce downregulation of the MHC-I or downregulation of antigen expression and upregulation of PD-L1 or other immunosuppressive molecules on tumor tissue. As a result, the destructive capacity of cytolytic cells (CD8+ T-cells and NK cells) is reduced.
Signaling pathways of checkpoint molecules and current therapeutics
A healthy immune system is one that can control T-cell stimulation and inflammation. Costimulatory molecules and immune checkpoint proteins work in harmony to prevent excessive inflammation and autoimmunity.
Chronic antigen stimulation induces coexpression of inhibitory receptors and is associated with T-cell hyporesponsiveness, termed “exhaustion”. The important role of inhibitory receptors in regulation of T-cell response in the context of cancer has recently been discovered.
Cancer behaves like a “self” protein, and immune checkpoints are activated to prevent inflammatory immunity developing against the “self”. Changing the balance of the immune system with checkpoint inhibition should allow it to be fully armed against cancer.
The major known inhibitory receptors implicated in downregulation of immune response are CTLA-4, PD-1, TIM-3, LAG-3, killer cell immunoglobulin-like receptor (KIR), and VISTA.15–33
Several therapeutic antibodies against these receptors have been developed, but anti-PD-1 and anti-PD-L1 antibodies are the most advanced in terms of clinical development in lung cancer.34
PD-1/PD-L1
The PD-1 receptor is an inhibitory receptor engaged by its ligands PD-L1 (also known as B7-H1 or CD274) and PD-L2 (also known as B7-DC or CD273).34 PD-1 is present in T activated cells, B-cells, monocytes, and NK cells. PD-L1 is expressed in several cells, such as tumor cells, some epithelial cells, lymphoid cells, myeloid cells, and APCs. Upon induction by cytokines such as IL-4, IL-10, and IFN-β, PD-L1 activates PD-1 on T-cells. PD-L1 is expressed in 27%–57% of non-small-cell lung cancer (NSCLC) and is located in the cell membrane or cytoplasm.34–36 PD-L1 can be expressed in tumor cells constitutively or as an acquired adaptive immunoresistance mechanism.37 PD-L2 is expressed mainly in APCs and some tumoral cells.
PD-1 activity occurs primarily within the tumor microenvironment, more peripherally than CTLA-4 action.38,39 PD-1 and its ligands, PD-L1 and PD-L2, like CTLA-4, also affect TCR and inhibit functional T-cell signaling. PD-1 later becomes operative in T-cell response once inflammatory cytokines have been released. PD-1/PD-L1 interactions limit inflammation and inhibit CTL activity. PD-1 activation inhibits CD8+ CTL proliferation, survival, and their effector function. It can also induce apoptosis of TILs and promote differentiation of CD4+ T-cells into Treg cells. PD-1 ligation by PD-L1 or PD-L2 recruits SHP2, resulting in membrane proximal decreases in TCR signaling by dephosphorylating the CD3-zeta chain (Figure 1).40 Interference with PD-L1/PD-1 signaling markedly inhibits TCR down-modulation leading to hyper-activated, proliferative CD8 T-cells, as assessed in in vitro and in vivo models. PD-L1 silencing accelerates antitumor immune responses and strongly potentiates dendritic cell (DC) antitumor capacities when combined with mitogen-activated kinase (MAPK) modulators that promote DC activation.41
Clinical trials with anti-PD-1/PD-L1 inhibitors in lung cancer
Lung inflammation due to chronic bronchitis or tobacco exposure increases production of TNF-α, IL-1, IL-6, and IL-8 by macrophages and decreases anti-inflammatory IL-10.42,43 TNF-α induces expression of NF-κB signaling and TWIST1, which leads to epithelial-to-mesenchymal transition which favors tumor growth and metastasis.44 Lung tumors also produce immunosuppressive factors such as IL-10, TGF-β, and the enzyme IDO that downregulate the expression of MHC-I or tumor-associated antigens, and upregulate PD-L1 expression or other inhibitory checkpoints.45–47
Results of early clinical trials including lung cancer patients treated with anti-PD-1 and anti-PD-L1 antibodies showed unexpected lasting responses in approximately 20% of patients.2,3 These results renewed interest in immunotherapy for treatment of lung cancer, and several drugs are now in preclinical and clinical development to block checkpoint-signaling molecules (Tables 1 and 2).
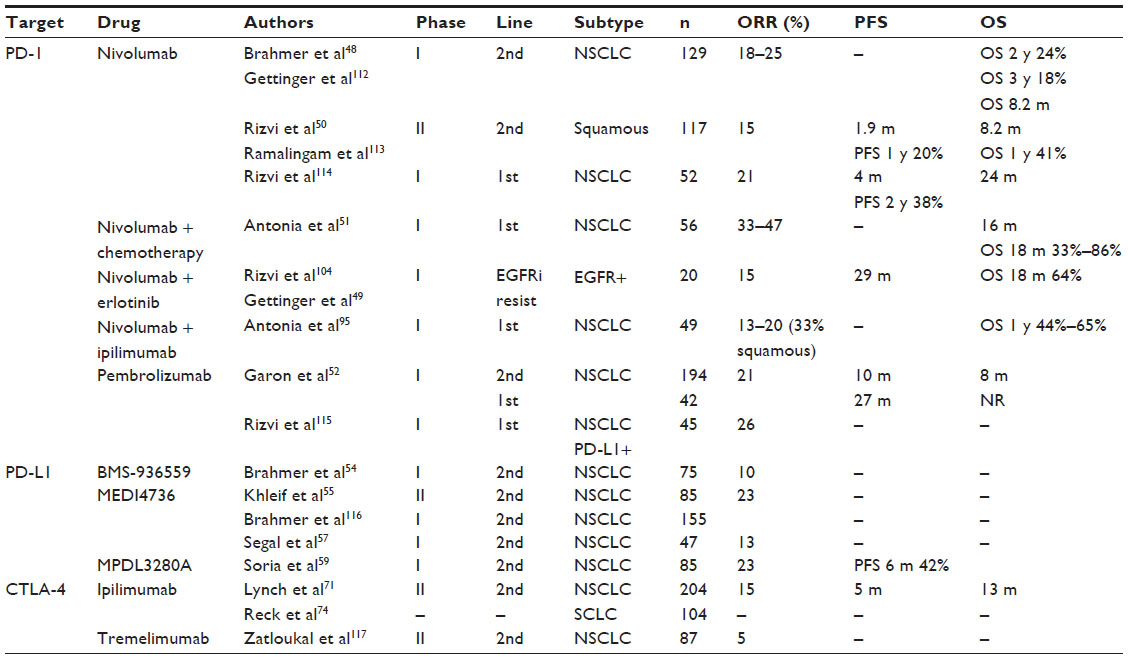 | Table 1 Results of clinical trials with anti-PD-1 and anti-PD-L1 antibodies |
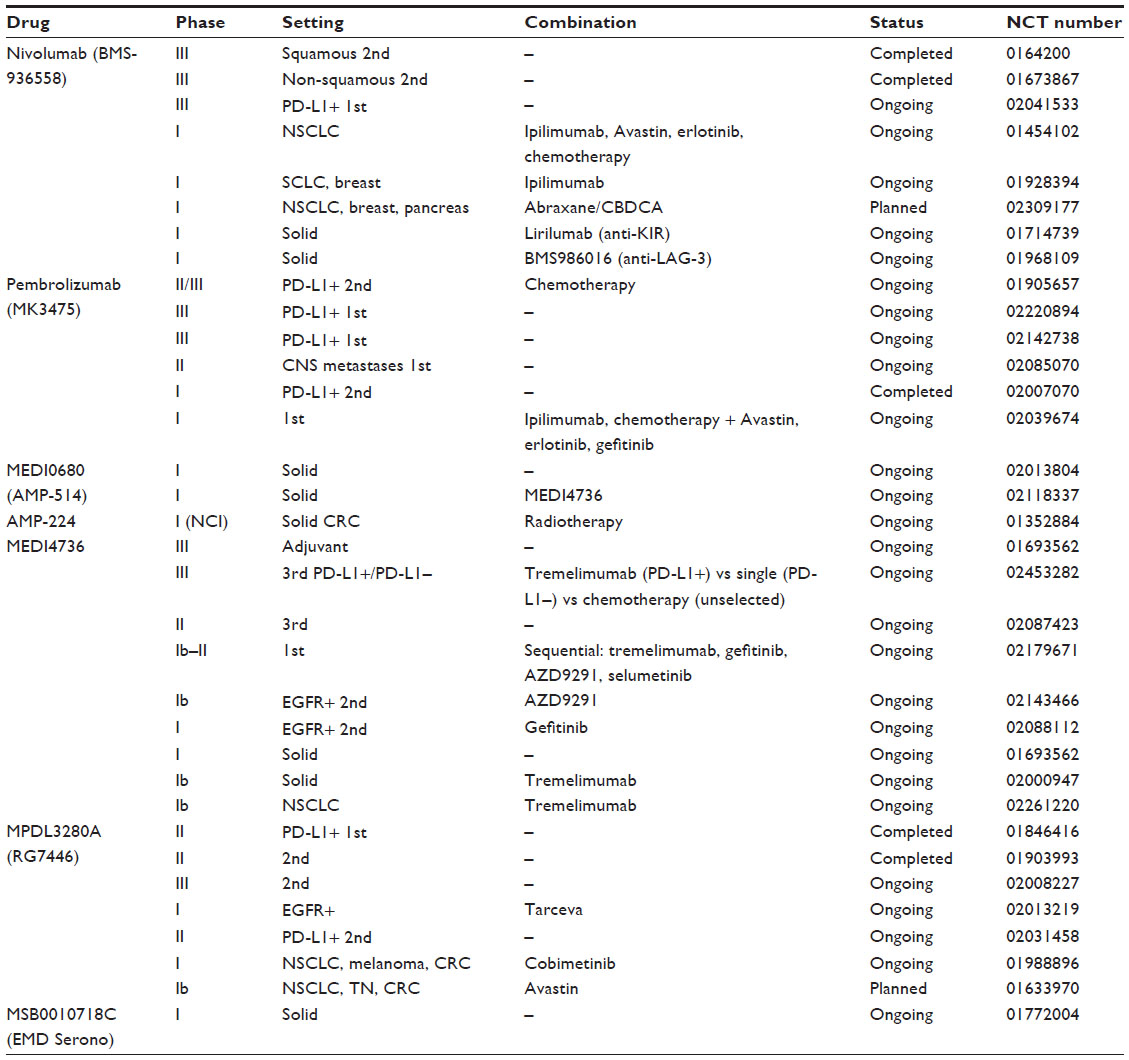 | Table 2 Ongoing clinical trials with anti-PD-1 and anti-PD-L1 drugs |
Anti-PD-1 drugs
Nivolumab
Nivolumab (BMS-936558) is a fully humanized Ig G4 monoclonal antibody from Bristol-Meyers Squibb (BMS) (New York, NY, USA). Results of a Phase I trial in 296 previously treated patients (including 122 NSCLC patients) were published in 2012 by Topalian et al.2 Nivolumab was administered every 2 weeks by intravenous infusion at different doses (0.1–10 mg/kg). Fourteen NSCLC (18%) patients of 76 evaluable for response had an objective response by RECIST 1.1 criteria, and responses were observed at all dose levels. Durable responses were seen both in non-squamous (seven of 57 patients, overall response rate [ORR] 12%) and squamous tumors (six of 18 patients, ORR 33%). The drug was well tolerated, with grade 3–4 toxic effects in 14% of patients.2 Long term follow-up of a Phase I trial of nivolumab in 129 heavily pretreated NCSLC patients showed an ORR of 14%–25%.48 Responses were rapid, with 50% of responders detected at first response evaluation (8 weeks after starting treatment). Median duration of response was 74 weeks (range, 6.1–133.9 weeks) and OS at 2 years was 14%.48 Updated results confirmed an OS rate at 3 years of 18%.49 The most common toxic effects were fatigue, anorexia, and diarrhea; grade 3–4 toxicity was found in 14% of patients, with pneumonitis in 7% of NSCLC patients.49
Phase III trials in NSCLC have now been completed. Recently, BMS communicated the OS advantage for nivolumab detected in the CheckMate 017 Phase III trial. This trial included 272 squamous NSCLC patients after prior progression to a platinum doublet-based chemotherapy regimen. Patients were randomized to nivolumab (3 mg/kg intravenously [iv] over 60 minutes every 2 weeks) versus standard of care, docetaxel (75 mg/m2 iv administered every 3 weeks). This trial included patients regardless of their PD-L1 status, and the primary endpoint was OS. In January 2015, the trial was stopped based on an assessment conducted by the independent Data Monitoring Committee which concluded that the study had met its endpoint, demonstrating superior OS in patients receiving nivolumab compared to docetaxel. The prespecified interim analysis was conducted when 199 events (86% of the planned number of events for final analysis) were observed (86 in the nivolumab arm and 113 in the docetaxel arm). Median OS was 9.2 months in the nivolumab arm (95% confidence interval [CI]: 7.3, 13.3) and 6 months in the docetaxel arm (95% CI: 5.1, 7.3). The hazard ratio was 0.59 (95% CI: 0.44, 0.79; P=0.00025), translating to a 41% reduction in the risk of death with nivolumab compared to docetaxel.
The safety profile of nivolumab in squamous NSCLC was established by CheckMate 063, a Phase II single-arm, open-label, multinational, multicenter trial of nivolumab administered as a single agent in patients with metastatic squamous NSCLC who had progressed after receiving a platinum-based therapy and at least one additional systemic treatment regimen (n=117).50 Patients received 3 mg/kg of nivolumab every 2 weeks. This trial included patients regardless of their PD-L1 status. The most common adverse reactions (reported in ≥20% of patients) were fatigue (50%), dyspnea (38%), musculoskeletal pain (36%), decreased appetite (35%), cough (32%), nausea (29%), and constipation (24%). Serious adverse reactions occurred in 59% of patients receiving nivolumab. The most frequent serious adverse reactions reported in ≥2% of patients were dyspnea, pneumonia, chronic obstructive pulmonary disease exacerbation, pneumonitis, hypercalcemia, pleural effusion, hemoptysis, and pain. Nivolumab was discontinued due to adverse reactions in 27% of patients. Twenty-nine percent of patients receiving nivolumab had a drug delay due to an adverse reaction. With at least 10 months of minimum follow-up for all patients, the study’s primary endpoint of confirmed ORR was 15% (17/117) (95% CI: 9, 22), all partial responses. The median time to onset of response was 3.3 months (range, 1.7–8.8 months) after the start of nivolumab treatment. Seventy-six percent of nivolumab responders (13/17 patients) had ongoing responses with durability of response ranging from 1.9+ to 11.5+ months; ten of these 17 (59%) patients had durable responses of 6 months or longer.50
Results from the Phase III trial of nivolumab in second-line non-squamous NSCLC are pending (NCT01673867).
Other studies are ongoing, such as the Phase III trial comparing nivolumab versus chemotherapy in first line in PD-L1-positive NSCLC patients (NCT02041533) and a Phase I multi-arm trial testing in NSCLC several combinations of nivolumab with chemotherapy, Avastin, ipilimumab, or erlotinib (CheckMate 012, NCT01454102). Preliminary results of the combination with platinum doublets demonstrated that, although tolerable, response rate was similar to that normally obtained with chemotherapy in the first-line setting (ORR 33%–47%). Median OS was 51–65 weeks with ongoing responses after 30 months (OS rate at 18 months was 86% in the arm of nivolumab at 5 mg/kg dose combined with carboplatin and Taxol).51
Pembrolizumab
Pembrolizumab (MK-3475), formerly named lambrolizumab, is a monoclonal anti-PD-1 antibody from Merck Sharp and Dohme (Kenilworth, NJ, USA). It has received FDA Breakthrough Therapy designation for lung cancer treatment, supported by data from a Phase Ib trial in previously treated patients. Results showed a median survival of 51 weeks and an ORR of 20%–25%. The drug was well tolerated, with 10% grade 2–3 adverse events.52 Update results from this study were also presented during the 2014 ESMO Meeting: in 236 patients (including 46 not previously treated), ORR was 21% (range, 16%–27%) and 26% in treatment-naïve patients.53
Based on these results, a randomized Phase II/III trial of two doses of pembrolizumab (2 mg/kg iv every 3 weeks and 10 mg/kg every 3 weeks) versus docetaxel (75 mg/m2 iv every 3 weeks) in patients with advanced PD-L1-positive NSCLC is being conducted (NCT01905657), as is a Phase I/II trial of pembrolizumab in combination with ipilimumab or chemotherapy (NCT02039674) and a Phase I trial in PD-L1-positive NSCLC (NCT02007070). Other trials of pembrolizumab in a first-line setting versus platinum-based chemotherapy are planned in PD-L1-positive NSCLC patients using a fixed dose of 200 mg iv every 3 weeks for up to 35 treatments (NCT02220894, NCT02142738).
Anti-PD-L1 drugs
BMS-936559
The second study presented in 2012 which renewed interest in immunotherapy for lung cancer tested the anti-PD-L1 antibody BMS-936559, a fully humanized Ig G4 monoclonal antibody. In 207 patients with advanced solid tumors, including 75 NSCLC patients, ORR was close to 10% (five of 49 evaluable patients, including one case of squamous cell lung cancer and four cases of non-squamous) and stable disease lasting at least 6 months in six (18%) patients. Response duration was 2.3–16.6 months (median duration not reached). However, development of BMS-936559 for cancer was halted due to BMS’ decision.54
MEDI4736
MEDI4736 (AstraZeneca plc, London, UK) is an antibody engineered with a triple mutation in the Fc domain to avoid Fc-mediated antibody-dependent cell-mediated cytotoxicity. Preliminary results from the Phase I study of patients with solid tumors reported clinical activity and durable disease stabilization in different tumor types, including NSCLC, with no dose-limiting toxicities or grade 3–4 treatment-related adverse events.55 In the Phase II trial, the drug was tested on 85 pretreated NSCLC patients. From 53 evaluable patients, 12 had an objective response (23%).55 At the 2014 ASCO Meeting, results from a Phase I multi-arm expansion study of MEDI4736 were presented. At that time, the dose-escalation phase had been completed for doses of 0.1–10 mg/kg every 2 weeks with extension to 15 mg/kg every 3 weeks. MEDI4736 was well tolerated at all tested doses, with no treatment-related serious adverse events such as colitis, hyperglycemia, or pneumonitis of any grade.56 Segal et al presented preliminary data from the ongoing study of MEDI4736 at a dosage of 10 mg/kg every 2 weeks for 1 year in 346 patients with solid tumors, including 143 with NSCLC. Median treatment duration was 8 weeks. As of May 18, 2014, there were very few (6%) grade 3–4 drug-related serious adverse events. Clinical activity was observed as early as 6 weeks, with maintenance for over 67 weeks and after finishing active therapy; the ORR rate in NSCLC was 13%.57 A Phase I trial is currently evaluating safety of doses of 0.1–10 mg/kg every 2 or 15 mg/kg every 3 weeks in NSCLC (NCT01693562). A Phase III trial is testing MEDI4736 (10 mg/kg iv every 2 weeks) in the adjuvant setting after chemoradiotherapy for unresectable stage III NSCLC (NCT02125461). In addition, other ongoing trials are: a Phase II trial in previously treated NSCLC (NCT02087423); a Phase Ib trial in NSCLC testing the combination of MEDI4736 with tremelimumab (NCT02000947); several Phase I trials of MEDI4736 in combination with small molecules (NCT02179671, NCT02143466); and a Phase III trial for pretreated NSCLC patients testing the combination of MEDI4736 with tremelimumab for PD-L1-negative patients and MEDI4736 as single agent for PD-L1-positive patients versus chemotherapy (NCT02352948).
MPDL3280A
MPDL3280A is an engineered IgG anti-PD-L1 antibody with modified Fc domain that prevents antibody-dependent cell-mediated cytotoxicity in other immune cells expressing PD-L1. Preliminary results of the Phase I study of MPDL3280A in pretreated patients with advanced NSCLC have been reported: a safety analysis has been presented from 85 patients included, and efficacy analysis from 53 patients. Response rate was 23% both in squamous and non-squamous NSCLC. Twenty-four-week PFS was 45% and median time to response 12 weeks; some responders had an early response at 6 weeks.58,59 Phase II studies in first line (NCT01846416) and second line (NCT01903993) have now been completed, and results are pending. Relevant ongoing trials are a Phase III trial testing MPDL3280A (1,200 mg iv every 3 weeks) versus docetaxel (75 mg/m2 iv every 3 weeks) (NCT02008227) after failure to chemotherapy, and a Phase I trial testing the combination with erlotinib in EGFR-positive patients (NCT02013219).
Predictive factors of response to anti-PD-1/PD-L1 antibodies
PD-L1 immunohistochemical expression
Some studies have found a significant correlation of the activity of anti-PD-1 and anti-PD-L1 antibodies with PD-L1 expression measured by immunohistochemistry (IHC) in tumor samples.
Results from these studies are difficult to interpret, since each pharmaceutical company uses its own antibody for IHC analysis. Some studies look at PD-L1 expression in cancer cells (such as those from BMS), while others focus on expression of PD-L1 in TILs (such as Roche antibodies).
Also, cut-off levels vary: most studies with nivolumab use 5% of membrane staining among more than 100 evaluable tumor cells and found positive PD-L1 tumors in 40%–50% of NSCLC patients. Studies with pembrolizumab mainly use 1% as a cut-off, but most also test other cut-off points for defining “negative”, “light positive”, and “intense positive” patients (over a cut-off of 50% of tumor cells and stroma). According to these criteria, there are 30% high intense positive PD-L1 lung cancer patients. Studies with MPDL3280A demonstrated that definition of positive PD-L1 patients using the criterion of 5% staining on TIL cells versus 5% of staining on tumor cells was a better predictive factor of response. Using this criterion, there are 25% positive lung cancer patients for PD-L1 expression (Table 3).
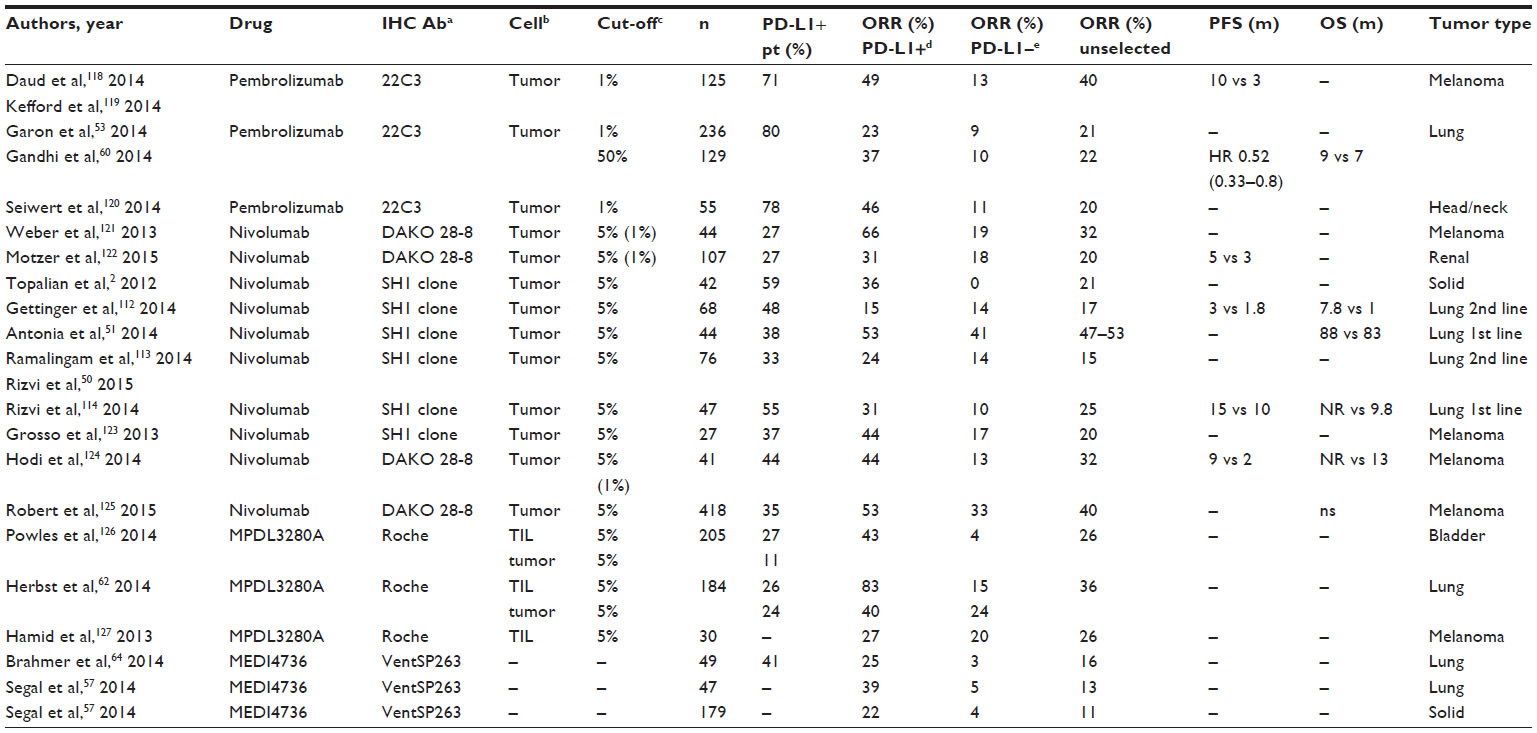 | Table 3 Correlation of PD-L1 expression by IHC analysis and anti-tumoral activity |
Another issue which complicates matters is that time of performing sample biopsy is not always reported in these studies. This could cause important bias, as PD-L1 expression changes during disease evolution (Table 3).60
Most studies performed in NSCLC looking at the predictive value of PD-L1 expression by IHC have demonstrated a higher response rate in PD-L1-positive patients, with a median response rate of 38% (range, 23%–83%) versus 7% (range, 0%–15%) in PD-L1-negative cases,52,55,57,59–64 but results are not definitive, and further studies are required. Some positive preliminary data have not yet been fully confirmed after including more patients in the analysis. For example, the first data from the Keynote 001 trial of pembrolizumab presented in 2013 showed, in 33 patients, ORR in nine PD-L1+ patients of 67% versus 4% in 24 PD-L1-negative cases.65 Updated results including 129 patients from the same Keynote 001 trial showed an ORR of 37% versus 10% for PD-L1-positive and -negative cases, respectively.53 More recent data from the same company, using a higher cut-off of 50% in 146 patients, demonstrated an ORR of 37% in 97 PD-L1-positive patients, versus 7% in PD-L1-negative, and median OS of 9.3 months versus 7.3 months, respectively.60
Correlation of PD-L1 expression with survival benefit is less clear, as longer follow-up is required. Although some Phase I studies have presented data on PD-L1 expression by IHC and a positive correlation with survival, others have not shown significant differences.53,60,63
The predictive value of PD-L1 expression by IHC is not the same in all tumor subtypes. For example, in melanoma, patients with PD-L1-negative tumors have a considerable chance of response. Several studies looking for a better predictive model are ongoing, and a better predictive model for response to anti-PD-1 drugs in melanoma has been described using a multi-marker panel, including also PD-1 expression and CD8 cell infiltration into the tumor and at the tumor margin.66,67
Smoking history
Tobacco is the primary cause of lung cancer; moreover, lung cancer related to tobacco smoking usually is more aggressive than lung cancer in nonsmokers and usually has mutations in non-targetable genes such as KRAS, but not in genes susceptible to treatment with very active target drugs such as EGFR or ALK inhibitors.
Tobacco smoke carcinogens interact with DNA and cause genetic changes that produce cancer development as well as numerous neoantigens that can be immunogenic.68,69 Several studies with PD-1 and PD-L1 drugs have shown a higher response rate in patients with a smoking history.
Results of pembrolizumab in 236 lung cancer patients demonstrated an ORR of 21% (range, 16%–27%); however, most patients had smoking history (ORR in former or current smokers was 27% versus 9% in never-smokers).52 Also, in a Phase I study of MPDL3280A in pretreated patients with advanced NSCLC, ORR was 23%, with an ORR in former/current smokers of 26% versus 10% in never-smokers.58,59 In a substudy of 88 patients treated in the Phase I study of nivolumab,2 ORR was significantly higher in former/current smokers (20/75, 27%, 95% CI: 17%–38%) versus minimal/never-smokers (0/13, 0%, 95% CI: 0%–25%) (P=0.034). Responders had significantly more tobacco exposure than nonresponders (median 50 pack-years versus 36 pack-years, P=0.036).49,70
Histological lung cancer subtypes
Malignant epithelial tumors of the lung are classified by the World Health Organization (WHO)/International Association for the Study of Lung Cancer (IASLC). There are two main subtypes: small-cell lung cancer (SCLC) and NSCLC. NSCLC is further classified into several subgroups; the most frequent are squamous, adenocarcinoma, large cell carcinoma, and sarcomatoid tumors.
Although higher activity of ipilimumab (an anti-CTLA-4 antibody) or anti-PD-1 or anti-PD-L1 drugs has been demonstrated in squamous lung cancer,71 this correlation was not observed in the clinical trials performed.
In the first trials, a trend toward a higher activity in squamous tumors led to development of specific trials for this subgroup. Afterward, this correlation was not further observed. Nevertheless, in squamous lung cancer with loss of PTEN and LKB1, some features that could preclude higher sensitivity to PD-1/PD-L1 blockade have been observed. These tumors have a high number of TILs with increased expression of PD-1, a high rate of tumor-associated neutrophils, few TAAMs, and only modest expression of TIM-3 and LAG-3.72 Another histological subtype with significant expression of PD-L1 is sarcomatoid NSCLC, with PD-L1 expressed in 69% of cases compared with 27% in non-sarcomatoid NSCLC.73
In SCLC, ipilimumab combined with chemotherapy has better results when used after two single cycles of chemotherapy than when it was combined from the first treatment cycle. Phased ipilimumab, concurrent ipilimumab, and control, respectively, were associated with median irPFS of 6.4, 5.7, and 5.3 months (P=0.03), and median OS of 12.9, 9.1, and 9.9 months.74 It could be interesting to analyze whether biological changes due to chemotherapy could improve the chances of responding to ipilimumab. Ipilimumab is currently being tested in Phase III and Phase II clinical trials in extensive-stage SCLC (NCT01450761, NCT01331525) and in one Phase II trial for limited-stage (NCT02046733). In addition, a Phase I trial is ongoing to test the combination of nivolumab plus ipilimumab in different tumor types including SCLC (NCT01928394).
Resistance to anti-PD-1/PD-L1 treatments
The majority of patients treated with anti-PD-1/PD-L1 drugs do not respond to treatment (called “primary resistance”). Also, there are patients who, after achieving a clinical response, finally develop resistance after a variable period of time (called “secondary acquired resistance”).
Mechanisms of resistance to anti-PD-1 oranti-PD-L1 antibodies are poorly understood.
Preexisting T-cell antitumor immunity has been hypothesized as a prerequisite for the activity of these drugs. Tumor PD-L1 expression correlates spatially with the presence of infiltrating CD8+ T-cells in melanoma, suggesting that tumor cells can upregulate PD-L1 in response to immune pressure and secretion of IFN-γ by CD8+ cells, a mechanism known as “adaptive immune resistance”.75,76 However, other tumors upregulate PD-L1 expression without infiltration by CD8 cells, as a result of other factors, such as EGFR mutations.77 These data suggest that the efficacy of checkpoint inhibitors may require the presence of an endogenous antitumor T-cell response. In fact, the augmentation of antitumor T-cell responses with vaccines, immune stimulatory agonist, or intratumoral oncolytic virus has been shown to improve responses to checkpoint inhibitors in murine models.78,79 The use of antibodies that are agonists of the stimulatory receptors of the TNF receptor family as such OX40 (CD134),80 4-1 BB (CD137), CD27, and CD30 is of particular interest. When activated, each of these receptors can enhance cytokine production and T-cell proliferation in response to T-cell receptor signaling.
There are multiple factors related to cancer cells or tumor microenvironment that can justify primary resistance. It has been demonstrated that tumors with a high number of mutations have a greater chance of achieving response to anti-PD-1 drugs or anti-CTLA-4 antibodies. Some tumor mutations are translated into neoantigens, rendering the tumor cells recognizable to the immune cells.68 Those tumors without these neoantigens could be resistant to anti-PD-1 or PD-L1 antibodies. Also, another factor of primary resistance could be the expression or coexpression of other inhibitory immune checkpoints as CTLA-4, B7-H3, LAG-3, IDO, and TIM-381 or the presence of tumor-infiltrating suppressor immune cells62 (MDSCs, macrophages, and fibroblast-activating protein+ [FAP+] mesenchymal cells).82,83 When these immune cells do not express PD-L1, the chance of responding to anti-PD-1/PD-L1 treatments is decreased. When the immune suppression is not related to PD-L1 expression on the immune infiltrating cells, blocking PD-L1 is not effective. Finally, tumors with activation of the TGF-β pathway are immunoresistant. In some studies, TGF-β activation, related to tumor hypoxia and Nanog (a stemness-associated transcription factor), correlates with preexisting and acquired immunoresistance.84
Different patterns of immune resistance based on the immune infiltration have been described in tumor biopsies: when tumors have no tumor-infiltrating immune cells, this is known as “immunological ignorance” when the infiltrating cells have minimal PD-L1 expression, this is called “nonfunctional immune response” and finally, when the infiltrating cells are only around the tumor, this is known as “excluded infiltrate”.62
Mechanisms involved in acquired secondary resistance are very poorly understood. Responses to anti-PD-1/PD-L1 drugs are durable, but longer follow-up is necessary. In melanoma patients, we have data about the long duration of response to anti-CTLA-4 antibodies, with a follow-up of more than 10 years without progression.85 Tumor cells or tumor-infiltrating immune cells could upregulate other checkpoint inhibitors or release immunosuppressive cytokines after an initial response to anti-PD-1/PD-L1 antibodies. Also, treatment could target tumor clones without MHC-I expression or cells with other defects in antigen presentation, which could be a cause of secondary resistance.
Future strategies
Checkpoint blockage combination
Although the function of checkpoints has been identified individually, nowadays it is well known that the coexpression of these molecules is common in tumor-specific T-cell lymphocytes. Preclinical and clinical studies show that the pathways activated by these different checkpoints are not redundant; therefore, the combination of inhibitors of different checkpoints could have a synergistic activity.86–93
Blockade of co-inhibitory molecules such as CTLA-4, PD-1, and LAG-3, or enhancement of costimulatory molecules, such as OX40, glucocorticoid-induced TNF receptor (GITR), and 4-1BB, can increase antitumor T-cell responses.
The combination of ipilimumab (anti-CTLA-4 antibody) and nivolumab (anti-PD-1 antibody) in melanoma patients showed a high response rate of close to 60% and 2-year survival of 80%.94 Results in lung cancer with the same combination were reported at the 2014 ASCO meetings. In 49 patients treated in first line, ORR was 13%–20% and 1-year OS was 44%–65%, with manageable toxicity.95
Other ongoing trials are testing the combination of several anti-PD-L1 antibodies with anti-CTLA-4 antibodies: a Phase Ib of MEDI3475 combined with tremelimumab (NCT02000947); a Phase I multi-arm trial of nivolumab plus ipilimumab (NCT01454102); a Phase I trial in SCLC and other tumor subtypes of nivolumab plus ipilimumab (NCT01928394); a Phase I study of lirilumab (BMS-986015, an anti-KIR antibody) in combination with nivolumab in patients with advanced solid tumors, including lung cancer (NCT01714739); a Phase I trial of BMS-986016 (an anti-LAG-3 antibody) with or without nivolumab for patients with solid tumors, including lung cancer (NCT01968109); and a Phase I trial of lirilumab in combination with ipilimumab for patients with selected advanced tumors, including lung cancer (NCT01750580) (Table 2).
Potential mechanisms leading to increased immunotherapy activity upon MAPK pathway blockade
Several molecular subtypes in NSCLC have been described; in adenocarcinomas, the most frequent alterations are mutations in KRAS, BRAF, EGFR, HER2, MET, FGFR1-2, and fusion genes involving ALK, ROS1, NRG1, neurotrophic tyrosine kinase receptor type 1 (NTRK1), and RET. In squamous cell carcinomas, different subtypes exist, such as those with mutations in genes of the PI3K pathway, in FGFR1-3m and in discoidin domain-containing receptor 2 (DDR2).40 There is a strong scientific rationale for sequentially or concurrently combining targeted drugs against these specific molecular alterations with immune checkpoint blockage.
Apoptosis and necrosis produced by target drugs on cancer cells result in release of tumor antigens that will presumably be available to DCs for cross-presentation.96 Reactivation of MAPK and PI3K signaling leads to c-Jun- and STAT3-dependent enhancement/promotion of PD-L1 expression. This likely involves AP-1-dependent enhancer and JAK3/STAT3-dependent PD-L1 promoter. Also, it has been shown that, in some cells, MEK inhibition and STAT3 inhibition could decrease PD-L1 expression depending on the different resistance mechanisms activated.97 It has been demonstrated that treatment with target drugs increases PD-L1 expression and TIL infiltration in responding tumors, suggesting that previous or concomitant treatment with target inhibitors could facilitate immune response to anti-PD-1 antibodies.98
EGFR-positive lung cancer has higher levels of PD-L1 expression than KRAS lung cancer. In animal models, activity of anti-PD-L1 antibodies is higher in EGFR-positive lung cancer.77 An Italian group analyzed human biopsies from 123 NSCLC patients and found 43 patients PD-1+, 68 PD-L1+, 56 EGFR mutant, and 29 KRAS mutant with a significant correlation of EGFR mutation and PD-L1 expression, and KRAS mutation with PD-1 expression. In addition, of 99 EGFR-positive patients treated with EGFR inhibitors, survival was better for those with PD-L1+ tumors.99 Others oncogenes such as HER2 and ALK harbor intrinsic immunogenicity which elicits CD4+ and CD8+ T-cell responses.100 For ALK-positive tumors, crizotinib has demonstrated an immunogenic cell death effect.101
Tumors responding to targeted therapies are enriched by tumor-propagating cells (TPCs). TPCs have the ability to self-renew, are slow cycling, and have stem cell antigen expression, such as Sca1+ or NGFR+.102 Most TPCs (Sca1+NGFR+) in lung cancer are PD-L1-positive (69%), whereas only 39% of Sca1+NGFR– or 32% of Sca1–NGFR+ are PD-L1+.103
A Phase I trial of nivolumab plus erlotinib in 20 EGFR+ resistant patients demonstrated ORR of 15% with median PSF of 29 months and an OS rate at 18 months of 64%. No toxic side effects were observed.104
Clinical trials testing the combination of EGFR inhibitors gefitinib and erlotinib or second-generation inhibitors such as AZ9291 are ongoing with several immune checkpoint inhibitors such as ipilimumab (NCT01998126), MEDI4736 (NCT02179671, NCT02143466), nivolumab (NCT01454102), MPDL3280A (NCT02013219), and tremelimumab (NCT02040064). Also, other targeted drugs like selumetinib or ALK inhibitors are being studied in combination with immunotherapy (NCT01998126, NCT02179671).
Lou et al reported a strong association between epithelial–mesenchymal transition status and immunophenotypes in NSCLC patients. These investigators analyzed two large datasets of lung cancer patients (the Cancer Genome Atlas [TCGA] and the PROSPECT database, University of Texas MD Anderson Cancer Center [Houston, TX, USA]) via gene expression profiling and found significant differences in immunophenotype between tumors with a mesenchymal versus epithelial phenotype. They found that mesenchymal tumors had higher levels of immune-activating and immunomodulatory molecules.105 Stemness-associated transcription factor Nanog is induced by hypoxia and expressed in several types of cancer. Nanog binds to the TGF-β promoter, modulating TGF-β transcription. When targeting Nanog, Tregs and macrophages are reduced, and CD8+ T effector cells increased.106,107 A close relation exists between the immunologic system, hypoxia, acquisition of stemness properties, and epithelial–mesenchymal transition induced by TGF-β. The combination of TGF-β inhibitors with anti-PD-1 or anti-PD-L1 antibodies may also be more active.
In lung cancer, other genes such as Brachyury are implicated in tumor stemness features and EGFR inhibitor resistance.108 Brachyury is a T-box transcription factor and a driver of epithelial–mesenchymal transition. High levels of Brachyury expression by protein and RNA expression analysis have been observed in lung cancer in 48% of adenocarcinomas and 25% of squamous cell carcinomas, mainly in EGFR inhibitor-resistant tumors. Active CD8+ Brachyury–specific T-cells can be expanded in vitro from peripheral blood mononuclear cells of prostate cancer patients.109 Other compounds with immunomodulatory properties are those which blockade the inhibitor apoptosis proteins (IAPs) such as the LCL161 drug. IAP antagonist sensitizes cells to TNF-α-mediated apoptosis and enhances cytokine secretion from T-cells. LCL161 plus TNF-α targeted by an adenovirus (AAVP-TNF-α) are synergistic in xenograft models.110,111
Conclusion
Immunotherapy is a promising approach for lung cancer. Its overall efficacy remains low according to response rate, but very high according to duration of responses. Identification of predictive biomarkers of clinical response will therefore strengthen its clinical value. In the era of personalized medicine, the development of immunotherapy will require better selection of patients and the clinical setting, and also finding the most active drug combinations.
The challenge for the future for this therapy to be successful is to achieve better understanding of the mechanisms underlying immunotherapy efficacy to help identify a predictive biomarker that is useful in the clinical setting.
Disclosure
The authors report no conflicts of interest in this work.
References
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363(8):711–723. | |
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. | |
Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28(19):3167–3175. | |
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–998. | |
Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–258. | |
Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell. 1992;71(7):1065–1068. | |
Chen L, Ashe S, Brady WA, et al. Costimulation of antitumor immunity by the B7 counterreceptor for the T lymphocyte molecules CD28 and CTLA-4. Cell. 1992;71(7):1093–1102. | |
Townsend SE, Allison JP. Tumor rejection after direct costimulation of CD8+ T cells by B7-transfected melanoma cells. Science. 1993; 259(5093):368–370. | |
Denkert C, Loibl S, Noske A, et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol. 2010;28(1):105–113. | |
Loi S, Sirtaine N, Piette F, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J Clin Oncol. 2013;31(7):860–867. | |
Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. | |
Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306. | |
Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–213. | |
Gros A, Robbins PF, Yao X, et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J Clin Invest. 2014;124(5):2246–2259. | |
Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med. 1991;174(3):561–569. | |
Salvi S, Fontana V, Boccardo S, et al. Evaluation of CTLA-4 expression and relevance as a novel prognostic factor in patients with non-small cell lung cancer. Cancer immunol Immunother. 2012;61(9):1463–1472. | |
Thompson CB, Allison JP. The emerging role of CTLA-4 as an immune attenuator. Immunity. 1997;7(4):445–450. | |
Walunas TL, Lenschow DJ, Bakker CY, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1(5):405–413. | |
Kearney ER, Walunas TL, Karr RW, et al. Antigen-dependent clonal expansion of a trace population of antigen-specific CD4+ T cells in vivo is dependent on CD28 costimulation and inhibited by CTLA-4. J Immunol. 1995;155(3):1032–1036. | |
Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182(2):459–465. | |
Krummel MF, Sullivan TJ, Allison JP. Superantigen responses and co-stimulation: CD28 and CTLA-4 have opposing effects on T cell expansion in vitro and in vivo. Int Immunol. 1996;8(4):519–523. | |
Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3(5):541–547. | |
Waterhouse P, Penninger JM, Timms E, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270(5238):985–988. | |
Chambers CA, Sullivan TJ, Allison JP. Lymphoproliferation in CTLA-4-deficient mice is mediated by costimulation-dependent activation of CD4+ T cells. Immunity. 1997;7(6):885–895. | |
Huang CT, Workman CJ, Flies D, et al. Role of LAG-3 in regulatory T cells. Immunity. 2004;21(4):503–513. | |
Gandhi MK, Lambley E, Duraiswamy J, et al. Expression of LAG-3 by tumor-infiltrating lymphocytes is coincident with the suppression of latent membrane antigen-specific CD8+ T-cell function in Hodgkin lymphoma patients. Blood. 2006;108(7):2280–2289. | |
Zhu C, Anderson AC, Schubart A, et al. The TIM-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005; 6(12):1245–1252. | |
Sabatos CA, Chakravarti S, Cha E, et al. Interaction of TIM-3 and TIM-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. 2003;4(11):1102–1110. | |
Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting TIM-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–2194. | |
Gao X, Zhu Y, Li G, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS One. 2012;7(2):e30676. | |
Zhuang X, Zhang X, Xia X, et al. Ectopic expression of TIM-3 in lung cancers: a potential independent prognostic factor for patients with NSCLC. Am J Clin Pathol. 2012;137(6):978–985. | |
Zhou P, Shaffer DR, Alvarez Arias DA, et al. In vivo discovery of immunotherapy targets in the tumour microenvironment. Nature. 2014; 506(7486):52–57. | |
Le Mercier I, Chen W, Lines JL, et al. VISTA regulates the development of protective antitumor immunity. Cancer Res. 2014;74(7):1933–1944. | |
Velcheti V, Schalper KA, Carvajal DE, et al. Programmed death ligand-1 expression in non-small cell lung cancer. Lab Invest. 2014; 94(1):107–116. | |
Chen YY, Wang LB, Zhu HL, et al. Relationship between programmed death-ligand 1 and clinicopathological characteristics in non-small cell lung cancer patients. Chin Med Sci J. 2013;28(3):147–151. | |
Chen YB, Mu CY, Huang JA. Clinical significance of programmed death-1 ligand-1 expression in patients with non-small cell lung cancer: a 5-year-follow-up study. Tumori. 2012;98(6):751–755. | |
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. | |
Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. | |
Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27(1):111–122. | |
Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14(8):535–546. | |
Karwacz K, Bricogne C, MacDonald D, et al. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol Med. 2011;3(10):581–592. | |
Freire J, Ajona D, de Biurrun G, et al. Silica-induced chronic inflammation promotes lung carcinogenesis in the context of an immunosuppressive microenvironment. Neoplasia. 2013;15(8):913–924. | |
O’Callaghan DS, O’Donnell D, O’Connell F, O’Byrne KJ. The role of inflammation in the pathogenesis of non-small cell lung cancer. J Thorac Oncol. 2010;5(12):2024–2036. | |
Li CW, Xia W, Huo L, et al. Epithelial-mesenchymal transition induced by TNF-α requires NF-κB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012;72(5):1290–1300. | |
So T, Takenoyama M, Mizukami M, et al. Haplotype loss of HLA class I antigen as an escape mechanism from immune attack in lung cancer. Cancer Res. 2005;65(13):5945–5952. | |
Shimizu K, Nakata M, Hirami Y, Yukawa T, Maeda A, Tanemoto K. Tumor-infiltrating Foxp3+ regulatory T cells are correlated with cyclooxygenase-2 expression and are associated with recurrence in resected non-small cell lung cancer. J Thorac Oncol. 2010;5(5):585–590. | |
Srivastava MK, Andersson Å, Zhu L, et al. Myeloid suppressor cells and immune modulation in lung cancer. Immunotherapy. 2012;4(3):291–304. | |
Brahmer J, Horn L, Antonia S, et al. Survival and long-term follow-up of the phase I trial of nivolumab (Anti-PD-1; BMS-936558; ONO-4538) in patients (pts) with previously treated advanced non-small cell lung cancer (NSCLC). Proc Am Soc Clin Oncol. 2013. | |
Gettinger S, Chow LQ, Borghaei H, et al. Safety and response with nivolumab (anti-PD-1; BMS-936558, ONO-4538) plus erlotinib in patients (pts) with epidermal growth factor receptor mutant (EGFR MT) advanced non-small cell lung cancer (NSCLC). Int J Radiat Oncol Biol Phys. 2014;90(5):S34–S35. | |
Rizvi NA, Mazières J, Planchard D, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 2015;16(3):257–265. | |
Antonia SJ, Brahmer JR, Gettinger S, et al. Nivolumab (anti-PD-1; BMS-936558, ONO-4538) in combination with platinum-based doublet chemotherapy (PT-DC) in advanced non-small cell lung cancer (NSCLC). Int J Radiat Oncol Biol Phys. 2014;90(5):S2. | |
Garon E, Balmanoukian A, Hamid O, et al. Preliminary safety and clinical activity of MK-3475 in previously treated patients (pts) with non-small cell lung cancer (NSCLC). Presented at: Annual Congress of the American Society of Clinical Oncology (ASCO); 2014. | |
Garon EB, Gandhi L, Rizvi N, et al. Antitumor activity of pembrolizumab (pembro; MK-3475) and correlation with programmed death ligand 1 (PD-L1) expression in a pooled analysis of patients (pts) with advanced non-small cell lung carcinoma (NSCLC). Ann Oncol. 2014; 25 (Supplement 5):v1–v41. | |
Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. | |
Khleif S, Lutzky J, Segal N, et al. MEDI4736, an anti-PD-L1 antibody with modified Fc domain: preclinical evaluation and early clinical results from a phase 1 study in patients with advanced solid tumors. Presented at: European Cancer Congress 2013; 2013. | |
Lutzky J, Antonia SJ, Blake-Haskins A, et al. A phase 1 study of MEDI4736, an anti–PD-L1 antibody, in patients with advanced solid tumors. J Clin Oncol. 2014;32:5s(Suppl; abstr 3001). | |
Segal NH, Antonia S, Brahmer J, et al. Preliminary data from a multi-arm expansion study of MEDI4736, an anti-PD-L1 antibody. J Clin Oncol. 2014;32:5s(Suppl; abstr 3002). | |
Spigel D, Gettinger S, Horn L, et al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic non-small cell lung cancer (NSCLC). Proc Am Soc Clin Oncol. 2013;31. | |
Soria JC, Cruz C, Bahleda R, et al. Clinical activity, safety, and biomarkers of a PD-L1 blockade in non-small cell lung cancer (NSCLC9: additional analyses from a clinical study of the engineered antibody MPDL3280A (anti-PDL1). Presented at: European Cancer Congress 2013; 2013. | |
Gandhi L, Balmanoukian A, Hui R, et al. MK-3475 (anti-PD-1 monoclonal antibody) for non-small cell lung cancer (NSCLC): antitumor activity and association with tumor PD-L1 expression. Presented at: Annual Congress of the American Association for Cancer Research 2014; 2014. | |
Gettinger SN, Shepherd FA, Antonia SJ, et al. First-line nivolumab (anti-PD-1; BMS-936558, ONO-4538) monotherapy in advanced NSCLC: safety, efficacy, and correlation of outcomes with PD-L1 status. J Clin Oncol. 2014;32:5s(Suppl; abstr 8024). | |
Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. | |
Brahmer JR, Horn L, Gandhi L, et al. Nivolumab (anti-PD-1, BMS-936558, ONO-4538) in patients (pts) with advanced non-small-cell lung cancer (NSCLC): survival and clinical activity by subgroup analysis. J Clin Oncol. 2014;32:5s(Suppl; abstr 8112). | |
Brahmer JR, Rizvi NA, Lutzky J, et al. Clinical activity and biomarkers of MEDI4736, an anti-PD-L1 antibody, in patients with NSCLC. J Clin Oncol. 2014;32:5s(Suppl; abstr 8021). | |
Garon E, Balmanoukian A, Hamid O, et al. Preliminary clinical safety and activity of MK-3475 mono-therapy for the treatment of previously treated patients with non-small cell lung cancer. IASLC 15th World Conference on Lung Cancer. 2013 Abstract MO18.02. | |
Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res. 2014;20(19):5064–5074. | |
Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014; 515(7528):568–571. | |
Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014; 371(23):2189–2199. | |
Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91(14):1194–1210. | |
Hellmann MD, Creelan BC, Woo K, et al. Smoking history and response to nivolumab in patients with advanced NSCLCs. Ann Oncol. 2014;25(Supplement 4):iv426–iv470. | |
Lynch TJ, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012;30(17):2046–2054. | |
Xu C, Fillmore CM, Koyama S, et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell. 2014;25(5):590–604. | |
Velcheti V, Rimm DL, Schalper KA. Sarcomatoid lung carcinomas show high levels of programmed death ligand-1 (PD-L1). J Thorac Oncol. 2013;8(6):803–805. | |
Reck M, Bondarenko I, Luft A, et al. Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol. 2013;24(1):75–83. | |
Taube JM, Anders RA, Young GD, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):127ra37. | |
Spranger S, Spaapen RM, Zha Y, et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013;5(200):200ra116. | |
Akbay EA, Koyama S, Carretero J, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3(12):1355–1363. | |
Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009;206(8):1717–1725. | |
Winograd R, Byrne KT, Evans RA, et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res. 2015;3(4):399–411. | |
Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. OX40 engagement depletes intratumoral Tregs via activating FcγRs, leading to antitumor efficacy. Immunol Cell Biol. 2014;92(6):475–480. | |
Feder-Mengus C, Wyler S, Hudolin T, et al. High expression of indoleamine 2,3-dioxygenase gene in prostate cancer. Eur J Cancer. 2008;44(15):2266–2275. | |
Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212–20217. | |
Zhu Y, Knolhoff BL, Meyer MA, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–5069. | |
Hasmim M, Noman MZ, Messai Y, et al. Cutting edge: hypoxia-induced Nanog favors the intratumoral infiltration of regulatory T cells and macrophages via direct regulation of TGF-β1. J Immunol. 2013;191(12):5802–5806. | |
Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from Phase II and Phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. Epub February 9, 2015. | |
Woo SR, Turnis ME, Goldberg MV, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–927. | |
Fourcade J, Sun Z, Benallaoua M, et al. Upregulation of TIM-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207(10):2175–2186. | |
Nirschl CJ, Drake CG. Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin Cancer Res. 2013;19(18):4917–4924. | |
Fourcade J, Sun Z, Pagliano O, et al. PD-1 and TIM-3 regulate the expansion of tumor antigen-specific CD8+ T cells induced by melanoma vaccines. Cancer Res. 2014;74(4):1045–1055. | |
Zhou Q, Munger ME, Veenstra RG, et al. Coexpression of TIM-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011;117(17):4501–4510. | |
Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti-TIM-3 antibody promotes T cell IFN-γ-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011;71(10):3540–3551. | |
Mittal D, Young A, Stannard K, et al. Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res. 2014;74(14):3652–3658. | |
Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Adenosine limits the therapeutic effectiveness of anti-CTLA4 mAb in a mouse melanoma model. Am J Cancer Res. 2014;4(2):172–181. | |
Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. | |
Antonia S, Gettinger S, Chow LQ, et al. Nivolumab and ipilimumab in first-line NSCLC: interim phase 1 results. Presented at: Annual Congress of the American Society of Clinical Oncology (ASCO); 2014. | |
Frederick DT, Piris A, Cogdill AP, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19(5):1225–1231. | |
Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, Hodi FS. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res. 2013;19(3):598–609. | |
Atefi M, Avramis E, Lassen A, et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res. 2014;20(13):3446–3457. | |
D’Incecco A, Andreozzi M, Ludovini V, et al. PD-L1 and PD-1 expression in molecularly selected non-small-cell lung cancer (NSCLC) patients. J Thorac Oncol. 2014;9(Supplement 9):S7–S52. | |
Ait-Tahar K, Barnardo MC, Pulford K. CD4 T-helper responses to the anaplastic lymphoma kinase (ALK) protein in patients with ALK-positive anaplastic large-cell lymphoma. Cancer Res. 2007;67(5):1898–1901. | |
Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012;11(3):215–233. | |
Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10(6):717–728. | |
Zheng Y, de la Cruz CC, Sayles LC, et al. A rare population of CD24(+)ITGB4(+)Notch(hi) cells drives tumor propagation in NSCLC and requires Notch3 for self-renewal. Cancer Cell. 2013;24(1):59–74. | |
Rizvi N, Chow LQ, Borghaei H, et al. Safety and response with nivolumab (anti-PD-1; BMS-936558, ONO-4538) plus erlotinib in patients (pts) with epidermal growth factor receptor mutant (EGFR MT) advanced NSCLC. J Clin Oncol. 2014;32:5s(Suppl; abstr 8022). | |
Lou Y, Diao L, Byers LA, et al. Association of epithelial-mesenchymal transition status with PD1/PDL1 expression and a distinct immunophenotype in non-small cell lung cancer: implications for immunotherapy biomarkers. J Clin Oncol. 2014;32:5s(Suppl; abstr 3018). | |
Mathieu J, Zhang Z, Zhou W, et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res. 2011;71(13):4640–4652. | |
Hasmim M, Noman MZ, Lauriol J, et al. Hypoxia-dependent inhibition of tumor cell susceptibility to CTL-mediated lysis involves NANOG induction in target cells. J Immunol. 2011;187(8):4031–4039. | |
Roselli M, Fernando RI, Guadagni F, et al. Brachyury, a driver of the epithelial-mesenchymal transition, is overexpressed in human lung tumors: an opportunity for novel interventions against lung cancer. Clin Cancer Res. 2012;18(14):3868–3879. | |
Hu Y, Mintz A, Shah SR, Quinones-Hinojosa A, Hsu W. The FGFR/MEK/ERK/brachyury pathway is critical for chordoma cell growth and survival. Carcinogenesis. 2014;35(7):1491–1499. | |
Yuan Z, Syrkin G, Adem A, et al. Blockade of inhibitors of apoptosis (IAPs) in combination with tumor-targeted delivery of tumor necrosis factor-α leads to synergistic antitumor activity. Cancer Gene Ther. 2013;20(1):46–56. | |
Knights AJ, Fucikova J, Pasam A, Koernig S, Cebon J. Inhibitor of apoptosis protein (IAP) antagonists demonstrate divergent immunomodulatory properties in human immune subsets with implications for combination therapy. Cancer Immunol Immunother. 2013;62(2):321–335. | |
Gettinger SN, Horn L, Gandhi L, et al. Long-term survival, clinical activity, and safety of nivolumab (anti-PD-1; BMS-936558, ONO-4538) in patients (pts) with advanced non-small cell lung cancer (NSCLC). Int J Radiat Oncol Biol Phys. 2014;90(5):S34. | |
Ramalingam SS, Mazières J, Planchard D, et al. Phase II study of nivolumab (anti-PD-1, BMS-936558, ONO-4538) in patients with advanced, refractory squamous non-small cell lung cancer. Int J Radiat Oncol Biol Phys. 2014;90(5):1266–1267. | |
Rizvi NA, Shepherd FA, Antonia SJ, et al. First-Line monotherapy with nivolumab (anti-PD-1; BMS-936558, ONO-4538) in advanced non-small cell lung cancer (NSCLC): safety, efficacy, and correlation of outcomes with PD-L1 status. Int J Radiat Oncol Biol Phys. 2014;90(5):S31. | |
Rizvi NA, Garon EB, Patnaik A, et al. Safety and clinical activity of MK-3475 as initial therapy in patients with advanced non-small cell lung cancer (NSCLC). J Clin Oncol. 2014;32:5s(Suppl; abstr 8007). | |
Brahmer J, Balmanoukian A, Goldberg S, et al. Development of MEDI4736, an anti-programmed cell death ligand 1 (PD-L1) antibody, as monotherapy or in combination with other therapies in the treatment of non-small cell lung cancer (NSCLC). J Immunother Cancer. 2014;2(Suppl 3):179. | |
Zatloukal P, Heo DS, Park K, et al. Randomized phase II clinical trial comparing tremelimumab (CP-675,206) with best supportive care (BSC) following first-line platinum-based therapy in patients (pts) with advanced non-small cell lung cancer (NSCLC). J Clin Oncol. 2009;27:15s(Suppl; abstr 8071). | |
Daud A, Hamid O, Ribas A, et al. Antitumor activity of the anti-PD-1 monoclonal antibody MK-3475 in melanoma: correlation of tumor PD-L1 expression with outcome. Presented at: AACR Annual Meeting 2014. | |
Kefford R, Ribas A, Hamid O, et al. Clinical efficacy and correlation with tumor PD-L1 expression in patients (pts) with melanoma (MEL) treated with the anti-PD-1 monoclonal antibody MK-3475. J Clin Oncol. 2014;32:5s(Suppl; abstr 3005). | |
Seiwert TY, Burtness B, Weiss J, et al. A phase Ib study of MK-3475 in patients with human papillomavirus (HPV)-associated and non-HPV–associated head and neck (H/N) cancer. J Clin Oncol. 2014;32:5s(Suppl; abstr 6011). | |
Weber JS, Kudchadkar RR, Yu B, et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol. 2013;31(34):4311–4318. | |
Motzer RJ, Rini BI, McDermott DF, et al. Nivolumab for metastatic renal cell carcinoma: results of a randomized Phase II trial. J Clin Oncol. 2015;33(13):1430–1437. | |
Grosso J, Horak CE, Inzunza D, et al. Association of tumor PD-L1 expression and immune biomarkers with clinical activity in patients (pts) with advanced solid tumors treated with nivolumab (anti-PD-1; BMS-936558; ONO-4538). J Clin Oncol. 2013;31(Suppl; abstr 3016). | |
Hodi FS, Sznol M, Kluger HM, et al. Long-term survival of ipilimumab-naive patients (pts) with advanced melanoma (MEL) treated with nivolumab (anti-PD-1, BMS-936558, ONO-4538) in a phase I trial. J Clin Oncol. 2014;32:5s(Suppl; abstr 9002). | |
Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–330. | |
Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515(7528):558–562. | |
Hamid O, Sosman JA, Lawrence DP, et al. Clinical activity, safety, and biomarkers of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic melanoma (mM). J Clin Oncol. 2013;31(Suppl; abstr 9010). |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.