")
Back to Journals » International Journal of Nanomedicine » Volume 15
Targeting Pancreatic Cancer Cells and Stellate Cells Using Designer Nanotherapeutics in vitro
Authors Elechalawar CK , Hossen MN, Shankarappa P, Peer CJ , Figg WD , Robertson JD, Bhattacharya R, Mukherjee P
Received 10 October 2019
Accepted for publication 15 January 2020
Published 13 February 2020 Volume 2020:15 Pages 991—1003
DOI https://doi.org/10.2147/IJN.S234112
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Chandra Kumar Elechalawar,1 Md Nazir Hossen,1 Priya Shankarappa,2 Cody J Peer,2 William D Figg,2 J David Robertson,3 Resham Bhattacharya,4 Priyabrata Mukherjee1,5
1Department of Pathology, The University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA; 2Clinical Pharmacology Program, National Cancer Institute, Bethesda, MD 20892, USA; 3Department of Chemistry and University of Missouri Research Reactor, University of Missouri, Columbia, MO 65211, USA; 4Department of Obstetrics and Gynecology, The University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA; 5Peggy and Charles Stephenson Cancer Center, The University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA
Correspondence: Priyabrata Mukherjee
Department of Pathology, The University of Oklahoma Health Sciences Center, 975 NE 10th Street, BRC-1409B, Oklahoma City, OK 73104, USA
Tel +1 405-271-1133
Fax +1 405-271-2472
Email [email protected]
Introduction and Objective: Pancreatic cancer (PC) is characterized by a robust desmoplastic environment, which limits the uptake of the standard first-line chemotherapeutic drug gemcitabine. Enhancing gemcitabine delivery to the complex tumor microenvironment (TME) is a major clinical challenge. Molecular crosstalk between pancreatic cancer cells (PCCs) and pancreatic stellate cells (PSCs) plays a critical role in desmoplastic reaction in PCs. Herein, we report the development of a targeted drug delivery system to inhibit the proliferation of PCCs and PSCs in vitro. Using gold nanoparticles as the delivery vehicle, the anti-EGFR antibody cetuximab (C225/C) as a targeting agent, gemcitabine as drug and polyethylene glycol (PEG) as a stealth molecule, we created a series of targeted drug delivery systems.
Methods: Fabricated nanoconjugates were characterized by various physicochemical techniques such as UV-Visible spectroscopy, transmission electron microscopy, HPLC and instrumental neutron activation analysis (INAA).
Results and Conclusion: Targeted gemcitabine delivery systems containing mPEG-SH having molecular weights of 550 Da or 1000 Da demonstrated superior efficacy in reducing the viability of both PCCs and PSCs as compared to their non-targeted counterparts. EGFR-targeted pathway was further validated by pre-treating cells with C225 followed by determining cellular viability. Taken together, in our current study we have developed a PEGylated targeted nanoconjugate ACG44P1000 that showed enhanced selectivity towards pancreatic cancer cells and pancreatic stellate cells, among others, for gemcitabine delivery. We will investigate the ability of these optimized conjugates to inhibit desmoplasia and tumor growth in vivo in our future studies.
Keywords: pancreatic cancer, gold nanoparticles, PEGylation, drug delivery
Introduction
Almost one-third of cancer patients in the US still die within 5 years of diagnosis; for pancreatic ductal adenocarcinoma (PDAC) deaths stubbornly remain closer to 90%. An estimated 56,770 new cases of and 45,750 deaths from PDAC are projected for 2019 in the US.1 Pancreatic cancer is characterized by early metastasis, high mortality and a dismal prognosis; median survival is only about 6 months.1,2 The most effective treatment is surgical resection of the tumor, however only about 20% of patients qualify. As a result, chemotherapy is the default treatments for most patients.3,4 Gemcitabine, a DNA synthesis inhibitor, is the first-line chemotherapy drug for PDAC although the results are disappointing (Daniel et al, n engl j med 369;18).5–8 Gemcitabine in combination with Abraxane (albumin-bound paclitaxel) significantly increases overall survival but is limited to advanced metastatic pancreatic cancer. Other drugs that have been used include nanoliposomal irinotecan, the FOLFIRINOX protocol, and other combinations (Andria et al, European Journal of Cancer 108 (2019) 78e87), however use of all available chemotherapeutic agents for PDAC remains limited by non-specific toxicity.9 Thus, novel methodologies are required to improve patent prognosis.
In PDAC patients, a stellate cell-associated desmoplastic tumor stroma and minimal blood flow inside the tumor limit gemcitabine penetration of the dense tumor tissue.10 This is compounded by poor bioavailability due to a short plasma half-life. Additionally, there is significant toxicity to healthy tissues due to the need for frequent dosing.11,12 Hence, new strategies are needed to improve chemotherapy for PDAC and improve patient outcomes. In this regard, targeted drug delivery systems (DDS) are a promising approach to address the current limitations; they can help achieve maximum drug concentrations within malignancies by enhancing bioavailability.
We have previously described an epidermal growth factor receptor (EGFR)-targeted gold nanoparticle DDS, called ACG44, for gemcitabine delivery to PDAC.13 We used ACG44 for targeted delivery of gemcitabine to pancreatic cancer cells using the monoclonal antibody cetuximab (C225) as the targeting ligand for EGFR. This strategy inhibited the growth of malignant pancreatic cancer cells both in vitro and in vivo in an orthotopic pancreatic cancer model. The EGFR protein is over-expressed in pancreatic cancer as well as other malignancies.14 And Cetuximab (C225) is a monoclonal antibody targeting EGFR that was approved by the FDA for the treatment of head and neck cancer and colorectal cancer in 2004.15
Another consideration in the nanotechnology and drug delivery field, in order to achieve effective drug concentrations at the tumor site, is the avoidance of biological barriers such as opsonization and rapid clearance. This can be achieved using the stealth-like properties that PEGylation imparts. In addition to avoidance of biological barriers, PEGylation can enhance uptake of drugs and nanoparticles into tumors through the enhanced permeability and retention (EPR) effect (i.e. leaky vasculature of the tumor),16 and overcome physical instability issues such as aggregation and drug leakage from colloidal nanoparticles in aqueous suspensions17 even after freeze-drying.
Based on these observations, we modified our gold nanoparticle ACG44 by PEGylating with PEG chains having a molecular weight of 2000; ACG44 was by conjugated with methoxy-PEG (2000)-SH. The resulting nanoconjugate, ACG44P2000, had a longer half-life (t1/2) and achieved greater plasma concentrations (Cmax) than the non-PEGylated nanoparticle following IP and IV injections in mice.18 The Cmax of ACG44P2000 was almost twenty times higher than that of ACG44 following IV administration clearly demonstrating the PEGylation of ACG44 increased circulation time of the nanoparticle in blood after IV injection and increased penetration of the mucus layer by the nanoparticle following IP injection.18
Though previous reports demonstrate that when nanoparticles are conjugated with lower molecular weight PEG chains adhesion to the mucus layer is minimal, preventing entanglement.19,20 For example, in the case of enzymes, PEGylation with lower molecular weight chains increased residual activity and solubility.21 Wang et al reported when polystyrene (PS) nanoparticles were PEGylated with 10-kDa molecular weight PEG (232 monomer chains) the resulting PS-PEG nanoparticles were immobilized in human cervicovaginal mucus; this they attributed to high PEG molecular weight and insufficient stealth coverage.22 Based on this we hypothesized that PEGylating ACG44 monomeric PEG chains of shorter length than the 46 monomer PEG-2000 would result in nanoconjugates with enhanced biodistribution characteristics.
Herein, we report the PEGylation of ACG44 with two lower molecular weight monomeric chains: PEG-550 (12 monomer chains) and PEG-1000 (23 monomer chains). The resulting ACG44P550 and ACG44P1000 nanoconjugates were assessed in in vitro cellular uptake and cytotoxicity studies using pancreatic cancer cells and cancer-associated fibroblast cells. After synthesis, PEGylated nanoconjugates ACG44P550 and ACG44P1000 were characterized by UV-Visible spectroscopy, hydrodynamic size (DLS), zeta potentials and TEM analysis. Next, we have assessed the efficacy of ACG44P550 and ACG44P1000 in in vitro level by cellular uptake and toxicity studies in EGFR overexpressing various pancreatic cancer cells PANC-1, AsPC-1, CAF-19 cells, and obtained results were compared with previously reported 46 monomeric PEG chains containing ACG44P2000. The results clearly demonstrated that 23 monomer PEG chains containing ACG44P1000 is more selective towards the pancreatic cancer cells after PEGylation of ACG44 than other chains (Although the reason for selectivity is not clear for now). We hereby strongly demonstrate PEG-1000 is considered as a suitable agent to stealth ACG44 nanoconjugate among other PEG molecules and we expect it will overcome the physical and biological instability barriers in in vivo and in further clinical studies. Our current study emphasizes the importance of PEGylating strategy with various molecular weight PEG monomeric chains to NPs or drugs for enhanced accumulation of tumors in cancers.
Materials and Methods
Synthesis of PEGylated Au-Antibody-Gemcitabine Conjugates
Synthesis of targeted nanoconjugates and their PEGylated derivatives was performed as previously described.18 Briefly, core nanoparticles were prepared by reducing 100mL of 0.1 mM tetrachloroauric acid trihydrate (HAuCl4) by the addition of 50mL of a freshly prepared aqueous solution containing 4.3 mg of sodium borohydride (NaBH4); the mixture was stirred constantly overnight at room temperature. Synthesized core gold nanoparticles were characterized by UV-Visible spectroscopy. Targeted (AC4) and non-targeted nanoconjugates (AI4) were prepared, respectively, by the addition of 4 μg/mL of the anti-EGFR antibody cetuximab (C225) or Immunoglobulin G (IgG) to the core nanoparticles followed by stirring for 1hr. In the next step, 4 μg/mL of gemcitabine was added and the mixture stirred for an additional hour to prepare ACG44 or AIG44. Finally, PEGylation is achieved by the addition of 5 μg/mL of m-PEG (550)-SH or m-PEG (1000)-SH with continued stirring one more hour. Ultracentrifugation was used to separate unbound antibodies, gemcitabine or m-PEGs from the mixture (Beckman ultra-centrifuge; 70.1Ti rotor; 25,000 rpm for 40 min at 10⁰C). After careful aspiration, all nanoconjugates yielded loose pellets. Gold concentrations in pellets were calculated by comparison of absorbance values before and after ultracentrifugation. Nanoconjugate sizes were measured by TEM and DLS. The physiological stabilities of nanoconjugates were analyzed by the addition of 150mM NaCl followed by incubation for 15mins and then measurement of absorbance.
Dynamic Light Scattering (DLS) Spectroscopy and Zeta Potentials
Hydrodynamic diameter (HD) and zeta potentials of all gemcitabine containing PEGylated nanoconjugates were measured using Zeta sizer instrument Nano ZS, Malvern Instruments Ltd equipped with LASER wavelength of 633 nm.
Cell Culture
The human pancreatic cancer cell lines PANC-1 (ATCC CRL-1469) and AsPC-1 (ATCC CRL-1482) and the cancer-associated fibroblast cell line (stellate cells) CAF-19 were used to assess the effects of nanoconjugate treatments. CAF-19 cells were a kind gift from Prof. Michael Goggins (John Hopkins, Baltimore, USA)23–27 All experiments performed in CAF-19 cells were approved by the institutional review committee. In order to investigate the toxicity of nanoconjugates in healthy pancreatic cells, we used human pancreatic duct epithelial (HPDE) cells. These were obtained from AddexBio (San Diego, CA). PANC-1 and CAF-19 cells were grown in Dulbecco’s Modified Eagle’s medium (Gibco) and AsPC-1 cells grown in Roswell Park Memorial Institute (RPMI 1640) medium; both media were supplemented with 10% fetal bovine serum and 1% Pen-Strep antibiotic solutions. HPDE cells were grown in Keratinocyte-SFM (Thermofisher Cat No. 107005042). All cells were maintained in a humidified atmosphere under 20% O2 and 5% CO2 at 37°C.
Transmission Electron Microscopy (TEM)
Samples were prepared as previously described.27 Briefly, 10 μL of freshly prepared gold nanoconjugates were coated on 300 Cu-mesh carbon-coated grids followed by drying with side wicking prior to TEM. For TEM analysis of in vitro cellular uptake, cells (AsPC-1, PANC-1 or CAF-19) were incubated with nanoconjugates (at 2μg/mL of gold equivalent) for 2hrs. Cells were then washed three times in PBS before trypsinization and centrifugation. Resulting pellets were suspended in fixative solution (4% Paraformaldehyde (EM grade) and 2% glutaraldehyde (EM grade) in 0.1M sodium cacodylate buffer) overnight at 4⁰C after which sections were prepared. After staining with lead citrate and uranyl acetate, sections were visualized using a Hitachi H7600 Transmission Electron Microscope at 80 kV equipped with a 2k X 2k.
Quantification of Gemcitabine Adsorbed by Gold Nanoconjugates
The amount of gemcitabine adsorbed to synthesized gold nanoconjugates was evaluated by performing HPLC of supernatants collected following ultracentrifugation after synthesis. Gemcitabine in the supernatants was measured using a validated UHPLC-MS/MS assay, with a calibration range of 5–10,000 ng/mL. Briefly, 100 μL of sample was mixed with 300 μL acetonitrile, and injected onto a Waters BEH BILIC® column, 2.1x100mm, 1.7μm to chromatographically separate gemcitabine from matrix. The column eluent was then directed into a Waters Quattro Micro-mass spectrometer operated in the positive MRM mode based on the mass transition of m/z 264 to 112.0.
Quantification of PEG Adsorbed by Gold Nanoconjugates
The amount of methoxy-PEG (550)-SH or methoxy-PEG (1000)-SH adsorbed to synthesized gold nanoconjugates was evaluated using ELISA methodology according to the manufacturer's protocol; PEG-ELISA kit (Enzo life sciences, Cat. No ADI-900-213). Supernatants collected after ultracentrifugation were analyzed to calculate unbound PEG subtraction from the initial given concentration.
In vitro Cellular Uptake Studies of ACG44P550 and ACG44P1000 Through Instrumental Neutron Activation Analysis (INAA)
Cells (AsPC-1, PANC-1 or CAF-1) were seeded in100mm dishes in complete media and allowed to incubate for 24hrs. On the following day when cells reached 30% confluence, they were treated with gold nanoconjugates in serum-free media (2 μg/mL gold equivalent) and incubated for 2hrs. The amount of gold taken up by cells was quantified in cell pellets recovered from dishes following three washes with PBS to remove unbound conjugates. After trypsinization and centrifugation cell pellets were processed for instrumental neutron activation analysis (INAA) to quantify gold uptake at the University of Missouri Research Reactor Center using previously described method.28
In vitro Toxicity Assays (MTT and Cyquant)
Toxicity of nanoconjugates in AsPC-1, PANC-1, and CAF-19 cells as well as in healthy pancreatic cells (HPDEC) was assessed using MTT assay and Cyquant proliferation assay. Cells were seeded in 96 well plates; AsPC-1, PANC-1 and HPDE at 1 ×104 cells per well and CAF-19 at 1 ×103 cells per well. Cells were grown in the specific media described above for 24 hrs. Cells were treated for 72 hrs with nanoconjugates AIG44, ACG44, AIG44P550, ACG44P550, AIG44P1000 andACG44P1000), pristine gemcitabine at 10 μM, 1 μM and 0.1 μM with respect to gemcitabine or an equimolar mixture of gemcitabine and C225. The amount of gold per treatment was calculated with respect to gemcitabine adsorbed from HPLC results. MTT assays were performed by adding the reagent in PBS (5 mg/mL) to a final concentration of 10% v/v. The Cyquant assay was performed according to the manufacturer’s protocol (Cat No. C35006). Absorbance and fluorescence readings were obtained using a BMG LABTECH plate reader.
In vitro Targeting Studies of ACG44P550 and ACG44P1000 Through EGFR Inhibitor Pretreatment
Assays were performed to determine if cellular uptake of nanoconjugates was EGFR dependent. For this, cells were first pretreated with 50μg/mL of Cetuximab (C225) and incubated for 2hrs. In the next step, cell culture medium was removed, replaced with fresh media containing nanoconjugates of 10 μM, 1μM and 0.1μM concentrations with respect to gemcitabine and allowed to incubate for another 72hrs. Toxicity inhibition was determined by MTT and Cyquant assays as previously described.
Results
Synthesis and Characterization of Gemcitabine Containing PEGylated and Non-PEGylated Gold Nanoconjugates
Synthesized nanoconjugates were characterized by dynamic light scattering (DLS), zeta potential (λ), absorbance maxima (λmax) using UV-visible spectroscopy (UV-vis), and transmission electron microscopy (TEM). Unmodified core 5nm nanoconjugates showed the characteristic surface plasma resonance (SPR) peak of spherical gold nanoparticles i.e., at λmax value of 512nm13,29 (Figure 1A). Conjugation of the anti-EGFR antibody cetuximab (C225) or a control Immunoglobulin G (IgG) with a concentration of 4 µg/mL to core 5nm nanoparticles increased the λmax from 512 to 518nm with a redshift in SPR peak indicating surface adsorption by C225 or IgG i.e., formation of targeted and non-targeted nanoconjugates Au-C225 (AC4) or Au-IgG (AI4) as previously reported.29 AC4 or AI4 were next conjugated to gemcitabine with a concentration of 4 µg/mL to generate targeted or non-targeted delivery systems (ACG44 or AIG44), respectively, Addition of gemcitabine caused further redshift in the SPR peak with an increase in λmax to 520 nm for both nanoconjugates demonstrating successful binding of gemcitabine to both.18 It is well known that under in vivo conditions maximal delivery of gemcitabine to the tumor requires an additional modification to prolong the circulation time of the nanoconjugates. We speculated that the PEG moiety would be the best candidate to enhance the circulation time and thus tumor-targeted gemcitabine delivery of our nanoconjugates. PEG coating would have two major effects: it would protect the nanoparticles from phagocytosis, and based on the EPR effect it would increase penetration of the tumor by the nanoconjugates. We fabricated PEGylated targeted-nanoconjugates using either methoxy-PEG (550)-SH or methoxy-PEG (1000)-SH. The fabrication was confirmed by a decrease in the absorbance with blue shift in the SPR band with a value of λmax 514nm. There was no difference in absorbance between ACG44 PEGylated with mPEG (550)-SH and that ACG44 PEGylated with mPEG (1000)-SH (Figure 1A).
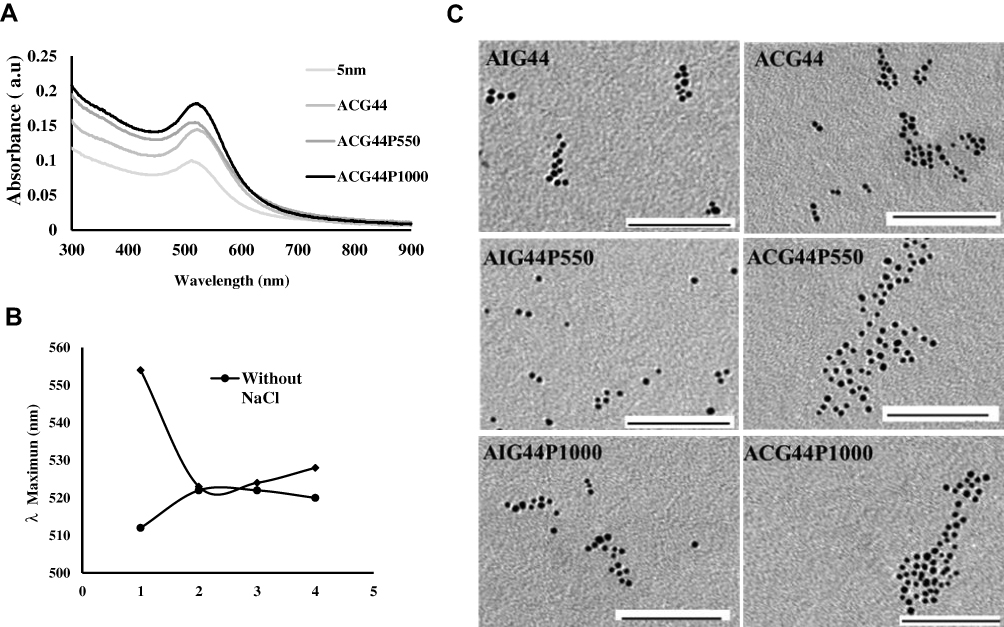 |
Figure 1 Physicochemical characterization of nanoconjugates. (A) UV-Visible spectrum of nanoconjugates and their absorbance maxima (λmax). (B) Stability studies of nanoconjugates in the presence of 150 mM NaCl. Minimum change in λmax values in the presence of 150 mM NaCl illustrating stability of targeted nanoconjugates: 1) 5nm , 2) ACG44, 3) ACG44P550, and 4) ACG44P1000. They are retaining their colloidal property under physiological conditions without any aggregation. (C) Transmission electron micrographs (TEM) of synthesized nanoconjugates were showing fine spherical shape with a diameter of ~5nm size. Scale bar 100 nm. |
The changes in λmax values with each conjugation step indicated gold nanoparticles were successfully and sequentially adsorbed with C225, gemcitabine and mPEG (550)-SH or m (PEG1000)-SH.
Moreover, it is also important that nanoparticles retain their fine colloidal properties in vivo. Blood contains large amounts of various proteins which can rapidly adsorb to a nanoparticle's surface and destabilize them by aggregation. In order to validate the stabilities of our nanoconjugates under physiological conditions, we incubated nanoparticles in 150mM of NaCl solution and determined aggregation by measuring absorbance values after 15 mins incubation. For the bare gold nanoparticles, there was a shift in absorbance maximum from 512 to 562 nm (Figure 1B) due to aggregation with NaCl addition resulting from the absence of surface protection. In contrast, there were minimal changes in λmax values for ACG44, ACG44P550 and ACG44P1000 (Figure 1B) after the addition of 150mM NaCl solution indicating their surface is protected by C225, gemcitabine or PEG moieties. These data demonstrated the stability of the nanoconjugates under physiological conditions. In addition, the hydrodynamic size (DLS) and λ-potential for both unmodified and modified nanoconjugates were determined. The core nanoparticles had a 6.5nm hydrodynamic size whereas the other targeted and non-targeted PEGylated conjugates were approximately 100–200 nm (Table 1). The zeta potentials for ACG44P550 and ACG44P1000 were – 9.33mV and – 16.6mV, respectively, values for the other conjugates are in Table 1. TEM analysis showed fine spherical shapes with an average size of 5nm for both modified and unmodified particles indicating that the shape of the nanoconjugate is retained after all conjugating steps till PEGylation (Figure 1C). The amount of gold by weight was kept constant in all conjugation steps. Conjugate adsorption to gold is probably due to Au-S/Au-NH2 bonding as reported previously.29 All small molecules, proteins and antibodies appear to bind to gold due to hydrophobic, electrostatic and covalent bonding.30
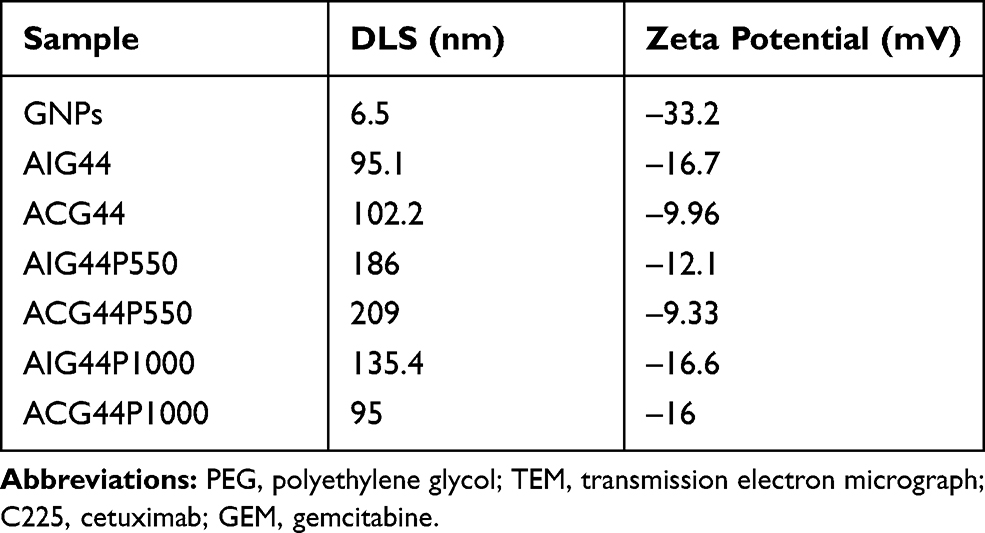 |
Table 1 Hydrodynamic Size (DLS) and ζ-Zeta Potentials of Synthesized Nanoconjugates |
Together, these results confirmed nanoconjugate synthesis, spherical morphological shape of the final nanoparticles, and their stability under physiological conditions.
Quantification of Unbound Gemcitabine (GEM) and PEG
The amount of gemcitabine bound to gold nanoconjugates was determined by HPLC analysis of supernatants after synthesis and subtraction of the unbound gemcitabine fraction from the amount added during synthesis. Nearly 25% of gemcitabine was bound to the ACG44P1000 nanoconjugate (Table S1). PEG binding to nanoconjugates was calculated by subtraction following PEG-ELISA assay of supernatants (Figure S1 and Table S2). Approximately 96–98% of both PEG 550-SH or PEG 1000-SH adsorbed to the nanoconjugates (Table S2). Importantly, gemcitabine was retained by the nanoconjugates following PEGylation; PEG addition did not affect gemcitabine that had already bound to nanoconjugates.
In vitro Uptake Studies
Having validated the physicochemical characteristics of our nanoconjugates, we evaluated their affects in vitro. First, we performed cellular uptake studies and compared with our previously reported nanoconjugate ACG44P2000. Briefly, pancreatic cancer cells PANC-1 and AsPC-1, and stellate cells CAF-19 were incubated with each nanoconjugate for 2hrs and following washing and trypsinization, resulted cell pellets were analyzed for both Instrumental and neutron activation analysis (INAA) and TEM analysis. TEM analysis showed the presence of the nanoconjugates ACG44P550 and ACG44P1000 in vesicular structures in all treated cells illustrating the internalization of nanoparticles (Figure 2A). UsingINAA analysis to quantify the cellular uptake efficacy of the nanoconjugates after treatments, we showed that the targeted nanoparticles ACG44, ACG44P550, ACG44P1000 and ACG44P2000 were taken up approximately 2 to 6 times more than the corresponding non-targeted IgG-conjugated nanoparticles (Figure 2B). Digital image picture of trypsinized cells captured after treatment with targeted nanoconjugates also confirmed enhanced cellular uptake of targeted nanoconjugates (Figure S2). This enhanced uptake by pancreatic cancer cells is due to selective uptake of the targeted nanoconjugates through overexpressed EGFR on their surface (Figure S3). Importantly, cellular uptake of ACG44P1000 was 50% and 20% more than the other PEGylated targeted nanoconjugates ACG44P550 andACG44P2000, respectively (Figure 2B), possibly due to greater selectivity for the PEG1000 chains by the cells. Cellular uptake of ACG44 was also high (Figure 2B), but ACG44P1000 is of greater interest since our intention was to make a better passive targeting PEGylated version of ACG44 with increased half-life and bioavailability, efficient accumulation around and penetration of the tumor, and excellent pharmacokinetic parameters in vivo.
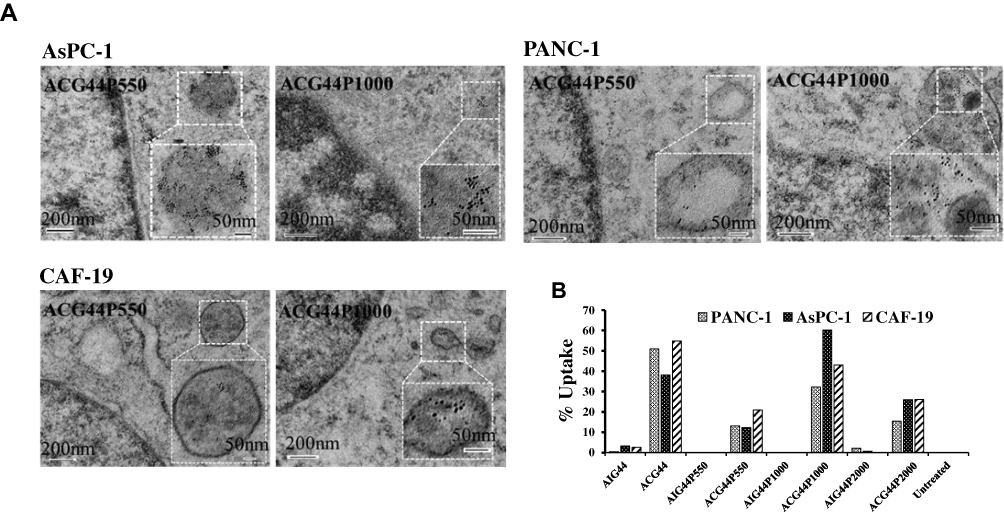 |
Figure 2 In vitro uptake studies of targeted nanoconjugates in AsPC-1, PANC-1 and CAF-19 cells after treatments. (A) TEM micrographs of cells AsPC-1, PANC-1 and CAF-19 after treatment with targeted conjugates are showing the presence of nanoconjugates in vesicular structures confirming their internalization. (B) Quantification of gold internalization by instrumental neutron activation analysis (INAA) after 2hrs of treatment. For both experiments, 2μg/mL gold treated to cells and incubated for 2hrs. After trypsinization, cell pellets were taken for analysis. |
Targeted Nanoconjugates Effectively Inhibit Proliferation of Pancreatic Cancer Cells
Having established in vitro cellular uptake we sought to determine the functional activity of our nanoconjugates in pancreatic cell lines PANC-1 and AsPC-1 and cancer-associated fibroblast CAF-19 cells. Cells were treated with nanoconjugates at 10 μM, 1 μM and 0.1 μM doses with respect to gemcitabine for 72hrs, and two assays used to assess toxicity the MTT cell viability assay and Cyquant proliferation assay. In the MTT assay, results demonstrated that growth inhibition was greater in cells treated with targeted nanoconjugates than their corresponding IgG-containing non-targeted counterparts, presumably due to the EGFR-targeted delivery of gemcitabine (Figure 3A) confirming the enhanced uptake results from INAA analysis in the previous experiment (Figure 2B). In these assays ACG44P1000 have the best response of all the conjugates, cell viability was 30% and 50% of that for untreated cells at the 10 μM and 1 μM concentrations, respectively (p <0.01). For the other nanoconjugates and gemcitabine alone 50% inhibition occurred at >1μM (p <0.01). Cyquant assay results also demonstrated that ACG44P1000 showed effective inhibition of cancer cell proliferation after treatment (Figure 3B). The greater toxicity with ACG44P1000 was likely due to enhanced gemcitabine delivery as shown in the cellular uptake data (Figure 2B). Importantly, toxicity from targeted nanoconjugates in treated healthy human pancreatic (HPDEC) cells was significantly less than that in the pancreatic cancer cells (Figure S4); this is due to low or basal level EGFR expression resulting in lower levels of intracellular gemcitabine. Non-targeted nanoconjugates retained some toxicity for the healthy pancreatic cells due to non-specific uptake. Free gemcitabine showed 80% and 60% killing at 10μM and 1μM, respectively, emphasizing its adverse toxicity to healthy pancreatic cells (Figure S4). This result also highlights the importance of targeted drug delivery systems. ACG44P1000 was significantly less toxic to healthy pancreatic cells (50% and 30% killing effect at 10μM and 1μM concentrations, respectively) than gemcitabine alone. Collectively, viability and proliferation assays (MTT and Cyquant) show ACG44P1000 to be the best candidate nanoconjugate among those tested to deliver gemcitabine specifically to pancreatic cancer cells with minimum toxicity to healthy pancreatic cells.
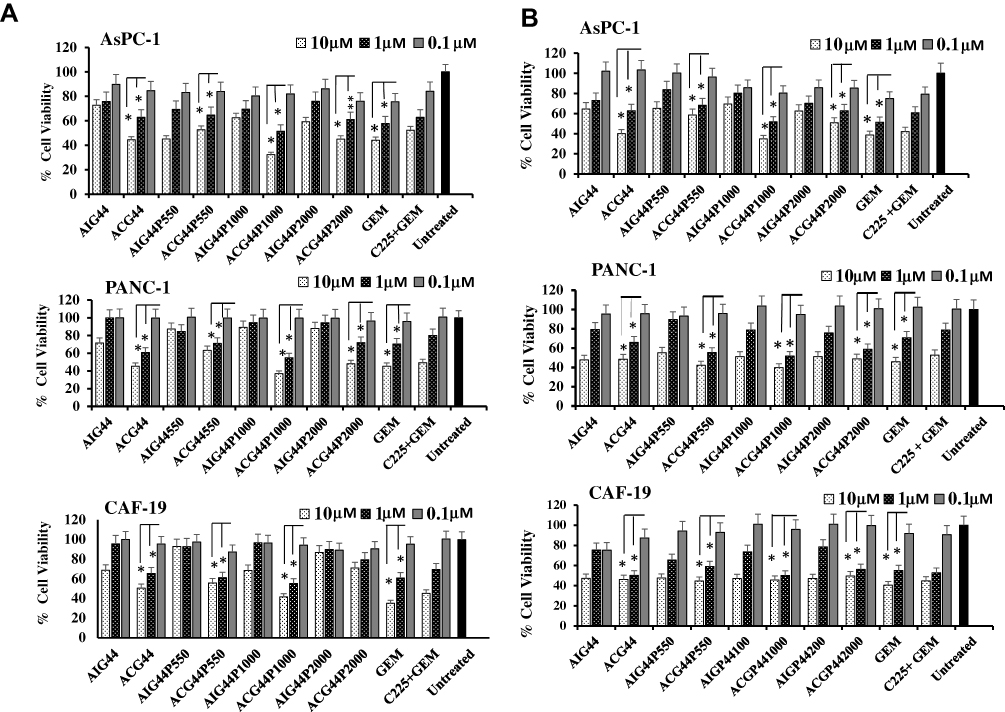 |
Figure 3 Toxicity study profile of synthesized nanoconjugates after treatments in AsPC-1, PANC-1 and CAF-19 cells. AsPC-1 or PANC-1 cells (1 x 104 cells per well), CAF-19 cells (1 x 103 cells per well) were seeded in 96 well plates. After 24hrs, cells were treated with nanoconjugates at 10 μM, 1μM and 0.1 μM concentrations with respect to gemcitabine and allowed for 72hrs incubation. Results were analyzed through (A) MTT and (B) Cyquant assays. * P ≤0.01, ** P ≤0.05 with respect to corresponding 0.1 μM treated cells. |
EGFR Selectivity Studies of Targeted Nanoconjugates
After evaluating the toxic effects of our newly synthesized targeted nanoconjugates we wanted to confirm the EGFR targeting ability role of Cetuximab – in conjugated targeted nanoparticles in PANC-1, AsPC-1 and CAF-19 cells. Since the monoclonal antibody cetuximab (C225) is selective for EGFR, we first blocked EGFR on cells by treating with cetuximab at a concentration of 50 μg/mL followed by treatment with targeted and non-targeted nanoconjugates with respect to gemcitabine at 10 μM, 1 μM and 0.1 μM and allowed to incubate for 72hrs. Toxicity was evaluated with MTT and Cyquant assays. Both assays (Figures 4 and 5) showed that toxicity was minimized in cells pretreated with cetuximab confirming the entry of cetuximab conjugated nanoconjugates through EGFR. Cetuximab pretreatment did not alter the effect of control IgG conjugated nanoconjugates. These observations clearly demonstrate gemcitabine delivery by targeted nanoconjugates through EGFR in pancreatic cancer cells.
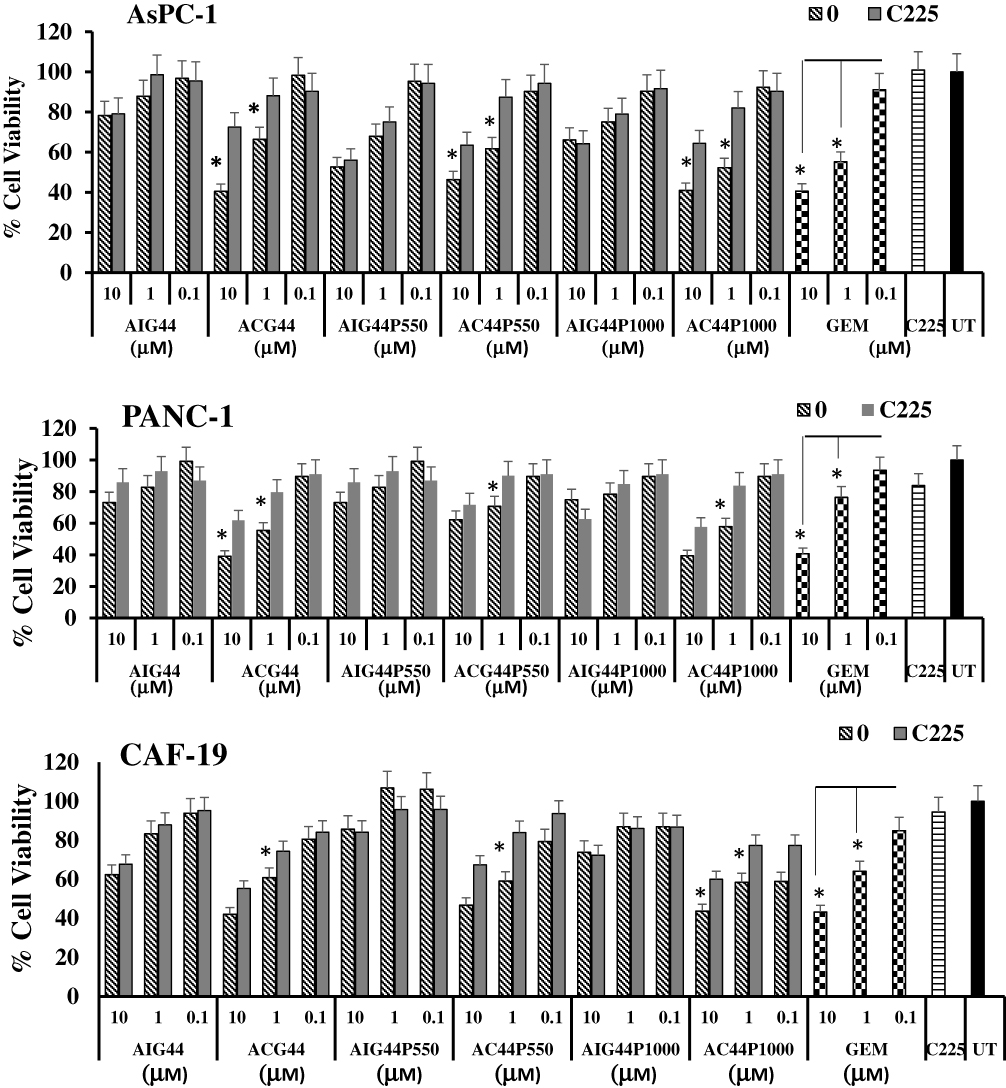 |
Figure 4 Receptor selectivity studies of nanoconjugates through MTT assay in treated AsPC-1, PANC-1 and CAF-19 cells. AsPC-1 or PANC-1 cells (1 x 104 cells per well) and CAF-19 cells (1 x 103 cells per well) were seeded in 96 well plates. After 24hrs, cells were pretreated with anti-EGFR antibody cetuximab (50 μg/mL) for 2hrs, followed treatments with gold nanoconjugates at 10 μM, 1μM and 0.1 μM concentrations with respect to gemcitabine and allowed for 72hrs incubation. Results were analyzed through MTT assay. * P ≤0.05 with respect to C225-pretreated cells. * P ≤0.01 with respect to corresponding 0.1 μM concentration in gemcitabine-treated cells. |
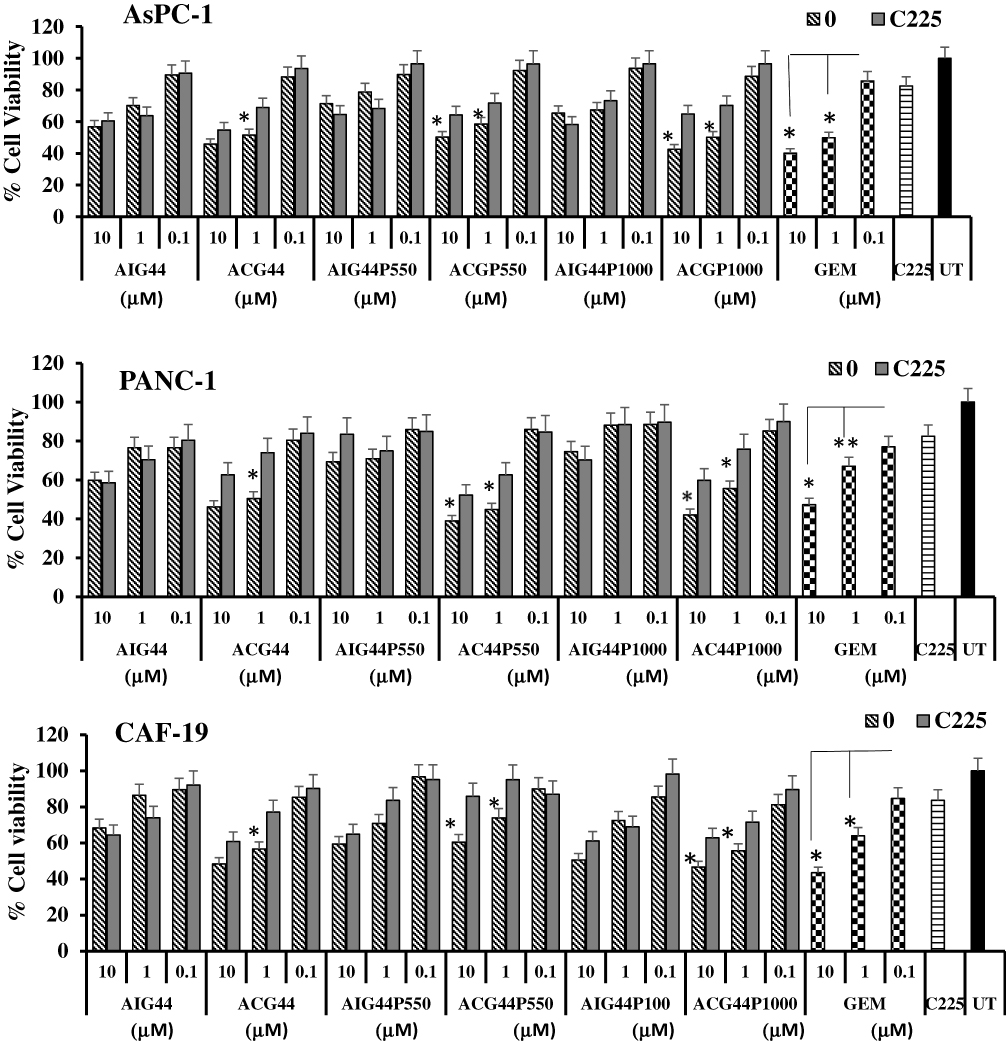 |
Figure 5 Receptor selectivity studies of nanoconjugates through Cyquant assay in treated AsPC-1, PANC-1 and CAF-19 cells. AsPC-1 or PANC-1 cells (1 x 104 cells per well) and CAF-19 cells (1 x 103 cells per well) were seeded in 96 well plates. After 24hrs, cells were pretreated with anti-EGFR antibody cetuximab (50 μg/mL) for 2hrs, followed by treatments with gold nanoconjugates at 10 μM, 1μM and 0.1 μM concentrations with respect to gemcitabine and allowed for 72hrs incubation. Results were analyzed through Cyquant proliferation assay. * P ≤ 0.05 with respect to C225-pretreated cells. * P =0.01, ** P ≤ 0.05 with respect to corresponding 0.1 μM concentration in gemcitabine-treated cells. |
Discussion
PEGylation of drugs and/or nanoparticles is a successful and important technology used in formulations to increase their half-life (t1/2) by bypassing non-specific immune responses from the mononuclear phagocytic system and reticuloendothelial system in vivo.31 To date, there are several FDA approved PEGylated drugs including Doxil (cancer), Peglntron (hepatitis C), and Mucagen (neovascular age-related macular degeneration) are in market and many others in active trails.32 Importantly, PEGylation reduces the amount or frequency of drug dosing by increasing plasma circulation time.18,33,34 Apart from these, PEGylation also has advantages over other molecules used to protect nanoparticles, such as glucose.35 For example, Stepien et al showed that glucose-coated iron oxide nanoparticles were rapidly recognized by macrophages, leading to early clearance from the circulation and decreased accumulation in mouse organs when compared to PEG-coated nanoparticles.35 This was explained by protein corona analysis ie, opsonic proteins predominated in the corona of the glucose nanoparticles whereas albumin enrichment characterized the PEG-coated version and enhanced their circulation time.35 PEG also minimizes adhesive interactions with the mucus membrane, thus overcoming an additional biological barrier. Also, important for the development of nanoparticle-based medications is the stability of the particle in aqueous solutions; physical instability issues including aggregation as well as drug leakage during storage are important considerations.36 Preferred storage conditions thus involve removal of water, and the preferred methodology is freeze-drying.37 But, some physical instability issues arise from the freeze-drying process. PEGylation can reduce these effects on nanoparticles and helps them retain their colloidal properties even after water removal. However, important characteristics impacting PEGylation are length of the PEG chain, grafting density, structure and conformation, and location of attachment to the nanoparticles; all of these can affect nanoparticle pharmacokinetics.38 Thus, it is very important and challenging to develop the appropriate PEGylated nanoparticles or drugs from bench to clinical trials in order to overcome both physicochemical and physiological barriers to their effectiveness.
We previously reported a targeted gold nanoparticle gemcitabine delivery system, ACG44, which efficiently regressed tumor in a mouse pancreatic orthotopic tumor model.13 In a follow-up study, we also showed that PEGylating ACG44 with a PEG-2000 chain enhanced bioavailability, plasma circulation and the ability to cross the mucus layer.18 However, PEGylation can itself create issues in cargo delivery; for example PEG chains can create steric hindrance that decreases drug uptake by target cells. The long-chain polymers can also prevent the recognition of receptors by their targeting ligands, a phenomenon termed the ‘PEG dilemma effect.’39 However, Fang et al, developed a cleavable PEGylation strategy for efficient cargo delivery and resolved this issue.40 Another option is PEGylating with short length polymers, which has distinct advantages over long polymeric chains. Short-chain PEGylated nanoparticles readily cross the mucus layer than those with longer chains.22,41 Wang et al, reported that polystyrene particles coated with PEG 2K polymers had reduced hydrophilic and electrostatic interactions with the mucus layer compared to PEG 10K-coated particles (Brownian motion is suppressed due to obstruction by the mucus layer; α= 0.83 for PEG 2k, α= 0.35 for PEG10K, where α =1 for unobstructed Brownian motion transport).42 Lower molecular weight PEG chains can also modulate or alter nanoparticle biodistribution; Yang et al reported that low molecular weight PEG chains with high grafting density decreased macrophage uptake of the coated nanoparticles due to reduced protein adsorption43 Based on these data we hypothesized that PEGylating ACG44 with shorter chains than PEG-2000 would improve its functionality further.
We synthesized short length PEGylated ACG44 nanoconjugates by conjugating ACG44 with mPEG (550)-SH or mPEG (1000)-SH. We assumed that the resulting conjugates would show significant improvements over either the non-PEGylated ACG44 or the PEG-2000-coated derivative.13 The synthesis and physicochemical characteristics for our nanoconjugates were confirmed prior to assessing their in vitro efficacy.
First cellular uptake assays demonstrated that the PEG1000 derivative, ACG44P1000, had the most favorable uptake ie, 2–6 times that of ACG44550 or ACG44P2000 depending on the pancreatic cell-line used in the assay. Although the non-PEGylated ACG44 also showed superior uptake, our goal was to develop PEGylated nanoconjugates that will have prolonged survival in the circulation while maximizing gemcitabine delivery to tumor cells. We also assessed cytotoxic effects; ACG44P1000 was the best nanoconjugate by this measure as well. All together, these data showed that the selectivity for pancreatic cancer cells is enhanced when ACG44 is fabricated with 23 monomeric PEG chains than other monomeric PEG chains. Importantly, the toxicity of ACG44 for healthy pancreatic cells was minimized by coating with PEG-1000. These results strongly demonstrated the importance of targeted delivery systems for gemcitabine delivery to pancreatic cancer cells. Further studies will seek to the effects on PEGylation with PEG-1000 of the biodistribution and efficacy of our targeted pancreatic cancer therapy ACG44 in vivo in mouse models of disease.
Conclusion
Taken together, we have developed an EGFR-targeted low molecular weight PEGylated nanoconjugate ACG44P1000 that shows enhanced cellular uptake and cytotoxicity to pancreatic cancer cells and stellate cells by delivering gemcitabine through EGFR. Since ACG44P1000 is associated with PEG chains we also expect good pharmacokinetics including good biodistribution profile and increased plasma circulation time that together will help in enhanced gemcitabine uptake in pancreatic desmoplastic tumors under in vivo settings for better tumor regression. The current study highlights the potential of a PEGylation strategy in nanoformulations with different PEG chains for combating pancreatic cancer. We anticipate that ACG44P1000 will be effective in planned in vivo models for inhibiting pancreatic tumor growth while also exhibiting minimum toxicity to healthy tissues.
Acknowledgments
This work was supported by National Institutes of Health Grants R01CA220237, R01CA136494, and R01CA213278 (to P.M.), and in part by the National Cancer Institute Cancer Center Support Grant P30CA225500 awarded to the University of Oklahoma Stephenson Cancer Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2019;69:7. doi:10.3322/caac.v69.1
2. Peixoto RD, Ho M, Renouf DJ, et al. Eligibility of Metastatic Pancreatic Cancer Patients for First-Line Palliative Intent nab-Paclitaxel Plus Gemcitabine Versus FOLFIRINOX. Am J Clin Oncol. 2017;40:507. doi:10.1097/COC.0000000000000193
3. McGuigan A, Kelly P, Turkington RC, Jones C, Coleman HG, McCain RS. Pancreatic cancer: a review of clinical diagnosis, epidemiology, treatment and outcomes. World J Gastroenterol. 2018;24:4846. doi:10.3748/wjg.v24.i43.4846
4. Conroy T, Desseigne F, Ychou M, et al.; Groupe Tumeurs Digestives of, U.; Intergroup, P. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N Engl J Med. 2011;364:1817. doi:10.1056/NEJMoa1011923
5. Hidalgo M. Pancreatic Cancer. N Engl J Med. 2010;362:1605. doi:10.1056/NEJMra0901557
6. Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, Gandhi V. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol. 1995;22:3.
7. Rathos MJ, Joshi K, Khanwalkar H, Manohar SM, Joshi KS. Molecular evidence for increased antitumor activity of gemcitabine in combination with a cyclin-dependent kinase inhibitor, P276-00 in pancreatic cancers. J Transl Med. 2012;10:161. doi:10.1186/1479-5876-10-161
8. Burris HA
9. Papadatos-Pastos D, Thillai K, Rabbie R, Ross P, Sarker D. FOLFIRINOX – a new paradigm in the treatment of pancreatic cancer. Expert Rev Anticancer Ther. 2014;14:1115. doi:10.1586/14737140.2014.957188
10. Whatcott CJ, Diep CH, Jiang P, et al. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin Cancer Res. 2015;21:3561. doi:10.1158/1078-0432.CCR-14-1051
11. Moysan E, Bastiat G, Benoit J-P. Gemcitabine versus Modified Gemcitabine: A Review of Several Promising Chemical Modifications. Mol Pharm. 2013;10:430. doi:10.1021/mp300370t
12. Yu X, Jin C, Xie C, et al. An in vitro and in vivo study of gemcitabine-loaded albumin nanoparticles in a pancreatic cancer cell line. Int J Nanomedicine. 2015;10:6825. doi:10.2147/IJN
13. Patra CR, Bhattacharya R, Wang E, et al. Targeted Delivery of Gemcitabine to Pancreatic Adenocarcinoma Using Cetuximab as a Targeting Agent. Cancer Res. 2008;68:1970. doi:10.1158/0008-5472.CAN-07-6102
14. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341. doi:10.1038/nrc1609
15. Wong S-F. Cetuximab: an epidermal growth factor receptor monoclonal antibody for the treatment of colorectal cancer. Clin Ther. 2005;27:684. doi:10.1016/j.clinthera.2005.06.003
16. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387.
17. Abdelwahed W, Degobert G, Stainmesse S, Fessi H. Freeze-drying of nanoparticles: formulation, process and storage considerations☆. Adv Drug Deliv Rev. 2006;58:1688. doi:10.1016/j.addr.2006.09.017
18. Kudgus RA, Walden CA, McGovern RM, Reid JM, Robertson JD, Mukherjee P. Tuning Pharmacokinetics and Biodistribution of a Targeted Drug Delivery System Through Incorporation of a Passive Targeting Component. Sci Rep. 2014;4:5669. doi:10.1038/srep05669
19. Lai SK, O’Hanlon DE, Harrold S, et al. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc Natl Acad Sci U S A. 2007;104:1482. doi:10.1073/pnas.0608611104
20. Lai SK, Wang -Y-Y, Wirtz D, Hanes J. Micro- and macrorheology of mucus. Adv Drug Deliv Rev. 2009;61:86. doi:10.1016/j.addr.2008.09.012
21. Morgenstern J, Baumann P, Brunner C, Hubbuch J. Effect of PEG molecular weight and PEGylation degree on the physical stability of PEGylated lysozyme. Int J Pharm. 2017;519:408. doi:10.1016/j.ijpharm.2017.01.040
22. Wang Y-Y, Lai SK, Suk JS, Pace A, Cone R, Hanes J. Addressing the PEG Mucoadhesivity Paradox to Engineer Nanoparticles that “Slip” through the Human Mucus Barrier. Angew Chem Int Ed Engl. 2008;47:9726. doi:10.1002/anie.v47:50
23. Yu J, Walter K, Omura N, et al. Unlike Pancreatic Cancer Cells Pancreatic Cancer Associated Fibroblasts Display Minimal Gene Induction after 5-Aza-2′-Deoxycytidine. PLoS One. 2012;7:e43456. doi:10.1371/journal.pone.0043456
24. Walter K, Omura N, Hong S-M, Griffith M, Goggins M. Pancreatic cancer associated fibroblasts display normal allelotypes. Cancer Biol Ther. 2008;7:882. doi:10.4161/cbt.7.6.5869
25. Walter K, Omura N, Hong S-M, et al. Overexpression of Smoothened Activates the Sonic Hedgehog Signaling Pathway in Pancreatic Cancer-Associated Fibroblasts. Clin Cancer Res. 2010;16:1781. doi:10.1158/1078-0432.CCR-09-1913
26. Hossen MN, Rao G, Dey A, Robertson JD, Bhattacharya R, Mukherjee P. Gold Nanoparticle Transforms Activated Cancer-Associated Fibroblasts to Quiescence. ACS Appl Mater Interfaces. 2019;11:26060. doi:10.1021/acsami.9b03313
27. Saha S, Xiong X, Chakraborty PK, et al. Gold Nanoparticle Reprograms Pancreatic Tumor Microenvironment and Inhibits Tumor Growth. ACS Nano. 2016;10:10636. doi:10.1021/acsnano.6b02231
28. Kudgus RA, Szabolcs A, Khan JA, et al. Inhibiting the Growth of Pancreatic Adenocarcinoma In Vitro and In Vivo through Targeted Treatment with Designer Gold Nanotherapeutics. PLoS One. 2013;8:e57522. doi:10.1371/journal.pone.0057522
29. Khan JA, Kudgus RA, Szabolcs A, et al. Designing Nanoconjugates to Effectively Target Pancreatic Cancer Cells In Vitro and In Vivo. PLoS One. 2011;6:e20347. doi:10.1371/journal.pone.0020347
30. Mukherjee P, Bhattacharya R, Bone N, et al. Potential therapeutic application of gold nanoparticles in B-chronic lymphocytic leukemia (BCLL): enhancing apoptosis. J Nanobiotechnology. 2007;5:4. doi:10.1186/1477-3155-5-4
31. Li SD, Huang L. Pharmacokinetics and Biodistribution of Nanoparticles. Mol Pharm. 2008;5:496. doi:10.1021/mp800049w
32. Swierczewska M, Lee KC, Lee S. What is the future of PEGylated therapies? Expert Opin Emerg Drugs. 2015;20:531. doi:10.1517/14728214.2015.1113254
33. Lipka J, Semmler-Behnke M, Sperling RA, et al. Biodistribution of PEG-modified gold nanoparticles following intratracheal instillation and intravenous injection. Biomaterials. 2010;31:6574. doi:10.1016/j.biomaterials.2010.05.009
34. Owens DE
35. Stepien G, Moros M, Perez-Hernandez M, et al. Effect of Surface Chemistry and Associated Protein Corona on the Long-Term Biodegradation of Iron Oxide Nanoparticles In Vivo. ACS Appl Mater Interfaces. 2018;10:4548. doi:10.1021/acsami.7b18648
36. Chacon M, Molpeceres J, Berges L, Guzman M, Aberturas MR. Stability and freeze-drying of cyclosporine loaded poly(d,l lactide–glycolide) carriers. Eur J Pharm Sci. 1999;8:99. doi:10.1016/S0928-0987(98)00066-9
37. Franks F. Freeze-drying of bioproducts: putting principles into practice. Eur J Pharm Biopharm. 1998;45:221. doi:10.1016/S0939-6411(98)00004-6
38. Steinmetz NF, Manchester M. PEGylated Viral Nanoparticles for Biomedicine: the Impact of PEG Chain Length on VNP Cell Interactions In Vitro and Ex Vivo. Biomacromolecules. 2009;10:784. doi:10.1021/bm8012742
39. Hatakeyama H, Akita H, Harashima H. The Polyethyleneglycol Dilemma: advantage and Disadvantage of PEGylation of Liposomes for Systemic Genes and Nucleic Acids Delivery to Tumors. Biol Pharm Bull. 2013;36:892. doi:10.1248/bpb.b13-00059
40. Fang Y, Xue J, Gao S, et al. Cleavable PEGylation: a strategy for overcoming the “PEG dilemma” in efficient drug delivery. Drug Deliv. 2017;24:22. doi:10.1080/10717544.2017.1388451
41. Yang M, Lai SK, Wang Y-Y, et al. Biodegradable Nanoparticles Composed Entirely of Safe Materials that Rapidly Penetrate Human Mucus. Angew Chem Int Ed Engl. 2011;50:2597. doi:10.1002/anie.201006849
42. Sahlin JJ, Peppas NA. Enhanced hydrogel adhesion by polymer interdiffusion: use of linear poly(ethylene glycol) as an adhesion promoter. J Biomater Sci Polym Ed. 1997;8:421. doi:10.1163/156856297X00362
43. Yang Q, Jones SW, Parker CL, Zamboni WC, Bear JE, Lai SK. Evading Immune Cell Uptake and Clearance Requires PEG Grafting at Densities Substantially Exceeding the Minimum for Brush Conformation. Mol Pharm. 2014;11:1250. doi:10.1021/mp400703d
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.