")
Back to Journals » Journal of Inflammation Research » Volume 17
Targeting Neutrophil Extracellular Traps in Gouty Arthritis: Insights into Pathogenesis and Therapeutic Potential
Authors Li C, Wu C, Li F, Xu W, Zhang X, Huang Y, Xia D
Received 18 January 2024
Accepted for publication 7 March 2024
Published 19 March 2024 Volume 2024:17 Pages 1735—1763
DOI https://doi.org/10.2147/JIR.S460333
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Cantao Li,* Chenxi Wu,* Fenfen Li, Wenjing Xu, Xiaoxi Zhang, Yan Huang, Daozong Xia
School of Pharmaceutical Sciences, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Daozong Xia, School of Pharmaceutical Sciences, Zhejiang Chinese Medical University, Baichuan Street, Fuyang District, Hangzhou, Zhejiang Province, People’s Republic of China, Email [email protected]
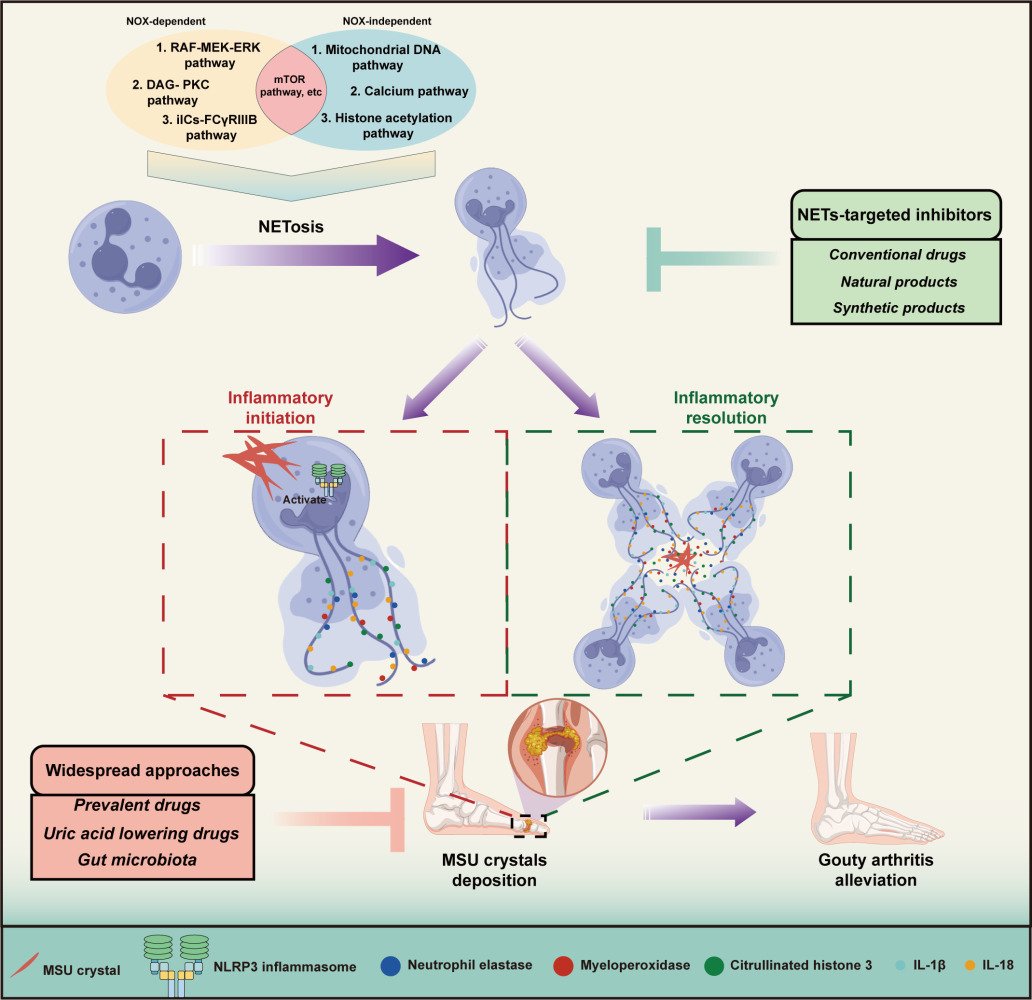
Abstract: Gouty arthritis (GA) is an immune-mediated disorder characterized by severe inflammation due to the deposition of monosodium urate (MSU) crystals in the joints. The pathophysiological mechanisms of GA are not yet fully understood, and therefore, the identification of effective therapeutic targets is of paramount importance. Neutrophil extracellular traps (NETs), an intricate structure of DNA scaffold, encompassing myeloperoxidase, histones, and elastases - have gained significant attention as a prospective therapeutic target for gouty arthritis, due to their innate antimicrobial and immunomodulatory properties. Hence, exploring the therapeutic potential of NETs in gouty arthritis remains an enticing avenue for further investigation. During the process of gouty arthritis, the formation of NETs triggers the release of inflammatory cytokines, thereby contributing to the inflammatory response, while MSU crystals and cytokines are sequestered and degraded by the aggregation of NETs. Here, we provide a concise summary of the inflammatory processes underlying the initiation and resolution of gouty arthritis mediated by NETs. Furthermore, this review presents an overview of the current pharmacological approaches for treating gouty arthritis and summarizes the potential of natural and synthetic product-based inhibitors that target NET formation as novel therapeutic options, alongside elucidating the intrinsic challenges of these inhibitors in NETs research. Lastly, the limitations of HL-60 cell as a suitable substitute of neutrophils in NETs research are summarized and discussed. Series of recommendations are provided, strategically oriented towards guiding future investigations to effectively address these concerns. These findings will contribute to an enhanced comprehension of the interplay between NETs and GA, facilitating the proposition of innovative therapeutic strategies and novel approaches for the management of GA.
Keywords: neutrophils, neutrophil extracellular traps, gouty arthritis, inflammation, NETs inhibitor
Graphical Abstract:
Introduction
Gouty arthritis (GA) is an inflammatory arthritis caused by abnormal monosodium urate (MSU) deposition that occurred in joints and peripheral tissues1 Ample evidence has suggested that the incidence and prevalence of GA are increasing around the world,2 bringing burdens on health systems.3 Patients with GA are characterized by high levels of serum uric acid (UA).4 experiencing bursts of symptoms and remission including intense pain, redness and swelling.5 Uric acid can accumulate in the bloodstream due to its excessive production or impaired renal excretion, ultimately resulting in the formation of needle-shaped crystals in the joints and adjacent tissues.6 Subsequently, the deposition of uric acid crystals in joints triggers an immune response as the body identifies the crystals as foreign substances, thereby initiating an inflammatory cascade that ultimately culminates in the distressing symptoms of gouty arthritis.7,8 Nevertheless, the underlying mechanisms behind GA onset and its interaction with the immune system are relatively vague.
Neutrophils are the most abundant immune cells in human and animal peripheral blood.9 As the first-line cells in immune system, neutrophils exhibit rapid responsiveness to both microbial and inflammatory cues emanating from damaged tissues. Neutrophils are swiftly mobilized to the site of injury to combat various pathogens, such as microcrystals.10 In homeostatic conditions, neutrophils are maintained at a low baseline density.11 However, when tissue damage or infection occurs, neutrophils assume critical roles as immune effectors, with their density increasing dramatically and their recruitment to the damaged site being promptly activated.12
Extracellular traps (ETs) are web-like structures containing DNA, histones, and various granule proteins like myeloperoxidase (MPO), released by activated neutrophils and other immune cells to capture and eliminate invading pathogens.13 Among them, neutrophils extracellular traps (NETs) have been implicated in different pathological conditions, such as GA.14 During GA development, NETs are released in response to MSU crystal stimulation, triggering an inflammatory cascade. It seems that inhibiting the formation of NETs, also called NETosis, appears as a feasible approach to alleviate GA.15 Several studies have also identified various factors that regulate the formation and degradation of NETs.16,17 However, evidence also showed that aggregated NETs (aggNETs), consisting of high-density of NETs, appeared to exert protecting role from inflammation via packing MSU crystals, and the mechanisms of which still remain elusive.18 Of note, targeting components from NETs offers new avenues for developing NET-based therapies for the GA treatment. In this review, we undertake a comprehensive review of the constituents of NETs and the diverse mechanisms underpinning NETosis, providing a detailed illustration. Additionally, we synthesize current knowledge on clinically available pharmacological interventions for GA and innovatively evaluate natural and synthetic product-based inhibitors that target NET formation as novel therapeutic options. This review aims to illuminate potential therapeutic strategies that leverage our understanding of NETs to combat GA effectively.
NET: The Special Network Structure Released by Neutrophil
NETs have gained a wide range of attention for their production, which is accompanied by the following inflammatory cascade. NETs are reticular skeletons composed of DNA released by neutrophils outside the cell, to which nuclear proteins (eg, histones), cytoplasmic proteins, granular proteins (eg, neutrophil elastase (NE), myeloperoxidase (MPO)), and antimicrobial peptides, etc., are attached (Figure 1). Most of these components exert different effects when they encounter different responses in the immune system.
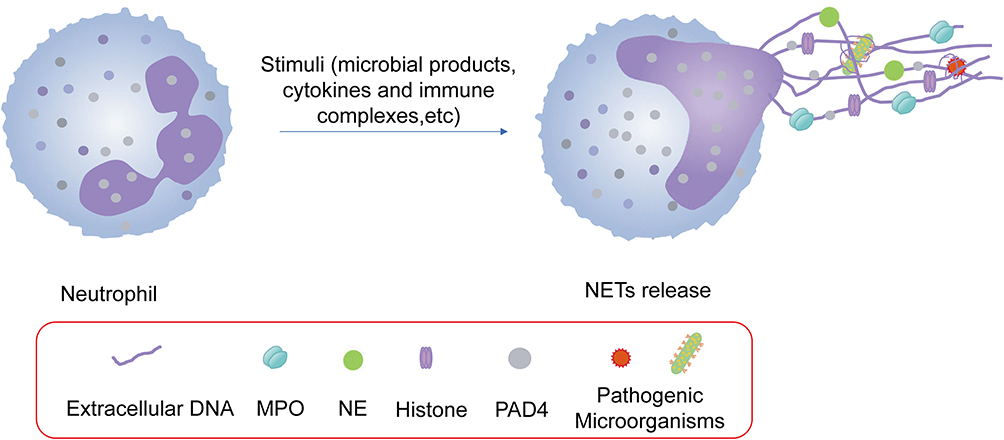 |
Figure 1 Neutrophils extracellular trap formation. After being stimulated by various inducers including microbial products, cytokines, immune complexes and etc, neutrophils undergo chromatin decondensation and nuclear membrane breakdown, leading to the release of NETs. NETs are mainly DNA-based skeleton structures whose surfaces are inlaid with histones, NE, MPO PAD4. Moreover, NETs are characterized by antibacterial effect through capturing and killing pathogenic microorganisms. |
The extracellular DNA in NETs is derived from the chromatin of the neutrophil nucleus, which is extruded from the cell and released into the extracellular space. The process of NETosis, characterized by the dissolution of the nuclear envelope, decondensation of chromatin and extrusion of DNA, is activated in response to various stimuli.19,20 As the major component of NETs, extracellular DNA is considered to be a damage-associated molecular patterns (DAMPs)21,22 that induces pro-inflammatory cascades.23 NETs have been implicated in the pathogenesis of various of inflammatory and autoimmune diseases, including sepsis, rheumatoid arthritis, and lupus.24,25 The DNA in NETs can activate different immune cells, such as macrophages and dendritic cells, leading to the release of pro-inflammatory cytokines and amplification of the inflammatory responses.26,27 A study28 has shown that the level of cell-free DNA in the plasma of patients with sepsis was positively correlated with mortality, meaning the higher the level of cell-free DNA, the higher the mortality rate of sepsis. Furthermore, DNA has been identified as a key factor in extending the lifespan of neutrophils. Notably, stimulation of neutrophils with mitochondrial DNA has been shown to enhance their activity.29 DNA’s significance as the fundamental framework of NETs is undeniable.
Histones are a group of highly basic and conserved proteins located in the nucleus and are a key component of the web-like structures that are released by activated neutrophils. Several histone subtypes, including histones H2, H3, and H4 constitute a complex with DNA called nucleosome.30 Histones, a key component of NETs, contribute significantly to the antimicrobial activity of NETs. Specifically, the positively charged amino acids in histones negatively interact with the charged cell membranes of microorganisms, promoting their adherence to the NETs and leading to subsequent elimination.31 Not only do histones possess antimicrobial properties, but they also exhibit toxic effects on the host, triggering pro-inflammatory responses upon their release from the nucleus into the extracellular space.32,33 The release of histones from dying cells, including from NETs, has been implicated in the pathogenesis of a range of inflammatory and autoimmune diseases, including sepsis, rheumatoid arthritis, and lupus.34 On the one hand, histones can be passively released through the necrotic program to exacerbate inflammatory responses. Moreover, histones can also be actively released during NETosis to exert an antibacterial effect.35 The role of histones as damage-associated molecular patterns (DAMPs) can trigger toll-like receptors (TLRs), activate the NLRP3 inflammasome, and induce calcium influx, leading to the initiation of inflammation in conditions, such as acute pancreatitis.36 In septic mice, histones can be expected to result in endothelial dysfunction, organ failure and even death.37 It must be mentioned that high mobility group box 1 (HMGB1) is a key component of NETs, where it plays an important role in immune response like histones. HMGB1 is a highly conserved nuclear protein present in most mammals. In its capacity as DAMPs, HMGB1 can promote the aggregation of neutrophils at sites of tissue damage and enhance inflammatory responses via interaction with other DAMPs (such as DNA) or pathogen-associated molecular patterns (PAMPs) (such as lipopolysaccharide (LPS)).38,39 HMGB1 containing disulfide bonds binds to TLR4, inducing cytokine production by macrophages and also promotes NETs formation by neutrophils.40 The relationship between histones and HMGB1 in the context of NETs is complex and involves both synergistic and antagonistic interactions.41 HMGB1 is known to promote the release of histones from neutrophils and to enhance their ability to bind to bacterial membranes, thereby increasing the efficacy of NETs in killing invading microorganisms.42 However, HMGB1 is able to exert both pro-inflammatory and anti-inflammatory effects, depending on the cellular and molecular context when histones begin to activate immune cells and promote inflammation.43,44
Peptidyl arginine deiminase 4 (PAD4) is a calcium-dependent enzyme that plays a critical role in NETs formation. During the process of NETosis, activated neutrophils undergo chromatin decondensation and nuclear membrane breakdown, allowing for the release of NETs composed of DNA, histones, and other proteins.45 The citrullination of histones by PAD4 is a crucial step in this process, as it alters their charge and structure, facilitating their release from chromatin and promoting the formation and stability of NETs.46,47 PAD4 has been implicated in the pathogenesis of a range of autoimmune and inflammatory diseases.48–50 Wesley et al demonstrated that mice lacking PAD4, a key enzyme involved in NETosis, exhibited a significant reduction in NET formation and pro-inflammatory cytokine production, leading to protection against acute kidney injury induced by renal ischemia/reperfusion.51 Renal function was restored to 48 hours after ischemia/reperfusion, whereas renal function in wild-type mice gradually deteriorated. The PAD-specific inhibitor YW3-56 was used for validation, which indicates that PAD4 plays a key role in ischemia/reperfusion-induced acute kidney injury. It is also a strategic point to differentiate apoptosis52 and necrosis.53,54
MPO is present in specific tissues within neutrophils, monocytes, and macrophages. The majority (95%) of MPO found in the bloodstream originates from neutrophils, making changes in its levels a reflection of alterations in neutrophil functionality. Some studies55,56 have found that MPO in NETs was biologically active and exhibited bactericidal ability after binding to DNA in NETs, where it generated hypochlorous acid and other reactive oxygen species (ROS) that contributed to the microbicidal activity of NETs.57 MPO is also involved in the modification of histones, which helps increase the antimicrobial activity of NETs.58 Simultaneously, by utilizing hydrogen peroxide (H2O2) and chloride ions, MPO has the ability to generate hypochlorite, which effectively contributes to the eradication of bacteria. This evidence demonstrates that MPO can enhance the bactericidal efficacy of NETs, while also causing tissue damage. Therefore, MPO may act as an antigen in the pathogenesis of anti-neutrophil cytoplasmic antibody-associated vasculitis.59 Meanwhile, MPO has emerged as a key player in the pathogenesis of numerous inflammatory conditions, such as vasculitis, systemic lupus erythematosus, and acute pancreatitis.60–62 By producing ROS and other substances, MPO can inflict tissue damage and exacerbate inflammation, thereby contributing to the pathophysiology of these diseases.63 Recent studies have shown that inhibiting MPO activity with a specific inhibitor is capable of suppressing the formation of NETs.57,64 Collectively, these findings underscore the importance of MPO as a critical component of NETs in mediating their antimicrobial activity, but also emphasize the need to regulate its activity to prevent its contribution to inflammatory disease pathogenesis.
Neutrophil elastase (NE) represents a serine protease variant prominently housed within the azurophilic granules of neutrophils, exerting a pivotal role as a fundamental constituent within NETs. The presence of both NE, as well as MPO, within neutrophils is notable, as these enzymes are extensively associated with the fiber network of NETs.65 During NETosis, translocation of NE to the nuclear membrane is required for chromatin decondensation, whereas MPO can bind to chromatin and enhance chromatin dedensification.66,67 In addition to degrade extracellular matrix proteins and inflammatory mediators such as cohesin, various membrane proteins and IL-8, etc., NE is capable of inhibiting tissue factors and promoting the formation of vascular fibrin.66,68,69 Studies have shown that the formation of NETs involved with MPO and NE depends on the different types of stimuli. Parker59 et al found that the generation of phorbol 12-myristate 13-acetate (PMA)-stimulated NETs required the involvement of MPO without the need for Staphylococcus aureus or E. coli. Conversely, Leishmania parasites70 induced the generation of NETs that required NE without the need for MPO and ROS. As a potent inducer of NETs, PMA is widely used in the research about NETs based on the different kinds of signaling pathways, enzymes, and ROS requirements.71 Another inducer of NETs is the MSU crystal. In gout, MSU crystal is the main precipitated form of urate in the blood, at the same time, is also an important participant in activating the neutrophils, which facilitates the release of NETs.72 It is worth noting that, unlike PMA, the release of NETs induced by MSU crystal is uncertain if the ROS involvement is needed. According to the in vivo research from Davidson and her companions,73 NET formation induced by MSU crystals was independent of ROS production. The same findings were also found in other studies. Tatsiy et al found that under the activation of MSU crystals, NETosis was found to be independent of endogenous ROS, but under the control of PAD4.74 Conversely, NETosis caused by MSU crystals in vitro was found to be in an ROS-dependent manner. What’s more, inhibiting ROS through different anti-oxidants restrained NETosis caused by MSU crystals.1 This controversial view deserves a deeper exploration, which may provide insights into the progression of NETosis in diseases.
The Formation of NETs: Vital NETosis and Suicidal NETosis
The formation process of NETs is called NETosis which is a novel type of programmed cell death distinguished from neutrophil apoptosis, necrosis and pyroptosis. The concept of NETosis was firstly purposed by Brinkman and his colleagues in 2004.75 They offered the opinion that neutrophils were activated and released extracellular meshwork, called NETs, that was decorated with granule proteins and chromatin, leading to degrade virulence factors and kill bacteria. Following an extensive span of nearly two decades devoted to research, there has been a discernible augmentation in the comprehension of NETosis, leading to a more comprehensive and profound knowledge of this biological process. The process of NETosis can be triggered by various stimuli, such as microbial products, cytokines, and immune complexes, and can occur via distinct mechanisms, mainly involving vital and suicidal NETosis that depend on the viability of neutrophils.19,45,76
The most striking difference between NETosis and other modes of death is the changes in the nucleus and the release of NETs. Upon stimulation, the nuclear membrane of neutrophils disintegrates into vesicles, while chromatin begins to deconcentrate. Then, antimicrobial peptides are released from intracellular particles to adhere to lost chromatin. Simultaneously, upon the disruption of the plasma membrane, a diverse array of intracellular components, including proteins, DNA, and other entities, are extruded into the extracellular milieu. Notably, this process does not entail the condensation of chromatin, commonly known as chromatin pyknosis. Although researches have proved that the formation of NETs improved the body’s defense mechanism, NETs are believed to be capable of causing the body to break the pro- and anti-inflammation balance.77,78 Apoptosis is cell shrinkage accompanied by DNA rupture and nucleus condensation. The whole cell forms apoptotic bodies through blebbing and other methods. Of note, the process of apoptosis does not cause a rupture of the plasma membrane and the release of NETs, which is the biggest difference between apoptosis and NETosis.79 Additionally, neither inflammatory mediators nor inflammatory responses occur in this process. Cell necrosis leads to the swelling and rupture of cells, including the processes like the swelling of various intracellular organelles such as the nucleus, the rupture of the plasma membrane, the release of cellular contents, and the triggering of inflammation.79 What distinguishes with NETosis is that in the process of cell necrosis, DNA is barely released. Fuchs et al80 performed live cell imaging of NETs to reveal the difference between NETosis, apoptosis and pyroptosis. Pyroptosis is typified by cellular shrinkage, accompanied by DNA fragmentation and degradation, which is similar with the apoptotic process.81 Pyroptosis is distinguished by cell necrosis accompanied by the process of cell swelling, rupturing and releasing intracellular inflammatory substances, which induces inflammation in the body without production and release of NETs structure.82,83
Vital NETosis, also known as non-lytic NETosis or “vivacious NETosis”, is a relatively new type of NETosis that was first described in 2011.84 Unlike suicidal NETosis, vital NETosis does not result in the death of the neutrophil, but rather, the release of NETs while the neutrophil remains alive and functional.85 During vital NETosis, the neutrophil undergoes significant morphological changes, including the formation of nuclear lobes and the extrusion of chromatin into the extracellular space, which is accompanied by the release of antimicrobial molecules such as myeloperoxidase and neutrophil elastase. Vital NETosis has been suggested to play a role in promoting wound healing and preventing excessive inflammation, as it can limit the spread of bacterial infections and promote tissue regeneration.86
On the other hand, suicidal NETosis, also known as lytic NETosis or “suicidal NETosis”, is a more well-known and characterized type of NETosis that results in the death of the neutrophil.87 Suicidal NETosis is distinguished by the rupture of the nuclear envelope of neutrophils, subsequently leading to the extracellular release of chromatin and granular proteins. This process is mediated by the activation of the NADPH oxidase complex, which produces ROS that lead to DNA damage and histone citrullination, as well as the activation of proteases and endonucleases that degrade the neutrophil’s nuclear and cellular components.88 Suicidal NETosis is important for the host defense against various pathogens, including bacteria, fungi, and viruses, but it can also contribute to tissue damage and the development of autoimmune and inflammatory diseases.89
In summary, vital and suicidal NETosis are two distinct forms of neutrophil death that contain the process of releasing extracellular traps to fight against pathogens. Vital NETosis is currently considered as a relatively new and less characterized process that enables neutrophils to remain viable and functional. In contrast, suicidal NETosis is a well-established mechanism that results in the death of neutrophils and is critical for host defense against infections.
The Underlying Mechanism of NETs Formation
According to the formation mechanism of NETs, it is mainly divided into three pathways: nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NADPH oxidase 2, NOX)-dependent, -independent and others (Figure 2).
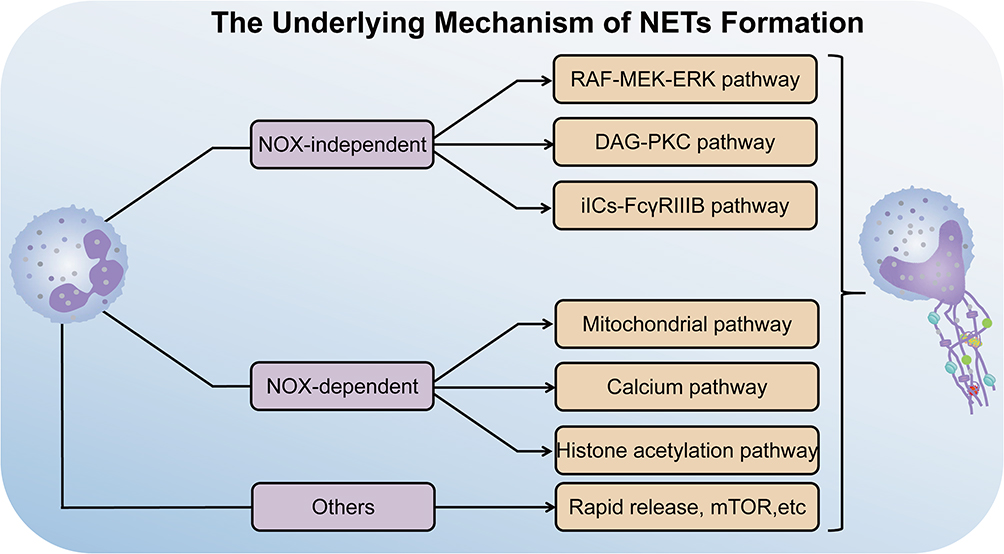 |
Figure 2 The underlying mechanism of NETs formation. The mechanism of NETs formation can be classified into NOX-dependent pathway, NOX-independent pathway and others. The NOX-dependent pathway to induce NETs formation contains RAF-MEK-ERK pathway, DAG-PKC pathway and iICs-FcγRIIIB Pathway. On the other hand, NETs formation is caused by NOX-independent pathway including the mitochondrial DNA pathway, Calcium pathway and histone acetylation pathway. In some special cases, NETs formation can be performed through neither NOX-dependent pathway nor NOX-independent pathway, as referred to rapid release of NETs formation. On the contrary, pathway like the mTOR pathway is involved in NETs formation by either NOX-dependent pathway or NOX-independent pathway. |
NOX-Dependent Formation of NETs
The NADPH oxidase complex is a critical enzyme involved in the generation of ROS that is necessary for the formation of NETs. It has been reported that NOX-dependent formation of NETs is a critical mechanism in the pathogenesis of various inflammatory and autoimmune diseases.90 The NOX-dependent pathway is the predominant mechanism involved in the production of NETs.80 Plenty of pro-inflammatory mediators can stimulate neutrophils to generate NETs, such as PMA, LPS, interlukin-6 (IL-6), IL-8, tumor necrosis factor alpha (TNF-ɑ), as well as bacteria, fungi, and chemicals.91 The activation of NOX by stimuli causes the generation of ROS, including O2, H2O2, and HOCl. In addition to killing microorganisms,92 ROS is capable of activating NE and MPO in neutrophils93 and interacting with the nucleus without restraint, in which serine proteases and NE cleave histones to promote chromatin decondensation.94,95 Soon afterwards, the nuclear membrane loses integrity, leading to the release of chromatin into the cytosol and subsequently the final form NETs.65,69 Here, the pathways involved in the NOX-dependent formation of NETs are illustrated in the following.
RAF-MEK-ERK Pathway
The rapidly accelerated fibrosarcoma (RAF)-mitogen-activated protein kinase (MEK)-extracellular signal-regulated kinase (ERK) pathway (RAF-MEK-ERK pathway) is one of the mitogen-activated protein kinase pathways. This signaling pathway exhibits the capacity to convey extracellular signals to the nucleus, facilitating interactions with specific transcription factors and subsequently eliciting context-dependent cellular responses.96 The RAF-MEK-ERK pathway can be activated by various stimuli, for example PMA and Fcγ receptors (FcγRIIIb), to induce NETosis.97 Some studies98,99 have shown that the RAF-MEK-ERK pathway is crucial in the process of PMA-induced NETs generation, while downregulating the expression of apoptotic protein Mcl-1 to prevent cell apoptosis. This downregulation can be blocked with protein kinase C (PKC), cRaf and MEK inhibitors.100 In addition, this pathway also acts as a vital role in NETs induced by unconventional stimuli. During the process of amoebiasis, neutrophils form the host are stimulated and NETs are induced by amoeba. Study has proved that selective inhibition on RAF and ERK exhibited prevention of E. histolytica induced NETs.101 The evidence strongly implies that RAF-MEK-ERK pathway serves as an upstream modulator of NADPH oxidase, thereby implicating its involvement in the formation of NETs through the activation of NADPH oxidase and up-regulation of anti-apoptotic proteins. While the study also showed that the inhibitor targeted NADPH could not block the E. histolytica-induced NETs formation,101 which might be explained by that ROS generated by trophozoites and processed by the extracellular MPO during the contact with neutrophils were of relatively importance for E. histolytica induced NETosis.102 These finding sheds light on the intricate regulatory mechanisms underlying NETosis and further underscores the interconnectedness of signaling pathways in orchestrating neutrophil functions.
DAG-PKC Pathway
Protein kinase C (PKC) is mainly distributed in the cytoplasm in an inactive state and is activated by diacylglycerol (DAG) and then transferred to the cell membrane to participate in the formation of NOX complex, leading to the increased generation of ROS and the sequent formation of NETs.103–105 The DAG-PKC pathway can be considered as an upstream signaling pathway for NOX-dependent formation of NETs.105 DAG mimicking PMA can stimulate neutrophils to form NETs,106 therefore, leading to the association between PKC signaling and NETs formation via NADPH oxidase-ROS pathway and PKC. Gray et al106 used LY333531 to inhibit PKCβ (LY333531 is a specific potent inhibitor of PKCβ that is a subtype of the PKC family), and the results showed the respiratory burst activated by NADPH oxidase was inhibited, and the formation of NETs was also suppressed. The findings of this study provide corroborative evidence for the involvement of the DAG-PKC pathway in the formation of NETs. Notably, among the PKC isoforms, PKCβ emerges as the primary regulatory isoform governing the process of NET generation.
iICs-FcγRIIIB Pathway
Immobilized immune complexes (iICs) are immune complexes that consist of autoantibodies and self-antigens, and Fc gamma receptor IIIb (FcγRIIIB) is a receptor expressed on neutrophils that binds to the Fc portion of IgG antibodies.107 Upon binding of iICs to FcγRIIIB receptors which presents on the surface of neutrophils, a signaling cascade is initiated, enhancing the induction of NETosis and subsequent formation of NETs in an ROS release-dependent manner.107 Alemán et al108 demonstrated that activation of FcγRIIIB receptors using specific antibodies effectively induces the generation of NETs, akin to the stimulatory effect observed with PMA. These findings highlight the iICs-FcγRIIIB pathway as a significant contributor to the mechanistic landscape of NET formation. Upon binding of iICs to FcγRIIIB, a plethora of downstream signaling cascades is triggered. Among these, the activation of protein kinases, particularly the Src family kinases, plays a pivotal role in phosphorylating and activating subsequent downstream effectors. Notably, this includes the guanine nucleotide exchange factor Vav1, which, upon activation, serves as a crucial mediator engaging the small GTPase Rac, thereby facilitating its activation. Consequently, Rac activation leads to the initiation of the NOX complex, ultimately enhancing the production of ROS. This robust ROS generation contributes significantly to the formation of NETs, thus substantiating the integral involvement of this signaling pathway in the NETosis process.107 Alongside the involvement of the Src family kinases and Vav1, a constellation of additional downstream signaling pathways is likely implicated in the activation of the NOX complex and subsequent ROS production. These pathways encompass the RAF-MEK-ERK pathway and the DAG-PKC pathway.100,109 These pathways can be activated by various signals, including cytokines, growth factors, and G protein-coupled receptors, and can activate the NOX complex and the subsequent formation of NETs.
NOX-Independent Formation of NETs
While the canonical pathway of NET formation involves the production of ROS by the NOX complex, NETs can also be formed through NOX-independent mechanisms. The exact signaling pathways for NOX-independent formation of NETs are still not fully understood, but several mechanisms have been proposed.110,111 Here are some of the proposed signaling pathways for NOX-independent formation of NETs.
Mitochondrial DNA (mtDNA) Pathway
The release of mtDNA can activate the immune system and contributes to the pathogenesis of several diseases, including autoimmune diseases, cancer, and cardiovascular diseases.112–114 It should be noted that mtDNA can serve as a DAMP to promote the formation of NETs.115 The pathway for mtDNA release involves several steps. Initially, a critical event in the cellular milieu involves the occurrence of mitochondrial permeability transition, culminating in the release of mitochondrial constituents, such as mtDNA, into the cytoplasmic compartment. This process significantly contributes to the intricate dynamics of cellular homeostasis and underscores the pivotal role played by mitochondria in orchestrating essential cellular functions. Second, the mtDNA is recognized by the cGAS-STING pathway, which triggers the production of type I interferons and other inflammatory cytokines. Finally, the mtDNA can directly activate the NLRP3 inflammasome, which leads to the production of IL-1β and other pro-inflammatory cytokines.116 According to Yousefi et al, their understanding suggests that the release of mtDNA is a regulated process that enables neutrophils to use their DNA as a weapon against microbes. In addition, it has been proposed that mitochondrial DNA may exert immunomodulatory effects on other immune cells.113,117 Furthermore, one of the responsible underlying mechanisms entails the belief that SIRT1 possesses the capacity to stimulate the initiation of mitochondrial permeability transition pore channels, facilitating the liberation of mitochondrial DNA and consequently giving rise to the formation of mitochondria-dependent vital NETs, as opposed to the conventional citrullinated histone H3-dependent NETs.118 It is implied that neutrophil-SIRT1-vital NET pathway may be a potential strategy to prevent tumor metastasis.
Calcium Pathway
The calcium pathway refers to the signaling cascade triggered by the release of intracellular calcium ions in response to stimuli such as LPS.119 In the context of NET formation, calcium signaling has been shown to activate the NOX-independent pathway. This pathway is triggered by the calcium-activated potassium channel of small conductance (SK channel), which is the major calcium activated potassium channel known to be present on neutrophils. Among the SK channel members (SK1, SK2 and SK3), SK3 is expressed predominantly on neutrophils and the knockdown of SK3 in differentiated HL-60 (dHL-60) cells reduces the NETs formation induced by ionomycin-mediated NOX-independent NETosis. Ionomycin is a natural calcium ionophore produced by a different gram-positive bacteria Streptomyces conglobatus and has proved the ability to induce NOX-independent NETosis.110 On the other hand, David et al raised the conclusion that NOX-independent NETosis caused by calcium ionophores required the mitochondrial ROS, which was regulated by the NOX2 enzyme.110 However, evidence also present the correlation between calcium pathway and NOX-dependent NETosis. Calcium is a vital regulator of NETosis, as it activated enzymes and signaling pathways that involve in the production of ROS, which are essential for NOX-dependent NETosis.19 One of the calcium dependent-enzymes is PAD4, which modified histones and chromatin decondensation.120 Also, calcium is deemed as a regulator of NADPH oxidase, the main source of ROS in neutrophils.121
Histone Acetylation Pathway
Histone acetylation is a post-translational modification that loosens the chromatin structure and affects gene expression.122 Histone deacetylases (HDACs) are enzymes that removes acetyl groups from histones and modulates chromatin condensation. According to the study,123 histone acetylation (particularly H4K8) and spontaneous NETosis at baseline were increased by agents that block HDAC activity and enhance histone acetylation, known as HDAC inhibitors. Also, the same situation happened to the NETosis induced by PMA, A23187, or LPS in an additive manner. In the further investigation, inhibition or knockdown of HDAC1, HDAC2, HDAC3, or HDAC6 increased histone acetylation and spontaneous NETosis at baseline. And the inhibition of HDAC was beneficial for the elevation of chromatin decondensation during NETosis induced by PMA. Interestingly, there was a performance that increased in mitochondrial ROS production and caspase-3 activation, both of which were associated with NOX-independent NETs formation, displayed by the inhibition of HDAC. What’s more, it is showed that the neutrophil death form was available to be switched from NETosis to apoptosis with HDAC inhibitors in dose-dependent manner.124 The evidence mentioned above implied that histone acetylation may be involved in NOX-independent NETs formation by multiple pathways and deserves attention of researchers in the field of NETs. Despite the researchers’ partial comprehension of the histone acetylation process, there is currently a limited repertoire of drugs targeting NETosis through modulation of histone acetylation. Consequently, there is still great potential for drug development in this specific domain.
Others
Apart from NOX-independent and NOX-dependent way for the formation of NETs, some methods are found to be classified into neither of them, as referred to rapid release of NETs formation. With the in-depth study of NETs, it has been found that neutrophils can continue to remain active after the clearance of NETs and come into play with antibacterial effects. This process only takes 5–60 minutes, which is different from the release of suicidal NOX-dependent NETosis or NOX-independent NETosis lasting more than 3 hours.125 At the end of this process, neutrophils eventually rupture and die. Yipp et al45,126 proposed that the formation of nuclear DNA in rapid NETosis was dependent on the binding of LPS. Under the action of gram-negative bacteria, generally, LPS binds to TLR4 on the platelet surface to generate NETs in a NOX-independent manner. For gram-positive bacteria, TLR2 and complement receptor 3 are required. After a few minutes of S. aureus stimulation, neutrophils were observed under a microscope to depolymerize their chromosomes, forming beads of DNA strands and nucleosomes. Late chromosomes are excreted in the form of nuclear vesicles, and neutrophils appear to be anucleate. Parts of chromatin and dense granules are excreted from cells by exocytosis to form NETs. The process mentioned above only takes 10 minutes. Anucleate neutrophils are still chemotactic, and this is manifested by hyperpolarization and poly pseudopodia crawling. They still have the function of degranulation and engulfing NETs released by bacteria. It is worth mentioning that according to the research, process of rapid release of NETs did not require the participation of NOX.127–129
On the other hand, mechanistic target of rapamycin (mTOR) signaling pathway is not solely included in the NOX-independent and NOX-dependent pathway, as it can potentially play a role in both pathways. mTOR is a serine/threonine protein kinase that regulates cellular growth, proliferation, and survival.130,131 mTOR signaling plays an important role in the regulation of autophagy, which is a conserved catabolic process that degrades cellular components and organelles, leading to NETs formation.132 Neutrophils are highly metabolically active cells that require significant amounts of energy to perform their functions.133
It has been reported that inhibiting the mTOR signaling pathway with rapamycin enhanced both autophagy and NETs formation, while activating the mTOR signaling pathway with insulin suppressed both processes in neutrophils isolated from health donors. And blocking autophagy with 3-MA or chloroquine showed a decrease in spontaneous NETs formation.134 It is suggested that spontaneous NETs formation is negatively regulated by the mTOR signaling pathway. However, a different trend in the relationship among mTOR, autophagy and NETs formation was found. Two β-lactam antibiotics, meropenem and ceftazidime/tazobactam, induced the activation of mTOR signaling pathway and inhibited autophagy in neutrophils and HL-60 cells. Moreover, they showed that blocking the mTOR signaling pathway with rapamycin or inhibiting β-lactamase with clavulanic acid attenuated the NETs formation induced by the antibiotics.135 Another study displayed that inhibiting mTOR pathway with rapamycin or Torin 1 enhanced autophagy and reduced NETs formation in PMA-stimulated neutrophils, while activating the mTOR pathway with insulin-suppressed autophagy and increased NETs formation in LPS-stimulated neutrophils. Also, blocking autophagy with 3-MA or bafilomycin A1 increased NETs formation in PMA-stimulated neutrophils, while enhancing autophagy with trehalose decreased NETs formation in LPS-stimulated neutrophils.136 Based on the aforementioned evidence, mTOR signaling may regulate NET formation by modulating autophagy, although the underlying mechanism appears to depend on the specific stimuli. Nonetheless, the exact mechanisms and implications of this regulation remain elusive and warrant further investigation.
In summary, various mechanisms do exist to achieve the purpose of NETs formation through specifical stimuli. It is supposed to be note that some pathways mentioned above possess the interaction with each other, rather than being independent, suggesting an objective and noticeable view that the classification used to distinguish either NOX-dependent pathway or NOX-independent pathway is relative.
Regulatory Role of NETs in GA Inflammation
GA is a common disease associated with inflammation. The main cause of GA can be attributable to a disorder of purine metabolism leading to excessive production or insufficient excretion of UA. GA attack usually occurs late at night accompanied by joint pain, swelling, redness and fever. Symptoms can cause discomfort or tingling in the joints and can get worse within 24 hours.5,137 In most cases, the acute manifestations of inflammation tend to subside spontaneously within a few days to weeks, without any notable residual effects. This observation suggests the existence of efficacious mechanisms that effectively mitigate acute inflammation.138
According to the pathogenesis of gout, there are 4 stages of gout, including asymptomatic hyperuricemia, acute gouty arthritis, intercritical gout and chronic tophaceous gout.139 GA is typically associated with the acute stage of gout. Nonetheless, there is a significant information that not all patients with gout will experience GA, and GA attacks will not only occur in the acute gouty arthritis stage but also in the chronic tophaceous gout stage.137,140 As reported that GA was the most commonly associated with the acute stage of gout and was the most common presenting symptom of gout causing inflammatory symptoms.141,142 Also, the chronic tophaceous gout stage is the most advanced stage of the disease and is characterized by the presence of tophi, which are deposits of urate crystals in the soft tissues with the less severe inflammation compared to the early stage.139 It is implied that there must be some physiological and pathological activity that acts as a regulator of inflammation.
Among different targets, NET is a crucial role in regulating inflammation.92,143 As is mentioned above, neutrophils can be activated through various mechanisms and pathways leading to the releasing of NETs. During the process of GA, stimuli like MSU crystals are beneficial for the recruitment of neutrophils and the generation of NETs, which is accompanied by the release of DAMPs such as histones, activate the immune system to release pro-inflammatory cytokines around joints resulting in an inflammatory environment.74,144,145 Herein, it is necessary to figure out the regulatory role of NETs in GA inflammation.
Inflammatory Initiation
GA is caused by the deposition of monosodium urate (MSU) crystals in joints, which triggers a strong inflammatory response. The MSU crystals are recognized by the innate immune system, which activates resident macrophages and recruits neutrophils to the joint resulting in the accompanying inflammation.146 When the crystals accumulate in the joint, they trigger an inflammatory response by activating the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome, a large protein complex that regulates the immune response. The activation of the NLRP3 inflammasome leads to the secretion of pro-inflammatory cytokines such as interleukin-1 beta (IL-1β) and IL-18, which causes the characteristic pain, redness, and swelling associated with gout. The activation of the NLRP3 inflammasome and subsequent release of IL-1β and IL-18 are triggered by innate immune cells like macrophages and dendritic cells, which recognize danger signals and respond by initiating the inflammatory response.147,148 During the process of GA, it is widely regarded that MSU stimulate macrophages to produce IL-1β and IL-18 via the NLRP3 inflammasome, which in turn leads to the recruitment of neutrophils and subsequent release of NETs. Meanwhile, neutrophils then undergo a process of activation and degranulation, leading to the release of their intracellular contents, including ROS, citrullinated histones, and granule enzymes, which together promote the formation of NETs.149 Also, neutrophils can release NLRP3 inflammasomes through a process called phagocytosis, which is the ingestion of foreign particles such as urate crystals.95,150 The urate crystals are recognized by the neutrophils and internalized through the formation of phagosomes. The phagosomes then fuse with lysosomes to form phagolysosomes, which contain enzymes and ROS that can degrade the urate crystals.151–154 This process results in the release of DAMPs, such as mitochondrial DNA and ATP, which can activate the NLRP3 inflammasome and bring about the generation of IL-1β and IL-18, ultimately promoting NET formation.18,155,156
NETs are deemed to be a vital role in initiating GA inflammation. Excessive formation of NETs is a sign of increased GA flare and a critical factor that contributes to GA pathology.157 NETs can cause tissue damage and trigger inflammation in GA in multiple ways. First, the histones and granular proteins from NETs are capable of activating the complement system and promoting the recruitment of other immune cells, such as monocytes and macrophages, to the site of inflammation.34,158,159 The recruited immune cells further propagate the inflammation by secreting pro-inflammatory cytokines, such as IL-1β, TNF-α, and IL-6.15,137,160 What’s more, NETs can directly foster inflammation by various pro-inflammatory molecules such as histones, DNA, and granule proteins that can activate the NLRP3 inflammasome.84,161 (Figure 3) For example, histones from NETs directly activate the NLRP3 inflammasome by disrupting lysosomal membranes and releasing cathepsins into the cytosol, which in turn activates the NLRP3 inflammasome that assembles with ASC and pro-caspase-1 followed by the step to cleave the pro-caspase-1 into caspase-1 and finally the release of IL-1β (Figure 3).33 Last but not least, NETs can cause mitochondrial damage, resulting in the release of mtDNA, which can activate the NLRP3 inflammasome in the diabetic wound.162,163 In the recent years, NETs have been treated as a potential target for the treatment of gout. Compelling evidence suggests that loganin exerts its inhibitory effects on NLRP3 inflammasomes by repressing mitochondrial stress in macrophages.164 However, similar finding has not been seen in studies of MSU crystals induced NETosis, leading to a valuable direction that needed explored. An interaction between macrophages and neutrophils has been used as a method for the treatment of gout. Ji Hye Jeong and his colleges proposed that synovial fluid macrophages were capable of clearing NETs by means of enhancing engulfment of MSU crystals without inducing any immunological response.15 Taken together, it is believed that NETs function as an intrinsic alarming that mainly initiates the inflammasome activation and elicits the inflammatory response in GA.
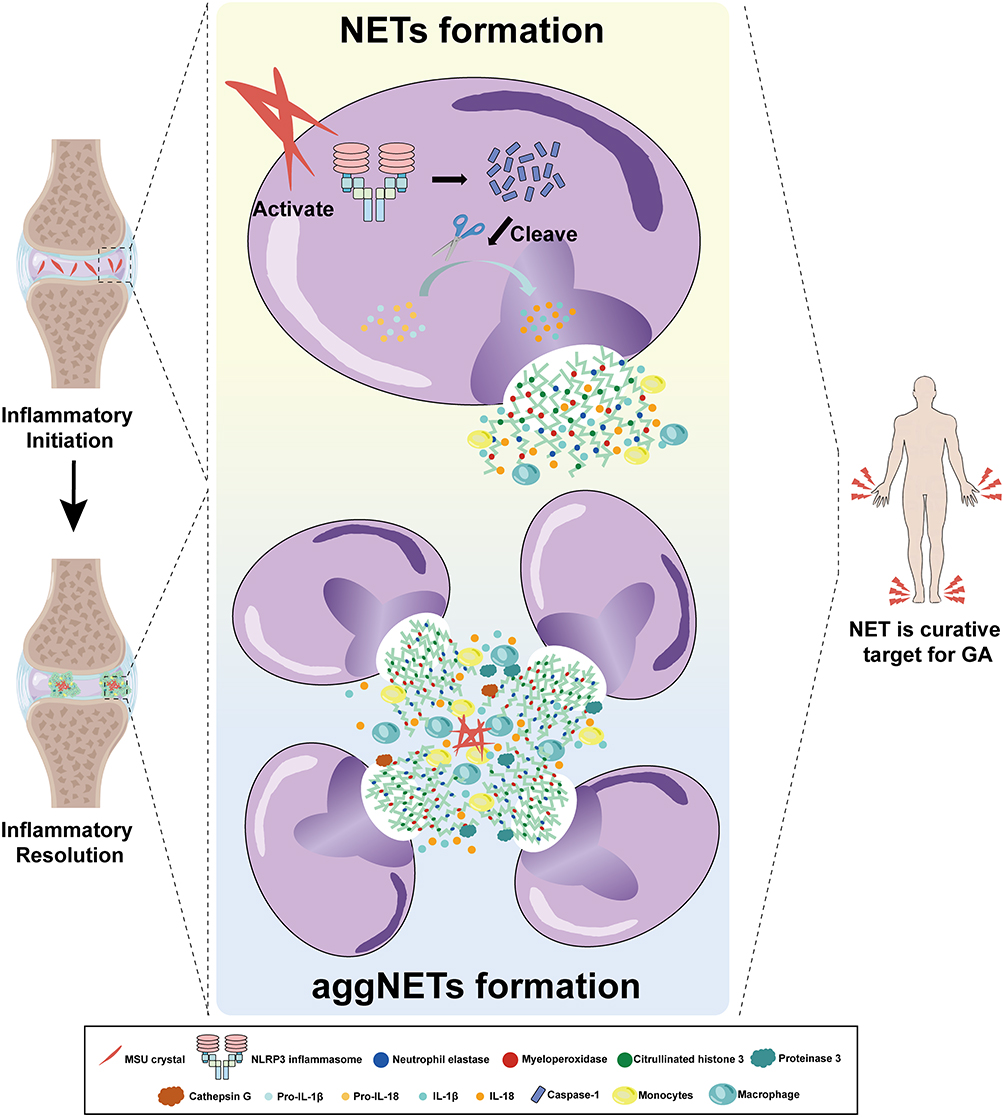 |
Figure 3 Regulatory role of neutrophil extracellular traps in gouty arthritis. MSU crystal deposition is formed at the joints due to high serum uric acid concentration, leading to the appearance of inflammatory response of neutrophils. During the early period of gouty arthritis, the NLRP3 inflammasome, which is activated by MSU crystals directly or indirectly, is responsible for turning pro-caspase-1 into the mature form of caspase-1 after the assembly of NLRP3. Pro-IL-1β and pro-IL-18 are transformed into IL-1β and IL-18 with the help of caspase-1, exhibiting pro-inflammatory effects and promoting the generation of NETs, which are accompanied by adherence to neutrophil elastases, myeloperoxidases and citrullinated histone 3. Additionally, macrophages and monocytes recruited by MSU crystals contribute to the generation of IL-1β and IL-18. With the recruitment of neutrophils, increasing numbers of neutrophils cause the aggregation of NETs, as referred to aggNETs. aggNETs manifest anti-inflammatory function by neutrophils serine proteases including NE, proteinase 3, cathepsin G and neutrophil serine proteases. At the same time, anti-inflammation is associated with the process of aggNETs engulfing and degrading MSU crystals. |
Inflammatory Resolution
In the chronic stage of gout, the symptoms that occurring at the beginning of gout are relieved and the inflammation around the joints is reduced.165 Inflammation during GA can expand without limitation, leading to joint necrosis and even death if there is no regulatory mechanism.166,167 There is no doubt that multiple mechanisms are utilized to prevent endless inflammation from happening. Evidence has showed that several key mechanisms are involved in inflammatory regression during gouty arthritis.157,168–170 Among them, NETs serve as a vital role in inflammation resolution in the form of aggregation, referred to as aggNETs. Here, we describe an inflammatory resolution that is regulated by NET.
The process of aggNETs formation begins when neutrophils are exposed to uric acid crystals. The crystals stimulate neutrophils to release NETs, which then aggregate and entrap the crystals within the web-like structure of the NETs.149 The aggregation of NETs and uric acid crystals leads to the formation of aggNETs that is capable of entrapping and degrading MSU crystals.149,171 In the research from Christine et al, neutrophils recruited to the site of inflammation and underwent oxidative burst caused by MSU crystals, which led to the formation of NETs.171 As neutrophils recruited to the site of inflammation, aggregation of NETs occurred under a high density of neutrophils and then resulted in the degradation of cytokines and chemokines. In another study, aggNETs displayed it anti-inflammation via the protection from antiproteases in vivo and in vitro.172 Actually, the process of manifesting anti-inflammatory function by aggNETs needs the involvement of neutrophil serine proteases (NSPs). As a kind of neutrophils granule, NSPs contain NE, proteinase 3 (PR3) and cathepsin G. In the context of gouty arthritis, NSPs are believed to contribute to the resolution of inflammation. It has been shown that neutrophils release elastase and PR3 together with aggNETs, which can then engulf and degrade MSU crystals, thus preventing further activation of the NLRP3 inflammasome and subsequent release of pro-inflammatory cytokines like IL-1β and IL-18 (Figure 3).173–175 Moreover, NSPs have been found to directly degrade and inactivate a range of pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6, and chemokines such as IL-8.171 Other than that, a report from Jasmin et al showed that the cytotoxic effect of histones on epithelial cells could be weakened by aggNETs’s function of sequestering and degrading histones, and the process of which required the participation of serine proteases, NE and PR3.176 The research suggested that the resolution of inflammation induced by histones resulted from degradation and detoxification of aggNETs. Collectively, aggNETs with the effector NSPs can exert anti-inflammatory effects through various mechanisms, including the degradation and inactivation of pro-inflammatory cytokines and chemokines. In recent times, the concept of NSP has received limited attention in the context of gout treatment. Researchers have instead directed their focus towards investigating distinct components of NSP for in-depth study and exploration. This shift in research emphasis signifies a renewed interest in understanding the intricate roles played by individual NSP constituents and their potential implications for therapeutic interventions in the management of gout and related inflammatory disorders.
Subsequently, the focus of attention falls on the formation of aggNETs, as its formation is currently viewed differently. It is believed that the release and degradation of inflammatory mediators is a dynamic balance.177 NSPs bonding to aggNETs cause the resolution of inflammation, which implies that the ability to degrade inflammatory mediators is greater than the ability to release them. According to the research by Jonas et al,172 peak generation and maximum supernatant concentrations of cytokines and chemokines appeared at a neutrophil density of 20–40×106/mL during the stimulation of MSU crystals. The study also found that the contents of inflammatory cytokines and chemokines began decreasing at higher densities of neutrophils with the help of NSPs. Moreover, maximum release of inflammatory mediators and chemokines occurred at a neutrophil density of 20–40×107/cm3 in arthritis mice model induced by MSU crystals, whereas the contents of the mediator reduced at higher density of neutrophils.172 The evidence mentioned above suggests the neutrophil induced inflammation is a self-limiting condition containing a dynamic process of releasing and degrading inflammatory mediator and chemokines, and more importantly, the formation of aggNETs seems to depend on the density of neutrophils. While in another study, the authors held the opinion that aggNETs formation depended on ROS in vivo and in vitro. In this study, it turned out that neutrophils had a weaker ability to degrade inflammatory mediators and generate aggNETs when compared to normal.171 In line with this, ROS-deficient Ncf1**mice which was characterized by the inability to produce ROS and represent mice model of CGD, showed a reduction in NETs aggregation and an increase in inflammatory mediators during the stimulation by MSU crystals when compared to air pouches of wild-type mice. It is assumed that the number of neutrophils only determines the size of the aggregates and then the aggNETs formation starts at 50 μg/mL of MSU crystals in a dose-dependent manner. Moreover, the level of neutrophils does not appear to be a critical factor, as NETs aggregation can still be induced by other stimuli such as ATP or lactoferrin, even at low neutrophil concentrations (5×106/mL).171 There exists uncertainty regarding the underlying mechanism for the formation of aggNETs that requires clarification.
NETs as Therapeutic Target for GA Treatment
The evidence we mentioned above strongly illustrates the close connection between GA and NETs. With the increasing research on GA in recent years, well-established anti-GA drugs and decoction have been widely used in the gout population.178–181 In addition to this, raising number of pharmaceutical agents are presently undergoing development to counteract GA in accordance with its pathogenesis. NETs have emerged as a pivotal target in the context of GA, and their inhibition has garnered considerable interest as an attractive therapeutic strategy for managing this condition. Consequently, an increasing array of drugs aimed at modulating NETs formation are currently under exploration and development, signifying the potential for novel and promising therapeutic avenues in GA treatment.
Clinical Approaches for GA
Prevalent Drugs
According to the American College of Rheumatology Guideline for the Management of Gout182 published in 2020, first-line drugs commonly used in clinical practice for GA attack involve colchicine, non-steroidal anti-inflammatory drugs (NSAID) and glucocorticoid. Colchicine has been a medicine widely used in GA for a long history.183,184 The incidence of adverse reactions is high since the therapeutic dose of colchicine is close to the toxic dose.185 For patients with acute gout attacks, it is recommended to accept colchicine treatment in the first dose of 1 mg, 0.5 mg after 1 h, and 0.5 mg after 12 h, with a frequency of once a day or twice a day.186 Small doses of colchicine combined with NSAID also are strategies recommended to cure acute GA attacks.187,188 Glucocorticoids are appropriate for the patients suffering GA attacks while having trouble with drug administration orally.182 In addition, lowering uric acid drugs, including allopurinol, benzbromarone and febuxostat are also vital treatments for gout. However, both advantages and disadvantages exist in drug administration. Studies have shown that side effects, including liver functional disorder and leukopenia, occur after taking allopurinol for a long time.189 Seriously, allopurinol could cause severe hypersensitivity in patients with a positive HLA⁃B*5801 allele.190 Benzbromarone is suitable for alleviating GA and hyperuricemia combined with acute GA, however, its unsatisfactory clinical efficacy limits its application.191 And clinical data suggested that a small number of patients might also be intolerable due to severe gastrointestinal adverse reactions, liver and kidney damage after benzbromarone administration.191,192 The use of febuxostat remains controversial because cardiovascular or all-cause mortality increased after long-term administration in adults over the age of 65, while the opposite result was proposed according to long-term cardiovascular safety of febuxostat compared with allopurinol in patients with gout.193,194 Surgery presents a comparatively lower occurrence of adverse effects on organs when contrasted with pharmacological interventions, rendering it a viable option that patients with gout are willing to consider. Also, surgical therapy is an effective approach to improve the quality of life of patients.195 The principal objective of minimally invasive arthroscopic surgery is to carefully insert a needle into the high-pressure region of the tophi, allowing for precise and closed cutting. This procedure facilitates the removal of excessive metabolites from the affected tissue and subsequently clears out the accumulated metabolites, resulting in the effective alleviation of joint cavity pressure. This meticulous approach accomplishes the immediate treatment goal with a focus on preserving tissue integrity and promoting patient recovery.196 Indeed, the widespread adoption and promotion of minimally invasive arthroscopic surgery are warranted due to its pronounced efficacy in facilitating rapid joint function recovery, minimizing trauma, and alleviating inflammation and pain. Consequently, patients experience a notable enhancement in their quality of life following the surgical intervention. Nevertheless, it is essential to acknowledge the inherent limitations of this surgical approach, particularly concerning the rehabilitation and overall quality of life of elderly patients. Exploring and addressing these limitations will be crucial in optimizing patient outcomes and ensuring the successful application of this surgical technique in the broader context of clinical practice.
Uric Acid Lowering Drugs
Uric acid-lowering drugs are a class of pharmaceutical agents specifically designed to mitigate elevated levels of uric acid in the bloodstream, a condition commonly associated with hyperuricemia and gout. Xanthine oxidase (XO) inhibitors represent a prominent class of pharmaceutical agents extensively employed as anti-gout drugs. XO is an enzyme that plays a critical role in the generation of uric acid, the end product of purine metabolism.197 Purines are nitrogenous bases found in DNA, RNA, and many cellular metabolites, and they can also be obtained from dietary sources such as meat and seafood.198–201 Purines are broken down into uric acid through a series of enzymatic reactions, with XO being the final enzyme involved in this process. XO catalyzes the oxidation of hypoxanthine to xanthine and then the oxidation of xanthine to uric acid, using molecular oxygen as the electron acceptor. This process leads to the ROS generation such as superoxide and hydrogen peroxide as byproducts, which can contribute to oxidative stress and damage in the body.202–204 Actually, XO inhibitors can be divided into three subsets including purines (such as allopurinol), non-purines (such as febuxostat) and natural inhibitors (such as quercetin).205 Allopurinol is a purine analog that acts as a suicide inhibitor of XO, meaning it irreversibly binds to the enzyme and prevents it from generating uric acid.206,207 Febuxostat, on the other hand, is a non-purine XO inhibitor that selectively targets the enzyme and has a longer half-life than allopurinol. Both drugs have been shown to effectively lower uric acid levels and prevent gout attacks.205 Nevertheless, the research of non-purine part of XO inhibitors attracts most attention because it possesses an interaction with several amino acids in the important domain called Mo-Pt that is a crucial domain for generating uric acid from substrates catalyzed by XO.208 It is necessary to note that a survey of the recent patent literature reveals that the predominant focus of research has been on the development of non-purine and natural XO inhibitors.205 To be added, different kinds of side effects like hypersensitivity can be caused by purines part of XO inhibitors.209–211 Besides, natural inhibitors have been a hot point of research for the gout treatment. Quercetin acts early on as a natural polyphenolic flavonoid compound and was first reported for its XO inhibitory effect in 1999.212 Recently, 1,4-dicaffeoylquinic acid, as a novel ingredient, is isolated from the leaves of Artemisia selengensis and is found to possess better XO inhibitory potential than allopurinol.213 Song and his companion proposed the davallialactone extracted from Sanghuangporus vaninii potently inhibited XO and possessed the potential to be developed into a medicine for gout.214 At the same time, another similar research raised the finding that the anti-gouty arthritis effect could be proved in vivo through an important medicinal and edible fungus in China, Phellinus igniarius which was constituted of davallialactone.215 Up to now, various kind of natural product exhibiting XO inhibitory activity have been found, leading to attract more and more attention from their potential for gout treatment.
Uricosuric agents can reduce serum uric acid levels in patients with hyperuricemia and gout by improving the clearance of uric acid in the kidneys. They achieved this by blocking the reabsorption of uric acid at the proximal tubule of the kidney via the inhibition of the urate transporter 1 (URAT1). The two main classes of uricosuric agents can be divided into probenecid and benzbromarone. Probenecid exerts its pharmacological action through competitive inhibition of URAT1, a urate transporter responsible for the reabsorption of uric acid in the proximal renal tubules. In contrast, benzbromarone exhibits dual inhibitory effects by targeting both URAT1 and xanthine oxidase, an enzyme crucially involved in the biosynthesis of uric acid. The distinct mechanisms of action of these two drugs hold significance in the context of hyperuricemia management and gout treatment, offering clinicians versatile options to modulate uric acid levels and mitigate the risk of gout-related complications. The suitable initial dose of probenecid was recommended for 250 mg for twice a day and the maximum dose could reach up to 1 mg twice a day.216,217 And benzbromarone’s administration had been specified at a dose of 100–200 mg/day for once a day.218–220 Other uricosuric agents are under investigation include sulfinpyrazone, losartan, and topiroxostat. There is possibility that association between each other may exist, such as aspirin and diuretics, which can reduce their effectiveness.221,222 In clinical practice, uricosuric agents are often used in combination with xanthine oxidase inhibitors, such as allopurinol or febuxostat, to achieve optimal control of serum uric acid levels.223,224 The selection of an appropriate therapeutic approach is contingent upon various factors, including the severity of hyperuricemia, the presence of comorbidities, and the individual patient’s response to treatment. These critical considerations underscore the necessity for personalized and tailored management strategies to optimize patient outcomes and ensure effective control of hyperuricemia and its associated complications.225,226 While uricosuric agents are generally well-tolerated, it is crucial to acknowledge that benzbromarone and probenecid may be associated with certain adverse effects. Benzbromarone has been linked to potential hepatic toxicity and cardiovascular risk, whereas probenecid may exhibit significant central nervous system toxicity and hypersensitivity syndrome, particularly when administered at higher dosages.227–231 These observations highlight the importance of vigilant monitoring and cautious dose adjustments to mitigate the risk of adverse reactions, ensuring the safe and effective utilization of these agents in the management of hyperuricemia and gout. As such, a balanced assessment of risks and benefits is essential to optimize patient outcomes and uphold safety in clinical practice.
Gut Microbiota
Due to the fact that the intestine is responsible for one third of uric acid excretion, gut microbiota possesses an important role in gout and hyperuricemia treatment because the function of eliminating uric acid cannot be ignored.232,233 The gut microbiota refers to the complex ecosystem of microorganisms that reside in the human gut, including bacteria, viruses, fungi, and protozoa. It plays a crucial role in regulating the immune system and maintaining host health. Several studies have shown that alterations in the gut microbiota may contribute to the development of gouty arthritis. For example, dysbiosis of gut microbiota has been associated with increased production of uric acid and inflammation, which are the key features of the disease.234,235 Modulating the gut microbiota through dietary interventions or probiotics has emerged as a potential therapeutic strategy for gouty arthritis. It has been reported that fisetin possessed the ability to decrease the content of uric acid through modulating the changes in the gut microbiota Bacteroides, and Firmicutes in hyperuricemia mice model.236 According to several cohort studies, evidence showed a higher risk of suffering from gout occurred to the people having western diet when compared to those of having Mediterranean diet.237 Prebiotic fiber and probiotics have been shown to increase the abundance of beneficial gut bacteria, such as Bifidobacterium and Lactobacillus, and decrease the abundance of pathogenic bacteria.238 These changes can be utilized for restoring a healthy gut microbiota and reducing inflammation as well as uric acid production.
In addition to dietary interventions, fecal microbiota transplantation (FMT) has been explored as a potential treatment for gouty arthritis. FMT involves the transfer of fecal material from a healthy donor to a recipient, with the aim of restoring a healthy gut microbiota. According to the reported findings, the administration of Qu-Zhuo-Tong-Bi decoction exhibited a notable impact on the gut microbiota composition, leading to a significant increase in the abundance of Allobaculum and Candidatus sacchairmonas. Concurrently, FMT from mice subjected to Qu-Zhuo-Tong-Bi decoction treatment demonstrated a beneficial effect in alleviating the hyperuricemia and gout-like condition in the Uox-KO mouse model.239 Similarly, another classic Chinese herbal medicine, Si-Miao decoction, exerted an improvement in repairing intestinal pathology and restored the abundance of phylum Proteobacteria and genus Helicobacter via FMT.240 A preliminary pilot study investigating the application of FMT in patients with gouty arthritis demonstrated encouraging findings. Notably, the study revealed noteworthy reductions in serum uric acid levels alongside evident improvements in clinical symptoms.241 These initial results warrant further investigation in larger-scale clinical trials to validate the therapeutic potential of FMT as a potential intervention in the management of hyperuricemia and gout-related manifestations. The observed positive outcomes underscore the need for more comprehensive and rigorous research to explore the potential role of FMT in ameliorating gouty arthritis and its associated metabolic abnormalities.
NETs-Targeted Inhibitors
Conventional Drugs
Even though the prevailing drugs used for the treatment of gout were not originally designed to specifically target NETs, advancements in comprehending their mechanisms of action have revealed their capacity to partially inhibit NETs formation. This emerging insight highlights an ancillary effect of these drugs and raising the importance of considering their broader impacts on neutrophil biology and inflammation, augmenting the potential for repurposing existing medications for the management of gout and related conditions. DNase (deoxyribonuclease) is an enzyme that occurs naturally in the body and is involved in the breakdown of DNA.242,243 It is produced by a variety of organisms, including bacteria, fungi, and animals, and still draws plenty of attraction even though it has been investigated in NETs study for years. Regarding the suppression of NETs, DNase is quite different from other NETs inhibitors since the focus of DNase is managed to extrude NETs instead of participating in the formation of NETs. The extracellular exposure of DNA in NETs makes them susceptible to degradation by DNase, which enzymatically cleaves the DNA backbone, resulting in the disassembly of the intricate web-like structures. This process facilitates the efficient clearance of NETs and their associated microorganisms, effectively curbing excessive inflammation and mitigating potential tissue damage. The breakdown of NETs by DNase plays a vital role in maintaining immune homeostasis and averting inflammatory overactivation, highlighting its significance as a regulatory mechanism in immune responses.244 The mechanism by which DNase degrades NETs involves the cleavage of the phosphodiester bonds that link the nucleotides in DNA. DNase complexes, composed of three enzymes including DNase I, DNase II, and DNase1L3, are able to bind to DNA molecules and cut the phosphodiester bonds between the nucleotides, leading to the degradation of the DNA backbone and the disassembly of the NETs.245 It’s worth noting that DNase is specific to extracellular DNA resulting in the expose of residual histones and neutrophils serine protease to host, possibly causing local inflammation and further tissue injury.246
Colchicine is a first-line drug highly recommended for the management of acute gout attacks, primarily owing to its exceptional therapeutic efficacy.182 Although colchicine was not originally intended to target NETs as a gout treatment, study had shown that colchicine impeded the production of NETs via reducing NOX2/ROS production and calcium influx.247 As early as 2018, scientific reports indicated that colchicine exerted an inhibitory effect on the ability of circulating neutrophils in the bloodstream of Behçet’s disease patients to form NETs.248 A recent study claimed that colchicine suppressed the production of NETs in patients with acute coronary syndrome post-percutaneous coronary intervention by restoring cytoskeletal dynamics.249 In addition, Apostolidou et al found that colchicine was capable of inhibiting the release of IL-1β in neutrophils and IL-1β activity in NETs.250 The evidence mentioned above suggest that the curative effect of colchicine on GA treatment may be targeting NETs.
As another member of first-line drug NSAIDs, ibuprofen is mainly used to treat rheumatic and rheumatoid arthritis.21,251 Victoria et al demonstrated that ibuprofen was able to alleviate the illness and inhibit NETs formation in bovine respiratory syncytial virus infection,252 which indicated the potential of ibuprofen in GA treatment by targeting NETs. At present, there are very few studies on the association between NSAIDs and NETs. It may be due to the clear pharmacological effects of NSAIDs and the lack of development potential.
Glucocorticoids are a class of medications that have potent anti-inflammatory and immunosuppressive effects.253,254 They have been used for many years for treating acute GA flares, as they can rapidly reduce pain and inflammation in affected joints.255 Glucocorticoids exert their therapeutic effects by modulating the immune response, leading to the suppression of inflammatory cytokine production, such as IL-1 and TNF-α, both of which play pivotal roles in the pathogenesis of GA. Additionally, glucocorticoids have been observed to impact NETs formation, further contributing to their anti-inflammatory properties and potential relevance in the management of GA. According to Amandine et al, glucocorticoids could reduce the formation of NETs in the lungs of the asthmatic horses either in vivo or in vitro.256 Despite the absence of specific literature reports on the effect of glucocorticoids in inhibiting NETs formation in the context of GA, the available evidence suggests that this mechanism holds the most plausible potential.
Natural Products
Natural products have been employed for medicinal applications spanning centuries, and their significance persists as a cornerstone in contemporary medicine. These biologically derived substances hold substantial therapeutic potential, contributing to the development of pharmaceutical agents and serving as a valuable resource for drug discovery and medical intervention. The enduring utilization of natural products underscores their enduring relevance and ongoing impact in addressing a diverse array of health conditions, highlighting their continued importance as an invaluable asset in the realm of modern healthcare practices.257
Resveratrol is a natural polyphenolic compound found in a variety of plant species, including grapes, peanuts, and berries. It contains numerous potential health benefits, including anti-inflammatory, antioxidant, and anti-aging properties.258 Resveratrol has been extensively studied for its potential effects on cardiovascular disease, diabetes, neurological disorders, and aging.258,259 According to the precious study, resveratrol showed the potential to cleave DNA in NETs and reduce the production of pro-inflammatory cytokines by neutrophils, suggesting its inhibitory effect on NETs formation.260 Additionally, evidence displayed that the patients with severe COVID-19 had an increased number of activated neutrophils that released NETs spontaneously, whereas the situation was improved by resveratrol by suppressing the formation of NETs.261 Recently, it is reported that resveratrol could improve GA in vitro and in vivo by inhibiting the activation of NLRP3 inflammasomes by triggering the Pink1/Parkin pathway to promote mitophagy.262 While the exact mechanisms linking resveratrol to gout and NETs are still under investigation, the cumulative evidence suggests that this natural compound holds therapeutic potential in managing both gout-related inflammation and the complex interplay of NETs in the pathogenesis of the disease.
Quercetin is a natural polyphenolic flavonoid and a kind of plant pigment found in various fruits, vegetables, grains and food sources, including onions, apples, berries, grapes, tea, and red wine.263 It is also available in supplement form. Pharmacologically, it has potent antioxidant and has anti-inflammatory properties.264,265 Clinical studies for the treatment of hyperuricemia showed that the growing content of plasma uric acid in healthy males could be significantly reduced by daily supplementation of quercetin (500 mg), for 4 weeks.266 Further study has shown the anti-GA effect of quercetin in vivo by suppressing the activation of the nuclear factor-κB (NF-κB) pathway and inflammasome.267 Moreover, recent study has revealed that quercetin exerted its ability to inhibit the activation and infiltration of neutrophils and suppress autophagy, thus led to the restrain of NETs formation in rheumatoid arthritis.268 The available evidence discussed above provides compelling indications of the inhibitory effect of quercetin on the process of GA. Therefore, conducting in-depth investigations into the use of quercetin as a treatment modality for GA represents a valuable avenue of study, with the potential to shed light on its clinical efficacy and mechanistic underpinnings.
Curcumin is a naturally occurring polyphenolic compound found in the rhizome of the turmeric plant, which has been used for centuries in traditional medicine for its anti-inflammatory, antioxidant, and anticancer properties. It is a bright yellow-orange pigment with a characteristic taste and odor, commonly used as a spice and food coloring agent.269 In recent years, curcumin has been investigated for its potential anti-GA properties. Study showed that curcumin was capable of improving the GA characteristics induced by MSU crystals, including joint swelling, inflammatory cell infiltration and MPO activity, and had influence on suppressing the NLRP3 activity and the activation NF-κB signaling pathway.270 Meanwhile, curcumin demonstrated its ability to inhibit the release of NETs induced by polybrominated diphenyl ethers. The underlying mechanism involved the modulation of ROS burst by interfering with Nrf2, a transcription factor central to the regulation of oxidative stress responses.271 Curcumin also can alleviate hepatic ischemia-reperfusion injury by inhibiting the formation of NETs resulting from the inhibition of MEK/ERK pathway.272 Therefore, targeting NETs may be one of the possible mechanisms of curcumin on the GA treatment. It should be also noted the fact that the application of curcumin is limited due to its rapid degradation, poor aqueous solubility, and low bioavailability.273,274 Modifying curcumin to strengthen the therapeutic effect becomes a strategy. For instance, curcumin-loaded tetrahedral framework nucleic acids were synthesized to deliver curcumin, which exhibited better drug stability, biocompatibility, ease of uptake, higher tissue utilization and a better anti-inflammatory effect when compared to free curcumin in vitro and in vivo.275 This is a worthwhile approach for other natural products that are not well bioavailable and thus limit their application.
Besides, another natural product from Andrographis paniculate called andrographolide possesses the potential to be a clinical drug for GA treatment. Recent research has proposed that andrographolide could attenuate the symptoms of rheumatoid arthritis by reducing the infiltration of neutrophils and NETosis in vivo. What’s more, andrographolide exhibited the ability in balancing NETosis and apoptosis. The NETosis induced by autophagy could be suppressed and the apoptosis induced by lipopolysaccharide-activated neutrophils was enhanced within the administration of andrographolide in vitro. These findings imply that andrographolide has considerable potential for being NETs inhibitor and further a strategy for GA treatment.276
In conclusion, utilizing natural products for the treatment of gouty arthritis by targeting NETs shows promising potential. The research and studies conducted in this area suggest that certain natural compounds possess anti-inflammatory and NETs-modulating properties, which could help alleviate the symptoms and progression of gouty arthritis. By targeting NETs, these natural products may disrupt the inflammatory cascade, reduce tissue damage, and decrease the frequency and intensity of gout flares. Furthermore, the use of natural products in gout treatment could offer a more holistic and potentially safer approach, as they are often associated with fewer side effects compared to conventional medications. However, it is important to acknowledge that more extensive research is needed to establish the efficacy, safety, and long-term benefits of these natural products in treating gouty arthritis via NETs targeting. Additionally, personalized treatment plans considering individual variations in response to natural compounds should be explored. Despite the promising results, natural products should not replace standard medical treatments for gouty arthritis. Instead, they can be considered as complementary therapies or adjuncts to conventional medications to enhance overall treatment outcomes.
Synthetic Products
Purinoceptor, also called P2 purinoceptor, is a category of nucleotide receptor. Among the families of purinergic receptors, the metabotropic receptors (P2Y) family is strongly associated with immune cell pathology and physiological activity. Under normal conditions,277 several P2Y receptors such as P2Y2, P2Y4 and P2Y6 receptors are involved in regulating the level of Ca2+, K+, Cl− and Na+. Whereas P2Y receptors act a vital role in immune cell recruitment, proliferation and differentiation.278 A specific inhibitor of the P2Y6 receptor called MRS2578 was used to verify the influence on neutrophil activation and aggNETs formation induced by gout associated MSU crystals. According to this research, the study revealed that suramin and PPADS, both recognized as general P2Y receptor blockers, as well as MRS2578, effectively inhibited the formation of NETs induced by MSU crystals. Attractively, even though it is the main receptors expressed in neutrophils, the P2Y2 receptor is barely involved in NETs generation due to the finding that the P2Y2 receptor antagonist called AR-C25118925XX had no influence on NETs release. On the other hand, the formation of aggNETs could be attributed to the P2Y6/store-operated calcium entry/IL-8 axis that plays a role in neutrophil migration.279 This mechanism was further elucidated in another study, Su-Hyun et al proposed that during the intracellular endosomal trafficking of the P2Y6 receptor induced by MSU crystals, neutrophil recruitment was enhanced on the basis of the IL-8 expression supported by endosome-dependent signaling.280 Numerous studies also reported the significant role of P2Y6 receptor expression on cells such as microglial and macrophage in different diseases.281,282 These evidences suggest a strong potential for MRS2578 to be further developed by targeting P2Y6 receptors for GA treatment. By selectively inhibiting or activating the P2Y6 receptor, at the same time, it may be possible to influence NET release and subsequently mitigate the inflammatory response and tissue damage characteristic of gout flares. This novel therapeutic strategy holds the potential to offer a more targeted and precise treatment option for gout patients, possibly leading to improved symptom management and disease outcomes.
LDC7559 is a newly discovered small molecule that has mighty potential to be developed. In accordance with Gabriel et al, LDC7559 was chosen through a chemical screening and had the ability to combined with gasdermin D (GSDMD) with rarely effect on the activity of MPO, NE and NOX. Besides, LDC7559 binding to GSDMS N terminus thus causing decrease in NETs formation suggested the specificity of LDC7559 to GSDMD. GSDMD, a pore-forming protein, serves as a crucial mediator of pyroptosis and plays a significant role in the process of NETosis. Its involvement in NETosis stems from its essential function in the release of chromatin structures into the extracellular space. GSDMD influences cell membrane stability and nuclear expansion, facilitating the extracellular release of chromatin and contributing to the formation of NETs.283 Also, inhibition of GSDMD became a therapeutical target to block NETs formation during sepsis.284 It has been reported that GSDMD-dependent NETs formation was associated with various stimuli such as LPS or cytosolic Gram-negative bacteria (Salmonella ΔsifA and Citrobacter rodentium)285 and PMA. To date, comprehensive research on the involvement of GSDMD in MSU-induced NETosis is yet to be established, presenting an area that significant attention. The current lack of evidence necessitates focused investigations to elucidate the potential role of GSDMD in the process of NETosis triggered by MSU.
Metformin is a synthetic product having a close connection with GA. While its chemical structure is based on a natural compound called guanidine, the compound itself is synthesized in a laboratory setting rather than being extracted from natural sources.286 Growing studies have found the effect of metformin on different diseases such as Covid-19 and gout.287,288 In various pathological conditions, metformin has been shown to directly impede the activity of the mitochondrial respiratory chain complex I. This specific inhibition results in a reduction of adenosine triphosphate (ATP) levels, concomitant with an elevation in adenosine monophosphate (AMP) concentrations. The altered AMP/ATP ratio subsequently activates adenosine monophosphate-activated protein kinase (AMPK), a pivotal metabolic regulator. This cascade of events, involving the inhibition of complex I and subsequent AMPK activation, constitutes a fundamental mechanism underlying Metformin’s therapeutic effects across diverse disease contexts.289,290 As an inhibitor of mTOR signaling, metformin resulted in the reduction of cell death and pro-inflammatory cytokines, which might be the main reason for the lower frequency of gout attacks occurred to patients with gout.291 Despite being discovered over fifty years ago, metformin still holds significant research value because metformin owns immunoregulating effects through suppressive effects on NETosis and NETs formation.292 In summary, the use of metformin as a potential inhibitor of NETs formation and NETosis shows promise in the treatment of GA. further research and clinical trials are necessary to fully understand the effectiveness and safety of metformin in targeting NETs during the GA process, paving the way for a more targeted and personalized treatment for individuals suffering from this debilitating condition.
Conclusions and Future Perspectives
In short, NET is a web like structure based on DNA accompanied with components such as NE, PAD4 and MPO. NETs can be stimulated and formed by various pathways in different diseases and perform distinct functions in response to immune system and inflammatory environment. During the process of GA, NETs exert both pro- and anti-inflammatory effects. With the stimulation of MSU crystals, inflammation onset caused by NETs is mainly induced by the activation of NLRP3 inflammasomes, leading to the release of mature inflammatory cytokines (IL-1β and IL-18). With increased inflammation and neutrophil recruitment, aggregation of NETs occurs causing the formation of aggNETs that contributes to the anti-inflammatory effect by engulfing and degrading MSU crystals together with the components such as NSPs. At the same time, we summarize the prevalent drugs for GA and describe NETs-targeted inhibitors naturally and synthetically that possess potential for development into clinical applications.
Neutrophil is a type of white blood cell with short lifespan and unable to expand or frozen in vitro. According to the research, neutrophils are more fragile than other blood cells and can only survive in vitro for up to 5 days maximum under certain condition.293,294 It is of great difficulty to conduct the research of NETs in-depth, and growing studies replace neutrophils with differentiated HL-60 cells via dimethyl sulfoxide or all-trans retinoic acid.295–297 HL-60 cells are differentiated human promyelocytic leukemia cells that can be used for laboratory research on blood cell formation and physiology. Nevertheless, it is of inefficiency for dHL-60 to generate NETs compared to neutrophils, and differences including the dynamics pattern of the cytoskeleton, endomembrane, phagocytosis abilities and calcium influx exist between dHL-60 and neutrophils.298–300 Considering current research, there exists an urgent need to optimize the in vitro conditions for neutrophil survival to extend their lifespan, or to develop a dHL-60 cell model that accurately mimics the physiological environment of neutrophils.
Even though the natural and synthetic products have their own advantages on inhibiting NETs, it is necessary to solve the challenges before being applied in the clinic. The integration of natural products in drug discovery has encountered difficulties in the form of practical hurdles for screening, isolation, characterization, and optimization. As a result, their utilization has decreased over time. In addition, natural products may possess a multiplicity of targets, which can lead to low bioavailability, poor pharmacokinetics, or toxic effects.257,301 On the other hand, synthetic products exhibit drawbacks including but not limited to high expense, instability, and potential immunogenicity. Synthetic compounds may also lack specificity and biological relevance toward NET-targeted inhibition. Moreover, they may elicit undesirable side effects or resistance mechanisms because of their interactions with other pathways or molecules.302 The evidence mentioned above lead to the attraction should be given to investigate more thoroughly the context-dependent functions and mechanisms of NETs in GA and different stages of gout, as well as their interactions with other immune cells or molecules. Additionally, future research on NETs-targeted inhibitors is also supposed to focus on evaluate more carefully the safety and efficacy of natural and synthetic products as NET-targeted inhibitors in preclinical and clinical studies, as well as their potential adverse effects on normal neutrophil functions and immune responses.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (grant number: 82074085), the Natural Science Foundation of Zhejiang Province (grant number: LQ22H280008) and the Postgraduate Scientific Research Fund of Zhejiang Chinese Medical University (grant number: 2022YKJ20). We appreciated the great technical support from the Public Platform of Medical Research Center, Academy of Chinese Medical Science, Zhejiang Chinese Medical University.
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Schorn C, Janko C, Krenn V., et al. Bonding the foe – nETting neutrophils immobilize the pro-inflammatory monosodium urate crystals. Front Immunol. 2012;3:376. doi:10.3389/fimmu.2012.00376
2. Dehlin M, Jacobsson L, Roddy E. Global epidemiology of gout: prevalence, incidence, treatment patterns and risk factors. Nat Rev Rheumatol. 2020;16(7):380–390. doi:10.1038/s41584-020-0441-1
3. Eun Y, Han K, Lee SW, et al. Altered risk of incident gout according to changes in metabolic syndrome status: a nationwide population-based cohort study of 1.29 million young men. Arthritis Rheumatol. 2022. doi:10.1002/art.42381
4. Ouyang X, Li NZ, Guo MX, et al. Active flavonoids from lagotis brachystachya attenuate monosodium urate-induced gouty arthritis via inhibiting tlr4/myd88/nf-kappab pathway and nlrp3 expression. Front Pharmacol. 2021;12:760331. doi:10.3389/fphar.2021.760331
5. Dalbeth N, Choi HK, Joosten LAB, et al. Gout. Nature reviews. Disease Primers. 2019;5(1):69. doi:10.1038/s41572-019-0115-y
6. Mandel NS, Mandel GS. Monosodium urate monohydrate, the gout culprit. J Am Chem Soc. 1976;98(8):2319–2323. doi:10.1021/ja00424a054
7. Hahn J, Knopf J, Maueroder C, et al. Neutrophils and neutrophil extracellular traps orchestrate initiation and resolution of inflammation. Clin Exp Rheumatol. 2016;34(4 Suppl 98):6–8.
8. Desai J, Kumar SV, Mulay SR, et al. Pma and crystal-induced neutrophil extracellular trap formation involves ripk1-ripk3-mlkl signaling. Eur j Immunol. 2016;46(1):223–229. doi:10.1002/eji.201545605
9. Rosales C. Neutrophil: a cell with many roles in inflammation or several cell types? Front Physiol. 2018;9:113. doi:10.3389/fphys.2018.00113
10. Yu FF, Yuan Y, Ao Y, et al. A new product of bilirubin degradation by h(2)o(2) and its formation in activated neutrophils and in an inflammatory mouse model. Biomolecules. 2022;12(9). doi:10.3390/biom12091237
11. Hassani M, Hellebrekers P, Chen N, et al. On the origin of low-density neutrophils. J Leukoc Biol. 2020;107(5):809–818. doi:10.1002/JLB.5HR0120-459R
12. Petri B, Sanz MJ. Neutrophil chemotaxis. Cell Tissue Res. 2018;371(3):425–436. doi:10.1007/s00441-017-2776-8
13. Gomez RM, Lopez Ortiz AO, Schattner M. Platelets and extracellular traps in infections. Platelets. 2021;32(3):305–313. doi:10.1080/09537104.2020.1718631
14. Mutua V, Gershwin LJ. A review of neutrophil extracellular traps (nets) in disease: potential anti-nets therapeutics. Clinical Reviews in Allergy & Immunology. 2021;61(2):194–211. doi:10.1007/s12016-020-08804-7
15. Jeong JH, Choi SJ, Ahn SM, et al. Neutrophil extracellular trap clearance by synovial macrophages in gout. Arthritis Res Ther. 2021;23(1):88. doi:10.1186/s13075-021-02472-4
16. Moran G, Uberti B, Quiroga J. Role of cellular metabolism in the formation of neutrophil extracellular traps in airway diseases. Front Immunol. 2022;13:850416. doi:10.3389/fimmu.2022.850416
17. Sprenkeler EGG, Tool ATJ, Henriet SSV, et al. Formation of neutrophil extracellular traps requires actin cytoskeleton rearrangements. Blood. 2022;139(21):3166–3180. doi:10.1182/blood.2021013565
18. Li Y, Cao X, Liu Y, et al. Neutrophil extracellular traps formation and aggregation orchestrate induction and resolution of sterile crystal-mediated inflammation. Front Immunol. 2018;9:1559. doi:10.3389/fimmu.2018.01559
19. Vorobjeva NV, Chernyak BV. Netosis: molecular mechanisms, role in physiology and pathology. Biochemistry. 2020;85(10):1178–1190. doi:10.1134/S0006297920100065
20. Rada B. Neutrophil extracellular traps and microcrystals. J Immunol Res. 2017;2017:2896380. doi:10.1155/2017/2896380
21. Atzeni F, Masala IF, Bagnasco M, et al. Comparison of efficacy of ketoprofen and ibuprofen in treating pain in patients with rheumatoid arthritis: a systematic review and meta-analysis. Pain Ther. 2021;10(1):577–588. doi:10.1007/s40122-021-00250-3
22. Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immun net. 2018;18(4):e27. doi:10.4110/in.2018.18.e27
23. Li Y, Berke IC, Modis Y. DNA binding to proteolytically activated tlr9 is sequence-independent and enhanced by DNA curvature. EMBO J. 2012;31(4):919–931. doi:10.1038/emboj.2011.441
24. Tan C, Aziz M, Wang P. The vitals of nets. J Leukoc Biol. 2021;110(4):797–808. doi:10.1002/JLB.3RU0620-375R
25. Masucci MT, Minopoli M, Del Vecchio S, et al. The emerging role of neutrophil extracellular traps (nets) in tumor progression and metastasis. Front Immunol. 2020;11:1749. doi:10.3389/fimmu.2020.01749
26. Morandini L, Avery D, Angeles B, et al. Reduction of neutrophil extracellular traps accelerates inflammatory resolution and increases bone formation on titanium implants. Acta Biomater. 2023. doi:10.1016/j.actbio.2023.05.016
27. Zou J, Zhao Z, Song X, et al. Elevated g-csf, il8, and hgf in patients with definite meniere’s disease may indicate the role of net formation in triggering autoimmunity and autoinflammation. Sci Rep. 2022;12(1):16309. doi:10.1038/s41598-022-20774-8
28. Dwivedi DJ, Toltl LJ, Swystun LL, et al. Prognostic utility and characterization of cell-free DNA in patients with severe sepsis. Critical Care. 2012;16(4):R151. doi:10.1186/cc11466
29. Bhagirath VC, Dwivedi DJ, Liaw PC. Comparison of the proinflammatory and procoagulant properties of nuclear, mitochondrial, and bacterial DNA. Shock. 2015;44(3):265–271. doi:10.1097/SHK.0000000000000397
30. Denning N-L, Aziz M, Gurien SD, et al. Damps and nets in sepsis. Front Immunol. 2019;10:2536. doi:10.3389/fimmu.2019.02536
31. Saffarzadeh M, Juenemann C, Queisser MA, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS One. 2012;7(2):e32366. doi:10.1371/journal.pone.0032366
32. Liu YW, Zhang J, Bi W, et al. Histones of neutrophil extracellular traps induce cd11b expression in brain pericytes via dectin-1 after traumatic brain injury. Neurosci Bull. 2022;38(10):1199–1214. doi:10.1007/s12264-022-00902-0
33. Allam R, Darisipudi MN, Tschopp J, et al. Histones trigger sterile inflammation by activating the nlrp3 inflammasome. Eur J Immunol. 2013;43(12):3336–3342. doi:10.1002/eji.201243224
34. Chen R, Kang R. Release and activity of histone in diseases. Cell Death Dis. 2014;5(8):e1370. doi:10.1038/cddis.2014.337
35. Szatmary P, Huang W, Criddle D, et al. Biology, role and therapeutic potential of circulating histones in acute inflammatory disorders. J Cell & Mol Med. 2018;22(10):4617–4629. doi:10.1111/jcmm.13797
36. Abrams ST, Zhang N, Manson J, et al. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med. 2013;187(2):160–169. doi:10.1164/rccm.201206-1037OC
37. Marsman G, Zeerleder S, Luken BM. Extracellular histones, cell-free DNA, or nucleosomes: differences in immunostimulation. Cell Death Dis. 2016;7(12):e2518. doi:10.1038/cddis.2016.410
38. Qu L, Chen C, Chen Y, et al. High-mobility group box 1 (hmgb1) and autophagy in acute lung injury (ali): a review. Med Sci Monit. 2019;25:1828–1837. doi:10.12659/MSM.912867
39. Chiang CY, Lin YJ, Weng WT, et al. Recuperative herbal formula jing si maintains vasculature permeability balance, regulates inflammation and assuages concomitants of “long-covid”. Biomed Pharmacother. 2023:163 114752. doi:10.1016/j.biopha.2023.114752
40. Kim SW, Lee JK. Role of hmgb1 in the interplay between netosis and thrombosis in ischemic stroke: a review. Cells. 2020;9(8). doi:10.3390/cells9081794
41. Zlatanova J, van Holde K. Linker histones versus hmg1/2: a struggle for dominance? Bioessays. 1998;20(7):584–588. doi:10.1002/(SICI)1521-1878(199807)20:7<584::AID-BIES10>3.0.CO;2-W
42. Tadie JM, Bae HB, Jiang S, et al. Hmgb1 promotes neutrophil extracellular trap formation through interactions with toll-like receptor 4. Am J Physiol Lung Cell Mol Physiol. 2013;304(5):L342–349. doi:10.1152/ajplung.00151.2012
43. Wang D, Xie Y, Peng HQ, et al. Lps preconditioning of msc-cm improves protection against hypoxia/reoxygenation-induced damage in h9c2 cells partly via hmgb1/bach1 signalling. Clin Exp Pharmacol Physiol. 2022;49(12):1319–1333. doi:10.1111/1440-1681.13714
44. Yasom S, Watcharanurak P, Bhummaphan N, et al. The roles of hmgb1-produced DNA gaps in DNA protection and aging biomarker reversal. FASEB Bioadv. 2022;4(6):408–434. doi:10.1096/fba.2021-00131
45. Yipp BG, Kubes P. Netosis: how vital is it? Blood. 2013;122(16):2784–2794. doi:10.1182/blood-2013-04-457671
46. Abrams ST, Zhang N, Dart C, et al. Human crp defends against the toxicity of circulating histones. J Iimmunol. 2013;191(5):2495–2502. doi:10.4049/jimmunol.1203181
47. Biron BM, Chung CS, O’Brien XM, et al. Cl-amidine prevents histone 3 citrullination and neutrophil extracellular trap formation, and improves survival in a murine sepsis model. J Innate Immunity. 2017;9(1):22–32. doi:10.1159/000448808
48. Liu X, Arfman T, Wichapong K, et al. Pad4 takes charge during neutrophil activation: impact of pad4 mediated net formation on immune-mediated disease. J Thromb Haemost. 2021;19(7):1607–1617. doi:10.1111/jth.15313
49. Huang H, Tohme S, Al-Khafaji AB, et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. 2015;62(2):600–614. doi:10.1002/hep.27841
50. Wiersma VR, Clarke A, Pouwels SD, et al. Galectin-9 is a possible promoter of immunopathology in rheumatoid arthritis by activation of peptidyl arginine deiminase 4 (pad-4) in granulocytes. Int J Mol Sci. 2019;20(16). doi:10.3390/ijms20164046
51. Raup-Konsavage WM, Wang YM, Wang WW, et al. Neutrophil peptidyl arginine deiminase-4 has a pivotal role in ischemia/reperfusion-induced acute kidney injury. Kidney Int. 2018;93(2):365–374. doi:10.1016/j.kint.2017.08.014
52. Neeli I, Khan SN, Radic M. Histone deimination as a response to inflammatory stimuli in neutrophils. J Iimmunol. 2008;180(3):1895–1902.
53. Wang Y, Li M, Stadler S, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184(2):205–213. doi:10.1083/jcb.200806072
54. Li P, Li M, Lindberg R, et al. Pad4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. 2010;207(9):1853–1862. doi:10.1084/jem.20100239
55. Parker H, Albrett AM, Kettle AJ, et al. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J Leukocyte Biol. 2012;91(3):369–376. doi:10.1189/jlb.0711387
56. Shearer HL, Kaldor CD, Hua H, et al. Resistance of streptococcus pneumoniae to hypothiocyanous acid generated by host peroxidases. Infect Immun. 2022;90(3):e00530–21. doi:10.1128/iai.00530-21
57. Bjornsdottir H, Welin A, Michaelsson E, et al. Neutrophil net formation is regulated from the inside by myeloperoxidase-processed reactive oxygen species. Free Radic Biol Med. 2015;89:1024–1035. doi:10.1016/j.freeradbiomed.2015.10.398
58. Yang J, Ge H, Poulton CJ, et al. Histone modification signature at myeloperoxidase and proteinase 3 in patients with anti-neutrophil cytoplasmic autoantibody-associated vasculitis. Clin Clin Epigenet. 2016;8:85. doi:10.1186/s13148-016-0251-0
59. Parker H, Dragunow M, Hampton B, et al. Requirements for nadph oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J Leukocyte Biol. 2012;92(4):841–849. doi:10.1189/jlb.1211601
60. Odobasic D, Holdsworth SR. Emerging cellular therapies for anti-myeloperoxidase vasculitis and other autoimmune diseases. Front Immunol. 2021;12:642127. doi:10.3389/fimmu.2021.642127
61. Yang X, Liu Y, Zhong C, et al. Total flavonoids of chrysanthemum indicum l inhibit acute pancreatitis through suppressing apoptosis and inflammation. BMC Complement Med Ther. 2023;23(1):23. doi:10.1186/s12906-023-03851-x
62. Reshetnyak T, Nurbaeva K, Ptashnik I, et al. Markers of netosis in patients with systemic lupus erythematosus and antiphospholipid syndrome. Int J Mol Sci. 2023;24(11). doi:10.3390/ijms24119210
63. Chen S, Chen H, Du Q, et al. Targeting myeloperoxidase (mpo) mediated oxidative stress and inflammation for reducing brain ischemia injury: potential application of natural compounds. Front Physiol. 2020;11:433. doi:10.3389/fphys.2020.00433
64. Kim DG, Kwon YM, Kang IS, et al. Taurine chloramine selectively regulates neutrophil degranulation through the inhibition of myeloperoxidase and upregulation of lactoferrin. Amino Acids. 2020;52(8):1191–1199. doi:10.1007/s00726-020-02886-5
65. Papayannopoulos V, Metzler KD, Hakkim A, et al. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677–691. doi:10.1083/jcb.201006052
66. Majewski P, Majchrzak-Gorecka M, Grygier B, et al. Inhibitors of serine proteases in regulating the production and function of neutrophil extracellular traps. Front Immunol. 2016;7:261. doi:10.3389/fimmu.2016.00261
67. Wang K, Liao Y, Li X, et al. Inhibition of neutrophil elastase prevents cigarette smoke exposure-induced formation of neutrophil extracellular traps and improves lung function in a mouse model of chronic obstructive pulmonary disease. Int Immunopharmacol. 2023:114 109537. doi:10.1016/j.intimp.2022.109537
68. Metzler KD, Goosmann C, Lubojemska A, et al. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during netosis. Cell Rep. 2014;8(3):883–896. doi:10.1016/j.celrep.2014.06.044
69. Martinod K, Witsch T, Farley K, et al. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J Thrombosis Haemostasis. 2016;14(3):551–558. doi:10.1111/jth.13239
70. O’Sullivan KM, Holdsworth SR. Neutrophil extracellular traps: a potential therapeutic target in mpo-anca associated vasculitis? Front Immunol. 2021;12:635188. doi:10.3389/fimmu.2021.635188
71. Rochael NC, Guimarães-Costa AB, Nascimento MTC, et al. Classical ros-dependent and early/rapid ros-independent release of neutrophil extracellular traps triggered by leishmania parasites. Sci Rep. 2015;5:18302. doi:10.1038/srep18302
72. Pascart T, Grandjean A, Capon B, et al. Monosodium urate burden assessed with dual-energy computed tomography predicts the risk of flares in gout: a 12-month observational study: msu burden and risk of gout flare. Arthritis Res Ther. 2018;20(1):210. doi:10.1186/s13075-018-1714-9
73. Davidsson L, Dahlstrand Rudin A, Sanchez Klose FP, et al. In vivo transmigrated human neutrophils are highly primed for intracellular radical production induced by monosodium urate crystals. Int J Mol Sci. 2020;21(11). doi:10.3390/ijms21113750
74. Tatsiy O, Mayer TZ, de Carvalho Oliveira V, et al. Cytokine production and net formation by monosodium urate-activated human neutrophils involves early and late events, and requires upstream tak1 and syk. Front Immunol. 2019;10:2996. doi:10.3389/fimmu.2019.02996
75. Brinkmann V, Reichard U, Goosmann C, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–1535. doi:10.1126/science.1092385
76. Swethakumar B, NaveenKumar SK, Girish KS, et al. The action of echis carinatus and naja naja venoms on human neutrophils; an emphasis on netosis. Biochim Biophys Acta Gen Subj. 2020;1864(6):129561. doi:10.1016/j.bbagen.2020.129561
77. Zhang Y, Peng R, Pei S, et al. Neutrophil extracellular traps are increased after extracorporeal membrane oxygenation support initiation and present in thrombus: a preclinical study using sheep as an animal model. Thromb Res. 2023:221 173–182. doi:10.1016/j.thromres.2022.10.019
78. Guillotin F, Fortier M, Portes M, et al. Vital netosis vs. Suicidal netosis during normal pregnancy and preeclampsia. Front Cell Dev Biol. 2022;10:1099038. doi:10.3389/fcell.2022.1099038
79. Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17(3):151–164. doi:10.1038/nri.2016.147
80. Fuchs TA, Abed U, Goosmann C, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–241.
81. Sollberger G. Approaching neutrophil pyroptosis. J Mol Biol. 2022;434(4):167335. doi:10.1016/j.jmb.2021.167335
82. Song M, Wang J, Sun Y, et al. Inhibition of gasdermin d-dependent pyroptosis attenuates the progression of silica-induced pulmonary inflammation and fibrosis. Acta Pharm Sin B. 2022;12(3):1213–1224. doi:10.1016/j.apsb.2021.10.006
83. Chauhan D, Demon D, Vande Walle L, et al. Gsdmd drives canonical inflammasome-induced neutrophil pyroptosis and is dispensable for netosis. EMBO Rep. 2022:e54277. doi:10.15252/embr.202154277
84. Mitroulis I, Kambas K, Chrysanthopoulou A, et al. Neutrophil extracellular trap formation is associated with il-1beta and autophagy-related signaling in gout. PLoS One. 2011;6(12):e29318. doi:10.1371/journal.pone.0029318
85. Berthelot JM, Le Goff B, Neel A, et al. Netosis: at the crossroads of rheumatoid arthritis, lupus, and vasculitis. Joint Bone Spine. 2017;84(3):255–262. doi:10.1016/j.jbspin.2016.05.013
86. Sabbatini M, Magnelli V, Reno F. Netosis in wound healing: when enough is enough. Cells. 2021;10(3). doi:10.3390/cells10030494
87. Masuda S, Nakazawa D, Shida H, et al. Netosis markers: quest for specific, objective, and quantitative markers. Clin Chim Acta. 2016;459:89–93. doi:10.1016/j.cca.2016.05.029
88. Azzouz D, Khan MA, Palaniyar N. Ros induces netosis by oxidizing DNA and initiating DNA repair. Cell Death Discov. 2021;7(1):113. doi:10.1038/s41420-021-00491-3
89. Huang J, Hong W, Wan M, et al. Molecular mechanisms and therapeutic target of netosis in diseases. MedComm. 2022;3(3):e162. doi:10.1002/mco2.162
90. Vorobjeva NV. Neutrophil extracellular traps: new aspects. Moscow Univ Biol Sci Bull. 2020;75(4):173–188. doi:10.3103/S0096392520040112
91. Papayannopoulos V, Zychlinsky A. Nets: a new strategy for using old weapons. Trends Immunol. 2009;30(11):513–521. doi:10.1016/j.it.2009.07.011
92. Fousert E, Toes R, Desai J. Neutrophil extracellular traps (nets) take the central stage in driving autoimmune responses. Cells. 2020;9(4). doi:10.3390/cells9040915
93. de Bont CM, Koopman WJH, Boelens WC, et al. Stimulus-dependent chromatin dynamics, citrullination, calcium signalling and ros production during net formation. BBA. 2018;1865(11 Pt A):1621–1629. doi:10.1016/j.bbamcr.2018.08.014
94. Volkman R, Ben-Zur T, Kahana A, et al. Myeloperoxidase deficiency inhibits cognitive decline in the 5xfad mouse model of Alzheimer’s disease. Front Neurosci. 2019;13:990. doi:10.3389/fnins.2019.00990
95. Manfredi AA, Ramirez GA, Rovere-Querini P, et al. The neutrophil’s choice: phagocytose vs make neutrophil extracellular traps. Front Immunol. 2018;9:288. doi:10.3389/fimmu.2018.00288
96. Roskoski R. Erk1/2 map kinases: structure, function, and regulation. Pharmacol Res. 2012;66(2):105–143. doi:10.1016/j.phrs.2012.04.005
97. Chen T, Li Y, Sun R, et al. Receptor-mediated netosis on neutrophils. Front Immunol. 2021;12:775267. doi:10.3389/fimmu.2021.775267
98. Pilsczek FH, Salina D, Poon KKH, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to staphylococcus aureus. J Iimmunol. 2010;185(12):7413–7425. doi:10.4049/jimmunol.1000675
99. Martinod K, Demers M, Fuchs TA, et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci USA. 2013;110(21):8674–8679. doi:10.1073/pnas.1301059110
100. Hakkim A, Fuchs TA, Martinez NE, et al. Activation of the raf-mek-erk pathway is required for neutrophil extracellular trap formation. Nature Chem Biol. 2011;7(2):75–77. doi:10.1038/nchembio.496
101. Fonseca Z, Diaz-Godinez C, Mora N, et al. Entamoeba histolytica induce signaling via raf/mek/erk for neutrophil extracellular trap (net) formation. Front Cell Infect Microbiol. 2018:8 226. doi:10.3389/fcimb.2018.00226
102. Diaz-Godinez C, Jorge-Rosas JF, Nequiz M, et al. New insights on netosis induced by entamoeba histolytica: dependence on ros from amoebas and extracellular mpo activity. Antioxidants (Basel). 2021;10(6). doi:10.3390/antiox10060974
103. Soroush F, Tang Y, Guglielmo K, et al. Protein kinase c-delta (pkcdelta) tyrosine phosphorylation is a critical regulator of neutrophil-endothelial cell interaction in inflammation. Shock. 2019;51(5):538–547. doi:10.1097/SHK.0000000000001247
104. Wang QJ. Pkd at the crossroads of dag and pkc signaling. Trends Pharmacol Sci. 2006;27(6):317–323. doi:10.1016/j.tips.2006.04.003
105. Neeli I, Radic M. Opposition between pkc isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front Immunol. 2013;4:38. doi:10.3389/fimmu.2013.00038
106. Gray RD, Lucas CD, MacKellar A, et al. Activation of conventional protein kinase c (pkc) is critical in the generation of human neutrophil extracellular traps. J Inflammation. 2013;10(1):12. doi:10.1186/1476-9255-10-12
107. Behnen M, Leschczyk C, Möller S, et al. Immobilized immune complexes induce neutrophil extracellular trap release by human neutrophil granulocytes via fcγriiib and mac-1. J Iimmunol. 2014;193(4):1954–1965. doi:10.4049/jimmunol.1400478
108. Alemán OR, Mora N, Cortes-Vieyra R, et al. Differential use of human neutrophil fcγ receptors for inducing neutrophil extracellular trap formation. J Immunol Res. 2016;2016:2908034. doi:10.1155/2016/2908034
109. Dwivedi N, Radic M. Citrullination of autoantigens implicates netosis in the induction of autoimmunity. Ann Rheum Dis. 2014;73(3):483–491. doi:10.1136/annrheumdis-2013-203844
110. Douda DN, Khan MA, Grasemann H, et al. Sk3 channel and mitochondrial ros mediate nadph oxidase-independent netosis induced by calcium influx. Proc Natl Acad Sci USA. 2015;112(9):2817–2822. doi:10.1073/pnas.1414055112
111. Awasthi D, Nagarkoti S, Sadaf S, et al. Glycolysis dependent lactate formation in neutrophils: a metabolic link between nox-dependent and independent netosis. Biochim Biophys Acta Mol Basis Dis. 2019;1865(12):165542. doi:10.1016/j.bbadis.2019.165542
112. Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the nalp3 inflammasome. Nat Immunol. 2011;12(3):222–230. doi:10.1038/ni.1980
113. Memon AA, Vats S, Sundquist J, et al. Mitochondrial DNA copy number: linking diabetes and cancer. Antioxid Redox Signal. 2022;37(16–18):1168–1190. doi:10.1089/ars.2022.0100
114. Dabravolski SA, Khotina VA, Sukhorukov VN, et al. The role of mitochondrial DNA mutations in cardiovascular diseases. Int J Mol Sci. 2022;23(2). doi:10.3390/ijms23020952
115. Singel KL, Grzankowski KS, Khan A, et al. Mitochondrial DNA in the tumour microenvironment activates neutrophils and is associated with worse outcomes in patients with advanced epithelial ovarian cancer. Br J Cancer. 2019;120(2):207–217. doi:10.1038/s41416-018-0339-8
116. Aarreberg LD, Esser-Nobis K, Driscoll C, et al. Interleukin-1beta induces mtdna release to activate innate immune signaling via cgas-sting. Mol Cell. 2019;74(4):801–815 e806. doi:10.1016/j.molcel.2019.02.038
117. Riley JS, Tait SW. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020;21(4):e49799. doi:10.15252/embr.201949799
118. Yang C, Wang Z, Li L, et al. Aged neutrophils form mitochondria-dependent vital nets to promote breast cancer lung metastasis. J Immunother Cancer. 2021;9(10). doi:10.1136/jitc-2021-002875
119. Zhang L, Wang L, Jiang J, et al. Lipopolysaccharides upregulate calcium concentration in mouse uterine smooth muscle cells through the t-type calcium channels. Int J Mol Med. 2015;35(3):784–790. doi:10.3892/ijmm.2014.2054
120. Thiam HR, Wong SL, Wagner DD, et al. Cellular mechanisms of netosis. Annu Rev Cell Dev Biol. 2020;36:191–218. doi:10.1146/annurev-cellbio-020520-111016
121. Hann J, Bueb JL, Tolle F, et al. Calcium signaling and regulation of neutrophil functions: still a long way to go. J Leukoc Biol. 2020;107(2):285–297. doi:10.1002/JLB.3RU0719-241R
122. Hamam HJ, Palaniyar N. Post-translational modifications in netosis and nets-mediated diseases. Biomolecules. 2019;9(8). doi:10.3390/biom9080369
123. Hamam HJ, Khan MA, Palaniyar N. Histone acetylation promotes neutrophil extracellular trap formation. Biomolecules. 2019;9(1). doi:10.3390/biom9010032
124. Hamam HJ, Palaniyar N. Histone deacetylase inhibitors dose-dependently switch neutrophil death from netosis to apoptosis. Biomolecules. 2019;9(5). doi:10.3390/biom9050184
125. Ahmad Mohamed Ali R, Mita D, Espulgar W, et al. Single cell analysis of neutrophils nets by microscopic lspr imaging system. Micromachines. 2019;11(1). doi:10.3390/mi11010052
126. Yipp BG, Petri B, Salina D, et al. Infection-induced netosis is a dynamic process involving neutrophil multitasking in vivo. Nature Med. 2012;18(9):1386–1393.
127. Nija RJ, Sanju S, Sidharthan N, et al. Extracellular trap by blood cells: clinical implications. Tissue Eng and Regener Med. 2020;17(2):141–153. doi:10.1007/s13770-020-00241-z
128. Alasmari SZ. Imaging of neutrophil extracellular traps (nets): visualization methods and outcomes. Biomed Res. Int. 2020;2020:4192745. doi:10.1155/2020/4192745
129. Hilscher MB, Shah VH. Neutrophil extracellular traps and liver disease. Semin Liver Disease. 2020;40(2):171–179. doi:10.1055/s-0039-3399562
130. Wang BT, Ducker GS, Barczak AJ, et al. The mammalian target of rapamycin regulates cholesterol biosynthetic gene expression and exhibits a rapamycin-resistant transcriptional profile. Proc Natl Acad Sci U S A. 2011;108(37):15201–15206. doi:10.1073/pnas.1103746108
131. Ramirez-Jarquin UN, Shahani N, Pryor W, et al. The mammalian target of rapamycin (mtor) kinase mediates haloperidol-induced cataleptic behavior. Transl Psychiatry. 2020;10(1):336. doi:10.1038/s41398-020-01014-x
132. Kim YC, Guan KL. Mtor: a pharmacologic target for autophagy regulation. J Clin Invest. 2015;125(1):25–32. doi:10.1172/JCI73939
133. Curi R, Levada-Pires AC, Silva EBD, et al. The critical role of cell metabolism for essential neutrophil functions. Cell Physiol Biochem. 2020;54(4):629–647. doi:10.33594/000000245
134. Guo Y, Gao F, Wang X, et al. Spontaneous formation of neutrophil extracellular traps is associated with autophagy. Sci Rep. 2021;11(1):24005. doi:10.1038/s41598-021-03520-4
135. Xie T, Duan Z, Sun S, et al. Beta-lactams modulate neutrophil extracellular traps formation mediated by mtor signaling pathway. Biochem Biophys Res Commun. 2021;534:408–414. doi:10.1016/j.bbrc.2020.11.067
136. Itakura A, McCarty OJ. Pivotal role for the mtor pathway in the formation of neutrophil extracellular traps via regulation of autophagy. Am J Physiol Cell Physiol. 2013;305(3):C348–354. doi:10.1152/ajpcell.00108.2013
137. Wu M, Tian Y, Wang Q, et al. Gout: a disease involved with complicated immunoinflammatory responses: a narrative review. Clin Rheumatol. 2020;39(10):2849–2859. doi:10.1007/s10067-020-05090-8
138. Yip RM, Cheung TT, So H, et al. The Hong Kong society of rheumatology consensus recommendations for the management of gout. Clin Rheumatol. 2023;42(8):2013–2027. doi:10.1007/s10067-023-06578-9
139. Ragab G, Elshahaly M, Bardin T. Gout: an old disease in new perspective - a review. J Adv Res. 2017;8(5):495–511. doi:10.1016/j.jare.2017.04.008
140. McQueen FM, Reeves Q, Dalbeth N. New insights into an old disease: advanced imaging in the diagnosis and management of gout. Postgrad Med J. 2013;89(1048):87–93. doi:10.1136/postgradmedj-2012-131000
141. Jafari-Nozad AM, Jafari A, Yousefi S, et al. Anti-gout and urate-lowering potentials of curcumin: a review from bench to beside. Curr Med Chem. 2023. doi:10.2174/0929867331666230721154653
142. Wang Q, Qiu H. Deubiquitinase usp16 induces gouty arthritis via drp1-dependent mitochondrial fission and nlrp3 inflammasome activation. Arthritis Res Ther. 2023;25(1):126. doi:10.1186/s13075-023-03095-7
143. Tang H, Tan C, Cao X, et al. Nfil3 facilitates neutrophil autophagy, neutrophil extracellular trap formation and inflammation during gout via redd1-dependent mtor inactivation. Front Med Lausanne. 2021;8:692781. doi:10.3389/fmed.2021.692781
144. Collison J. Msu crystals at-tak! Nat Rev Rheumatol. 2019;15(11):638. doi:10.1038/s41584-019-0316-5
145. Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immun net. 2018;18(4):e27. doi:10.4110/in.2018.18.e27
146. So AK, Martinon F. Inflammation in gout: mechanisms and therapeutic targets. Nat Rev Rheumatol. 2017;13(11):639–647. doi:10.1038/nrrheum.2017.155
147. Zhao Q, Xia N, Xu J, et al. Pro-inflammatory of prdm1/sirt2/nlrp3 axis in monosodium urate-induced acute gouty arthritis. J Innate Immun. 2023. doi:10.1159/000530966
148. Wang HY, Lin X, Huang GG, et al. Atranorin inhibits nlrp3 inflammasome activation by targeting asc and protects nlrp3 inflammasome-driven diseases. Acta Pharmacol Sin. 2023;44(8):1687–1700. doi:10.1038/s41401-023-01054-1
149. Desai J, Steiger S, Anders HJ. Molecular pathophysiology of gout. Trends Mol Med. 2017;23(8):756–768. doi:10.1016/j.molmed.2017.06.005
150. Pieterse E, Jeremic I, Czegley C, et al. Blood-borne phagocytes internalize urate microaggregates and prevent intravascular netosis by urate crystals. Sci Rep. 2016;6:38229. doi:10.1038/srep38229
151. Shirahama T, Cohen AS. Ultrastructural evidence for leakage of lysosomal contents after phagocytosis of monosodium urate crystals. A mechanism of gouty inflammation. Am J Pathol. 1974;76(3):501–520.
152. Nauseef WM. Proteases, neutrophils, and periodontitis: the net effect. J Clin Invest. 2014;124(10):4237–4239. doi:10.1172/JCI77985
153. Nguyen JA, Yates RM. Better together: current insights into phagosome-lysosome fusion. Front Immunol. 2021;12:636078. doi:10.3389/fimmu.2021.636078
154. Emmerson BT, Cross M, Osborne JM, et al. Ultrastructural studies of the reaction of urate crystals with a cultured renal tubular cell line. Nephron. 1991;59(3):403–408. doi:10.1159/000186599
155. Amini P, Stojkov D, Felser A, et al. Neutrophil extracellular trap formation requires opa1-dependent glycolytic atp production. Nat Commun. 2018;9(1):2958. doi:10.1038/s41467-018-05387-y
156. Franklin BS, Mangan MS, Latz E. Crystal formation in inflammation. Annu Rev Immunol. 2016;34:173–202. doi:10.1146/annurev-immunol-041015-055539
157. Euler M, Hoffmann MH. The double-edged role of neutrophil extracellular traps in inflammation. Biochem Soc Trans. 2019;47(6):1921–1930. doi:10.1042/BST20190629
158. Soehnlein O, Zernecke A, Eriksson EE, et al. Neutrophil secretion products pave the way for inflammatory monocytes. Blood. 2008;112(4):1461–1471. doi:10.1182/blood-2008-02-139634
159. Soehnlein O, Lindbom L, Weber C. Mechanisms underlying neutrophil-mediated monocyte recruitment. Blood. 2009;114(21):4613–4623. doi:10.1182/blood-2009-06-221630
160. So A. Gout in the spotlight. Arthritis Res Ther. 2008;10(3):112. doi:10.1186/ar2396
161. Zheng SC, Zhu XX, Xue Y, et al. Role of the nlrp3 inflammasome in the transient release of il-1beta induced by monosodium urate crystals in human fibroblast-like synoviocytes. J Inflamm (Lond). 2015;12:30. doi:10.1186/s12950-015-0070-7
162. Liu D, Yang P, Gao M, et al. Nlrp3 activation induced by neutrophil extracellular traps sustains inflammatory response in the diabetic wound. Clin Sci. 2019;133(4):565–582. doi:10.1042/CS20180600
163. Pereira CA, Carlos D, Ferreira NS, et al. Mitochondrial DNA promotes nlrp3 inflammasome activation and contributes to endothelial dysfunction and inflammation in type 1 diabetes. Front Physiol. 2019;10:1557. doi:10.3389/fphys.2019.01557
164. Choi N, Yang G, Jang JH, et al. Loganin alleviates gout inflammation by suppressing nlrp3 inflammasome activation and mitochondrial damage. Molecules. 2021;26(4):1071. doi:10.3390/molecules26041071
165. Butt JH, Docherty KF, Claggett BL, et al. Association of dapagliflozin use with clinical outcomes and the introduction of uric acid-lowering therapy and colchicine in patients with heart failure with and without gout: a patient-level pooled meta-analysis of dapa-hf and deliver. JAMA Cardiol. 2023;8(4):386–393. doi:10.1001/jamacardio.2022.5608
166. Steiger S, Harper JL. Mechanisms of spontaneous resolution of acute gouty inflammation. Curr Rheumatol Rep. 2014;16(1):392. doi:10.1007/s11926-013-0392-5
167. Wei H, Liu B, Yin C, et al. Electroacupuncture improves gout arthritis pain via attenuating ros-mediated nlrp3 inflammasome overactivation. Chin Med. 2023;18(1):86. doi:10.1186/s13020-023-00800-1
168. Cobo I, Cheng A, Murillo-Saich J, et al. Monosodium urate crystals regulate a unique jnk-dependent macrophage metabolic and inflammatory response. Cell Rep. 2022;38(10):110489. doi:10.1016/j.celrep.2022.110489
169. Cronstein BN, Terkeltaub R. The inflammatory process of gout and its treatment. Arthritis Res Ther. 2006;8 Suppl 1(Suppl 1):S3. doi:10.1186/ar1908
170. Biasizzo M, Kopitar-Jerala N. Interplay between nlrp3 inflammasome and autophagy. Front Immunol. 2020;11:591803. doi:10.3389/fimmu.2020.591803
171. Schauer C, Janko C, Munoz LE, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. 2014;20(5):511–517. doi:10.1038/nm.3547
172. Hahn J, Schauer C, Czegley C, et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J. 2019;33(1):1401–1414. doi:10.1096/fj.201800752R
173. Qiao CY, Li Y, Shang Y, et al. Management of gout-associated msu crystals-induced nlrp3 inflammasome activation by procyanidin b2: targeting il-1beta and cathepsin b in macrophages. Inflammopharmacology. 2020;28(6):1481–1493. doi:10.1007/s10787-020-00758-8
174. Zhou Y, Chen Y, Zhong X, et al. Lipoxin a4 attenuates msu-crystal-induced nlrp3 inflammasome activation through suppressing nrf2 thereby increasing txnrd2. Front Immunol. 2022;13:1060441. doi:10.3389/fimmu.2022.1060441
175. Chen C, Wang J, Liang Z, et al. Monosodium urate crystals with controlled shape and aspect ratio for elucidating the pathological progress of acute gout. Biomater Adv. 2022:139 213005. doi:10.1016/j.bioadv.2022.213005
176. Knopf J, Leppkes M, Schett G, et al. Aggregated nets sequester and detoxify extracellular histones. Front Immunol. 2019;10(2176). doi:10.3389/fimmu.2019.02176
177. Abdulkhaleq LA, Assi MA, Abdullah R, et al. The crucial roles of inflammatory mediators in inflammation: a review. Vet World. 2018;11(5):627–635. doi:10.14202/vetworld.2018.627-635
178. Li C, Wang C, Guo Y, et al. Research on the effect and underlying molecular mechanism of cangzhu in the treatment of gouty arthritis. Eur J Pharmacol. 2022:927 175044. doi:10.1016/j.ejphar.2022.175044
179. Yang J, Chen G, Guo TW, et al. Simiao wan attenuates monosodium urate crystal-induced arthritis in rats through contributing to macrophage m2 polarization. J Ethnopharmacol. 2021:275 114123. doi:10.1016/j.jep.2021.114123
180. Chen R, Li F, Zhou K, et al. Component identification of modified sanmiao pills by uplc-xevo g2-xs qtof and its anti-gouty arthritis mechanism based on network pharmacology and experimental verification. J Ethnopharmacol. 2023:311 116394. doi:10.1016/j.jep.2023.116394
181. Rocha MP, Oliveira DP, de Oliveira VLS, et al. Ouratea spectabilis and its biflavanone ouratein d exert potent anti-inflammatory activity in msu crystal-induced gout in mice. Planta Med. 2023;89(7):718–728. doi:10.1055/a-2009-9809
182. FitzGerald JD, Dalbeth N, Mikuls T, et al. 2020 American college of rheumatology guideline for the management of gout. Arthritis Care Res (Hoboken). 2020;72(6):744–760. doi:10.1002/acr.24180
183. Kwon KW, Kim LH, Kang SM, et al. Host-directed anti-mycobacterial activity of colchicine, an anti-gout drug, via strengthened host innate resistance reinforced by the il-1beta/pge(2) axis. Br J Pharmacol. 2022;179(15):3951–3969. doi:10.1111/bph.15838
184. Oh YJ, Lee YJ, Lee E, et al. Cancer risk in Korean patients with gout. Korean J Intern Med. 2022;37(2):460–467. doi:10.3904/kjim.2020.259
185. Jiang Q, Li S, Yang Z, et al. experience sharing on treatment of an acute colchicine poisoning patient. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2020;32(12):1526–1529. doi:10.3760/cma.j.cn121430-20201018-00676
186. Latourte A, Pascart T, Flipo RM, et al. 2020 recommendations from the French society of rheumatology for the management of gout: management of acute flares. Joint Bone Spine. 2020;87(5):387–393. doi:10.1016/j.jbspin.2020.05.001
187. McKenzie BJ, Wechalekar MD, Johnston RV, et al. Colchicine for acute gout. Cochrane Database Syst Rev. 2021;8:CD006190. doi:10.1002/14651858.CD006190.pub3
188. Morris I, Varughese G, Mattingly P. Colchicine in acute gout. BMJ. 2003;327(7426):1275–1276. doi:10.1136/bmj.327.7426.1275
189. Stamp LK, Day RO, Yun J. Allopurinol hypersensitivity: investigating the cause and minimizing the risk. Nat Rev Rheumatol. 2016;12(4):235–242. doi:10.1038/nrrheum.2015.132
190. Stamp LK, Dalbeth N. Prevention and treatment of gout. Nat Rev Rheumatol. 2019;15(2):68–70. doi:10.1038/s41584-018-0149-7
191. Chen RJ, Chen MH, Chen YL, et al. Evaluating the urate-lowering effects of different microbial fermented extracts in hyperuricemic models accompanied with a safety study. Journal of Food and Drug Analysis. 2017;25(3):597–606. doi:10.1016/j.jfda.2016.07.003
192. Kaufmann P, Torok M, Hanni A, et al. Mechanisms of benzarone and benzbromarone-induced hepatic toxicity. Hepatology. 2005;41(4):925–935. doi:10.1002/hep.20634
193. White WB, Saag KG, Becker MA, et al. Cardiovascular safety of febuxostat or allopurinol in patients with gout. N Engl J Med. 2018;378(13):1200–1210. doi:10.1056/NEJMoa1710895
194. Mackenzie IS, Ford I, Nuki G, et al. Long-term cardiovascular safety of febuxostat compared with allopurinol in patients with gout (fast): a multicentre, prospective, randomised, open-label, non-inferiority trial. Lancet. 2020;396(10264):1745–1757. doi:10.1016/S0140-6736(20)32234-0
195. Carcione J, Bodofsky S, LaMoreaux B, et al. Beyond medical treatment: surgical treatment of gout. Curr Rheumatol Rep. 2020;23(1):1. doi:10.1007/s11926-020-00969-6
196. Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, et al. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15(2):135–147. doi:10.1038/nrm3737
197. Glantzounis GK, Tsimoyiannis EC, Kappas AM, et al. Uric acid and oxidative stress. Curr Pharm Des. 2005;11(32):4145–4151. doi:10.2174/138161205774913255
198. Mancinelli R, Fano-Illic G, Pietrangelo T, et al. Guanosine-based nucleotides, the sons of a lesser god in the purinergic signal scenario of excitable tissues. Int J Mol Sci. 2020;21(5). doi:10.3390/ijms21051591
199. Nelson KL, Voruganti VS. Purine metabolites and complex diseases: role of genes and nutrients. Curr Opin Clin Nutr Metab Care. 2021;24(4):296–302. doi:10.1097/MCO.0000000000000764
200. Rees F, Hui M. M Doherty Optimizing current treatment of gout. Nat Rev Rheumatol. 2014;10(5):271–283. doi:10.1038/nrrheum.2014.32
201. Kumar R, Joshi G, Kler H, et al. Toward an understanding of structural insights of xanthine and aldehyde oxidases: an overview of their inhibitors and role in various diseases. Med Res Rev. 2018;38(4):1073–1125. doi:10.1002/med.21457
202. Ojha R, Singh J, Ojha A, et al. An updated patent review: xanthine oxidase inhibitors for the treatment of hyperuricemia and gout (2011-2015). Expert Opin Ther Pat. 2017;27(3):311–345. doi:10.1080/13543776.2017.1261111
203. Stockert AL, Shinde SS, Anderson RF, et al. The reaction mechanism of xanthine oxidase: evidence for two-electron chemistry rather than sequential one-electron steps. J Am Chem Soc. 2002;124(49):14554–14555. doi:10.1021/ja027388d
204. Kumar R, Darpan S. Xanthine oxidase inhibitors: a patent survey. Expert Opin Ther Pat. 2011;21(7):1071–1108. doi:10.1517/13543776.2011.577417
205. Singh JV, Bedi PMS, Singh H, et al. Xanthine oxidase inhibitors: patent landscape and clinical development (2015-2020). Expert Opin Ther Pat. 2020;30(10):769–780. doi:10.1080/13543776.2020.1811233
206. Tamta H, Thilagavathi R, Chakraborti AK, et al. 6-(n-benzoylamino)purine as a novel and potent inhibitor of xanthine oxidase: inhibition mechanism and molecular modeling studies. J Enzyme Inhib Med Chem. 2005;20(4):317–324. doi:10.1080/14756360500112326
207. Kalra S, Jena G, Tikoo K, et al. Preferential inhibition of xanthine oxidase by 2-amino-6-hydroxy-8-mercaptopurine and 2-amino-6-purine thiol. BMC Biochem. 2007;8:8. doi:10.1186/1471-2091-8-8
208. Shi C, Zhou Z, Chi X, et al. Recent advances in gout drugs. Eur J Med Chem. 2023;245(Pt 1):114890. doi:10.1016/j.ejmech.2022.114890
209. Stamp LK, Taylor WJ, Jones PB, et al. Starting dose is a risk factor for allopurinol hypersensitivity syndrome: a proposed safe starting dose of allopurinol. Arthritis Rheum. 2012;64(8):2529–2536. doi:10.1002/art.34488
210. Sattui SE, Gaffo AL. Treatment of hyperuricemia in gout: current therapeutic options, latest developments and clinical implications. Ther Adv Musculoskelet Dis. 2016;8(4):145–159. doi:10.1177/1759720X16646703
211. Jordan A, Gresser U. Side effects and interactions of the xanthine oxidase inhibitor febuxostat. Pharmaceuticals (Basel). 2018;11(2). doi:10.3390/ph11020051
212. Nagao A, Seki M, Kobayashi H. Inhibition of xanthine oxidase by flavonoids. Biosci Biotechnol Biochem. 1999;63(10):1787–1790. doi:10.1271/bbb.63.1787
213. Lu JM, Yao Q, Chen C. 3,4-dihydroxy-5-nitrobenzaldehyde (dhnb) is a potent inhibitor of xanthine oxidase: a potential therapeutic agent for treatment of hyperuricemia and gout. Biochem Pharmacol. 2013;86(9):1328–1337. doi:10.1016/j.bcp.2013.08.011
214. Song J, Wang Z, Chi Y, et al. Anti-gout activity and the interaction mechanisms between sanghuangporus vaninii active components and xanthine oxidase. Bioorg Chem. 2023;133:106394. doi:10.1016/j.bioorg.2023.106394
215. Li H, Zhang X, Gu L, et al. Anti-gout effects of the medicinal fungus phellinus igniarius in hyperuricaemia and acute gouty arthritis rat models. Front Pharmacol. 2021;12:801910. doi:10.3389/fphar.2021.801910
216. Pui K, Gow PJ, Dalbeth N. Efficacy and tolerability of probenecid as urate-lowering therapy in gout; clinical experience in high-prevalence population. J Rheumatol. 2013;40(6):872–876. doi:10.3899/jrheum.121301
217. Kasper IR, Juriga MD, Giurini JM, et al. Treatment of tophaceous gout: when medication is not enough. Semin Arthritis Rheum. 2016;45(6):669–674. doi:10.1016/j.semarthrit.2016.01.005
218. Reinders MK, van Roon EN, Jansen TL, et al. Efficacy and tolerability of urate-lowering drugs in gout: a randomised controlled trial of benzbromarone versus probenecid after failure of allopurinol. Ann Rheum Dis. 2009;68(1):51–56. doi:10.1136/ard.2007.083071
219. Perez-Ruiz F, Calabozo M, Fernandez-Lopez MJ, et al. Treatment of chronic gout in patients with renal function impairment: an open, randomized, actively controlled study. J Clin Rheumatol. 1999;5(2):49–55. doi:10.1097/00124743-199904000-00003
220. Perez-Ruiz F, Alonso-Ruiz A, Calabozo M, et al. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia. A pathogenic approach to the treatment of primary chronic gout. Ann Rheum Dis. 1998;57(9):545–549. doi:10.1136/ard.57.9.545
221. Takahashi S, Moriwaki Y, Yamamoto T, et al. Effects of combination treatment using anti-hyperuricaemic agents with fenofibrate and/or losartan on uric acid metabolism. Ann Rheum Dis. 2003;62(6):572–575. doi:10.1136/ard.62.6.572
222. Milionis HJ, Kakafika AI, Tsouli SG, et al. Effects of statin treatment on uric acid homeostasis in patients with primary hyperlipidemia. Am Heart J. 2004;148(4):635–640. doi:10.1016/j.ahj.2004.04.005
223. Abhishek A. New urate-lowing therapies. Curr Opin Rheumatol. 2018;30(2):177–182. doi:10.1097/BOR.0000000000000476
224. Perez-Ruiz F, Dalbeth N. Combination urate-lowering therapy in the treatment of gout: what is the evidence? Semin Arthritis Rheum. 2019;48(4):658–668. doi:10.1016/j.semarthrit.2018.06.004
225. Richette P, Doherty M, Pascual E, et al. 2016 updated eular evidence-based recommendations for the management of gout. Ann Rheum Dis. 2017;76(1):29–42. doi:10.1136/annrheumdis-2016-209707
226. Perez-Ruiz F, Calabozo M, Pijoan JI, et al. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Rheum. 2002;47(4):356–360. doi:10.1002/art.10511
227. Orhan IE, Deniz FSS. Natural products and extracts as xantine oxidase inhibitors - a hope for gout disease? Curr Pharm Des. 2021;27(2):143–158. doi:10.2174/1381612826666200728144605
228. Sawada S, Kajiyama K, Shida H, et al. Cardiovascular risk of urate-lowering drugs: a study using the national database of health insurance claims and specific health checkups of Japan. Clin Transl Sci. 2023;16(2):206–215. doi:10.1111/cts.13439
229. Wang H, Wang W, Gong B, et al. Glutathione conjugation and protein adduction derived from oxidative debromination of benzbromarone in mice. Drug Metab Dispos. 2019;47(11):1281–1290. doi:10.1124/dmd.119.087460
230. Jiang B, Zhao Y, Cao J, et al. Synthesis and preliminary biological evaluation of naproxen-probenecid conjugate for central nervous system (cns) delivery. Pak J Pharm Sci. 2021;34(6):2197–2203.
231. Lee D, Kim JK, Han Y, et al. Antihyperuricemic effect of dendropanax morbifera leaf extract in rodent models. Evid Based Complement Alternat Med. 2021;2021:3732317. doi:10.1155/2021/3732317
232. Grelska A, Sharan D, Light SH. Purine-ifying uric acid by gut microbes. Cell Chem Biol. 2023;30(7):706–708. doi:10.1016/j.chembiol.2023.06.022
233. Liu X, Ke L, Lei K, et al. Antibiotic-induced gut microbiota dysbiosis has a functional impact on purine metabolism. BMC Microbiol. 2023;23(1):187. doi:10.1186/s12866-023-02932-8
234. Lin X, Shao T, Huang L, et al. Simiao decoction alleviates gouty arthritis by modulating proinflammatory cytokines and the gut ecosystem. Front Pharmacol. 2020;11:955. doi:10.3389/fphar.2020.00955
235. Wei J, Zhang Y, Dalbeth N, et al. Association between gut microbiota and elevated serum urate in two independent cohorts. Arthritis Rheumatol. 2022;74(4):682–691. doi:10.1002/art.42009
236. Ren Q, Cheng L, Guo F, et al. Fisetin improves hyperuricemia-induced chronic kidney disease via regulating gut microbiota-mediated tryptophan metabolism and aryl hydrocarbon receptor activation. J Agric Food Chem. 2021;69(37):10932–10942. doi:10.1021/acs.jafc.1c03449
237. Rai SK, Fung TT, Lu N, et al. The dietary approaches to stop hypertension (dash) diet, western diet, and risk of gout in men: prospective cohort study. BMJ. 2017:357 j1794. doi:10.1136/bmj.j1794
238. Holscher HD. Dietary fiber and prebiotics and the gastrointestinal microbiota. Gut Microbes. 2017;8(2):172–184. doi:10.1080/19490976.2017.1290756
239. Song S, Fan M, Wen X, et al. Integrated network pharmacology and gut microbiome analysis to reveal the mechanism of qu-zhuo-tong-bi decoction against hyperuricemia and gout. J Ethnopharmacol. 2023;316:116736. doi:10.1016/j.jep.2023.116736
240. Lin X, Wang M, He Z, et al. Gut microbiota mediated the therapeutic efficiency of simiao decoction in the treatment of gout arthritis mice. BMC Complement Med Ther. 2023;23(1):206. doi:10.1186/s12906-023-04042-4
241. Xie WR, Yang XY, Deng ZH, et al. Effects of washed microbiota transplantation on serum uric acid levels, symptoms, and intestinal barrier function in patients with acute and recurrent gout: a pilot study. Dig Dis. 2022;40(5):684–690. doi:10.1159/000521273
242. Dong W, Liu Y, Hou J, et al. Nematodes degrade extracellular antibiotic resistance genes by secreting dnase ii encoded by the nuc-1 gene. Environ Sci Technol. 2023. doi:10.1021/acs.est.3c03829
243. Huang W, Wen L, Tian H, et al. Self-propelled proteomotors with active cell-free mtdna clearance for enhanced therapy of sepsis-associated acute lung injury. Adv Sci (Weinh). 2023:e2301635. doi:10.1002/advs.202301635
244. Hosseinnejad A, Ludwig N, Wienkamp AK, et al. Dnase i functional microgels for neutrophil extracellular trap disruption. Biomater Sci. 2021;10(1):85–99. doi:10.1039/d1bm01591e
245. Angeletti A, Volpi S, Bruschi M, et al. Neutrophil extracellular traps-dnase balance and autoimmunity. Cells. 2021;10(10). doi:10.3390/cells10102667
246. Kolaczkowska E, Jenne CN, Surewaard BG, et al. Molecular mechanisms of net formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. 2015;6:6673. doi:10.1038/ncomms7673
247. Li YW, Chen SX, Yang Y, et al. Colchicine inhibits nets and alleviates cardiac remodeling after acute myocardial infarction. Cardiovasc Drugs Ther. 2022. doi:10.1007/s10557-022-07326-y
248. Safi R, Kallas R, Bardawil T, et al. Neutrophils contribute to vasculitis by increased release of neutrophil extracellular traps in behcet’s disease. J Dermatol Sci. 2018;92(2):143–150. doi:10.1016/j.jdermsci.2018.08.010
249. Vaidya K, Tucker B, Kurup R, et al. Colchicine inhibits neutrophil extracellular trap formation in patients with acute coronary syndrome after percutaneous coronary intervention. J Am Heart Assoc. 2021;10(1):e018993. doi:10.1161/JAHA.120.018993
250. Apostolidou E, Skendros P, Kambas K, et al. Neutrophil extracellular traps regulate il-1beta-mediated inflammation in familial Mediterranean fever. Ann Rheum Dis. 2016;75(1):269–277. doi:10.1136/annrheumdis-2014-205958
251. Khan D, Qindeel M, Ahmed N, et al. Development of an intelligent, stimuli-responsive transdermal system for efficient delivery of ibuprofen against rheumatoid arthritis. Int J Pharm. 2021:610 121242. doi:10.1016/j.ijpharm.2021.121242
252. Mutua V, Cavallo F, Gershwin LJ. Neutrophil extracellular traps (nets) in a randomized controlled trial of a combination of antiviral and nonsteroidal anti-inflammatory treatment in a bovine model of respiratory syncytial virus infection. Vet Immunol Immunopathol. 2021;241 110323. doi:10.1016/j.vetimm.2021.110323
253. Barnes PJ. Glucocorticosteroids. Handb Exp Pharmacol. 2017;237:93–115. doi:10.1007/164_2016_62
254. Cohn LA. Glucocorticosteroids as immunosuppressive agents. Semin Vet Med Surg Small Anim. 1997;12(3):150–156. doi:10.1016/s1096-2867(97)80026-6
255. Terkeltaub R. Update on gout: new therapeutic strategies and options. Nat Rev Rheumatol. 2010;6(1):30–38. doi:10.1038/nrrheum.2009.236
256. Vargas A, Boivin R, Cano P, et al. Neutrophil extracellular traps are downregulated by glucocorticosteroids in lungs in an equine model of asthma. Respir Res. 2017;18(1):207. doi:10.1186/s12931-017-0689-4
257. Islam MS. Natural products and disease prevention, relief and treatment. Nutrients. 2022;14(12). doi:10.3390/nu14122396
258. Galiniak S, Aebisher D. D Bartusik-Aebisher Health benefits of resveratrol administration. Acta Biochim Pol. 2019;66(1):13–21. doi:10.18388/abp.2018_2749
259. Malaguarnera L. Influence of resveratrol on the immune response. Nutrients. 2019;11(5). doi:10.3390/nu11050946
260. Vargas JE, Souto AA, Pitrez PM, et al. Modulatory potential of resveratrol during lung inflammatory disease. Med Hypotheses. 2016;96:61–65. doi:10.1016/j.mehy.2016.09.023
261. de Souza Andrade MM, Leal VNC, Fernandes IG, et al. Resveratrol downmodulates neutrophil extracellular trap (net) generation by neutrophils in patients with severe covid-19. Antioxidants (Basel). 2022;11(9). doi:10.3390/antiox11091690
262. Fan W, Chen S, Wu X, et al. Resveratrol relieves gouty arthritis by promoting mitophagy to inhibit activation of nlrp3 inflammasomes. J Inflamm Res. 2021;14:3523–3536. doi:10.2147/JIR.S320912
263. Polera N, Badolato M, Perri F, et al. Quercetin and its natural sources in wound healing management. Curr Med Chem. 2019;26(31):5825–5848. doi:10.2174/0929867325666180713150626
264. Yang D, Wang T, Long M, et al. Quercetin: its main pharmacological activity and potential application in clinical medicine. Oxid Med Cell Longev. 2020;2020:8825387. doi:10.1155/2020/8825387
265. Alizadeh SR, Savadkouhi N, Ebrahimzadeh MA. Drug design strategies that aim to improve the low solubility and poor bioavailability conundrum in quercetin derivatives. Expert Opin Drug Discov. 2023;1–16. doi:10.1080/17460441.2023.2241366
266. Shi Y, Williamson G. Quercetin lowers plasma uric acid in pre-hyperuricaemic males: a randomised, double-blinded, placebo-controlled, cross-over trial. Br J Nutr. 2016;115(5):800–806. doi:10.1017/S0007114515005310
267. Ruiz-Miyazawa KW, Staurengo-Ferrari L, Mizokami SS, et al. Quercetin inhibits gout arthritis in mice: induction of an opioid-dependent regulation of inflammasome. Inflammopharmacology. 2017. doi:10.1007/s10787-017-0356-x
268. Yuan K, Zhu Q, Lu Q, et al. Quercetin alleviates rheumatoid arthritis by inhibiting neutrophil inflammatory activities. J Nutr Biochem. 2020;84:108454. doi:10.1016/j.jnutbio.2020.108454
269. Gu Y, Zhu Y, Deng G, et al. Curcumin analogue ai-44 alleviates msu-induced gouty arthritis in mice via inhibiting cathepsin b-mediated nlrp3 inflammasome activation. Int Immunopharmacol. 2021;93:107375. doi:10.1016/j.intimp.2021.107375
270. Chen B, Li H, Ou G, et al. Curcumin attenuates msu crystal-induced inflammation by inhibiting the degradation of ikappabalpha and blocking mitochondrial damage. Arthritis Res Ther. 2019;21(1):193. doi:10.1186/s13075-019-1974-z
271. Ye S, Li S, Ma Y, et al. Curcumin hinders pbde-47-induced neutrophil extracellular traps release via nrf2-associated ros inhibition. Ecotoxicol Environ Saf. 2021:225 112779. doi:10.1016/j.ecoenv.2021.112779
272. Zhu C, Shi S, Jiang P, et al. Curcumin alleviates hepatic ischemia-reperfusion injury by inhibiting neutrophil extracellular traps formation. J Invest Surg. 2023;36(1):2164813. doi:10.1080/08941939.2022.2164813
273. Silvestre F, Santos C, Silva V, et al. Pharmacokinetics of curcumin delivered by nanoparticles and the relationship with antitumor efficacy: a systematic review. Pharmaceuticals (Basel). 2023;16(7). doi:10.3390/ph16070943
274. Huang M, Zhai BT, Fan Y, et al. Targeted drug delivery systems for curcumin in breast cancer therapy. Int J Nanomed. 2023;18:4275–4311. doi:10.2147/IJN.S410688
275. Zhang M, Zhang X, Tian T, et al. Anti-inflammatory activity of curcumin-loaded tetrahedral framework nucleic acids on acute gouty arthritis. Bioact Mater. 2022;8:368–380. doi:10.1016/j.bioactmat.2021.06.003
276. Li X, Yuan K, Zhu Q, et al. Andrographolide ameliorates rheumatoid arthritis by regulating the apoptosis-netosis balance of neutrophils. Int J Mol Sci. 2019;20(20). doi:10.3390/ijms20205035
277. Semenova S, Shatrova A, Vassilieva I, et al. Adenosine-5’-triphosphate suppresses proliferation and migration capacity of human endometrial stem cells. J Cell Mol Med. 2020;24(8):4580–4588. doi:10.1111/jcmm.15115
278. Le Duc D, Schulz A, Lede V, et al. P2y receptors in immune response and inflammation. Adv Immunol. 2017:136 85–121. doi:10.1016/bs.ai.2017.05.006
279. Sil P, Hayes CP, Reaves BJ, et al. P2y6 receptor antagonist mrs2578 inhibits neutrophil activation and aggregated neutrophil extracellular trap formation induced by gout-associated monosodium urate crystals. J Immunol. 2017;198(1):428–442. doi:10.4049/jimmunol.1600766
280. Shin SH, Jeong JH. 1-palmitoyl-2-linoleoyl-3-acetyl-rac-glycerol (plag) mitigates monosodium urate (msu)-induced acute gouty inflammation in balb/c mice. Front Immunol. 2020;11:710. doi:10.3389/fimmu.2020.00710
281. Wen RX, Shen H, Huang SX, et al. P2y6 receptor inhibition aggravates ischemic brain injury by reducing microglial phagocytosis. CNS Neurosci Ther. 2020;26(4):416–429. doi:10.1111/cns.13296
282. Nagai J, Lin J. A Boyce Macrophage p2y6 receptor signaling selectively activates nfatc2 and suppresses allergic lung inflammation. J Immunol. 2022;209(12):2293–2303. doi:10.4049/jimmunol.2200452
283. Sollberger G, Choidas A, Burn GL, et al. Gasdermin d plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3(26). doi:10.1126/sciimmunol.aar6689
284. Silva CMS, Wanderley CWS, Veras FP, et al. Gasdermin d inhibition prevents multiple organ dysfunction during sepsis by blocking net formation. Blood. 2021;138(25):2702–2713. doi:10.1182/blood.2021011525
285. Chen KW, Monteleone M, Boucher D, et al. Noncanonical inflammasome signaling elicits gasdermin d-dependent neutrophil extracellular traps. Sci Immunol. 2018;3(26). doi:10.1126/sciimmunol.aar6676
286. Hernandez-Velazquez ED, Alba-Betancourt C, Alonso-Castro AJ, et al. Metformin, a biological and synthetic overview. Bioorg Med Chem Lett. 2023;86:129241. doi:10.1016/j.bmcl.2023.129241
287. Kim JW, Choe JY, Park SH. Metformin and its therapeutic applications in autoimmune inflammatory rheumatic disease. Korean J Intern Med. 2022;37(1):13–26. doi:10.3904/kjim.2021.363
288. Mw MC. Metformin as a potential treatment for covid-19. Expert Opin Pharmacother. 2023;24(10):1199–1203. doi:10.1080/14656566.2023.2215385
289. Feng J, Wang X, Ye X, et al. Mitochondria as an important target of metformin: the mechanism of action, toxic and side effects, and new therapeutic applications. Pharmacol Res. 2022:177 106114. doi:10.1016/j.phrs.2022.106114
290. Dong Y, Qi Y, Jiang H, et al. The development and benefits of metformin in various diseases. Front Med. 2023;17(3):388–431. doi:10.1007/s11684-023-0998-6
291. Vazirpanah N, Ottria A, van der Linden M, et al. Mtor inhibition by metformin impacts monosodium urate crystal-induced inflammation and cell death in gout: a prelude to a new add-on therapy? Ann Rheum Dis. 2019;78(5):663–671. doi:10.1136/annrheumdis-2018-214656
292. Wang H, Li T, Chen S, et al. Neutrophil extracellular trap mitochondrial DNA and its autoantibody in systemic lupus erythematosus and a proof-of-concept trial of metformin. Arthritis Rheumatol. 2015;67(12):3190–3200. doi:10.1002/art.39296
293. Kolman JP, Pagerols Raluy L, Muller I, et al. Net release of long-term surviving neutrophils. Front Immunol. 2022;13:815412. doi:10.3389/fimmu.2022.815412
294. Simon SI, Kim MH. A day (or 5) in a neutrophil’s life. Blood. 2010;116(4):511–512. doi:10.1182/blood-2010-05-283184
295. Han NR, Ko SG, Park HJ, et al. Hydrogen sulfide downregulates oncostatin m expression via pi3k/akt/nf-kappab signaling processes in neutrophil-like differentiated hl-60 cells. Antioxidants (Basel). 2023;12(2). doi:10.3390/antiox12020417
296. Bai F, Fan C, Lin X, et al. Hemin protects uvb-induced skin damage through inhibiting keratinocytes apoptosis and reducing neutrophil infiltration. J Photochem Photobiol B. 2023:238 112604. doi:10.1016/j.jphotobiol.2022.112604
297. Kwiatkowski P, Tabis A, Sobolewski P, et al. Enhancement of neutrophil chemotaxis by trans-anethole-treated staphylococcus aureus strains. PLoS One. 2023;18(4):e0284042. doi:10.1371/journal.pone.0284042
298. Guo Y, Gao F, Wang Q, et al. Differentiation of hl-60 cells in serum-free hematopoietic cell media enhances the production of neutrophil extracellular traps. Exp Ther Med. 2021;21(4):353. doi:10.3892/etm.2021.9784
299. Manda-Handzlik A, Bystrzycka W, Wachowska M, et al. The influence of agents differentiating hl-60 cells toward granulocyte-like cells on their ability to release neutrophil extracellular traps. Immunol Cell Biol. 2018;96(4):413–425. doi:10.1111/imcb.12015
300. Thiam HR, Wong SL, Qiu R, et al. Netosis proceeds by cytoskeleton and endomembrane disassembly and pad4-mediated chromatin decondensation and nuclear envelope rupture. Proc Natl Acad Sci USA. 2020;117(13):7326–7337. doi:10.1073/pnas.1909546117
301. Atanasov AG, Zotchev SB, Dirsch VM, et al. Natural products in drug discovery: advances and opportunities. Nat Rev Drug Discov. 2021;20(3):200–216. doi:10.1038/s41573-020-00114-z
302. Wang L, Wang N, Zhang W, et al. Therapeutic peptides: current applications and future directions. Signal Transduct Target Ther. 2022;7(1):48. doi:10.1038/s41392-022-00904-4
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.