Back to Journals » Drug Design, Development and Therapy » Volume 16
Targeting mTOR Complex 2 in Castration-Resistant Prostate Cancer with Acquired Docetaxel Resistance
Authors Huang Y, Zhai Y ![]() , Wu M, Chang C, Luo J, Hong D, Zhao Q, Dai Y, Liu J
, Wu M, Chang C, Luo J, Hong D, Zhao Q, Dai Y, Liu J ![]()
Received 10 June 2022
Accepted for publication 24 October 2022
Published 4 November 2022 Volume 2022:16 Pages 3817—3828
DOI https://doi.org/10.2147/DDDT.S376474
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Manfred Ogris
Yujie Huang,1 You Zhai,1 Meijia Wu,1 Chengdong Chang,2 Jindan Luo,3 Dongsheng Hong,1 Qingwei Zhao,1 Yao Dai,4 Jian Liu1
1Research Center for Clinical Pharmacy, Zhejiang Provincial Key Laboratory for Drug Evaluation and Clinical Research, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, People’s Republic of China; 2Department of Pathology, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, People’s Republic of China; 3Department of Urology, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, People’s Republic of China; 4Department of Radiation Oncology, College of Medicine, University of Florida, Gainesville, FL, USA
Correspondence: Jian Liu, Research Center for Clinical Pharmacy, Zhejiang Provincial Key Laboratory for Drug Evaluation and Clinical Research, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, People’s Republic of China, Tel +86-571-87236560, Fax +86-571-87236531, Email [email protected] Yao Dai, Department of Radiation Oncology, College of Medicine, University of Florida, Gainesville, FL, USA, Tel +1-352-273-8237, Fax +1-352-273-8252, Email [email protected]
Purpose: Mammalian Target of rapamycin (mTOR) plays a central role in regulating cell growth, proliferation, and cell cycle. The key component of mTORC2 is highly expressed in docetaxel-resistant prostate cells. However, the underlying molecular effects on prostate cells remain unclear.
Methods: A docetaxel-resistant human prostate cell line (PC-3/DTX) was constructed to investigate the role of mTORC2 in docetaxel resistance. The lentivirus was transfected into cells to knock down the expression of Rictor, and cell viability was measured by Cell Counting Kit 8 (CCK-8). Flow cytometry was used to analyze the cell cycle, and the changes in related signal cascades were assessed by immunohistochemistry (IHC) staining and Western blot.
Results: Docetaxel showed the lowest IC50 (50% inhibitory concentration) in PC-3/DTX cells with sh-RNA. Decreased Rictor expression resulted in a larger proportion of arrested cells in the G0/G1 phase in PC-3/DTX cells. The IC50 values of the AZD8055 group were lower than in the Rapamycin group when treated with docetaxel again. Furthermore, a larger proportion of PC-3/DTX cells were arrested in the G0/G1 phase in the AZD8055 group compared to the Rapamycin group. The IHC results of the prostate cancer tissues from a CRPC patient revealed the over expression of Rictor only, while Raptor expression was unaffected.
Conclusion: We investigated the role of mTORC2 signaling on the acquired docetaxel -resistant PC-3 cells to identify potential methods for clinical treatment. MTORC2 expression is essential for docetaxel drug resistance of PC-3 cells. The mTORC1/2 inhibitor AZD8055 caused more significant disruption of mTORC2 kinase activity than the mTORC1 inhibitor Rapamycin, which lead to decreased docetaxel-mediated resistance. Therefore, reversing docetaxel resistance, may become a therapeutic option in the treatment of mCRPC patients.
Keywords: prostate cancer cell, docetaxel, drug resistance, mTORC2, reverse
Introduction
Female breast cancer, lung cancer, prostate cancer, and nonmelanoma skin cancer have the highest prevalence and deaths of all malignancies worldwide.1 Prostate cancer has the largest average annual percentage change in incidence rate among men in the United States.2
Prostate cancer is one of the most common malignant tumors in men, accounting for the second highest incidence of male cancer in the world, and has become the fastest growing cancer among Chinese men. Compared with European and American countries, the incidence of prostate cancer in China is lower, but the proportion of prostate cancer deaths worldwide is far more higher than that in Western countries.2,3 Its incidence is rapidly rising in Asia, where it was traditionally considered an uncommon tumor. Androgen deprivation therapy (ADT) has emerged as the first-line treatment for advanced prostate cancer patients. Although this treatment is highly effective initially, the disease eventually progresses and develops into castration-resistant prostate cancer (CRPC) within a few years. The incidence of prostate cancer is still increasing in China and the United States due to limited treatment options.4 Metastatic CRPC (mCRPC) is a major clinical challenge and the major cause of mortality.5 In patients with mCRPC, the first-generation taxane docetaxel was standard of care and demonstrated a survival benefit. However, docetaxel has a high toxicity profile, and nearly 50% of patients fail to respond to docetaxel treatment. Moreover, the responsive patients eventually develop disease progression within 1 year of treatment.6,7 Salvage treatment options for docetaxel-resistant mCRPC include a chemotherapeutic agent (cabazitaxel) and next-generation androgen receptor (AR) targeting agents (abiraterone, enzalutamide), although none of them is curative. Given the high AR heterogeneity in late-stage prostate cancer, AR-targeting drugs may not be effective in tumors with low AR expression.8 Indeed, AR amplification is found in less than 30% of mCRPC.9 Therefore, alternative intervention strategies are clearly needed aimed to benefit the subset of mCRPC patients with weak or absent AR.
The mammalian Target of rapamycin (mTOR) plays an important role in regulating cell growth, proliferation, cell cycle and other aspects.10 mTOR signaling overactivation is closely related to the occurrence, development, and drug resistance of prostate cancer, which is an important target for tumor treatment.11 The upstream mTOR signaling pathway PTEN/PI3K/AKT plays a pivotal regulatory role in promoting the occurrence and development of prostate cancer cells.12 Among them, PTEN loss or mutation is the primary cause of mTOR signaling pathway overactivation in prostate cancer.13 The incidence of prostate cancer and tumor metastasis is 40% and 70%, respectively. Furthermore, over-activated mTOR has been found to be associated with the progression and poor prognosis of prostate cancer.14,15 Notably, 80% of mCRPC cases demonstrated PTEN loss,16 suggesting a high incidence in highly malignant and metastatic cancers. In the preclinical studies, mice with PTEN gene knockout eventually developed to CRPC.8 Moreover, consistent evidence revealed a strong activation of the Akt/mTOR signaling, downstream of PTEN, in advanced prostate cancer, especially in CRPC.15 Therefore, the mTOR signaling pathway plays an important role in prostate cancer, highlighting its potential as a therapeutic intervention target to explore the mechanism of CRPC resistance and improve drug chemotherapy sensitivity.
There are two different complexes of mTOR, namely mTORC1 and mTORC2. Raptor and Rictor are two key proteins that make up mTORC1 and mTORC2, respectively. The activation of mTORC1 lead to hyperphosphorylation of its downstream ribosome p70S6 kinase (pS6K1) and 4E-binding protein 1 (4E-BP1), which regulate protein transcription and cell proliferation.17 On the other hand, the mTORC2 complex, phosphorylates Akt directly, thereby regulating cytoskeletal rearrangement, cell survival and migration.
Docetaxel is a microtubule stabilizer that can inhibit microtubule disassembly, leading to G2/M cell cycle arrest and several forms of cell death, such as apoptosis and mitotic catastrophe.18 However, various mechanisms have been proposed in an attempt to explain drug resistance and the related unsatisfactory clinical outcomes in patients. For example, the activation of compensatory pro-survival signaling pathways independent of the AR is one potential mechanism.19 Several preclinical studies have shown that docetaxel induces an increase in Akt phosphorylation (p-Akt) at S473, a direct downstream target of mTORC2, in prostate cancer cells.20 Our group previously demonstrated strong mTORC2 activity and downstream signaling pathways in the docetaxel-resistant cell line, PC‑3/DTX cells.21 The PC-3/DTX cells overexpressed Rictor proteins, which are subunits specific to mTORC2.
This finding suggested that the mTORC2 signaling pathway may serve an important role in the regulation of acquired docetaxel resistance in CRPC cells. The current study further examines the impact of mTORC2 signaling on acquired docetaxel resistance in PC-3 cells and protein expression on tissue specimens from patients with metastatic castration resistance. More importantly, the application of dual mTORC1/2 kinase inhibitors to reverse acquired chemoresistance should be investigated to explore potential intervention strategies for mCRPC patients with docetaxel failure.
Materials and Methods
Cell Line Culture
The PC-3 human prostate cancer cell line was purchased from the American Type Culture Collection, Manassas, VA, USA (CRL-1435) and cultured in F-12 medium (Gibco, UK) with 10% fetal bovine serum (Gibco, Australia), streptomycin (100 U/mL; Gibco, Eggenstein, Germany) and penicillin (100 U/mL; Gibco, Eggenstein, Germany). The docetaxel-resistant PC-3 cells (PC-3/DTX) were induced and constructed the same way as in our previously published article.21 PC-3/DTX cells were also cultured under the same conditions. The cells were incubated in 5% CO2 at 37°C.
Chemicals and Antibodies
Docetaxel was purchased from Sanofi. Rapamycin (S1039) and AZD8055 (S1555) were purchased from Selleck Chemicals. The primary antibodies anti-Rictor (cat. no. 2114), anti-Raptor (cat. no. 2280T) and anti-GAPDH (cat. no. 5174T) for Western blot were purchased from Cell Signaling Technology (Danvers, MA). The antibodies, anti-Rictor (cat. no. 70374) and anti-Raptor (cat. no. 40768) for IHC were obtained from Abcam (Cambridge, USA). The Cell Counting Kit-8 (cat. no. C0037) and cell cycle and apoptosis analysis kit (cat. no. C1052) were obtained from the Beyotime Institute of Biotechnology (Suzhou, Jiangsu, China).
Cell Viability Measurement
Cell proliferation was measured by Cell Counting Kit-8 (CCK-8, Beyotime Institute of Biotechnology Suzhou, Jiangsu, China) to compare the cell viability. Cells were seeded in triplicate at a density of 2.0×103 cells/well on 96-well plates and were incubated in a 95% humidified atmosphere with 5% CO2 at 37°C for 24 h. Subsequently, 10 µL CCK-8 solution was added to each well and further incubated for 1 h at 37°C. A microplate spectrophotometer (ELX808, Bioteck, Germany) was used to measure the absorption at 450 nm. The measurement was repeated every day until day 5 to obtain the growth curves. Resistance indices (RIs) were calculated with the ratio of the 50% inhibitory concentration (IC50) values of cells in each group.
Western Blot
The protein expression of Rictor and Raptor were measured by Western blot. Protein concentration was assayed using the Bio-Rad protein kit (Beyotime Institute of Biotechnology, Haimen, Jiangsu, China). Briefly, 10 µg of proteins extracted from PC-3/DTX cells after drug treatment were loaded per well on a sodium dodecyl sulfate-polyacrylamide gel with 10% resolution and 4% stacking gel. The gel was treated in running buffer with 60V for 40 min and 120V for 90 min. Subsequently, proteins were transferred onto 0.45 µm pore-sized positively-charged nylon polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA) and blocked with 5% dry milk in PBS with 0.1% Tween-20 at room temperature for 1 h. The membranes were rinsed with PBST (0.1% Tween-20) 3 times and then incubated with anti-Rictor (dilution 1:1000), anti-Raptor (dilution 1:1000) and anti-GAPDH (dilution 1:1000) antibodies overnight at 4°C. The membranes were then incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG(H+L) secondary antibodies (Beyotime Institute of Biotechnology, Suzhou, Jiangsu, China; 1:1000; cat. no. A0208) in TBS-T for 1 h at room temperature. Enhanced chemiluminescence was used to observe the results.
shRNA and Lentiviral Transductions
The human Rictor-specific shRNA vectors and the sh-NC were purchased from Obio Technology Corp. (Shanghai, China). Three short hairpin RNAs (shRNA) against the human Rictor gene were constructed in pLKD-CMV- EGFP-2A-Puro vectors to generate stable knockdown PC-3/DTX cells (#1: GCACTGATACTCTCTGCAT, #2: CCATCTGTCTCTCTCCAAA, #3: GCAACC AACTGAGTGCAAT). The lentivirus packaging and transduction were conducted in accordance with the manufacture’s instructions. The transfection efficiency was examined by using a fluorescence microscope (Nikon, ECLIPSE TS100, Tokyo, Japan) after 72h of transfection. Cells stably expressing sh-Rictor were enriched by puromycin (ST551, Beyotime Institute of Biotechnology, Suzhou, Jiangsu, China) selection for positive clones. The infected cells were cultured in F-12 medium with puromycin (6 µg/mL), which was reduced to 2 µg/mL after one week and maintained in a culture medium for further experiments.
Immunohistochemistry (IHC) Staining
Human tissues of primary prostate cancer (PPC) and tissues prior to castration-resistant prostate cancer (CRPC) development were collected and cut into paraffin sections. The expression of Rictor and Raptor in the samples were detected by immunohistochemistry staining using anti-Rictor (dilution 1:500, Abcam 70374, Cambridge, USA) and anti-Raptor (dilution 1:100, Abcam 40768, Cambridge, USA) antibodies, respectively. The sections were deparaffinized with xylene, rehydrated in an alcohol gradient, immersed in 3% H2O2, and then incubated with primary antibodies at 4°C overnight. The processed sections were incubated with a secondary antibody using the ABC kit (Vector Laboratories. Burlingame, CA, USA) for 1h at room temperature. The resultant signals were visualized by diaminobenzidine reaction and counterstained with hematoxylin. The number of positive cells was analyzed from 3 random high-power fields from each slide. Sections without primary antibodies and the same concentration of secondary antibodies served as a negative control.
Cell-Cycle Analysis
The cell cycle was analyzed by the cell cycle and apoptosis analysis kit (C1052, Beyotime Institute of Biotechnology, Suzhou, Jiangsu, China). Different cell groups were harvested, washed and fixed in 70% ethanol overnight. According to the manufacture’s instructions the fixed cells were washed and resuspended at a density of 2×105 cells/mL in 500 µL binding buffer. The cells were then stained with propidium iodide (PI) and RNase A and incubated at room temperature for 30 mins. Next, 500 µL PBS was added to the cells and analyzed by a flow cytometer (BD‑FACS‑Calibur; BD Biosciences, USA). The PI fluorescence was monitored at 488nm. The percentage of cells across different cell cycles was analyzed with Modifit software.
Statistical Analysis
All statistical analyses were performed using SPSS software (19). Quantitative data were described as mean ± standard deviation (SD). All in vitro experiments were conducted at least in triplicate. One-way analysis of variance (ANOVA) was performed for statistical comparison among groups, followed by Fisher’s post hoc analysis. In this study, p<0.05 was considered statistically significant.
Results
Knockdown of Rictor Inhibits Proliferation of PC-3/DTX Cells
Our previous experiment,21 revealed that the expression of Rictor may be correlated with the docetaxel drug resistance of PC-3 cells. A significantly higher expression of Rictor was observed in PC-3 docetaxel-resistant cells (PC-3/DTX cells) than in PC-3 cells. Therefore, we hypothesized that Rictor play an essential role in prostate cancer cell growth and proliferation. To validate this, 3 shRNA vectors (sh-1, sh-2 and sh-3) that suppressed the expression of Rictor in PC-3/DTX cells were constructed, and the RNAi - mediated knockdown was verified by Western blotting (Figure 1A and B). CCK-8 assays revealed that downregulation of Rictor significantly inhibited cell proliferation after 72h of incubation (Figure 1C and D). Figure 1 illustrates a decrease in cell proliferation with the downregulated expression of Rictor by sh-RNA transfection. In contrast, PC-3/DTX cells transfection with sh-NC revealed no significant difference with PC-3/DTX cells.
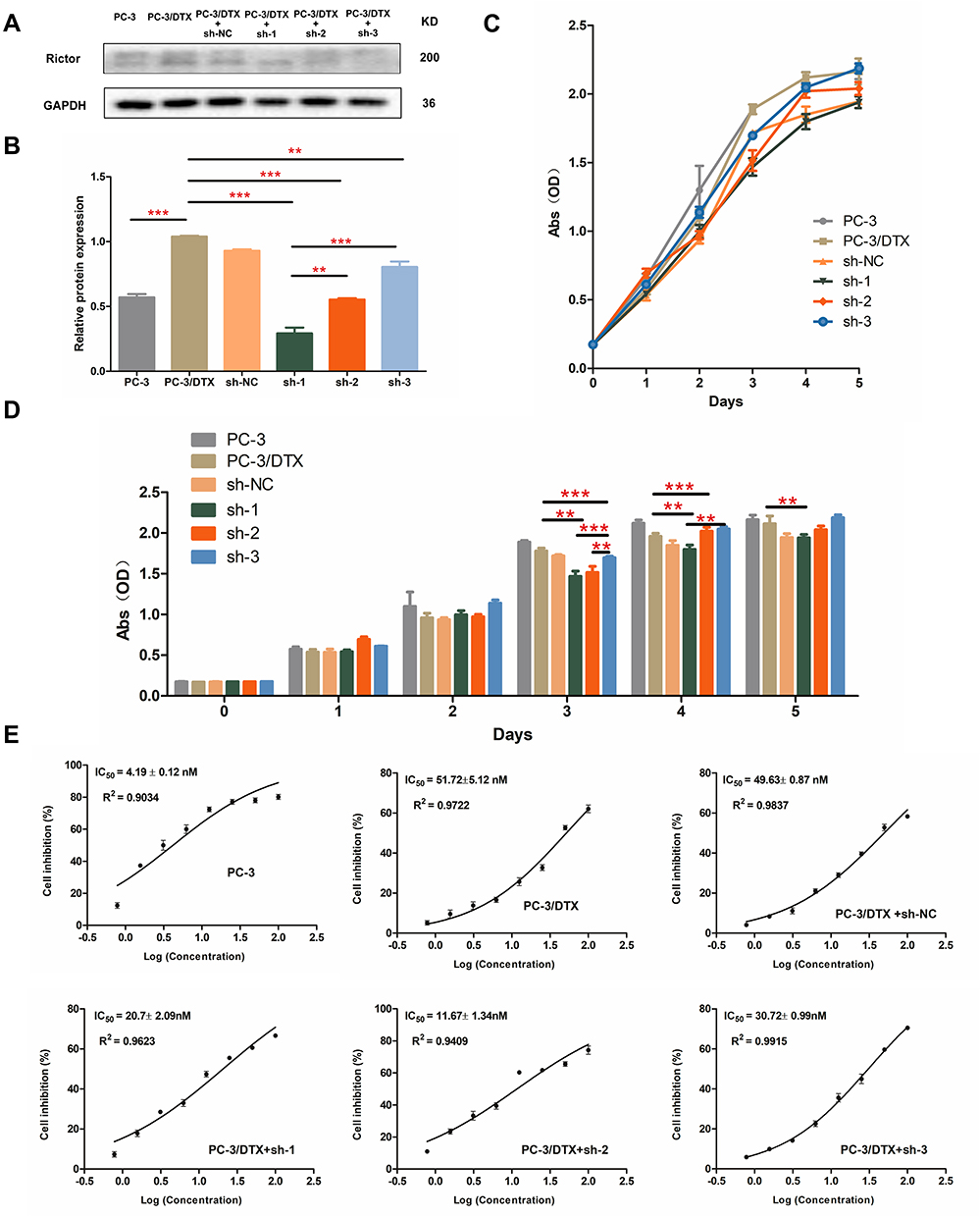 |
Figure 1 shRNA mediated Rictor knockdown inhibits PC-3/DTX cell proliferation. (A) Protein expression of Rictor in cells transfected with shRNA plasmids or mock vector, GAPDH served as a loading control. (B) Quantification of the protein expression levels of Rictor. (C) The growth curves of six kinds of cells. Cell proliferation was decreased in sh-Rictor cells assessed by CCK-8 assay. Data are expressed as the mean ± standard deviation. n=3. (D) Quantification of cell proliferation between different groups. (E) IC50 was detected by CCK-8. Docetaxel-resistant PC-3 cells transfected with shRNA plasmids or mock vector, and then treated with increasing concentrations of Docetaxel. Data are presented as the mean ± SD of independent experiments performed in triplicate. **P<0.01, ***P<0.001 vs control. Abbreviations: CCK-8, cell counting kit-8; PC-3/DTX, docetaxel-resistance PC-3 cells; IC50, 50% inhibitory concentration. |
The cell growth curves are presented in Figure 1C and D, with no significant differences among the 6 groups within 2 days. After 2 days, slowest growth was noted in PC-3/DTX cells transfected with Rictor knockdown sh-RNA compared to PC-3 cells or PC-3/DTX cells. Cells proliferation was decreased more significantly in cells treated with sh-1 or sh-2 than in cells treated with sh-3. These findings suggest that Rictor knockdown could inhibit the proliferation of PC-3/DTX cells.
The CCK-8 assay was used to detect the docetaxel sensitivity of PC-3 cells, PC-3/DTX cells, PC-3/DTX cells with sh-NC, PC-3/DTX cells with sh-1, PC-3/DTX cells with sh-2 and PC-3/DTX cells with sh-3 (Figure 1E). The 50% inhibitory concentration of docetaxel for each group was 4.19±0.12 nM, 51.72±5.12 nM, 49.63±0.87 nM, 20.7±2.09 nM, 11.67±1.34 nM and 30.72±0.99 nM, respectively. Notably, the IC50 of PC-3/DTX cells with sh-RNA were all lower than the sh-NC blank group or PC-3/DTX cells without sh-RNA transfected. In addition, cells treated with sh-1 or sh-2 turned to reversed the docetaxel drug resistance to a greater extent. These results indicate that Rictor might be associated with cellular docetaxel resistance. Inhibition of Rictor may increase the sensitivity of the cells to docetaxel.
Knockdown of Rictor Mediates Cell Cycle Arrest in PC-3/DTX Cells
To investigate the mechanism by which Rictor knockdown inhibits cell proliferation, flow cytometry was used to assess the effect of Rictor on cell cycle progression. The distribution of PC-3, PC-3/DTX cells, PC-3/DTX cells with sh-NC, PC-3/DTX cells with sh-1, PC-3/DTX cells with sh-2 and PC-3/DTX cells in the G0/G1 phase were 50.29±1.72%, 58.08±1.41%, 58.93±0.41%, 65.13±0.32%, 67.65±0.88%, 58.82±0.66%, and cells in S phase were 32.91±4.98%, 19.62±2.4%, 21.38±0.74%, 16.37±0.91%, 14.59±1.43%, 18.34±1.25%, respectively. As shown in Figure 2, the cells treated with sh-1 and sh-2 demonstrated a significant decrease in the percentage of cells in the S phase, but a significant increase in the percentage of cells in the G0/G1 phase compared with the control group. Cells in the G2/M phase were 16.8±3.27%, 22.3±1.09%, 19.68±0.68%, 18.51±0.61%, 17.76±0.55%, 22.85±0.91%, respectively. Moreover, greater inhibition of Rictor resulted in a greater proportion of cells arrested in the G0/G1 phase compared with the sh-1/sh-2 group and sh-3 group. Groups with sh-RNA Rictor knockdown also revealed fewer cells in the G2/M phase.
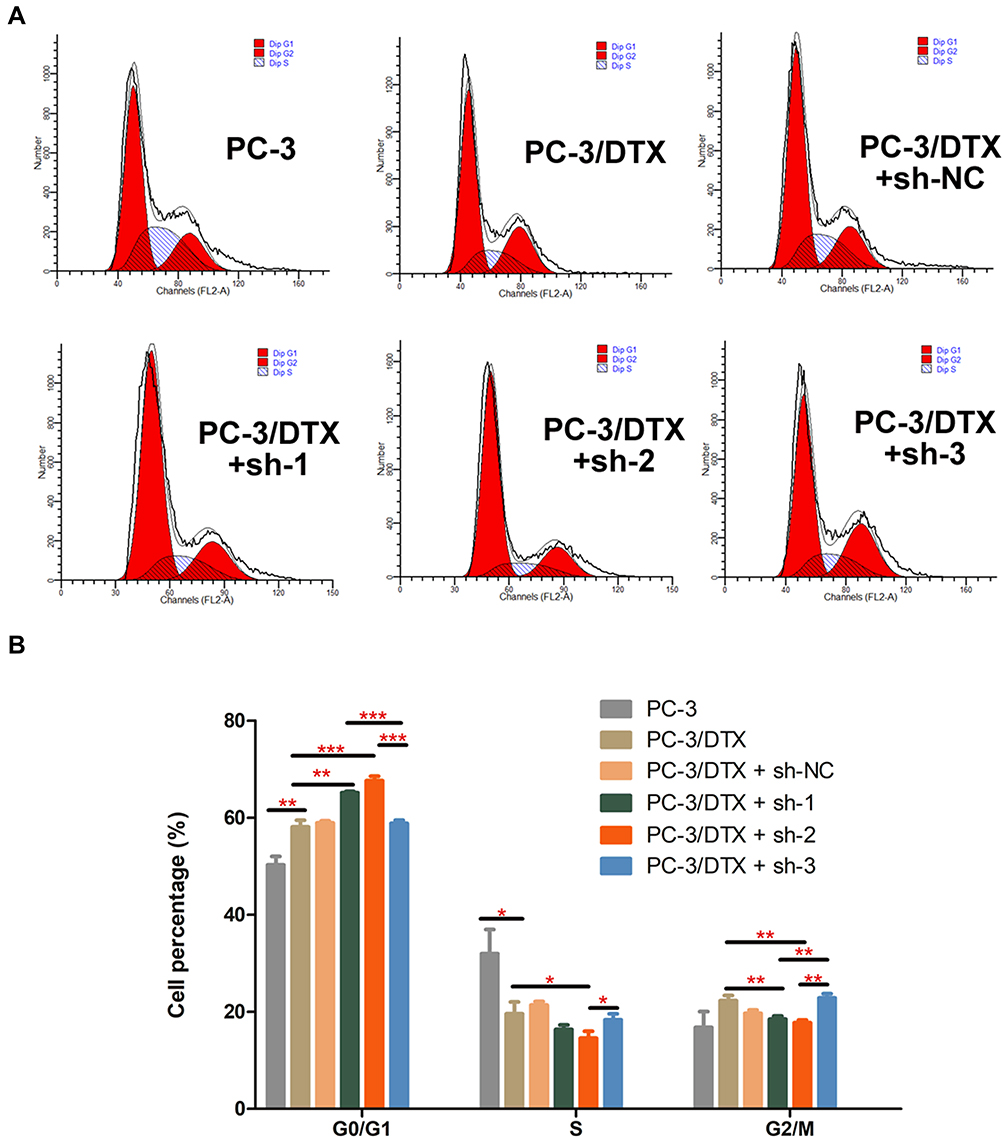 |
Figure 2 Rictor knockdown promotes cell cycle arrest. (A) Cell cycle progression analysis by flow cytometry showed G0/G1 phase arrest in sh-Rictor cells. (B) Quantification of the cell cycle, Data are presented as the mean ± SD of independent experiments performed in triplicate. *P<0.05, **P<0.01, ***P<0.001 vs control. |
Docetaxel-Resistance Reversal Effect of Rapamycin and AZD8055 on Cell Proliferation and Cell Cycle
Since mTOR signaling functions as a central regulator of cell proliferation and survival, the effect of mTORC1 inhibitor (Rapamycin) and mTORC1, mTORC2 inhibitor (AZD8055) on docetaxel-resistance PC-3 cells were detected. First, the IC50 of PC-3/DTX cells treated with Rapamycin and AZD8055 were evaluated with the CCK-8 assay. The IC50 for Rapamycin was 1.496±0.030 μM, while the IC50 for AZD8055 was 0.2265±0.017μM (Figure 3A). The PC-3/DTX cells were treated with Rapamycin and AZD8055 at the IC50 concentration for 48h, and then turned into a normal culture. As a result, a significant decrease in Raptor protein expression was observed in the cells cultured with Rapamycin and AZD8055. Compared with the Rapamycin group and the control group, cells cultured with AZD8055 resulted in a significantly lower expression of Rictor (Figure 3B). Furthermore, the sensitivity of these cells to different concentrations of docetaxel treatment was also tested after cultured with Rapamycin or AZD8055 for 48h. Co-culture with different concentrations of docetaxel for 24h revealed a stronger inhibition of cell proliferation in the AZD8055 group compared to the Rapamycin group (Figure S1).
 |
Figure 3 Docetaxel-resistance reversal effect of Rapamycin and AZD8055 on cell proliferation. (A) IC50 was detected by cell counting kit-8 (CCK-8). Docetaxel-resistant PC-3 cells treated with increasing concentrations of Rapamycin or AZD8055 respectively. (B) Protein expression of Rictor and Raptor of PC-3/DTX cells treated with Rapamycin or AZD8055 at the concentration of IC50 for 48h respectively. (C) After the 48h treatment of Rapamycin or AZD8055 at the concentration of IC50, cells then treated with increasing concentrations of Docetaxel, and CCK-8 assay was used to test the change of IC50 to docetaxel between groups. Data are presented as the mean ± standard deviation of independent experiments performed in triplicate. **P<0.01, ***P<0.001 vs control. |
The 50% inhibitory concentration of docetaxel in the Rapamycin group and AZD 8055 group was 28.15±0.16 nM and 18.25±0.13 nM, respectively. The cells cultured with Rapamycin or AZD8055 showed higher drug sensitivity than control cells, with the AZD8055 group including a higher docetaxel sensitivity (Figure 3C).
The change in cell cycle was also detected by flow cytometry (Figure 4). The distribution of PC-3/DTX cells cultured with Rapamycin and AZD8055 in the G0/G1 phase were 65.72±0.84%, 67.92±0.70%, and cells in the S phase were 16.56±1.17%, 9.93±2.32%, respectively. Compared with PC-3 cells and PC-3/DTX cells, after 48h cultured with Rapamycin and AZD8055 induced a significant increase in the number of cells in the G0/G1 phase, indicating cells were blocked in the G1 phase. Consequently, the number of cells in the S phase also decreased.
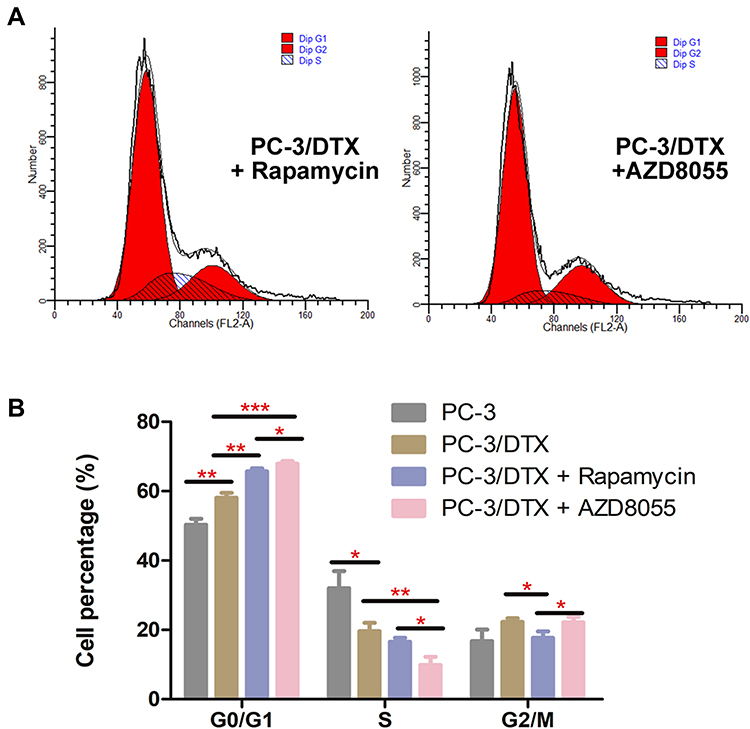 |
Figure 4 Docetaxel-resistance reversal effect of Rapamycin and AZD8055 on cell cycle. (A) Cell cycle progression analysis by flow cytometry showed G0/G1 phase arrest in Rapamycin and AZD8055 group cells. (B) Quantification of the cell cycle, Data are presented as the mean ± standard deviation of independent experiments performed in triplicate. *P<0.05, **P<0.01, ***P<0.001 vs control. |
Increased of Rictor Was Associated with Docetaxel-Resistant Prostate Cancer Tissue
PPC specimens and pre-CRPC tissues were analyzed to investigate whether docetaxel drug resistance in human prostate cancer tissue was regulated by mTORC2. IHC staining revealed tumor regions with high levels of expression of Rictor and Raptor. Higher expression levels of Rictor were found in the CRPC tissues than in PPC tissues. In contrast, the Raptor expression showed no significant difference (Figure 5). It can be inferred that Rictor is associated with docetaxel drug resistance. The current proposal seeks to examine the impact of mTORC2 signaling on the acquired docetaxel resistant phenotype in preclinical CRPC models.
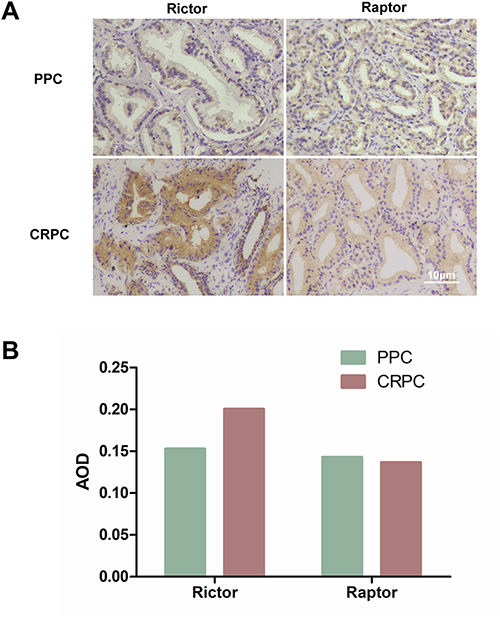 |
Figure 5 Increased of Rictor was associated with docetaxel-resistant prostate cancer tissue. (A) The levels of Rictor and Raptor in PPC and CRPC tissues were detected using immunohistochemistry (×200). (B) Expression of Rictor and Raptor in PPC and CRPC tissue (AOD value). Abbreviations: PPC, primary prostate cancer; CRPC, castration-resistant prostate cancer; AOD, average optical density. |
Discussion
mTOR is a central controller of cell growth and metabolism and is a key regulator of the growth and proliferation of tumor cells.22 The complex mTOR is formed by mTORC1 and mTORC2, which are completely different in structure and function. MTORC2 include backbone protein Rictor and mammalian stress-activated map kinase interacting protein 1 (mSIN1), and is involved in cell metabolism.23 Interestingly, only mTORC2 is essential for the development of prostate cancer in the absence of PTEN, but is not necessary for normal prostate epithelial cell function.12 P-Akt, a direct downstream target of mTORC2, exhibited greater expression in prostate cancer cells that have developed docetaxel resistance following long-term drug exposure.20 These phenomena indicate that mTORC2 can specifically promote the occurrence and development of prostate cancer, but does not induce hyperplasia in normal prostate tissue. Furthermore, the mTORC2 pathway may be a causal factor for docetaxel resistance in CRPC cells a putative mechanism that has not been investigated previously.
In our previous study, resistant models were developed by chronically treating CRPC mouse cells with docetaxel that were previously sensitive to the drug, which induced the activation of mTORC2 downstream signaling pathways. The current study investigates the impact of mTORC2 on the acquired docetaxel resistance, and evaluates novel dual mTORC1/2 inhibitors to overcome such chemoresistance.
Firstly, mTOR-associates proteins were genetically modified to explore whether mTOR complexes are required for docetaxel-mediated resistance. PC-3/DTX cells were transfected with shRNA of Rictor to stably knock down mTORC2.24 PC-3/DTX cells transfected with Rictor knockdown sh-RNA grew significantly more slowly than PC-3 cells or PC-3/DTX cells. The results suggested that Rictor knockdown could inhibit the proliferation of PC-3/DTX cells. The lower IC50 of PC-3/DTX cells with sh-RNA indicated that Rictor might be associated with cellular docetaxel resistance. Inhibition of Rictor may increase docetaxel sensitivity. G0/G1 phase arrest and apoptosis with Rictor knockdown in PC-3/DTX cells is expected to restore sensitivity of PC-3/DTX cells to docetaxel by mTORC1/2 inhibitor in vitro. Moreover, Rictor inhibition turned to be positive collection with the reverse of docetaxel drug resistant.
Then we sought to evaluate the therapeutic potential of mTOR inhibitors to overcome docetaxel-mediated resistance in vitro. The PC-3/DTX cells were first cultured with Rapamycin and AZD8055 at the IC50 concentration and then treated with docetaxel, lower IC50 values were observed in the AZD8055 group than in the rapamycin group. Nevertheless, the IC50 values were both lower than the control. The data revealed that mTOR was involved in cells drug resistance. Cells from the AZD8055 group demonstrated higher docetaxel sensitivity. The role of mTORC2 in the induction of cell cycle arrest was further explored by using AZD8055 and rapamycin. Flow cytometric analysis showed that PC-3/DTX cells treated with AZD8055 and rapamycin were arrested in the G0/G1 phase of the cell cycle. The cell percentage of cell in the G0/G1 phase was significantly higher in the AZD8055 group than in the rapamycin group. These data suggest that targeting mTORC1/2 resulted in a significantly higher level of G0/G1-phase arrest compared to mTORC1 only.
Finally, the IHC results of the prostate cancer tissues from a CRPC patient revealed that only Rictor not Raptor was higher expressed. This result confirmed our previous in vitro study that mTORC2 was significantly associated with docetaxel resistance.
This study provides new evidence for developing effective treatments for high- risk/metastatic prostate cancer. Currently, second-line strategies for high-risk mCRPC patients who have developed docetaxel resistance are limited to chemotherapy and new AR-targeting agents. Given the fact that AR is highly heterogeneous in the late-stage prostate cancer, AR-targeting drugs may not be effective in tumors with low or null AR.25 Therefore, alternative interventions are clearly needed to benefit even a subset of mCRPC patients with weak AR in tumors. The current study is designed to understand whether mTORC2 signaling activation serves as an alternative mechanism for docetaxel-mediated resistance in CRPC models with PTEN loss, another common molecular feature in mCRPC, irrespective of AR status. More importantly, we seek to evaluate whether dual mTORC1/2 kinase inhibitors have the potential to reverse such acquired chemoresistance. The current investigation offers valuable evidence the development of effective intervention strategies for mCRPC patients with docetaxel failure.
Conclusion
Our study demonstrated that mTORC2 was required for docetaxel drug resistance of PC-3 cells. Dual mTOR kinase inhibitors are superior to Rapamycin in circumventing docetaxel-mediated resistance by disrupting mTORC2 kinase activity. Targeting mTORC2 could provide an effective therapeutic strategy in the treatment of mCRPC patients with docetaxel resistance.
Abbreviations
mTOR, mammalian target of rapamycin; PC-3/DTX, docetaxel-resistance PC-3 cells; mCRPC, metastatic castration-resistant prostate cancer; ADT, androgen deprivation therapy; IHC, immunohistochemistry; IC50, 50% inhibitory concentration; CCK-8, Cell Counting Kit-8; RI, resistance index; PPC, primary prostate cancer; PI, propidium iodide; SD, standard deviation; AOD, average optical density; mSIN1, mammalian stress-activated map kinase interacting protein 1; AR, androgen receptor; ADT, androgen deprivation therapy.
Ethics Approval
This study followed the principles of the Helsinki Declaration. All procedures carried out on human tissues were approved by the Experimental Human Ethics Committee of The First Affiliated Hospital, Zhejiang University School of Medicine. The informed consent was exempted with the approval of the ethics committee for the reason as follows:
- The biological specimens used in this study were discarded specimens obtained in previous clinical diagnosis and treatment;
- The risk to subjects in this study is not greater than the minimum risk;
- Waiving informed consent will not adversely affect the rights or health of the subject;
- The subjects were orally informed before the study and did not reject the use of discarded samples;
- The confidentiality of subjects’ privacy and identifying information is guaranteed.
Funding
Supported by the National Natural Science Foundation of China (Grants no:81702862); National Major Scientific and Technological Special Project for Significant New Drugs Development during the Thirteenth Five-year Plan Period 2020 ZX 09201-003.
Disclosure
The authors declared that no conflicts of interest exist for this article.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7–33. doi:10.3322/caac.21708
3. Culp MB, Soerjomataram I, Efstathiou JA, Bray F, Jemal A. Recent global patterns in prostate cancer incidence and mortality rates. Eur Urol. 2020;77(1):38–52. doi:10.1016/j.eururo.2019.08.005
4. Xia C, Dong X, Li H, et al. Cancer statistics in China and United States, 2022: profiles, trends, and determinants. Chin Med J. 2022;135(5):584–590. doi:10.1097/CM9.0000000000002108
5. Henríquez I, Roach M, Morgan TM, et al. Current and emerging therapies for Metastatic Castration-Resistant Prostate Cancer (mCRPC). Biomedicines. 2021;9(9):1247. doi:10.3390/biomedicines9091247
6. Seruga B, Ocana A, Tannock IF. Drug resistance in metastatic castration-resistant prostate cancer. Nat Rev Clin Oncol. 2011;8(1):12–23. doi:10.1038/nrclinonc.2010.136
7. Xinxing L, Yang F, Chen D, et al. Quercetin reverses docetaxel resistance in prostate cancer via androgen receptor and PI3K/Akt signaling pathways. Int J Biol Sci. 2020;16(7):1121–1134. doi:10.7150/ijbs.41686
8. Mulholland DJ, Tran LM, Li Y, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19(6):792–804. doi:10.1016/j.ccr.2011.05.006
9. Massard C, Fizazi K. Targeting continued androgen receptor signaling in prostate cancer. Clin Cancer Res. 2011;17(12):3876–3883. doi:10.1158/1078-0432
10. Tian T, Li X, Zhang J. Mtor signaling in cancer and mtor inhibitors in solid tumor targeting therapy. Int J Mol Sci. 2019;20(3):755. doi:10.3390/ijms20030755
11. Xu Z, Han X, Ou D, et al. Targeting Pi3k/Akt/Mtor-mediated autophagy for tumor therapy. Appl Microbiol Biotechnol. 2020;104(2):575–587. doi:10.1007/s00253-019-10257-8
12. Guertin DA, Stevens DM, Saitoh M, et al. Mtor complex 2 is required for the development of prostate cancer induced by PTEN loss in mice. Cancer Cell. 2009;15(2):148–159. doi:10.1016/j.ccr.2008.12.017
13. Sun J, Li S, Wang F, Fan C, Wang J. Identification of key pathways and genes in PTEN mutation prostate cancer by bioinformatics analysis. BMC Med Genet. 2019;20(1):191. doi:10.1186/s12881-019-0923-7
14. Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22. doi:10.1016/j.ccr.2010.05.026
15. Morgan TM, Koreckij TD, Corey E. Targeted therapy for advanced prostate cancer: inhibition of the Pi3k/Akt/Mtor pathway. Curr Cancer Drug Targets. 2009;9(2):237–249. doi:10.2174/156800909787580999
16. Lunardi A, Ala U, Epping MT, et al. A co-clinical approach identifies mechanisms and potential therapies for androgen deprivation resistance in prostate cancer. Nat Genet. 2020;52(10):1132. doi:10.1038/s41588-020-0701-7
17. Jhanwar-Uniyal M, Wainwright JV, Mohan AL, et al. Diverse signaling mechanisms of mtor complexes: Mtorc1 and Mtorc2 in forming a formidable relationship. Adv Biol Regul. 2019;72:51–62. doi:10.1016/j.jbior.2019.03.003
18. Singh SK, Banerjee S, Acosta EP, Lillard JW, Singh R. Resveratrol induces cell cycle arrest and apoptosis with docetaxel in prostate cancer cells via a P53/ P21waf1/Cip1 and P27kip1 pathway. Oncotarget. 2017;8(10):17216–17228. doi:10.18632/oncotarget.15303
19. Galletti G, Leach BI, Lam L, Tagawa ST. Mechanisms of resistance to systemic therapy in metastatic castration-resistant prostate cancer. Cancer Treat Rev. 2017;57:16–27. doi:10.1016/j.ctrv.2017.04.008
20. Kosaka T, Miyajima A, Shirotake S, Suzuki E, Kikuchi E, Oya M. Long-term androgen ablation and docetaxel up-regulate phosphorylated akt in castration resistant prostate cancer. J Urol. 2011;185(6):2376–2381. doi:10.1016/j.juro.2011.02.016
21. Liu J, Huang Y, Zhu D, et al. Establishment and characterization of a docetaxel-resistant human prostate cancer cell line. Oncol Lett. 2020;20(5):230. doi:10.3892/ol.2020.12093
22. Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mtor signaling network for cancer therapy. J Clin Oncol. 2009;27(13):2278–2287. doi:10.1200/JCO.2008.20.0766
23. Wenxiang F, Hall MN. Regulation of mTORC2 signaling. Genes. 2020;11(9):1045. doi:10.3390/genes11091045
24. Bian YH, Xu J, Zhao WY, et al. Targeting Mtorc2 component Rictor inhibits cell proliferation and promotes apoptosis in gastric cancer. Am J Transl Res. 2017;9(9):4317–4330.
25. Li Q, Deng Q, Chao HP, et al. Linking prostate cancer cell ar heterogeneity to distinct castration and enzalutamide responses. Nat Commun. 2018;9(1):3600. doi:10.1038/s41467-018-06067-7
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.