")
Back to Journals » Cancer Management and Research » Volume 10
Targeting energy metabolism to eliminate cancer cells
Authors Shahruzaman SH, Fakurazi S, Maniam S
Received 6 March 2018
Accepted for publication 13 April 2018
Published 31 July 2018 Volume 2018:10 Pages 2325—2335
DOI https://doi.org/10.2147/CMAR.S167424
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Antonella D'Anneo
Shazwin Hani Shahruzaman, Sharida Fakurazi, Sandra Maniam
Department of Human Anatomy, Faculty of Medicine and Health Sciences, Universiti Putra Malaysia, Serdang, Selangor Darul Ehsan, Malaysia
Abstract: Adaptive metabolic responses toward a low oxygen environment are essential to maintain rapid proliferation and are relevant for tumorigenesis. Reprogramming of core metabolism in tumors confers a selective growth advantage such as the ability to evade apoptosis and/or enhance cell proliferation and promotes tumor growth and progression. One of the mechanisms that contributes to tumor growth is the impairment of cancer cell metabolism. In this review, we outline the small-molecule inhibitors identified over the past decade in targeting cancer cell metabolism and the usage of some of these molecules in clinical trials.
Keywords: cancer metabolism, hypoxia, glycolysis, oxidative phosphorylation
Introduction
Cancer is one of the leading causes of morbidity and mortality worldwide, with approximately 14 million new cases and 8.2 million cancer-related deaths reported in 2012.1 It is a major disease burden worldwide and will be a prominent health issue globally. The inability of cells to respond to stress and to repair damage underlies many forms of cancer.
Lung, liver, stomach, and bowel cancers are the most common causes of cancer death worldwide, accounting for nearly half of all cancer death. Lung and breast cancers were reported as the most common cancers diagnosed in men and women, respectively.2 Interestingly, in more developed countries (as defined by the United Nations which includes all regions of Europe, Northern America, Australia/New Zealand, and Japan), prostate and lung cancers are the leading cause of cancer death in men and women, respectively.2 The regional imparity in the mix of cancers is driven largely by the availability of improved treatment and technological progress in early detection of tumor.3 Cancer is also regarded differently in different settings, by which in high-income countries, it is regarded as a preventable and often curable disease. However, in many low- and middle-income countries, it is regarded as a painful death sentence. According to the World Health Organization, it is expected that the number of annual new cases to rise by 70% for the next 20 years.
Past research has identified many oncogenes and tumor suppressors that are frequently altered in human tumors. These altered genes affect key signaling pathways that govern the cell cycle and thus have the potential to be therapeutic targets. Multiple classes of chemotherapy drugs are used, mainly in combination, to target cancer cells more effectively. In recent years, it has become apparent that differences in the genetic background of cancer patients result in varying responses to chemotherapy. This has led to an increased research focus on pharmacogenomics. Cancer pharmacogenomics relates to the study of germline genetic variants that contribute to a chemotherapy-induced phenotype.4
A fundamental characteristic of cancer cells is their ability to sustain indefinite cycles of proliferation by deregulating the release of growth signals. Growth-promoting signals govern entry and progression of cells through the cell cycle, thereby ensuring the homeostasis of cell number and maintenance of normal tissue architecture and function. In 2000, Hanahan and Weinberg outlined the acquired capability of “hallmarks of cancer,” which includes the ability to sustain growth signals, avoid growth suppressors, invade and metastasize to other tissues, induce angiogenesis, resist cell death, and proliferate indefinitely.5 In 2011, these hallmarks were updated to include the ability of cancer cells to maintain the tumor microenvironment.6 Among the ways, cancer cells achieve this by metabolic reprogramming that involves altering energy metabolism in order to fuel their growth and division.6 Mitochondria have long been recognized as the powerhouses of the cell and have recently received recognition as a potential target for therapeutic intervention of cancer.
In contrast to normal undifferentiated cells, which mainly rely on oxidative phosphorylation (OXPHOS) to produce energy for cellular processes, cancer cells in the hypoxic microenvironment rely on glycolysis as their primary energy source. The phenomenon in which cancer cells preferentially use glycolysis to metabolize glucose in the presence of oxygen was identified by Otto Warburg and was later known as aerobic glycolysis. At that time, Warburg hypothesized that the switch to energy production by glycolysis in cancer cells was due to irreversible mitochondrial damage leading to permanent impairment of aerobic respiration.7 However, this hypothesis was disputed as most cancer cells retain functional mitochondria. Although aerobic glycolysis is often found in malignant tumors, OXPHOS still contributes to energy production in cancers and may play a major role in energy production in some cancers.8 Nonetheless, the “Warburg effect” is one of the best studied metabolic phenotype of cancer cells.
In this review, we focus on the research conducted over the past decade in identifying potential therapeutic targets of cell metabolism mainly in cancer. We summarize published in vivo, in vitro, and clinical studies of new evidences and novel small molecules that have implications in regulating cancer cell metabolism.
The OXPHOS system in mitochondria
Functionally, mitochondria are the key players in cellular adenosine triphosphate (ATP) production, fatty acid oxidation, heme biosynthesis, apoptosis induction, heat generation, and calcium homeostasis. Mitochondria are best known for their role in ATP production, which is performed via OXPHOS. The mitochondrial OXPHOS system is embedded in the mitochondrial inner membrane (MIM) and represents the final step in the conversion of nutrients to energy by catalyzing the generation of ATP. Carbon fuels are oxidized in the citric cycle to yield high-energy electrons.9 This electron motive force is converted into a proton motive force, and this, in turn, is converted into a phosphoryl transfer potential. The flow of electrons from nicotinamide adenine dinucleotide (NADH) or flavin adenine dinucleotide (FADH) to oxygen through protein complexes located in the MIM leads to the pumping of protons out of the matrix. The resulting uneven distribution of protons generates a pH gradient and transmembrane electrical potential that creates the proton motive force. ATP is synthesized when protons flow back to the mitochondrial matrix through ATP synthase (complex V).10 The membrane potential is essential for other mitochondrial functions such as mitochondrial protein import and is used to trigger molecular changes that alter mitochondrial behavior in response to mitochondrial dysfunction.9 Induction of cell death is influenced by changes in the amount of ATP generated by the mitochondria, and this death may occur in various ways depending on the level of ATP in the cell. In several cell types, it has been shown that a change in the amount of ATP production is sufficient in itself to initiate apoptosis.10,11 The intrinsic apoptotic pathway is initiated following the activation of caspase-9, which requires Apaf-1. In the absence of apoptotic stimuli, Apaf-1 exists in a monomeric form. In response to apoptotic stimuli, cytochrome c activates Apaf-1 by binding to its C-terminus. Concurrently, ATP bound to the ATPase domain of Apaf-1 is hydrolyzed, and this promotes Apaf-1 oligomerization.
Interestingly, Izyumov et al reported a two-third decrease of the initial intracellular ATP level in HeLa cells, which was achieved by using 2-deoxyglucose (DOG) and OXPHOS inhibitors, and was found to induce apoptosis.12 In contrast, a severe depletion of ATP (>70%) using staurosporine (protein kinase inhibitor) in human T cells resulted in a switch in response of cells to typical apoptotic stimuli from apoptosis to necrosis.12 Collectively, these data suggest that cell death is elicited under a compromised ATP level. Apoptosis inactivates PARP-1, whereas necrosis causes PARP-1 overactivation, which consumes large amount of NAD+ resulting in massive ATP depletion.10,13 Perhaps modulating in vivo ATP availability might be an important strategy in inducing cell death and preventing tumor growth and progression.
OXPHOS as a therapeutic target
Aerobic glycolysis has long been accepted as one of the pathways utilized by proliferative cancer cells to meet their bioenergetics demand, which is essential in tumorigenesis. Elevated mitochondria-derived reactive oxygen species (ROS), a by-product of electron transport chain activity, regulate both tumor cell proliferation and quiescence and promote metastasis14 and genomic instability.15 This review discusses on the current small-molecule inhibitors, typically less than 900 Da16 that are developed to target the mitochondrial OXPHOS in cancer cells (Table 1).
 | Table 1 List of OXPHOS drugs and their functions Abbreviations: OXPHOS, oxidative phosphorylation; ATP, adenosine triphosphate; 3-NP, 3-nitropropionic acid. |
Uncouplers transport protons into mitochondrial matrix via ATP-synthase-independent pathway. Hence, this reduces the proton motive force across the MIM and decreases the mitochondrial ROS production. Mitochondrial uncouplers such as FCCP not only inhibit mitochondrial membrane potential but may also affect the membrane potential and cell volume.17 Kenwood et al reported a bona fide mitochondria uncoupler, BAM15 that has preference for protonophore activity at the mitochondria but not at the plasma membrane. The exact mechanism of BAM15 remains unclear. However, BAM15 was capable in increasing mitochondrial respiration in the presence of an ATP synthase inhibitor, providing evidence of BAM15 as a mitochondrial uncoupler.18 It was suggested that the effect is due to the structural preference of BAM15 for the lipid composition of the inner mitochondrial membrane.18
A high-throughput screening of small-molecule library using luciferase-driven hypoxia-inducible factor (HIF)-1 reporter HCT116 cells under hypoxic condition led to the identification of BAY 87-2243. BAY 87-2243, a class of aminoalkyl-substituted compounds, was initially found to inhibit HIF-1α and HIF-2α protein accumulation without affecting HIF target gene expression levels.17 Further investigations revealed that BAY 87-2243 inhibited in vitro and in vivo mitochondrial complex I activity which prevented ROS-mediated HIF-1 inhibition.19,20 It was also noted that BAY 87-2243 restored prolyl hydroxylase enzyme activity, a negative regulator of HIF-1, and thus, a reduced level of HIF-1 was observed.19 Combination of B-RAF inhibitor that upregulates OXPHOS and BAY 87-2243 was shown to induce augmented tumor regression in nude mice-bearing B-RAF mutant melanoma xenografts.20 This evidence provides a promising future for combination of targeted therapies to prevent resistance to either agent.
A biochemical absorbance-based high-throughput study was used to identify the small-molecule inhibitor of complex IV in mitochondrial extracts. The current available complex IV inhibitors either bind to the allosteric site18 or inhibit the copper-dependent activity21 within complex IV, which disrupts the electron transport chain. Tissues with high dependence on aerobic respiration, such as the central nervous system and heart, are affected upon treatment with these inhibitors. ADDA, 5(1-[2-(1-adamantyl)ethoxy]-3-(3,4-dihydro-2(1H)-isoquinolinyl)-2-propanol hydrochloride]), was found to possess a significant in vitro and mouse xenograft model (nude mouse, rear flank) antiglioblastoma multiforme activity by specifically noncompetitively inhibiting complex IV activity.22 Of note, chemoresistance to temozolomide in glioblastoma multiforme is associated with an increased level of complex IV activity.23
Glycolysis
Both normal and cancer cells utilize glucose and glutamine as sources of energy and also for biosynthesis of macromolecules. The high rate of glucose metabolism in cancer cells is facilitated by an increase in glucose transport by one or more isoenzymes of the glucose transporters (GLUT 1–4).19 Glucose molecules are phosphorylated by hexokinases to form glucose-6-phosphate. Glucose-6-phosphate will either generate pyruvate or be fed into the pentose phosphate pathway (PPP) to generate ribose-5-phosphate and nicotinamide adenine dinucleotide phosphate (NADPH). Glutamine, on the other hand, is converted into glutamate in the cytosol by the enzyme glutaminase-1. Glutamate is then transported to the mitochondria, where it is converted into α-ketoglutarate. α-Ketoglutarate that enters the tricarboxylic acid (TCA) cycle results in the generation of malate and citrate. Glutamine serves as a source of carbon to replenish the TCA cycle, produces glutathione, and is a precursor to nucleotide and lipid synthesis via reductive carboxylation.24
The inappropriate proliferation of cancer cells requires high energy, and the cells generate ATP to satisfy the biomass production. The enhanced glycolytic rate in cancer cells is required not only to meet the need for energy but also to maintain the level of glycolytic intermediates needed for biosynthesis of macromolecules. The biosynthetic activities required for proliferating cancer cells involve high rates of nucleotide synthesis, amino acid synthesis, and lipogenesis. Glycolysis provides essential molecules for both nucleotide and amino acid syntheses. The PPP consists of the oxidative generation of NADPH and nucleotide biosynthesis. The oxidation of glucose-6-phosphate generates NADPH, and the non-oxidative interconversion of phosphorylated sugars is required for nucleotide biosynthesis.
Glycolysis inhibitors
The accelerated glycolysis rate in cancer cells is associated with the overexpression of pathway enzymes, transporters, and isoenzymes with different regulatory properties.25 Interestingly, overexpression of glycolytic enzymes was noted in almost 70% of human cancers.26 The key enzymes involved in glycolysis are hexokinase, phosphofructokinase, and pyruvate kinase (PK). Inhibition of glycolysis severely impairs the ATP generation and renders cancer cells to be highly dependent on this metabolic pathway for survival. Table 2 depicts small-molecule inhibitors of glycolysis along with their mechanisms emphasized on current molecular targets.
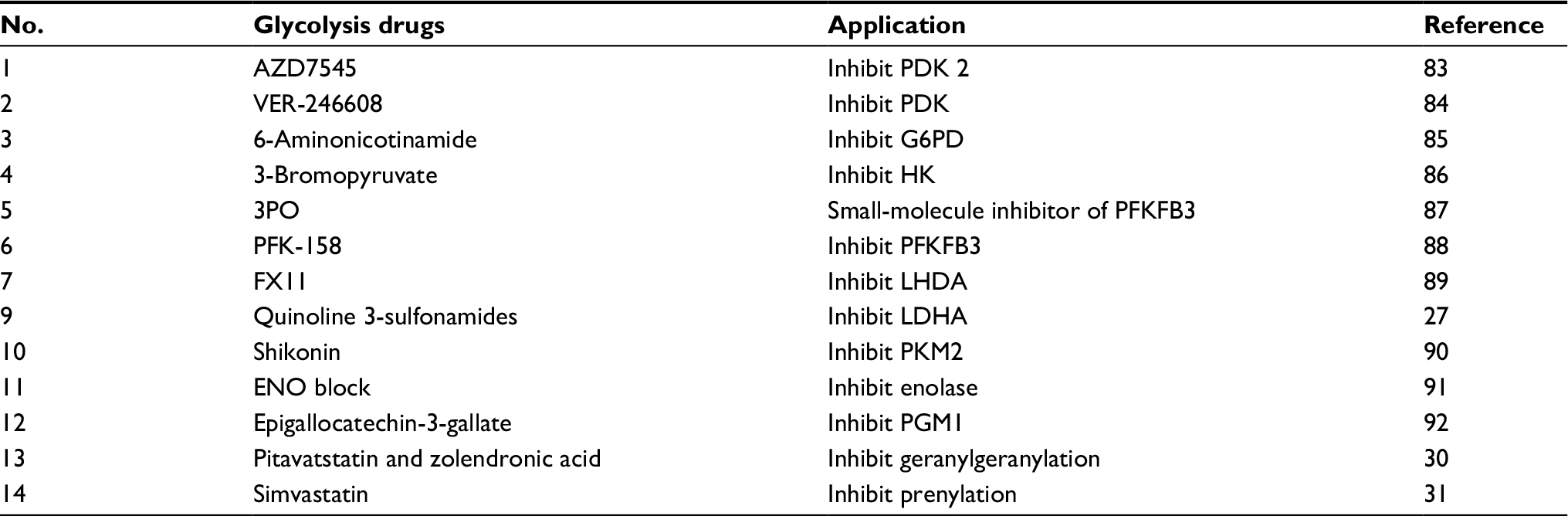 | Table 2 List of glycolysis drugs and their functions Abbreviations: 3PO, 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one; LDHA, lactate dehydrogenase A. |
The generation of pyruvate is an important metabolic control point in cellular metabolism as it can be converted to either lactate and NAD+ by lactate dehydrogenase A (LDHA), which is important for continuation of glycolysis, or acetyl coenzyme A (CoA) by pyruvate dehydrogenase (PDH) for entry to glycolysis and mitochondrial respiration. NADH-competitive selective inhibitor of LDHA, quinolone-3-sulfonamide, was developed through high-throughput screening and leads optimization.27 However, this compound was reported to possess an unacceptable pharmacokinetic profile that prevents it to be further investigated in in vivo models.27
Acetyl CoA is involved in the synthesis of several lipid building blocks, which includes mevalonate. Protein farnesylation and geranylgeranylation are collectively known as prenylation, a lipid posttranslational modification that facilitates cell membrane anchoring and is required for oncoprotein transformation.28 Statins are lipid-lowering drugs that block cholesterol biosynthesis by inhibiting 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the enzyme that catalyzes the rate-limiting step in the mevalonate pathway.27 The role of statins in modulating the mevalonate pathway to exert the anticancer activity was reported as early as 2005.29 In a recent study, mevalonate inhibitors, pitavatstatin and zolendronic acid, synergistically prevented geranylgeranylation, which resulted in inducing apoptosis in a panel of ovarian cancer cell lines.30 Simvastatin exerted an antiproliferative effect in renal cancer cells through cholesterol deprivation and prenylation-associated mechanisms.31
AZD3965 is a potent selective inhibitor of monocarboxylate transporter 1 (MCT1) found in cell lines displaying MCT1 and shows higher sensitivity in hypoxia.32 Recent studies have demonstrated that AZD3965 inhibits lactate transport and cell growth in cancer cells which lack MCT4 protein.32–34 AZD3965 is currently tested in Phase I clinical trial.
The most promising drug-targeting cancer cell metabolism to date is 3-bromopyruvate, which was first discovered in the 1960s, but its anticancer properties were discovered in 2001.15 3-Bromopyruvate, an alkylating agent, is a broad-spectrum inhibitor of multiple metabolic enzymes. It was shown to dampen ATP generation and inhibits multiple metabolic targets such as hexokinase 2 (HK 2), 3-phosphoglycerate kinase (PGK), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), lactate dehydrogenase (LDH), pyruvate dehydrogenase complex (PDC), succinate dehydrogenase (SDH), α-ketoglutarate dehydrogenase, isocitrate dehydrogenase (IDH), glyoxalases 1 and 2, and serine hydroxyethyltransferase.35–39 Currently, no clinical trials have been approved for 3-bromopyruvate even though two reports have already appeared describing the use of this inhibitor in volunteer patients with cancer.40,41
Small-molecule activators of glycolysis
PK is a rate-limiting glycolytic enzyme that catalyzes the final step of glycolysis, which produces pyruvate and ATP.42 PK muscle isozyme, PKM2, is expressed in tissues with anabolic functions and is found both in cancer and in normal tissues. In the majority of cancer cells, the expression of PKM2 is increased, which suggests that PKM2 may be an attractive target for cancer therapy.43 PKM2 is expressed in essentially all human cancers, and efforts have been made to use PKM2 as a cancer biomarker.44
ML265 is a potent PKM2 activator that significantly reduces the tumor size in 7-week mouse xenograft without showing signs of acute toxicity.45 ML265 was found to activate PKM2 in pervanadate-treated cells, a condition known to inhibit PKM2 activity through accumulation of phosphotyrosine peptides.45 ML285, another small-molecule activator of PKM2, was found to reduce the shunt of glucose through the PPP. This sequentially allows the synthesis of NADPH, which is needed for generating reduced glutathione and required for ROS detoxification. ML285 protects the enzyme from oxidation by ROS and results in sensitization to oxidative stress and increased apoptosis.46 Activation of PKM2 promotes tumor suppression in tumor growth in vivo.47–49 PA-12 stimulates the PK activity of recombinant PKM2 and effectively suppresses both anchorage-dependent and anchorage-independent growth of lung cancer cells in the nonessential amino acid-depleted medium.50
Activation of AMP-activated protein kinase (AMPK) is required to facilitate the oxidative metabolism of non-glucose substrates, specially glutamine and lactate, to maintain cell survival. BL-AD008, a small-molecule activator of AMPK/ZIPK, showed antiproliferative activities toward cervical cancer cells. It induced apoptosis by targeting AMPK/ZIPK in cervical cancer.51 AMPK direct activator MT 63-78 exerted growth inhibitory effects in prostate cancer cells in vitro as well as in xenograft models by lipogenesis inhibition.52 Table 3 shows the lists of small-molecule activators in glycolysis.
 | Table 3 List of small-molecule activators of glycolysis Abbreviation: AMPK, AMP-activated protein kinase. |
Hypoxia targeting small-molecule inhibitors
Adaptive metabolic response toward a low oxygen environment is essential to maintain rapid tumor proliferation and progression.53 The vascular network that surrounds the tumor creates an intermittent hypoxia, which plays a crucial role in cancer development, along with mitochondrial transformation associated with treatment failure.54 When oxygen demand exceeds supply, HIF is switched on.55 HIF is a heterodimer comprising α and β subunits that translocate into the nucleus.56 Mammalian cells respond to hypoxia by implementing changes in gene expression controlled by the hypoxia-inducible factor 1 (HIF-1) transcription factor.57,58 Three different genes have been identified that encode the subunits of HIF: HIF1α, HIF2α, and HIF3α. All three HIFα subunits heterodimerized with HIF-1β subunit are subjected to posttranslational regulations dictated by the oxygen concentration in the environment.59 In hypoxia, HIF-1α heterodimerized with the constitutively expressed HIF-1β [the aryl hydrocarbon receptor nuclear transporter (Arnt)] and translocated to the nucleus, where it bound to hypoxia response elements (HREs) in the promoters of various genes.55 The interaction between HIF-1α and HIF-1β is critical in the process of tumor survival. A hypoxic state is important for tumor growth to start. It is thought that HIF-1 expression (HIF-1α and HIF-1β) controls the initiation of tumor growth and could be important in affecting the antitumor growth by its changing growth to be more malignant in a hypoxic state.60 Therefore, therapeutic targeting of hypoxia in cancer has the potential to improve treatment efficacy.
Resistance to therapy in the presence of hypoxia was noted as early as the 1920s, with the first clinical implications observed in tumors in the lung that exhibited resistance to ionizing radiation. Under hypoxic conditions, DNA-damaging free radicals are rapidly reduced, thereby avoiding DNA damage. HIF-1 plays a central role in tumor pathology and is a target for treatment and therapy.61–65 To date, the small-molecule inhibitors that target HIF mainly decrease its protein levels, DNA-binding, or transactivation.66,67 Table 4 lists various small molecules that target the hypoxic microenvironment in cancer.
 | Table 4 List of HIF drugs and their functions Abbreviations: HIF, hypoxia-inducible factor; 2ME2, 2-methoxyestradiol; GLUT1, glucose transporter 1. |
Many different agents that specifically target HIF have been investigated for their role during hypoxia over the past few decades. PX-478 (S-2-amino-3-[4V-N,N,-bis(2-chloroethyl)amino]phenyl propionic acid N-oxide dihydrochloride) is known to suppress constitutive and hypoxia-induced levels of HIF-1α in various cancer cells, which include ovarian, colon, prostate, breast, renal, pancreatic, and lung.68 It was noted to show marked tumor regression and growth delay in tumor xenografts with high level of HIF-1α.69 This HIF-1α suppressor was shown to be selective in lowering HIF-1α and inhibiting its activity in multiple levels.70 PX-478 has completed its Phase I clinical trial as an oral agent in lymphoma and advanced solid tumors.
Another small-molecule inhibitor that targets HIF1a is EZN-2968. EZN-2968 is an RNA antagonist composed of locked nucleic acid-based oligonucleotide that specifically binds and inhibits the expression of HIF-1α mRNA.69,70 A pilot study of EZN-2968 in patients with refractory solid tumors showed a reduction in HIF-1α mRNA and its target genes in tumor biopsies.71 Third Phase I clinical trial has been recently initiated in patients with hepatocellular carcinoma to demonstrate the proof of mechanism of EZN-2968.
A library of aryloxy acetylaminobenzoic acid scaffold-focused screening followed by lead optimization for the identification of HIF-1α inhibitor resulted in the identification of IDF-11774, which was later shown to inhibit the expression of HIF-1α target genes. IDF-11774 inhibits angiogenesis, suppresses HIF-1α refolding, and stimulates proteosomal-mediated HIF-1α-degradation.72 Additionally, IDF-11774 was shown to decrease the glucose-dependent energy metabolism.72 This promising small-molecule inhibitor modulates both hypoxic regulators and glycolysis to prevent cancer cell growth.
Summary and future perspectives
Cancer metabolism is one of the oldest areas of research in cancer biology which pioneers from the identification of tumor suppressors and oncogenes. With explosion of research in cancer cell metabolism in the last decade, it is almost impossible to highlight all the findings in one review. In this review, we have discussed the various small molecules that were discovered in the last decade that are involved in cell metabolism, mainly glycolysis, OXPHOS, and hypoxia (Figure 1). The advancement of current technologies especially in high-throughput studies allows rapid identification of compounds with selected and specific targets. The ability of these small molecules to render cell metabolism is supported by the impaired growth of tumor in vivo and/or in vitro.
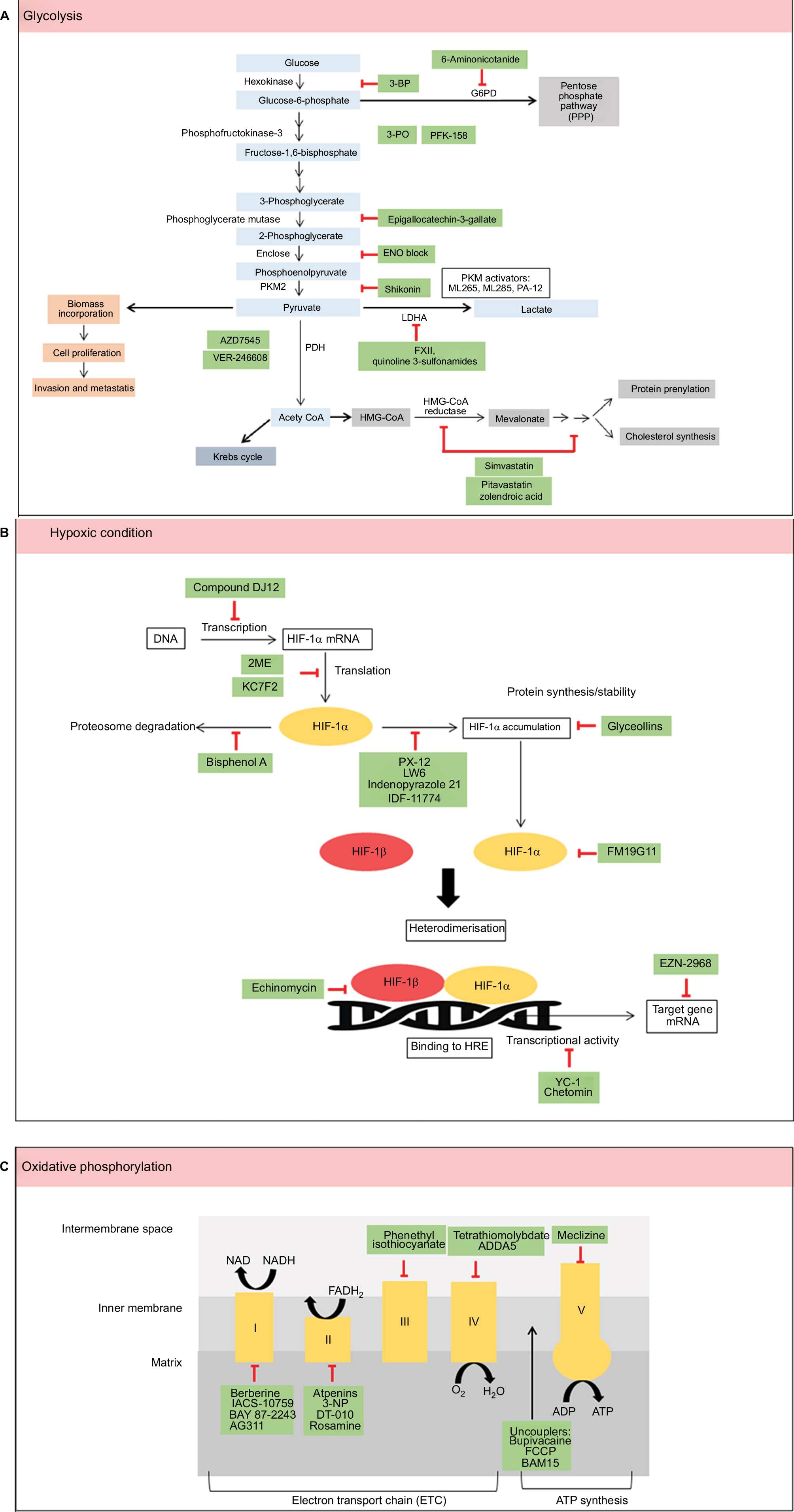 | Figure 1 Summary of metabolic pathways (A–C) and small molecules that modulate these pathways (green). Abbreviation: HIF, hypoxia-inducible factor. |
Rewiring metabolism in cancer cells plays a pivotal role in tumor survival, invasion, proliferation, and resistance to anticancer therapies. The use of cell metabolism small-molecule inhibitor in cancer treatment imposes several challenges. Several drugs that impair cell metabolism have demonstrated the modest effect in cancer therapy. This is most likely due to the complexity of the metabolic pathways in cancer. Many pathways and transcriptional factors associated with cancer metabolism are also involved in maintaining the physiological process of the body. For example, the normal tissues of brain, retinae, and testis use glucose as their main energy source. Impairment of glycolysis that limits the glucose supply may potentially be toxic to these tissues. Metabolic plasticity and heterogeneity in cancer cells drive tumor growth, mediate metastasis, and contribute to treatment resistance. Hence, the efficacy of cancer metabolism therapy requires careful evaluation. Inhibiting individual enzymes or blocking single pathways seldom leads to effective cancer treatment. Therefore, the combined approach of targeting cellular metabolism in conjunction with the use of chemotherapeutic drugs may provide a promising strategy to overcome therapeutic resistance and therefore aid in cancer therapy. In conclusion, the concept of cancer metabolism provides more targets for cancer treatment. Undoubtedly, like other targeted therapies being developed, therapies directed to cancer metabolism will be most effective if the orchestrations of metabolic alterations in cancer cells to support growth, proliferation, and differentiation are well understood.
Acknowledgments
We thank the Universiti Putra Malaysia (Programme Grant: Putra Grant GP-IPM/2014/9445100) for supporting this work. SHS was supported by Graduate Research Fellowship from Universiti Putra Malaysia and MyBrain program from Malaysian Ministry of Higher Education.
Disclosure
The authors report no conflicts of interest in this work.
References
Stewart BW, Wild CP. World Cancer Report 2014. Lyon, France: WHO; 2014:1–2. | ||
WHO [webpage on the Internet]. Fact Sheets by Cancer; 2012 [cited June 2017]. Available from: http://globocan.iarc.fr. Accessed June 7, 2018. | ||
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Wheeler HE, Maitland ML, Dolan ME, Cox NJ, Ratain MJ. Cancer pharmacogenomics: strategies and challenges. Nat Rev Genet. 2013;14(1):23–34. | ||
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Warburg O. On the origin of cancer cells on the origin of cancer. Source Sci New Ser. 1956;123(123):309–314. | ||
Zu XL, Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun. 2004;313(3):459–465. | ||
Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148(6):1145–1159. | ||
Nicotera P, Leist M, Ferrando-May E. Intracellular ATP, a switch in the decision between apoptosis and necrosis. Toxicol Lett. 1998;102-103(8):139–142. | ||
Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57(10):1835–1840. | ||
Izyumov DS, Avetisyan AV, Pletjushkina OY, et al. “Wages of fear”: transient threefold decrease in intracellular ATP level imposes apoptosis. Biochim Biophys Acta Bioenerg. 2004;1658(1–2):141–147. | ||
Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM. P53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell. 2012;149(7):1536–1548. | ||
Brown NS, Bicknell R. Hypoxia and oxidative stress in breast cancer. Oxidative stress: its effects on the growth, metastatic potential and response to therapy of breast cancer. Breast Cancer Res. 2001;3(5):323–327. | ||
Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014;2(1):17. | ||
Zhao L, Ren TH, Wang DD. Clinical pharmacology considerations in biologics development. Acta Pharmacol Sin. 2012;33(11):1339–1347. | ||
To MS, Aromataris EC, Castro J, Roberts ML, Barritt GJ, Rychkov GY. Mitochondrial uncoupler FCCP activates proton conductance but does not block store-operated Ca2+ current in liver cells. Arch Biochem Biophys. 2010;495(2):152–158. | ||
Kenwood BM, Weaver JL, Bajwa A, et al. Identification of a novel mitochondrial uncoupler that does not depolarize the plasma membrane. Mol Metab. 2014;3(2):114–123. | ||
Ellinghaus P, Heisler I, Unterschemmann K, et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med. 2013;2(5):611–624. | ||
Schöckel L, Glasauer A, Basit F, et al. Targeting mitochondrial complex I using BAY 87-2243 reduces melanoma tumor growth. Cancer Metab. 2015;3(1):11. | ||
Kim KK, Abelman S, Yano N, et al. Tetrathiomolybdate inhibits mitochondrial complex IV and mediates degradation of hypoxia-inducible factor-1α in cancer cells. Sci Rep. 2015;5:14296. | ||
Oliva CR, Markert T, Ross LJ, et al. Identification of small molecule inhibitors of human cytochrome c oxidase that target chemoresistant glioma cells. J Biol Chem. 2016;291(46):24188–24199. | ||
Oliva CR, Nozell SE, Diers A, et al. Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain. J Biol Chem. 2010;285(51):39759–39767. | ||
Cluntun AA, Lukey MJ, Cerione RA, Locasale JW. Glutamine metabolism in cancer: understanding the heterogeneity. Trends Cancer. 2017;3(3):169–180. | ||
Marín-Hernández A, Gallardo-Pérez JC, Rodríguez-Enríquez S, Encalada R, Moreno-Sánchez R, Saavedra E. Modeling cancer glycolysis. Biochim Biophys Acta Bioenerg. 2011;1807(6):755–767. | ||
Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84(6):1014–1020. | ||
Billiard J, Dennison JB, Briand J, et al. Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab. 2013;1(1):19. | ||
Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer. 2011;11(11):775–791. | ||
Cafforio P, Dammacco F, Gernone A, Silvestris F. Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis. 2005;26(5):883–891. | ||
Abdullah MI, Abed MN, Richardson A. Inhibition of the mevalonate pathway augments the activity of pitavastatin against ovarian cancer cells. Sci Rep. 2017;7(1):8090. | ||
Woschek M, Kneip N, Jurida K, Marzi I, Relja B. Simvastatin reduces cancerogenic potential of renal cancer cells via geranylgeranyl pyrophosphate and mevalonate pathway. Nutr Cancer. 2016;68(3):420–427. | ||
Polański R, Hodgkinson CL, Fusi A, et al. Activity of the monocarboxylate transporter 1 inhibitor AZD3965 in small cell lung cancer. Clin Cancer Res. 2014;20(4):926–937. | ||
Bola BM, Chadwick AL, Michopoulos F, et al. Inhibition of monocarboxylate transporter-1 (MCT1) by AZD3965 enhances radiosensitivity by reducing lactate transport. Mol Cancer Ther. 2014;13(12):2805–2816. | ||
Doherty JR, Yang C, Scott KEN, et al. Blocking lactate export by inhibiting the myc target MCT1 disables glycolysis and glutathione synthesis. Cancer Res. 2014;74(3):908–920. | ||
Azevedo-Silva J, Queirós O, Baltazar F, et al. The anticancer agent 3-bromopyruvate: a simple but powerful molecule taken from the lab to the bedside. J Bioenerg Biomembr. 2016;48(4):349–362. | ||
Pedersen PL. 3-Bromopyruvate (3BP) a fast acting, promising, powerful, specific, and effective “small molecule” anti-cancer agent taken from labside to bedside: introduction to a special issue. J Bioenerg Biomembr. 2012;44(1):1–6. | ||
Shoshan MC. 3-Bromopyruvate: targets and outcomes. J Bioenerg Biomembr. 2012;44(1):7–15. | ||
Jardim-Messeder D, Moreira-Pacheco F. 3-Bromopyruvic acid inhibits tricarboxylic acid cycle and glutaminolysis in HepG2 cells. Anticancer Res. 2016;36(5):2233–2241. | ||
Paiardini A, Tramonti A, Schirch D, et al. Differential 3-bromopyruvate inhibition of cytosolic and mitochondrial human serine hydroxymethyltransferase isoforms, key enzymes in cancer metabolic reprogramming. Biochim Biophys Acta. 2016;1864(11):1506–1517. | ||
El Sayed SM, Baghdadi H, Zolaly M, Almaramhy HH, Ayat M, Donki JG. The promising anticancer drug 3-bromopyruvate is metabolized through glutathione conjugation which affects chemoresistance and clinical practice: an evidence-based view. Med Hypotheses. 2017;100:67–77. | ||
Ko YH, Verhoeven HA, Lee MJ, Corbin DJ, Vogl TJ, Pedersen PL. A translational study “case report” on the small molecule “energy blocker” 3-bromopyruvate (3BP) as a potent anticancer agent: from bench side to bedside. J Bioenerg Biomembr. 2012;44(1):163–170. | ||
Israelsen WJ, Vander Heiden MG. Pyruvate kinase: function, regulation and role in cancer. Semin Cell Dev Biol. 2015;43:43–51. | ||
Dong G, Mao Q, Xia W, et al. PKM2 and cancer: the function of PKM2 beyond glycolysis (review). Oncol Lett. 2016;11(3):1980–1986. | ||
Mazurek S. Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol. 2011;43(7):969–980. | ||
Walsh MJ, Brimacombe KR, Anastasiou D, et al. [database on the Internet]. ML265: A Potent PKM2 Activator Induces Tetramerization and Reduces Tumor Formation and Size in a Mouse Xenograft Model; 2010. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23905203. Accessed June 7, 2018. | ||
Brimacombe KR, Anastasiou D, Hong BS, et al. ML285 affects reactive oxygen species’ inhibition of pyruvate kinase M2. Probe Reports from the NIH Molecular Libraries Program. Vol. 50. Bethesda, MD: National Center for Biotechnology Information (US); 2010:1–18. | ||
Anastasiou D, Yu Y, Israelsen WJ, et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol. 2012;8(10):839–847. | ||
Kung C, Hixon J, Choe S, et al. Small molecule activation of pkm2 in cancer cells induces serine auxotrophy. Chem Biol. 2012;19(9):1187–1198. | ||
Parnell KM, Foulks JM, Nix RN, et al. Pharmacologic activation of PKM2 slows lung tumor xenograft growth. Mol Cancer Ther. 2013;12(8):1453–1460. | ||
Kim DJ, Park YS, Kim ND, et al. A novel pyruvate kinase M2 activator compound that suppresses lung cancer cell viability under hypoxia. Mol Cells. 2015;38(4):373–379. | ||
Fu L, Zhang S, Zhang L, et al. Systems biology network-based discovery of a small molecule activator BL-AD008 targeting AMPK/ZIPK and inducing apoptosis in cervical cancer. Oncotarget. 2015;6(10):8071–8088. | ||
Zadra G, Photopoulos C, Tyekucheva S, et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol Med. 2014;6(4):519–538. | ||
McDonald PC, Chafe SC, Dedhar S. Overcoming hypoxia-mediated tumor progression: combinatorial approaches targeting pH regulation, angiogenesis and immune dysfunction. Front Cell Dev Biol. 2016;4:27. | ||
Tang C, Yu J. Hypoxia-inducible factor-1 as a therapeutic target in cancer. J Gastroenterol Hepatol. 2013;28(3):401–405. | ||
Cavadas MA, Nguyen LK, Cheong A. Hypoxia-inducible factor (HIF) network: insights from mathematical models. Cell Commun Signal. 2013;11(1):42. | ||
Peng G, Liu Y. Hypoxia-inducible factors in cancer stem cells and inflammation. Trends Pharmacol Sci. 2015;36(6):374–383. | ||
Shen C, Nettleton D, Jiang M, Kim SK, Powell-Coffman JA. Roles of the HIF-1 hypoxia-inducible factor during hypoxia response in Caenorhabditis elegans. J Biol Chem. 2005;280(21):20580–20588. | ||
Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40(2):294–309. | ||
Dengler VL, Galbraith MD, Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol. 2015;49(1):1–15. | ||
Choi SH, Chung AR, Kang W, et al. Silencing of hypoxia-inducible factor-1β induces anti-tumor effects in hepatoma cell lines under tumor hypoxia. PLoS One. 2014;9(7):e103304. | ||
Tsai Y-P, Wu K-J. Hypoxia-regulated target genes implicated in tumor metastasis. J Biomed Sci. 2012;19(1):102. | ||
Ward C, Langdon SP, Mullen P, et al. New strategies for targeting the hypoxic tumour microenvironment in breast cancer. Cancer Treat Rev. 2013;39(2):171–179. | ||
Hu F, Shi L, Mu R, et al. Hypoxia-inducible factor-1α and interleukin 33 form a regulatory circuit to perpetuate the inflammation in rheumatoid arthritis. PLoS One. 2013;8(8):8–14. | ||
Li G, Lu W, Wu X, et al. Admission hypoxia-inducible factor 1α levels and in-hospital mortality in patients with acute decompensated heart failure. BMC Cardiovasc Disord. 2015;15:79. | ||
Shen H, Maki CG. Pharmacologic activation of p53 by small-molecule MDM2 antagonists. Curr Pharm Des. 2011;17(6):560–568. | ||
Devine T, Dai M-S. Targeting the ubiquitin-mediated proteasome degradation of p53 for cancer therapy. Curr Pharm Des. 2013;19(18):3248–3262. | ||
Welsh S, Williams R, Kirkpatrick L, Paine-Murrieta G, Powis G. Antitumor activity and pharmacodynamic properties of PX-478, an inhibitor of hypoxia-inducible factor-1alpha. Mol Cancer Ther. 2004;3(3):233–244. | ||
Koh MY, Spivak-Kroizman T, Venturini S, et al. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1. Mol Cancer Ther. 2008;7(1):90–100. | ||
Greenberger LM, Horak ID, Filpula D, et al. A RNA antagonist of hypoxia-inducible factor-1alpha, EZN-2968, inhibits tumor cell growth. Mol Cancer Ther. 2008;7(11):3598–3608. | ||
Borsi E, Perrone G, Terragna C, et al. Hypoxia inducible factor-1 alpha as a therapeutic target in multiple myeloma. Oncotarget. 2014;5(7):1779–1792. | ||
Jeong W, Rapisarda A, Park SR, et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1α), in patients with refractory solid tumors. Cancer Chemother Pharmacol. 2014;73(2):343–348. | ||
Ban HS, Kim BK, Lee H, et al. The novel hypoxia-inducible factor-1α inhibitor IDF-11774 regulates cancer metabolism, thereby suppressing tumor growth. Cell Death Dis. 2017;8(6):e2843. | ||
Turner N, Li JY, Gosby A, et al. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate amp-activated protein kinase and improve insulin action. Diabetes. 2008;57(5):1414–1418. | ||
Protopopova M, Madhavi B, Jennifer B, et al. IACS-10759: a novel OXPHOS inhibitor which selectively kill tumors with metabolic vulnerabilities. Cancer Res. 2015;75:4380. | ||
Bastian A, Matsuzaki S, Humphries KM, et al. AG311, a small molecule inhibitor of complex I and hypoxia-induced HIF-1α stabilization. Cancer Lett. 2017;388:149–157. | ||
Miyadera H, Shiomi K, Ui H, et al. Atpenins, potent and specific inhibitors of mitochondrial complex II (succinate-ubiquinone oxidoreductase). Proc Natl Acad Sci U S A. 2003;100(2):473–477. | ||
Huang LS, Sun G, Cobessi D, et al. 3-Nitropropionic acid is a suicide inhibitor of mitochondrial respiration that, upon oxidation by complex II, forms a covalent adduct with a catalytic base arginine in the active site of the enzyme. J Biol Chem. 2006;281(9):5965–5972. | ||
Wang L, Zhang X, Cui G, et al. A novel agent exerts antitumor activity in breast cancer cells by targeting mitochondrial complex II. Oncotarget. 2016;7(22):1–11. | ||
Lim SH, Wu L, Kiew LV, Chung LY, Burgess K, Lee HB. Rosamines targeting the cancer oxidative phosphorylation pathway. PLoS One. 2014;9(3):e82934. | ||
Xiao D, Powolny AA, Moura MB, et al. Phenethyl isothiocyanate inhibits oxidative phosphorylation to trigger reactive oxygen species-mediated death of human prostate cancer cells. J Biol Chem. 2010;285(34):26558–26569. | ||
Gohil VM, Sheth SA, Nilsson R, et al. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol. 2010;28(3):249–255. | ||
Cela O, Piccoli C, Scrima R, et al. Bupivacaine uncouples the mitochondrial oxidative phosphorylation, inhibits respiratory chain complexes I and III and enhances ROS production: results of a study on cell cultures. Mitochondrion. 2010;10(5):487–496. | ||
Morrell JA, Orme J, Butlin RJ, Roche TE, Mayers RM, Kilgour E. AZD7545 is a selective inhibitor of pyruvate dehydrogenase kinase 2. Biochem Soc Trans. 2003;31(Pt 6):1168–1170. | ||
Moore JD, Staniszewska A, Shaw T, et al. VER-246608, a novel pan-isoform ATP competitive inhibitor of pyruvate dehydrogenase kinase, disrupts Warburg metabolism and induces context-dependent cytostasis in cancer cells. Oncotarget. 2014;5(24):12862–12876. | ||
Varshney R, Dwarakanath BS, Jain V. Radiosensitization by 6-aminonicotinamide and 2-deoxy-D-glucose in human cancer cells. Int J Radiat Biol. 2005;81(5):397–408. | ||
Ganapathy-Kanniappan S, Kunjithapatham R, Geschwind JF. Anticancer efficacy of the metabolic blocker 3-bromopyruvate: specific molecular targeting. Anticancer Res. 2013;33(1):13–20. | ||
Clem B, Telang S, Clem A, et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther. 2008;7(1):110–120. | ||
Telang S, O’Neal J, Tapolsky G, et al. Identification of a PFKFB3 inhibitor suitable for phase I trial testing that synergizes with the B-Raf inhibitor vemurafenib. Cancer Metab. 2014;2(Suppl 1):14. | ||
Le A, Cooper CR, Gouw AM, et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 2010;107(5):2037–2042. | ||
Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene. 2011;30(42):4297–4306. | ||
Jung DW, Kim WH, Park SH, et al. A unique small molecule inhibitor of enolase clarifies its role in fundamental biological processes. ACS Chem Biol. 2013;8(6):1271–1282. | ||
Li X, Tang S, Wang Q-Q, et al. Identification of epigallocatechin-3- gallate as an inhibitor of phosphoglycerate mutase 1. Front Pharmacol. 2017;8:1–9. | ||
Sujobert P, Poulain L, Paubelle E, et al. Co-activation of AMPK and mTORC1 induces cytotoxicity in acute myeloid leukemia. Cell Rep. 2015;11(9):1446–1457. | ||
Kung AL, Zabludoff SD, France DS, et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell. 2004;6(1):33–43. | ||
Sapra P, Kraft P, Pastorino F, et al. Potent and sustained inhibition of HIF-1α and downstream genes by a polyethyleneglycol-SN38 conjugate, EZN-2208, results in anti-angiogenic effects. Angiogenesis. 2011;14(3):245–253. | ||
Terzuoli E, Puppo M, Rapisarda A, et al. Aminoflavone, a ligand of the aryl hydrocarbon receptor (AhR), inhibits HIF-1α expression in an AhR-independent fashion. Cancer Res. 2011;70(17):6837–6848. | ||
Kubo T, Maezawa N, Osada M, Katsumura S, Funae Y, Imaoka S. Bisphenol A, an environmental endocrine-disrupting chemical, inhibits hypoxic response via degradation of hypoxia-inducible factor 1alpha (HIF-1alpha): structural requirement of bisphenol A for degradation of HIF-1alpha. Biochem Biophys Res Commun. 2004;318(4):1006–1011. | ||
Yeo E-J, Chun Y-S, Cho Y-S, et al. YC-1: a potential anticancer drug targeting hypoxia-inducible factor 1. J Natl Cancer Inst. 2003;95(7):516–525. | ||
Jones DT, Harris AL. Identification of novel small-molecule inhibitors of hypoxia-inducible factor-1 transactivation and DNA binding. Mol Cancer Ther. 2006;5(9):2193–2202. | ||
Narita T, Yin S, Gelin CF, et al. Identification of a novel small molecule HIF-1α translation inhibitor. Clin Cancer Res. 2009;15(19):6128–6136. | ||
Moreno-Manzano V, Rodríguez-Jiménez FJ, Aceña-Bonilla JL, et al. FM19G11, a new hypoxia-inducible factor (HIF) modulator, affects stem cell differentiation status. J Biol Chem. 2010;285(2):1333–1342. | ||
Welsh SJ, Williams RR, Birmingham A, Newman DJ, Kirkpatrick DL, Powis G. The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1alpha and vascular endothelial growth factor formation. Mol Cancer Ther. 2003;2(3):235–243. | ||
Lee K, Kang JE, Park SK, et al. LW6, a novel HIF-1 inhibitor, promotes proteasomal degradation of HIF-1α via upregulation of VHL in a colon cancer cell line. Biochem Pharmacol. 2010;80(7):982–989. | ||
Minegishi H, Fukashiro S, Ban HS, Nakamura H. Discovery of indenopyrazoles as a new class of hypoxia inducible factor (HIF)-1 inhibitors. ACS Med Chem Lett. 2013;4(2):297–301. | ||
Lee SH, Jee JG, Bae JS, Liu KH, Lee YM. A group of novel HIF-1α inhibitors, glyceollins, blocks HIF-1α synthesis and decreases its stability via inhibition of the PI3K/AKT/mTOR pathway and Hsp90 binding. J Cell Physiol. 2015;230(4):853–862. | ||
Mabjeesh NJ, Escuin D, LaVallee TM, et al. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003;3(4):363–375. | ||
Kong D, Park EJ, Stephen AG, et al. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005;65(19):9047–9055. | ||
Nguyen TD, Jin X, Lee JH, et al. Abietane diterpenes from Salvia miltiorrhiza inhibit the activation of hypoxia-inducible factor-1. J Nat Prod. 2007;70(7):1093–1097. | ||
Wallace EM, Rizzi JP, Han G, et al. A small-molecule antagonist of HIF2α is efficacious in preclinical models of renal cell carcinoma. Cancer Res. 2016;76(18):5491–5500. | ||
Chen W, Hill H, Christie A, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature. 2016;539(7627):112–117. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.