Back to Journals » OncoTargets and Therapy » Volume 11
Targeting DTL induces cell cycle arrest and senescence and suppresses cell growth and colony formation through TPX2 inhibition in human hepatocellular carcinoma cells
Authors Chen YC, Chen IS, Huang GJ, Kang CH, Wang KC, Tsao MJ, Pan HW
Received 27 July 2017
Accepted for publication 11 January 2018
Published 21 March 2018 Volume 2018:11 Pages 1601—1616
DOI https://doi.org/10.2147/OTT.S147453
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Yu-Chia Chen,1,* I-shu Chen,1,* Guan-Jin Huang,2 Chi-hsiang Kang,1 Kuo-Chiang Wang,1 Min-Jen Tsao,3 Hung-Wei Pan4,5
1Department of Surgery, Division of General Surgery, Kaohsiung Veterans General Hospital, Kaohsiung, Taiwan; 2Department of Pathology, National Chung Kung University Hospital, Tainan, Taiwan; 3Department of General Surgery, Zuoying Branch of Kaohsiung Armed Forces General Hospital, Kaohsiung, Taiwan; 4Department of Medical Education and Research, Kaohsiung Veterans General Hospital, Kaohsiung, Taiwan; 5Department of Applied Chemistry, National Pingtung University, Pingtung, Taiwan
*These authors contributed equally to this work
Background: Hepatocellular carcinoma (HCC) has an increasing incidence and high mortality. Surgical operation is not a comprehensive strategy for liver cancer. Moreover, tolerating systemic chemotherapy is difficult for patients with HCC because hepatic function is often impaired due to underlying cirrhosis. Therefore, a comprehensive strategy for cancer treatment should be developed. DTL (Cdc10-dependent transcript 2) is a critical regulator of cell cycle progression and genomic stability. In our previous study, the upregulation of DTL expression in aggressive HCC correlated positively with tumor grade and poor patient survival. We hypothesize that targeting DTL may provide a novel therapeutic strategy for liver cancer. DTL small interference RNAs were used to knock down DTL protein expression.
Methods: A clonogenic assay, immunostaining, double thymidine block, imaging flow cytometry analysis, and a tumor spheroid formation assay were used to analyze the role of DTL in tumor cell growth, cell cycle progression, micronucleation, ploidy, and tumorigenicity.
Results: Our results demonstrated that targeting DTL reduced cell cycle regulators and chromosome segregation genes, resulting in increased cell micronucleation. DTL depletion inhibited liver cancer cell growth, increased senescence, and reduced tumorigenesis. DTL depletion resulted in the disruption of the mitotic proteins cyclin B, CDK1, securin, seprase, Aurora A, and Aurora B as well as the upregulation of the cell cycle arrest gene p21. A rescue assay indicated that DTL should be targeted through TPX2 downregulation for cancer cell growth inhibition. Moreover, DTL silencing inhibited the growth of patient-derived primary cultured HCC cells.
Conclusion: Our study results indicate that DTL is a potential novel target gene for treating liver cancer through liver cancer cell senescence induction. Furthermore, our results provide insights into molecular mechanisms for targeting DTL in liver cancer cells. The results also indicate several other starting points for future preclinical and clinical studies on liver cancer treatment.
Keywords: DTL, cell cycle, senescence, TPX2, hepatocellular carcinoma
Introduction
DTL (Cdc10-dependent transcript 2; also known as CDT2, DCAF2, L2DTL, and RAMP) is a DDB1- and CUL4-associated factor (DCAF) that enables CRL4 to form CRL4Cdt2, a critical regulator of cell cycle progression and genome stability.1 Human L2DTL is homologous to lethal (2) denticleless, l(2)dtl, initially identified in Drosophila melanogaster. Drosophila l(2)dtl is so designated because embryos with homozygous mutations of the gene lack ventral denticle belts and are lethal.2 The DTL protein is a nuclear-matrix-associated protein and is downregulated during the retinoic acid-induced neuronal differentiation of NT2 cells; hence, it is designated as RAMP.3
DTL belongs to the family of WD40 repeat-containing DCAF proteins that work as substrate receptors for CRL4 ubiquitin ligases. DTL is conserved from nematodes to humans and plays fundamental roles in the regulation of the S phase of the cell cycle by regulating the degradation of replication licensing (CDT1), cell cycle control (p21), and chromatin modification (SET8) for destruction by CUL4-based E3 ligases (CRL4) under normal and stress conditions.4,5 DTL expression is also elevated in human breast and gastric cancers and in various cell lines derived from these primary tumors.6,7 In vitro, DTL could promote the growth of mammary epithelial and gastric cancer cells, and the silencing of DTL by small interference RNA (siRNA) significantly impaired the growth of these cells by inducing defects in chromosomal segregation and cytokinesis as well as apoptosis.6,7 DTL depletion alone could induce apoptotic death in all tested human cancer cell lines; however, it did not affect nontransformed human cells.8 These studies provide insights into how elevated CDT2 expression levels may provide tumors with a proliferative advantage. Therefore, we hypothesize that investigating the biological function of DTL targeting and its downstream molecules can provide novel therapeutic strategies for liver cancer.
In our previous study, the expression of DTL was frequently upregulated in aggressive hepatocellular carcinoma (HCC), and its level correlated positively with tumor grade and poor patient survival.9 DTL is a nuclear protein and a novel component of centrosome proteins throughout the cell cycle and participates in cell cycle progression.9 Although DTL silencing suppressed tumor cell growth and invasion capability, cDNA microarray analysis indicated that DTL was involved in chromosome segregation, DNA replication, and cell division.9 However, other mechanisms of this protein in cancer cell proliferation are now being identified, and further understanding of the functional relevance of DTL may facilitate the design of therapeutic approaches for HCC. In this paper, we present the first evidence regarding mechanisms for targeting DTL in liver cancer cells. Targeting DTL arrested cell cycle progression; caused genomic instability events, such as DNA damage, multinuclearity, and increased senescence; and suppressed the tumorigenesis of HCC cells. DTL depletion resulted in the disruption of the mitotic proteins cyclin B, CDK1, securin, seprase, Aurora A, and Aurora B as well as the upregulation of the cell cycle arrest gene p21. The results of a rescue assay indicated that targeting DTL inhibited cancer cell growth through TPX2 downregulation.
Materials and methods
Cell culture
Human liver cancer cells (Sk-Hep-1) were cultured in RPMI1640 medium supplemented with 10% fetal bovine serum (FBS). A human retinal pigment epithelial cell line (ARPE-19) was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM):F12 medium supplemented with 10% FBS. The Sk-Hep-1 cell line was kindly provided by Prof Hey-Chi Hsu, Graduate Institute of Pathology, College of Medicine, National Taiwan University, Taipei, Taiwan.10–12 The ARPE-19 cell line was kindly provided by Dr Wen-Huei Chang, Department of Applied Chemistry, National Pingtung University, Pingtung, Taiwan.
Reverse-transcription polymerase chain reaction (RT-PCR)
We used RT-PCR assays for the semiquantitative analysis of DTL mRNA, as described previously.9,11,13,14 The mRNA encoding S26 ribosomal protein (RPS26) was used as an internal control for initial RNA loading. PCR was performed using the SelectCycler Thermal Cycler SBT9600 (Select BioProducts, Edison, NJ, USA). The quantification of mRNA expression levels (quantitative RT-PCR) was performed in an automated DNA thermal cycler, the StepOnePlus Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA). The human beta-actin gene (ACTB) was used as an internal control for quantitative RT-PCR. Primers used for target amplification are listed in Table 1. All quantitative PCR data were calculated and expressed using the 2−ΔΔCt method and then converted into fold changes.
 | Table 1 Primers for RT-PCR and quantitative RT-PCR analysis |
Western blotting
Protein samples (60 μg) were separated through sodium dodecyl sulfate polyacrylamide gel electrophoresis (10% gels) and transferred to nitrocellulose membranes (Amersham Pharmacia Biotech, Buckinghamshire, UK). The membranes were then incubated with primary and secondary antibodies, and immunoreactive signals were detected using an ECL kit (Amersham Pharmacia Biotech).
Antibodies
The anti-DTL antibody was produced using GST–L2DTL protein, as previously described.9 The following antibodies were used: cyclin A, cyclin B1, cyclin E, CDK1, and CDK2 (Thermo Fisher Scientific); β-actin, separase, securin, and Aurora B (Abcam, Cambridge, UK); TPX2 (Sigma-Aldrich Co., St Louis, MO, USA); Aurora A (Leica, Milton Keynes, UK); and NDC80 (Proteintech, Rosemont, IL, USA).
siRNA and knockdown of gene expression
The DTL siRNA oligonucleotide pool (1: 5′-TCAAATCAGTGGAGCTCAC-3′ and 2: 5′-GTGTGTGAAGAGTTGTAAC-3′) was synthesized by Sigma-Aldrich Co. All transient transfections of the DTL siRNA oligonucleotide pool at a final concentration of 50 nM were performed using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s protocol.
Cell proliferation, colony formation, and anchorage-independent (spheroid) growth
The initial cell density for DTL siRNA transfection was 1.5×105 cells per 2-mL suspension. For cell proliferation analysis, 1,000 living cells were plated on 96-well plates after transfection with 20 nM siRNA oligonucleotide pool. Luminescence units indicating cell growth were determined at 0, 1, 2, and 3 days by using the CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI, USA). For colony formation assays, 2,500 cells were seeded in six-well plates and incubated for 2 weeks. The colonies were then fixed with 2% formaldehyde and stained with 0.5% crystal violet. Photographs were obtained, and the number of colonies in each well was counted. For spheroid formation assays, 1,000 living cells were seeded in an ultra-low-attachment 96-well microplate (Corning Incorporated, Corning, NY, USA), and cell spheroids were visualized under a microscopic low-power field. Spheroid sizes (n=12) were measured using ImageJ software. The experiments and readings were repeated and analyzed using the two-sided Student’s t-test.
Expression vector and stable transfection
RT-PCR-amplified full-length TPX2 cDNA was subcloned into the expression vector pEGFP-C1 (Takara Bio USA, Mountain View, CA, USA). HCC cells (Sk-Hep-1) were grown in DMEM. We used the Lipofectamine 3000 reagent (Thermo Fisher Scientific) for transfection. Cells stably expressing EGFP-TPX2 were obtained by culturing HCC cells in a medium with G418 (800 μg/mL) for more than 2 weeks. EGFP-H2B (human histone H2B protein) was used as the control vector.
Cell cycle analysis (imaging flow cytometry)
Cells were harvested and washed with phosphate-buffered saline (PBS) and then fixed in 0.5% formaldehyde for 1 h at 0°C. After a second wash with PBS, the cells were resuspended in 50% ethanol in PBS for 1 h at −20°C. The cells were washed at least once with cold PBS and then resuspended in 300–500 μL of PI/Triton X-100 staining solution (10 mL of 0.1% [v/v] Triton X-100; Sigma-Aldrich Co.) in PBS and 0.40 mL of 500 mg/mL 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich Co.). Data were acquired using the NucleoCounter NC-3000 machine (Chemometec, Allerød, Denmark) and Nucleoview NC-3000 software for automated imaging flow cytometry analysis. The cell cycle distribution was determined using the Nucleoview NC-3000 software.
Immunofluorescence staining
Cells cultured on coverslips were fixed in 4% paraformaldehyde in 1× PBS for 10 min at room temperature. After being washed in PBS, the cells were incubated with 1× PBST (0.2% Triton X-100 in 1× PBS) for 20 min and then incubated in 5% FBS blocking solution for 30 min. The cells were incubated with rhodamine-phalloidin for specifically labeling actin filaments at 37°C for 30 min. After being washed with 1× PBST three times for 5 min each, the coverslips were incubated with DAPI (2.5 μg/mL) for 2 min and rewashed in 1× PBST three times for 5 min. The cells were mounted in 90% glycerol in Tris buffer (pH 10.0) and observed using a fluorescence microscope.
Comet assays
Neutral comet assays were performed using a CometAssay kit (Trevigen, Gaithersburg, MD, USA) in accordance with the manufacturer’s instructions. Images were acquired using an Olympus IX81F-3 fluorescence microscope mounted with an Olympus DP70 digital camera. The comet tail lengths were measured using ImageJ software.
Senescence-associated β-galactosidase assay
Senescence-associated β-galactosidase (SA-β-gal), a widely used marker for identifying senescent cells, was detected according to the manufacturer’s protocol (Senescence Cells Histochemical Staining Kit; Sigma-Aldrich Co.). Briefly, cells were fixed in 4% paraformaldehyde and stained with β-gal staining solution containing X-Gal (1 mg/mL), citric acid/sodium phosphate (pH 6.0, 40 mM), potassium ferrocyanide (5 mM), potassium ferricyanide (5 mM), NaCl (150 mM), and MgCl2 (2 mM). The plates were incubated in a dry incubator and monitored every 2 h for the development of blue color. The reaction was then inhibited by the addition of 50% glycerol solution and then photographed under a light microscope. Twelve photos (10× magnification) of the cells from different fields were examined for β-gal positivity to quantify the senescent cells in each treatment.
Patient-derived primary HCC cells
Surgically resected HCC samples were obtained from Kaohsiung Veterans General Hospital. All patients provided written informed consent for the use of tissue samples in this research. Our study was approved by the Ethics Committee of Kaohsiung Veterans General Hospital (IRB protocol: VGHKS13-CT3-009). To establish a primary HCC culture, single-cell suspensions from HCC specimens were generated using a dissociator. Briefly, the tumor tissue was washed and minced using fine scissors into fragments of 1×1×1 mm3 and placed in the GentleMACS Dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany). The single-cell suspensions were subjected to red blood cell (RBC) cell lysis in RBC lysis buffer (Qiagen NV, Venlo, the Netherlands). Then, the cells were seeded into an ultra-low-attachment 24-well microplate (Corning Incorporated), and spheroids formed after 14–21 days in defined media. The spheroids were then seeded into a six-well culture plate. When a monolayer of primary tumor cells was formed, the cells were detached using trypsin/EDTA and resuspended in defined media for primary HCC cells,15 which contained DMEM:F12 (1:1) medium supplemented with 15 ng/mL basic fibroblast growth factor, 20 ng/mL epidermal growth factor, 2 mM/L L-glutamine, 4 U/L insulin growth factor, and B27 supplement (1:50; Sigma-Aldrich Co.). The cells were cultured in a humidified atmosphere at 37°C and 7% CO2.
Statistical analysis
For all bar plots, two-sided t-tests were used to determine the significance of the data with a normal distribution and equal variances. A box plot was plotted using SigmaPlot.v10, with the bottom and top of the box corresponding to the 25th and 75th percentiles, respectively, and whiskers representing data points. The gray line extending through the box plot indicates the mean value and the black line within the box plot represents the median value. Groups with P-values of less than an alpha of 0.05 were considered significantly different.
Results
Silencing DTL inhibits liver cancer cell growth
To investigate the role of DTL in liver cancer cell proliferation and to evaluate DTL functions in Sk-Hep-1 cells, we synthesized two commercial siRNAi oligonucleotides for DTL. RT-PCR analysis indicated that the DTL transcript was downregulated by DTL siRNA oligonucleotides (Figure 1A). The depletion of DTL protein expression completely inhibited anchorage-dependent cell growth, as observed in the clonogenic formation assay (Figure 1B), and anchorage-independent cell growth, as observed in the spheroid formation assay (Figure 1C), in Sk-Hep-1 liver cancer cells. These results indicate that DTL can be a therapeutic target for liver cancer.
 | Figure 1 Silencing of DTL inhibits cell growth and reduces tumorigenesis in human liver cancer cell lines. |
Changes in cell fitness caused by DTL depletion were reflected by changes in cell cycle regulation and feature genes of chromosome segregation
DTL depletion reduced the proportion of cells in the G2/M phase. By contrast, inhibiting DTL expression increased the proportion of sub-G1 cells (cell death, ~11%; Figure 2A). We further elucidated the role of DTL in the mitosis of liver cancer cells. Morphological examination indicated that DTL-depleted cells were enlarged and flattened. The number of micronucleated Sk-Hep-1 cells was increased compared with that of control RNAi-oligonucleotide-transfected cells (Figure 2B). The mitosis phase of Sk-Hep-1 cells was produced by synchronization in nocodazole medium and these cells were released into standard cell culture medium for 30 minutes. Morphological examination indicated that DTL-depleted cells had DNA lagging abnormal architecture of spindle and centrosome bodies (Figure S1A). The number of mitotic cells was decreased compared with that of control RNAi-oligonucleotide-transfected cells (Figure S1B) and they had decreased γ-tubulin (Figure S1C). This phenomenon indicated the occurrence of mitotic defects and consequent genomic instability. We further characterized molecular changes accompanying the depletion at the DTL level. The Western blot assay showed that the G2/M phase proteins that are required in chromosome segregation (cyclin B1, cyclin B2, CDK1, securin, and separase), spindle pole organization (TPX2 and NDC80), and kinetochore attachment (Aurora A and Aurora B) decreased after DTL depletion in liver cancer cells. Because of the observed inhibitory effect on cell growth, we evaluated the expression of cyclin E, cyclin A, and CDK2, which are crucial initiators of DNA replication in the S phase. We found that the expression of these proteins decreased after the silencing of DTL (Figure 2C). To define the role of DTL depletion in genomic instability, we assessed DNA damage in Sk-Hep-1 cells transfected with siRNAs targeting DTL. DNA damage was measured using a comet assay (a single-cell gel electrophoresis assay). Undamaged DNA remained in the nucleoid and was observed as the comet head, whereas damaged DNA migrated through the agarose gel and formed a comet tail. We measured the tail length and found a significant increase in the mean tail length in DTL-depleted cells (Figure 2D), indicating that DTL inhibition increased DNA damage.
 | Figure 2 DTL depletion increased the proportion of sub-G1 cells, micronucleation, and DNA damage. |
DTL depletion arrested cell cycle progression and senescence
To elucidate the biological role of DTL in cell cycle progression in liver cancer cells, we analyzed the dynamics of cell cycle progression in DTL-depleted cells. The cell cycle of Sk-Hep-1 cells was synchronized at the G1/S boundary by a double thymidine block, and the cells were then released into nocodazol medium for arresting cell cycle progression in the G2/M phase (Figure 3A). Through the time courses of cell cycle progression, we found that DTL-depleted cells did not progress from the G1/S to the G2/M phase and observed a cell cycle progression arrest event (Figure 3B and C). Consistently, cell cycle arrest in G2/M to G1/S phase progression when DTL depletion Sk-Hep-1 cells released into standard cell culture medium from nocodazole block (Figure S2). Cellular senescence is a stable form of cell cycle arrest that limits the proliferative potential of cells.16,17 Senescent cells have well-defined features, which include cell cycle arrest, and morphological changes, such as enlarged and flattened cells with enlarged nuclei.17–19 Our results demonstrated that DTL depletion resulted in enlarged and flattened cells, cell cycle arrest, and DNA damage. Hence, in the β-galactosidase assay, DTL-depleted cells exhibited strong positive β-gal staining (senescent cells, ~73%; Figure 3D). Moreover, the collective expression of SA-β-gal, p21 (a potent cell cycle inhibitor), and interleukin (IL)-8 (a gene of the senescence-associated secretary phenotype [SASP]) is known as the cellular senescence signature.17 The quantitative RT-PCR assay indicated the upregulation of p21 and IL-8 mRNA (Figure 3E).
 | Figure 3 Cell cycle progression arrest by DTL depletion. |
DTL downregulation in cell cycle arrest caused by contact inhibition
When cells reach a high density, they stop proliferating and can stay in cell cycle arrest caused by contact inhibition. To elucidate the biological role of DTL in the G1 phase of the cell cycle in liver cancer cells, we analyzed DTL expression during G1 phase cell cycle arrest by using growth contact inhibition treatment. We plated Sk-Hep-1 cells at a regular density. After 6 days, the cells reached high-density confluence and were completely contact inhibited. The contact-inhibited cells were arrested in the G1 phase, as observed through flow cytometry (Figure 4A). By using RT-PCR in the linear range and quantitative RT-PCR, we observed that DTL mRNA expression decreased upon entry into G1 phase arrest at day 6, and TPX2 had an expression pattern similar to that of DTL (Figure 4B and C). Consistently, DTL and TPX2 had a similar pattern in protein expression (Figure 4D).
 | Figure 4 DTL downregulation in cell cycle arrest caused by contact inhibition. |
TPX2 is a downstream molecule of DTL in liver cancer cell growth
In our previous study, targeting TPX2 suppressed the tumorigenesis of HCC cells, arrested cell cycle progression, increased genomic instability, and led to liver cancer cell senescence.20 Moreover, DTL-silenced cells exhibited TPX2 downregulation. DTL and TPX2 had concordant expression in G1-phase-arrested cells. To further evaluate whether TPX2 is a downstream protein of DTL, we stably expressed EGFP-TPX2 protein in Sk-Hep-1 cells. The EGFP-TPX2 stable expression cell line was used for DTL depletion treatment and the EGFP-H2B cell line was used as a control. TPX2 and EGFP-TPX2 protein levels were measured using Western blot analysis (Figure 5A). Cell growth inhibition was diminished by the ectopic expression of EGFP-TPX2. DTL silencing reduced cell growth inhibition (Figure 5B), increased the number of large tumor spheroids (Figure 5C), and reduced senescence (Figure 5D) in EGFP-TPX2 cells compared with EGFP-H2B cells. These results indicate that TPX2 is a downstream molecule of DTL and contributes to tumorigenesis in liver cancer cells.
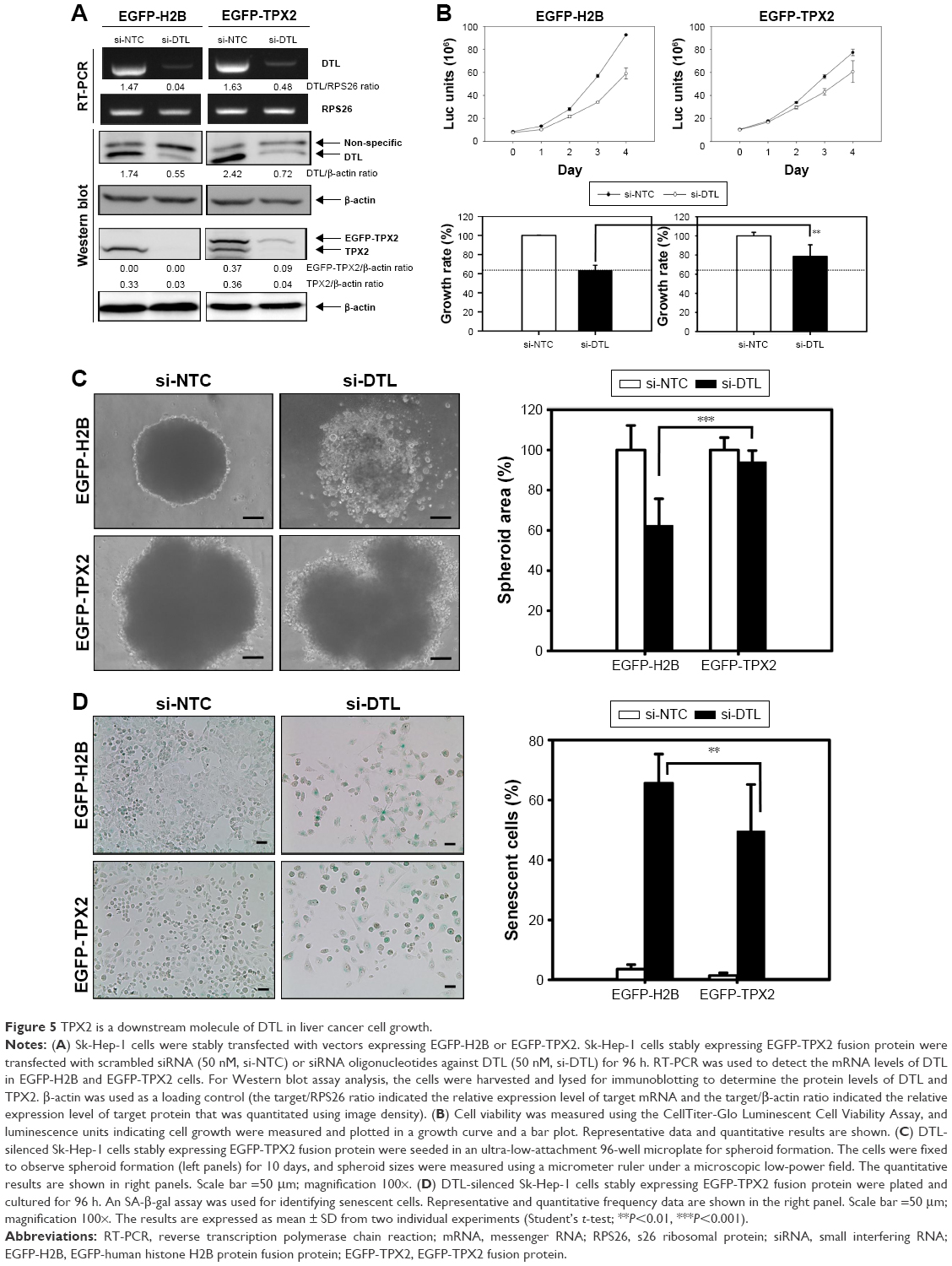 | Figure 5 TPX2 is a downstream molecule of DTL in liver cancer cell growth. |
Therapeutic potential of targeting DTL in HCC
To investigate the therapeutic potential of targeting DTL in HCC, we performed the suspension culture process for patient-derived primary cultured HCC cells (PDC-HCCs; Figure 6A). Eight types of PDC-HCCs were cultured in our laboratory and named VGHKS-#-3T-L, 21T, 25T, 71T, 72T, 80T, 89T, and 90T (Figure 6B). By using RT-PCR in the linear range, we measured the mRNA expression levels of DTL and TPX2 for study targets. The hepatocyte-linked genes hepatocyte growth factor and albumin were expressed in these PDC-HCCs, but not in ARPE19 cells (Figure 6C). DTL depletion caused cell growth inhibition in three of the four examined PDC-HCC types, of which two (72T and 90T) had cell growth inhibition of more than 40% (Figure 6D). Moreover, DTL-silenced cells exhibited TPX2 protein downregulation in two PDC-HCC types with growth inhibition (ie, 72T and 90T) (Figure 6E), but not in the 21T and 25T PDC-HCCs. These results indicate that targeting DTL has therapeutic potential for HCC. However, the cell cycle proportions were not influenced in 72T and 90T cells in si-DTL transfection (Figure S3).
 | Figure 6 Therapeutic potential of targeting DTL in HCC. |
Discussion
HCC has an increasing incidence and high mortality. Treatment options are limited if the disease is not diagnosed in its early stage.21 The natural course of the disease is aggressive but not always predictable. Surgical operation is the most effective treatment for cure; however, this is not a comprehensive strategy in liver cancer.22 In addition, tolerability to chemotherapy is a concern in the cirrhotic population, and the complications of cirrhosis, such as impaired liver function and thrombocytopenia, have rendered a substantial proportion of patients not suitable for chemotherapy.23,24 Because of the lack of success in treating advanced HCC with traditional modalities of radiation and systemic chemotherapy, studies have recently focused on developing targeted therapies.25 In this study, our results indicated the therapeutic potential of targeting DTL in liver cancer treatment.
Rereplication and G2 cell cycle arrest were observed in DTL-depleted cells.5 DTL promotes the degradation of Set8 during the S phase of the cell cycle.26 Inactivation of DTL-mediated Set8 degradation results in DNA damage, induction of p53 gene and p53-transactivated proapoptotic genes, delayed progression through the G2 phase of the cell cycle due to activation of the G2/M checkpoint, and repression of E2F1-dependent gene transcription.26,27 Knockdown of DTL by siRNA resulted in enhanced G2 arrest, p53 and p21 induction, and reduced cell proliferation in colon cancer cells.28 DTL-dependent destruction of Set8 in the S phase preserves genomic stability by preventing aberrant chromatin compaction during DNA synthesis.26 These results demonstrate a central role of DTL-dependent cell cycle regulation in the maintenance of a stable epigenetic state essential for cell viability. In our study, we observed that targeting DTL stopped cell cycle progression; reduced cell cycle regulation and mitosis progression; caused genomic instability events, such as DNA damage and micronuleation; and led to growth inhibition in Sk-Hep-1 liver cancer cells. Moreover, DTL depletion led to cell growth inhibition in PDC-HCCs. These results indicated that targeting DTL has therapeutic potential in HCC.
Cellular senescence is a stress response that entails an irreversible cessation of mitotic activity. The senescence response is a potent tumor suppressor mechanism.29,30 Senescence can be induced by different stimuli, such as telomere attrition, DNA damage, chromatin perturbations, and oncogene activation. These triggers initiate signaling cascades that activate p53–p21 and/or p16INK4a–pRB tumor suppressor pathways, which promote proliferation arrest and the senescence response.31 Senescent cells also develop the SASP and secrete multiple proinflammatory molecules (cytokines, chemokines, growth factors, and proteases),30 such as proinflammatory cytokines (IL-1α, IL-1β, IL-6, and IL-8).30,31 The senescent hepatocyte gene signature includes changes in cell cycle regulation, morphology, inflammation, signal transduction, metabolism, and stellate cell activation.17 A study on the senescent hepatocyte gene signature reported that major downregulated genes in liver cancer cell senescence were cell cycle regulation genes.17 Genes involved in cell cycle phases beyond the G1/S transition, including the S phase, DNA replication, and cytokinesis, were downregulated, indicating G1/S cell cycle arrest in senescent liver cancer cells.9 In our study, DTL depletion caused DNA damage; genomic instability; downregulation of cyclin B2, Aurora B, and NDC80 proteins; and an increase in p21 and IL-8 mRNA expression in liver cancer cells, finally resulting in cancer cell senescence.
TPX2 is a protein required for spindle assembly, particularly for spindle pole organization, in cells.32,33 Furthermore, TPX2 silencing causes cell cycle arrest,34–37 inhibits cell proliferation,35–37 and induces cell apoptosis35,36 in several cancer types. In our previous study, targeting TPX2 arrested cell cycle progression, increased genomic instability and cell senescence, and suppressed tumorigenesis in HCC and prostate cancer cells.20,38 These results indicated that TPX2 is an important factor for tumor cell growth. In our study, the TPX2 rescue assay demonstrated that DTL depletion suppressed tumorigenesis and increased senescence through TPX2 inhibition in liver cancer cells. Downregulation of TPX-2 expression by DTL depletion was observed in si-DTL and si-DTL EGFP-TPX-2 cells. This phenomenon indicated that DTL depletion led to TPX2 protein degradation not only in endogenous TPX2 but also in epigenetic TPX2 protein. However, as described in the introduction, DTL belongs to the family of WD40 repeat-containing DCAF proteins that work as substrate receptors for CRL4 ubiquitin ligases and promotes the ubiquitylation of proteins in S phase and after DNA damage. Our previous study showed that the downregulation of TPX2 increased DNA damage in liver cancer cells. Taken together, these results suggested that DTL may influence TPX2 stabilization after DNA damage.
Conclusion
Our study results indicated that DTL is a potential novel target gene for treating liver cancer through cancer cell senescence induction. Furthermore, our results provide insights into molecular mechanisms for targeting DTL in liver cancer cells. The results of this study also indicate several other starting points for future preclinical and clinical studies on liver cancer treatment.
Acknowledgments
This study was supported by grants from Kaohsiung Veterans General Hospital, Kaohsiung, Taiwan, Republic of China (VGHKS104-121, VGHKS105-137, VGHKS106-155 to Hung-Wei Pan) and the Zuoying Branch of Kaohsiung Armed Forces General Hospital, Kaohsiung, Taiwan (ZAFGH 102-10 to Min-Jen Tsao). We acknowledge Wallace Academic Editing for editing this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Abbas T, Dutta A. CRL4Cdt2: master coordinator of cell cycle progression and genome stability. Cell Cycle. 2011;10(2):241–249. | ||
Kurzik-Dumke U, Neubauer M, Debes A. Identification of a novel Drosophila melanogaster heat-shock gene, lethal(2)denticleless [l(2)dtl], coding for an 83-kDa protein. Gene. 1996;171(2):163–170. | ||
Cheung WM, Chu AH, Chu PW, Ip NY. Cloning and expression of a novel nuclear matrix-associated protein that is regulated during the retinoic acid-induced neuronal differentiation. J Biol Chem. 2001;276(20):17083–17091. | ||
Rossi M, Duan S, Jeong YT, et al. Regulation of the CRL4(Cdt2) ubiquitin ligase and cell-cycle exit by the SCF(Fbxo11) ubiquitin ligase. Mol Cell. 2013;49(6):1159–1166. | ||
Huh J, Piwnica-Worms H. CRL4(CDT2) targets CHK1 for PCNA-independent destruction. Mol Cell Biol. 2013;33(2):213–226. | ||
Li J, Ng EK, Ng YP, et al. Identification of retinoic acid-regulated nuclear matrix-associated protein as a novel regulator of gastric cancer. Br J Cancer. 2009;101(4):691–698. | ||
Ueki T, Nishidate T, Park JH, et al. Involvement of elevated expression of multiple cell-cycle regulator, DTL/RAMP (denticleless/RA-regulated nuclear matrix associated protein), in the growth of breast cancer cells. Oncogene. 2008;27(43):5672–5683. | ||
Olivero M, Dettori D, Arena S, Zecchin D, Lantelme E, Di Renzo MF. The stress phenotype makes cancer cells addicted to CDT2, a substrate receptor of the CRL4 ubiquitin ligase. Oncotarget. 2014;5(15):5992–6002. | ||
Pan HW, Chou HY, Liu SH, Peng SY, Liu CL, Hsu HC. Role of L2DTL, cell cycle-regulated nuclear and centrosome protein, in aggressive hepatocellular carcinoma. Cell Cycle. 2006;5(22):2676–2687. | ||
Peng SY, Lai PL, Pan HW, Hsiao LP, Hsu HC. Aberrant expression of the glycolytic enzymes aldolase B and type II hexokinase in hepatocellular carcinoma are predictive markers for advanced stage, early recurrence and poor prognosis. Oncol Rep. 2008;19(4):1045–1053. | ||
Lin SY, Pan HW, Liu SH, et al. ASPM is a novel marker for vascular invasion, early recurrence, and poor prognosis of hepatocellular carcinoma. Clin Cancer Res. 2008;14(15):4814–4820. | ||
Lin ZZ, Jeng YM, Hu FC, et al. Significance of Aurora B overexpression in hepatocellular carcinoma. Aurora B overexpression in HCC. BMC Cancer. 2010;10:461. | ||
Pan HW, Ou YH, Peng SY, et al. Overexpression of osteopontin is associated with intrahepatic metastasis, early recurrence, and poorer prognosis of surgically resected hepatocellular carcinoma. Cancer. 2003;98(1):119–127. | ||
Lee YC, Pan HW, Peng SY, et al. Overexpression of tumour-associated trypsin inhibitor (TATI) enhances tumour growth and is associated with portal vein invasion, early recurrence and a stage-independent prognostic factor of hepatocellular carcinoma. Eur J Cancer. 2007;43(4):736–744. | ||
Tomuleasa C, Soritau O, Rus-Ciuca D, et al. Isolation and characterization of hepatic cancer cells with stem-like properties from hepatocellular carcinoma. J Gastrointest Liver Dis. 2010;19(1):61–67. | ||
Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8(9):729–740. | ||
Aravinthan A, Shannon N, Heaney J, Hoare M, Marshall A, Alexander GJ. The senescent hepatocyte gene signature in chronic liver disease. Exp Gerontol. 2014;60:37–45. | ||
Narita M. Cellular senescence and chromatin organisation. Br J Cancer. 2007;96(5):686–691. | ||
Mehta IS, Figgitt M, Clements CS, Kill IR, Bridger JM. Alterations to nuclear architecture and genome behavior in senescent cells. Ann N Y Acad Sci. 2007;1100:250–263. | ||
Hsu CW, Chen YC, Su HH, et al. Targeting TPX2 suppresses the tumorigenesis of hepatocellular carcinoma cells resulting in arrested mitotic phase progression and increased genomic instability. J Cancer. 2017;8(8):1378–1394. | ||
Vitale A, Peck-Radosavljevic M, Giannini EG, et al. Personalized treatment of patients with very early hepatocellular carcinoma. J Hepatol. 2017;66(2):412–423. | ||
Best J, Schotten C, Theysohn JM, et al. Novel implications in the treatment of hepatocellular carcinoma. Ann Gastroenterol. 2017;30(1):23–32. | ||
Samonakis DN, Kouroumalis EA. Systemic treatment for hepatocellular carcinoma: still unmet expectations. World J Hepatol. 2017;9(2):80–90. | ||
Chan SL, Yeo W. Targeted therapy of hepatocellular carcinoma: present and future. J Gastroenterol Hepatol. 2012;27(5):862–872. | ||
Marks EI, Yee NS. Molecular genetics and targeted therapy in hepatocellular carcinoma. Curr Cancer Drug Targets. 2016;16(1):53–70. | ||
Centore RC, Havens CG, Manning AL, et al. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol Cell. 2010;40(1):22–33. | ||
Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol Cell. 2010;40(1):9–21. | ||
Song B, Wang Y, Titmus MA, et al. Molecular mechanism of chemoresistance by miR-215 in osteosarcoma and colon cancer cells. Mol Cancer. 2010;9:96. | ||
Naylor RM, van Deursen JM. Aneuploidy in Cancer and Aging. Ann Rev Genetics. 2016;50:45–66. | ||
Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Ann Rev Pathol. 2010;5:99–118. | ||
Loaiza N, Demaria M. Cellular senescence and tumor promotion: is aging the key? Biochim Biophys Acta. 2016;1865(2):155–167. | ||
Wittmann T, Wilm M, Karsenti E, Vernos I. TPX2, a novel xenopus MAP involved in spindle pole organization. J Cell Biol. 2000;149(7):1405–1418. | ||
Garrett S, Auer K, Compton DA, Kapoor TM. hTPX2 is required for normal spindle morphology and centrosome integrity during vertebrate cell division. Curr Biol. 2002;12(23):2055–2059. | ||
Chang H, Wang J, Tian Y, Xu J, Gou X, Cheng J. The TPX2 gene is a promising diagnostic and therapeutic target for cervical cancer. Oncol Rep. 2012;27(5):1353–1359. | ||
Zhang L, Huang H, Deng L, et al. TPX2 in malignantly transformed human bronchial epithelial cells by anti-benzo[a]pyrene-7,8-diol-9,10-epoxide. Toxicology. 2008;252(1–3):49–55. | ||
Yang X, Liu G, Xiao H, et al. TPX2 overexpression in medullary thyroid carcinoma mediates TT cell proliferation. Pathol Oncol Res. 2014;20(3):641–648. | ||
Liu HC, Zhang GH, Liu YH, et al. TPX2 siRNA regulates growth and invasion of esophageal cancer cells. Biomed Pharmacother. 2014;68(7):833–839. | ||
Pan HW, Su HH, Shu CW, Huang GJ, Wu TT. Targeted TPX2 increases chromosome missegregation and suppresses tumor cell growth in human prostate cancer. Onco Targets Ther. 2017;10:3531–3543. |
Supplementary materials
 | Figure S1 DTL depletion led to DNA lagging and reduced the mitotic cells and γ-tubulin protein expression during mitosis. |
 | Figure S2 Cell cycle progression arrest by DTL depletion in nocodazole block and release. |
 | Figure S3 DTL depletion did not influence the cell cycle proportions in 72T and 90T primary HCC cells. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.