")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Targeting delivery of Radix Ophiopogonis polysaccharide to ischemic/reperfused rat myocardium by long-circulating macromolecular and liposomal carriers
Authors Wang L, Yao C, Wu F, Lin X, Shen L, Feng Y
Received 28 May 2015
Accepted for publication 14 August 2015
Published 10 September 2015 Volume 2015:10(1) Pages 5729—5737
DOI https://doi.org/10.2147/IJN.S89445
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
LiNa Wang,1,2,* ChunXia Yao,1,* Fei Wu,2 Xiao Lin,1,2 Lan Shen,1 Yi Feng2
1College of Chinese Materia Medica, 2Engineering Research Center of Modern Preparation Technology of TCM of Ministry of Education, Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: Drug delivery to ischemic myocardium is an enormous challenge. This work aimed to characterize cardiac delivery behaviors of mono-polyethylene glycosylated (PEGylated) conjugates and long-circulating liposomes (L-Lps) with Radix Ophiopogonis polysaccharide (ROP) as drug. The results showed that compared to native ROP, 32-, 52-, and 45-fold increases in blood half-life were achieved by 20-kDa PEG mono-modified ROP (P20k-R), 40-kDa PEG mono-modified ROP (P40k-R), and ROP-loaded L-Lp, respectively. With comparable blood pharmacokinetics, ROP-loaded L-Lp showed both significantly higher targeting efficacy and drug exposure in infarcted myocardium than P40k-R. With regard to P20k-R, both its targeting efficacy and its level in infarcted myocardium at 3 hours postdose were comparable to P40k-R, but its level in blood and myocardium reduced obviously faster. As a whole, the results indicate that both loading in L-Lps and mono-PEGylation are effective in targeting drug to ischemic myocardium, but the former appears to induce stronger effects.
Keywords: PEGylation, liposome, myocardial targeting, Radix Ophiopogonis polysaccharide, fluorescent imaging
Introduction
The ischemic heart disease is one of the leading causes of death and disability worldwide.1 When an acute episode of coronary obstruction or spasm happened, blood flow in affected myocardia significantly decreased immediately. This still happened after reperfusion due to microvascular obstruction caused by many factors.2 With ischemia continued, perfusion is partially restored by angiogenesis;3 however, the endothelium of these newly formed blood vessels was shown recently to be highly intact, making its permeability to macromolecules and nanosized carrier systems rather low.4,5 All these together make the efficient drug delivery to the ischemic local challenging. On the other hand, many antimyocardial ischemic drugs and adjuvants exhibit a short blood circulation half-life and/or poor solubility in blood, which further limits their accumulation in the ischemic region.6–8 Furthermore, several compounds, like adenosine and vascular endothelial growth factor, have been shown to induce severe systemic side effects.6,9 Therefore, strategies that do not require direct intramyocardial injection of therapeutic molecules for efficiently delivering drugs to and retaining them in the damaged myocardium to limit the extent of ischemia/reperfusion (IR) injury are needed.10,11 This is just the case for Radix Ophiopogonis polysaccharide (ROP, a natural water-soluble fructan with a molecular weight of ~5 kDa). ROP was found having a unique antimyocardial ischemic activity via 1) protecting myocardial cells from damage by inhibiting the production of free radicals caused by myocardial ischemia and eliminating the oxygen radicals and 2) promoting the formation of microvessels in ischemic zones.12–14 However, its pretty short blood half-life (~0.4 hour) and low target distribution limit to a large extent its efficacy and clinical applications.15,16
Presently, there are two main strategies for targeting parenteral drugs to ischemic myocardium by the ischemia-induced enhanced permeability and retention (EPR) effect17,18 alone or in combination with certain active and/or physicochemical targeting mechanisms.19–22 One comprises physical entrapment of native drugs in various nanosized insoluble drug delivery systems (eg, liposomes, micelles, nanoparticles, microbubbles, cell ghosts). The other involves chemical modification of native drugs through covalent linkage with hydrophilic polymers such as polyethylene glycol (PEG), poly-L-glutamic acid, and dextran.
Among nanosized insoluble drug delivery systems, liposomes should be a promising one for hydrophilic drugs in terms of their safety, formulation flexibility, selective delivery to special organs, and long-confirmed “plug and seal” effect to augment the membrane repair process.23–25 To combat their immediate uptake and clearance by the reticuloendothelial system as well as their relatively low stability in vivo, PEG-coated long-circulating liposomes (L-Lps) have been developed and widely used.4,25,26 However, the application of liposomes to polysaccharide-based drugs is still quite limited.
Among polymers for drug conjugation, PEG is the most successful and promising one. Up to now, there have already been nine PEGylated protein drugs and one PEGylated oligonucleotide drug in the market.27,28 By PEG conjugation, the physicochemical and biopharmaceutical properties of drugs can be greatly improved, thus resulting in enhancement of therapeutic efficacy and reduction in side effects. However, there are yet few reports on using PEG conjugation as a strategy for cardiac drug delivery. Previously, ROP was successfully mono-PEGylated to markedly reduce the injection-administered frequency without sacrificing the therapeutic efficacy.29
To our best knowledge, there is no report on direct comparison between the aforementioned two strategies about their cardiac distribution and retention behaviors. In light of the earlier discussion, this work therefore aimed to characterize cardiac delivery behaviors of PEG conjugates and L-Lps with ROP as drug. This study was carried out in ischemic/reperfused rats via the fluorescent imaging, through which infarcted and noninfarcted areas in each left ventricular transverse slice were accurately assayed in parallel.
Materials and methods
Materials and animals
ROP was self-made according to a previous report.30 Linear amino-terminated polyethylene glycol methyl ether (mPEG-NH2) hydrochlorides with molecular weights of 20 and 40 kDa were purchased from Jenkem Technology Co., Ltd. (Beijing, People’s Republic of China). Fluorescein isothiocyanate (FITC) was purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Distearyl phosphatidylethanolamine-PEG2000 (DSPE-PEG2000), fully hydrogenated soybean phosphatidylcholine (HSPC), and cholesterol were purchased from Shanghai Advanced Vehicle Technology Co., Ltd (Shanghai, People’s Republic of China). All other chemicals were of reagent grade and purchased from commercial sources.
Male Sprague Dawley rats, weighing 200–250 g, were supplied by the Lab Animal Center of Shanghai University of Traditional Chinese Medicine (Shanghai, People’s Republic of China). They were kept in an environmentally controlled breeding room for at least 4 days before starting the experiments and were fed with standard laboratory food and given water ad libitum. The Animal Ethical Experimentation Committee of Shanghai University of Traditional Chinese Medicine (People’s Republic of China), according to the requirements of the National Act on the Use of Experimental Animals, approved all procedures of the animal experiments.
Preparation and characterization of FITC-labeled mono-PEGylated ROPs (FP-R)
The mono-PEGylated ROPs were synthesized through a moderate coupling reaction between amino-terminated mPEG and hydroxyl-activated ROP.29 Characterization of the conjugates was carried out by anthrone–sulfuric acid colorimetry coupled high-performance gel permeation chromatography and 1H NMR.29 To determine their levels in biosamples, the conjugates were prelabeled with FITC before administration.31 The amounts of FITC conjugated were determined by a Hitachi F-4500 fluorescence spectrophotometer (Tokyo, Japan) and were calculated in terms of the fluorescence intensity.
Preparation and characterization of L-Lps loaded with FITC-labeled ROP (FR)
L-Lps, which are composed of HSPC, DSPE-PEG2000, and cholesterol at a molar ratio of 4.68:3:0.37, were prepared by the reversed phase evaporation method. In detail, HSPC (104.4 mg) and cholesterol (33 mg) were dissolved in 10 mL of a chloroform and n-hexane mixture (1:1, v/v) in a round-bottomed flask, followed by the addition of 2 mL of 100 mg/mL FR in phosphate-buffered saline (PBS) (pH 7.4). The mixture obtained was sonicated in a bath sonicator for 5 minutes to produce stable and fine emulsion droplets, and then DSPE-PEG2000 (29.4 mg) in 1 mL of organic phase was added. The emulsion was submitted to evaporation under vacuum to remove organic solvents, allowing the formation of lipid vesicles. Following the addition of a small amount of PBS, the system was vigorously vortexed until the formation of a homogeneous and stable liposome suspension, which was then transferred to 5 mL volumetric flask and diluted with PBS to volume. Finally, the liposomes were down-sized by probe sonication for 10 minutes under ice bath. The nonencapsulated FR was separated from the liposome-encapsulated FR by ultracentrifugation (60,000 rpm, 30 minutes, 4°C). The precipitate was resuspended with PBS for pharmacokinetics and cardiac distribution studies.
Mean size, size distribution, and zeta potential of liposomes prepared were measured using dynamic light scattering with a computerized Malvern Autosizer Nano ZS90 inspection system (Malvern Instruments, Malvern, UK). The measurements were performed in triplicate.
Rat model of myocardial IR injury
Myocardial IR was induced in male rats as described before.32 In brief, following being anesthetized by intraperitoneal injection of 10% chloral hydrate, rats were intubated and mechanically ventilated. Left intercostal thoracotomy was performed in the intercostal space to expose the heart. Myocardial ischemia was induced by transient ligation of the left coronary artery for 30 minutes and restoration of perfusion thereafter. The ischemic condition was verified by evidence of immediate changes including sudden pallor and distinct dilatation and paralysis of the affected portion of the left ventricle. Finally, the thorax was closed.
Measurement of infarct sizes
Infarct sizes of myocardium were measured after triphenyltetrazolium chloride (TTC) staining. In detail, the heart was harvested and rinsed with normal saline. The excised left ventricle was frozen at -20°C for 30 minutes and then sectioned from apex to base into ~2 mm slices. The slices were incubated in a 1% solution of buffered TTC (pH 7.4) at 37°C for 15 minutes and then fixed in 10% formaldehyde. The slices were photographed at the next day by digital camera. The infarct region (TTC unstained) was isolated from the rest cardiac tissues, which were stained red by TTC. The infarct and normal tissues were weighed, respectively, and the infarct size was expressed as a percentage of mass of the left ventricle.
Plasma pharmacokinetics
Each drug was administered intravenously to rats at a same molar dose of 4 μmol/kg. Blood samples (~400 μL at a time) were collected into heparin-contained tubes from the orbital sinus at the specified time points and then immediately centrifuged at 4,000 rpm for 10 minutes. The separated plasma was frozen at −20°C until assay. To a 100 μL portion of each plasma sample that did not contain liposomes, 40 μL of 1 M perchloric acid was added to precipitate plasma proteins. After centrifugation at 10,000 rpm for 2 minutes, the 80-μL supernatant of the sample was transferred to another clean tube and neutralized using 30 μL of 1 M NaOH. For plasma samples containing liposomes, each 100 μL of plasma was added with 100 μL of 20% Triton X-100 and heated at 60°C for 10 minutes, and then 40 μL of 1 M perchloric acid was added. After centrifugation, the 140-μL supernatant was transferred to another clean tube and neutralized using 23.5 μL of 1 M NaOH. With another centrifugation, 50 μL of the supernatant was taken out and diluted with PBS (pH 7.4) before determining spectrofluorimetrically at λex 495 nm and λem 515 nm. The pharmacokinetic parameters were determined by noncompartmental analysis using WinNonlin® Professional 6.2.
Cardiac distribution
Rats with myocardial ischemia were injected with each drug at the same molar dose of 4 μmol/kg via the tail vein and were sacrificed at 0.167, 3, or 12 hours postdose. The hearts were harvested and rinsed with normal saline. The excised left ventricle was frozen at −20°C for 30 minutes and then sectioned from apex to base into ~2 mm slices, which were photographed at Em = DsRed and Ex =500 nm by fluorescent imaging (Lumina XR; PerkinElmer Inc., Waltham, MA, USA). After that, the heart slices were immediately submitted to TTC staining to demarcate the infarcted and noninfarcted myocardium. The quantitative analysis was performed using the Lumina II living Image 4.3 software.
Data analysis
Data were presented as means ± standard deviations. Statistical analyses were assessed using Student’s t-test. Statistically significant differences were indicated by P-values of <0.05.
Results
Characterization of P-R, FP-R, and FR-loaded L-Lp
In Table 1, the calculated apparent molecular masses and PEG-grafting number for the conjugates are listed. As anticipated, with quite narrow molecular weight distribution, both of the conjugates were mono-PEGlyated. To direct comparison among data obtained by fluorescent imaging, the molar fluorescent ratios of FP20k-R and FP40k-R to FR were determined, which were calculated to be 1.15 and 0.737, respectively.
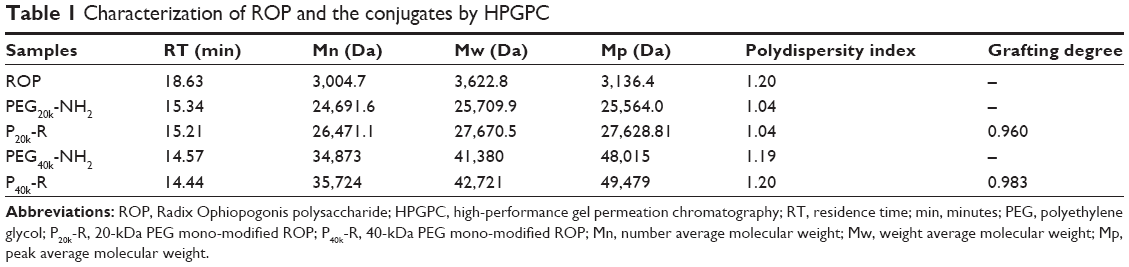 | Table 1 Characterization of ROP and the conjugates by HPGPC |
As measured by the dynamic light scattering, FR-loaded L-Lps had the size of 171.7±7.7 nm with polydispersity index being 0.114±0.071 (n=3). At pH 7.4, they had a negative charge of −1.41±0.45 mV and the ROP loading was 77.73±1.45 mg/mmol (n=3). The in vitro drug leakage test showed that FR-loaded L-Lps were pretty stable in rat plasma with the cumulative FR releases being only 7.37%±0.34%, 12.52%±0.70%, and 17.89%±0.51% at 3, 8, and 24 hours, respectively (n=3).
Plasma pharmacokinetics
In this work, pharmacokinetics of drugs was evaluated in rats following intravenous administration at a same molar dose of 4 μmol/kg, which is the effective dose of ROP, P20k-R, and P40k-R.13,29 As clearly shown in Table 2 and Figure 1, both PEG conjugation and loading in L-Lp improved the pharmacokinetic behaviors of ROP remarkably. Compared to FR, 43-, 24-, and 32-fold increases in area under the curve (AUC), mean residence time (MRT), and t1/2 were achieved by FP20k-R, respectively. Approximately two to three times further increases in these pharmacokinetic parameters were observed for both FP40k-R and FR-loaded L-Lp, compared to FP20k-R. There is no significant difference in the parameters between FP40k-R and FR-loaded L-Lp, although according to the values, FP40k-R showed slightly prolonged blood retention compared to FR loaded in L-Lp. At 3 hours, the residues of approximately 24%, 38%, and 41% of the original dose in blood were observed for FP20k-R, FP40k-R, and FR-loaded L-Lp, respectively. The values at 12 hours were 5%, 19%, and 22%, respectively. Without the help of any carrier, approximately 93% and 99% of ROP have already been eliminated from the blood at 10 and 60 minutes postdose, respectively.
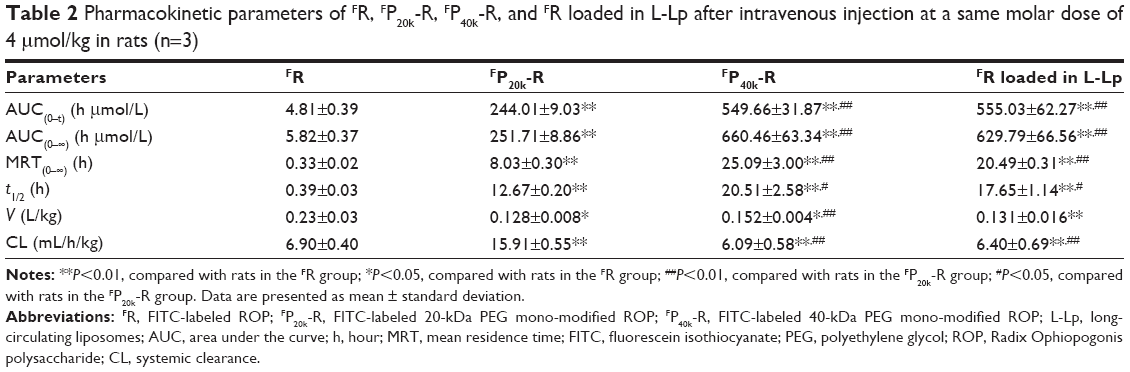 | Table 2 Pharmacokinetic parameters of FR, FP20k-R, FP40k-R, and FR loaded in L-Lp after intravenous injection at a same molar dose of 4 μmol/kg in rats (n=3) |
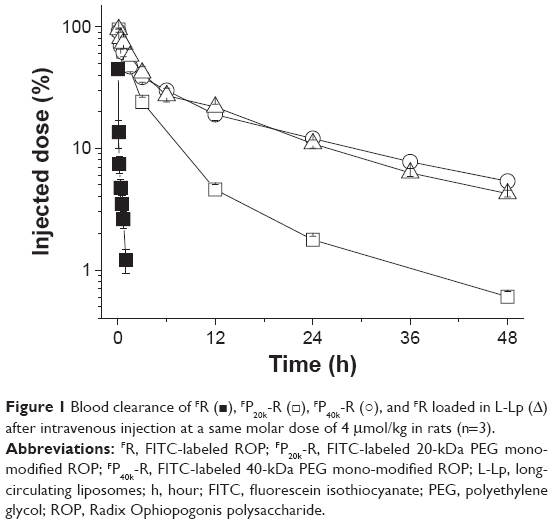 | Figure 1 Blood clearance of FR (◼), FP20k-R (□), FP40k-R (○), and FR loaded in L-Lp (Δ) after intravenous injection at a same molar dose of 4 μmol/kg in rats (n=3). |
Validation of the fluorescent imaging
As shown in Table 3 and Figures 2 and 3, the feasibility of quantitative analysis of cardiac drug distribution by the ex vivo bioluminescene imaging was validated. First, the episode of IR itself did not cause any meaningful change in the fluorescent intensity of background. At 3 and 12 hours after reperfusion, the fluorescent intensity ratios of infarcted and noninfarcted myocardium to normal myocardium were all close to 1 (Table 3), which also indicated that the background fluorescence intensity of myocardium was nearly constant in the rats used. Second, uneven drug distribution can be clearly seen in myocardial ischemic rats, but not in normal rats (Figure 2). More importantly, the areas with darker color coincided well with the infarcted zones revealed by the histochemical staining, indicating the preferential distribution of drugs in the infarcted zones (Figure 3). Finally, significantly higher than background fluorescence intensities were achieved by drugs, which, together with the nearly constant background value, makes the quantitative analysis feasible (Table 3).
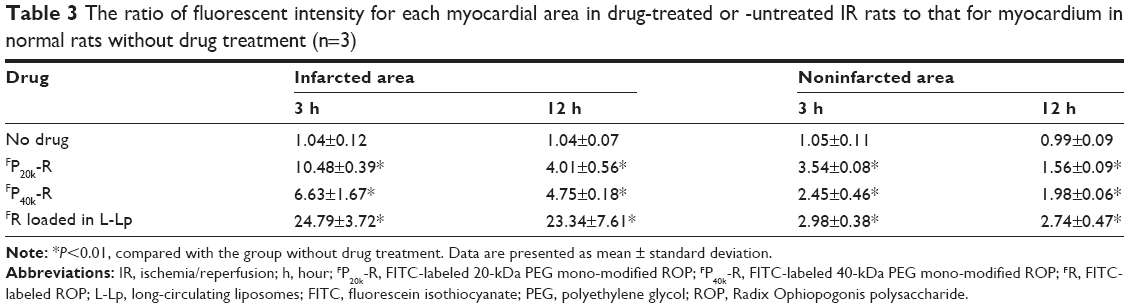 | Table 3 The ratio of fluorescent intensity for each myocardial area in drug-treated or -untreated IR rats to that for myocardium in normal rats without drug treatment (n=3) |
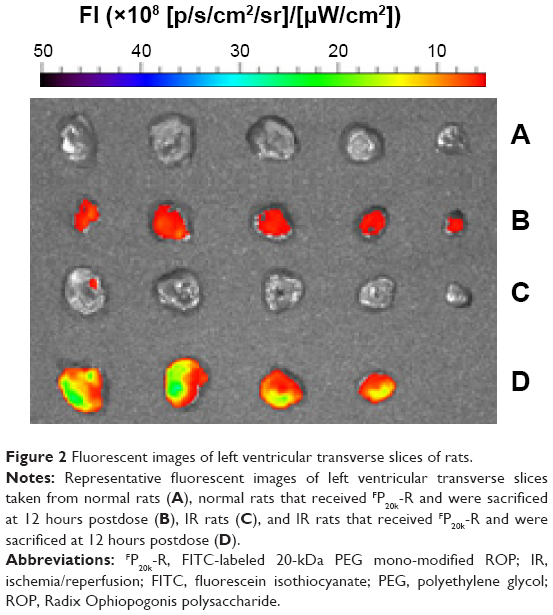 | Figure 2 Fluorescent images of left ventricular transverse slices of rats. |
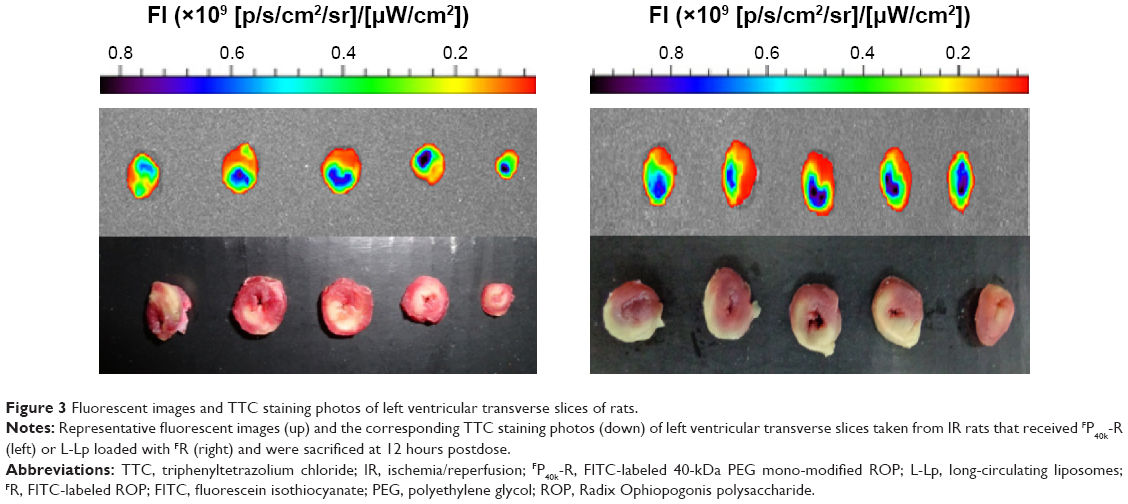 | Figure 3 Fluorescent images and TTC staining photos of left ventricular transverse slices of rats. |
Cardiac distribution
The infarct sizes in IR rats after 3, 12, and 24 hours reperfusion were 36.4%±2.3%, 39.9%±8.6%, and 34.5%±7.7%, respectively (n=3–4 for each time point), indicating that the infarct sizes should be time-independent during these periods. The representative photographs of transverse slices following the TTC staining were shown in Figure 3.
Figure 4 shows changes in drug level with time postdose in both infarcted and noninfarcted myocardium of IR rats. It can be clearly seen that drug levels were several times higher in infarcted myocardium than in noninfarcted myocardium at all the time points studied. Coincided with the fast clearance from blood, FR cannot be detected in myocardium at 3 hours postdose. FP20k-R and FP40k-R levels in both infarcted and noninfarcted myocardium at 3 hours postdose were comparable, being approximately 68% and 96% of those of FR at 10 minutes postdose, respectively. However, FP40k-R showed prolonged retention in myocardium compared to FP20k-R. With an approximately fourfold higher level in blood, FP40k-R levels in infarcted and noninfarcted myocardium were approximately twofold and threefold higher than those of FP20k-R at 12 hours postdose, respectively. Intriguingly, approximately threefold and fourfold higher than FP40k-R drug levels were observed for FR-loaded L-Lp in infarcted myocardium at 3 and 12 hours postdose, respectively, although their levels in blood and noninfarcted myocardium were almost the same at both the time points.
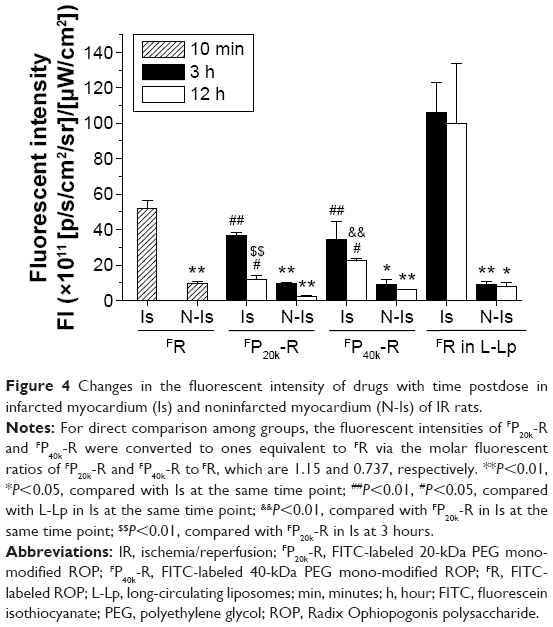 | Figure 4 Changes in the fluorescent intensity of drugs with time postdose in infarcted myocardium (Is) and noninfarcted myocardium (N-Is) of IR rats. |
Figure 5 shows the infarcted myocardium targeting efficacy of each case, which was calculated as the ratio of drug fluorescence intensity in the infarcted myocardium to that in the noninfarcted myocardium of the same slice. Approximately, all the values at 3 hours postdose are comparable to those at 12 hours postdose, suggesting that this parameter might have no remarkable time dependence. It is interesting to note that there is no increase in targeting efficacy after PEG conjugation. The values for FP20k-R and FP40k-R at 3 and 12 hours postdose, which range from 3.74 to 5.37, only approach the value of 5.40 for FR itself at 10 minutes postdose. However, an ~2.4-fold increase was achieved by loading FR in L-Lp.
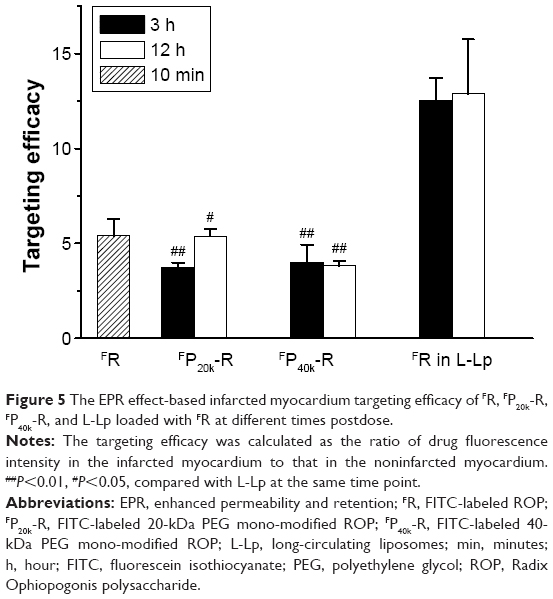 | Figure 5 The EPR effect-based infarcted myocardium targeting efficacy of FR, FP20k-R, FP40k-R, and L-Lp loaded with FR at different times postdose. |
Discussion
The EPR effect has been demonstrated to occur in some pathological areas such as tumors33–35 and ischemic myocardium.4,6,18,36 Size dependence is the most prominent feature of the effect. That is to say, the EPR effect-based passive targeting depends to a large degree on the size of drug or carriers. It was observed that PEG-coated liposomes (mean diameter 134±21 nm; zeta potential −2.3±1.1 mV) were capable of accumulating approximately threefold higher (P<0.05) in the border (risk areas) and fivefold higher (P<0.05) in the infarcted myocardium than in normal heart tissues at 3 hours postdose in a rat model of IR.6 PEG-PE micelles with a size of 7–20 nm and zeta potential of approximately −18 mV were demonstrated to passively accumulate in the infarcted myocardium with efficiency approximately eightfold higher (P<0.001) than in the normal heart tissues at 3 hours postdose in a rabbit model of IR.18 In a recent study, the accumulation behaviors of micelles and liposomes were characterized in mouse models of both acute and chronic myocardial infarction.4,11 It was found that micelles (~15 nm) permeated the entire infarcted area, which renders them very suited for the local delivery of cardioprotective or antiremodeling drugs. In comparison, liposomes (~100 nm) displayed slower and more restricted extravasation from the vasculature and are therefore an attractive vehicle for the delivery of pro-angiogenic drugs.4
Compared to nanosized insoluble carriers, polymeric drugs with the size order of several to tens of nanometers were only limitedly studied on this aspect. Given at a same molar dose, 20 and 40 kDa PEGs were reported to distribute ~1.5-fold and ~1.9-fold more in ischemic mouse hearts caused by isoprenaline injection than in normal ones, respectively.37 In addition, with a comparable AUC0.5–24h, 20-kDa PEG exhibited a quicker distribution to and elimination from myocardium, while 40-kDa PEG achieved a smoother cardiac level-time profile. This suggests that conjugates with higher molecular weight might be more suitable for a drug with narrow therapeutic window or severe cardiac toxicity. However, since PEG levels were measured by treating the whole heart that is composed of not only ischemic myocardium but also normal ones, the targeting efficacy was underestimated to a certain degree in the study. A targeting efficacy of ~2 was also reported for both native ROP16 and 20-kDa PEG mono-modified ROP,38 but the value was underestimated with the same reason.
To our best knowledge, there is no report on direct comparison between the aforementioned two strategies about their cardiac distribution and retention behaviors, which is exactly the aim of this study. As a whole, the results obtained suggest that loading in L-Lp is a better strategy for delivering ROP to the infarcted myocardium than mono-PEGylation in terms of both accumulation and retention behaviors. Approximately, a trend similar to the aforementioned, that is, nanosized insoluble carriers could achieve a higher targeting efficacy than polymeric conjugation, was also observed in this study. Compared to ROP-loaded L-Lp, the 2.4–3.4 times lower targeting efficacy and the 2.9–8.6 times lower drug exposure in infarcted myocardium for P20k-R and P40k-R are believed to be due to the soluble, linear, and flexible structure of the conjugates, which made the conjugates 1) spread evenly in blood rather than precipitate onto the luminal surface of the blood vessel endothelium at blood flow-reduced or -stopped ischemic areas, 2) get entangled with molecules and structures in its pathway to interstitial spaces, and 3) be able to diffuse through the normal vessel endothelium and extracellular space by a “snake-like” movement.
In addition, both the targeting efficacy values got here for L-Lp and mono-PEGylated ROPs are higher than those previously reported. For L-Lp, the factors that cause the difference might include but not limited to carrier size, size distribution, zeta potential, formulation, and in vivo stability, among which size should be the most prominent one. With larger diameter (171.7±7.7 nm), distribution of the L-Lp we prepared in normal myocardium should be further limited, thus causing a higher targeting efficacy of ~12.7. Besides, difference in the severity of cardiac damage should also be a contributing factor. Considering that 1) the increased vascular permeability caused by the IR injury persisted for at least 48 hours and gradually recovered within 2 weeks39 and 2) the therapeutic interventions proven to reduce the infarct size in both experimental and clinical models were often administered at the time of myocardial reperfusion;40 each drug investigated here was injected via the tail vein immediately after the IR surgery finished. The ischemic degree was proven to be severe, and the average infarct size within 3–24 hours reperfusion was 37.2%±6.7% (n=10). As to mono-PEGylated ROPs, the difference should be caused by the different methodology used. Isoprenaline injection leads to small and scattered infarct areas in the whole heart, making the isolation of infarcted myocardium from normal ones impossible.41 In comparison, transient coronary artery ligation used in this study causes a large infarct zone mainly in the left ventricle, and thanks to the use of fluorescent imaging, infarcted and noninfarcted areas in each left ventricular transverse slice can be assayed separately. As a result, the targeting efficacy was determined to be ~4.5 for P20k-R and P40k-R without underestimation. In addition, it was reported that choral hydrate, which was used as anesthetic in the preparation of rat model of myocardial IR injury in the study, could inhibit the respiratory and vasomotor centers of medulla oblongata, and, thus, cause decreases in both the basic metabolic rate and the blood circulation rate of rats. This might influence drug distribution to some degree. However, considering that the targeting efficacy is a ratio, the effect on it might be offset by the simultaneous drug decrease in both infarcted and noninfarcted areas.
Conclusion
This study demonstrated cardiac delivery characteristics of ROP-loaded L-Lp and mono-PEGylated ROPs (P20k-R and P40k-R) via a verified fluorescent imaging method. With comparable blood pharmacokinetics, ROP-loaded L-Lp showed both significantly higher targeting efficacy and drug exposure in infarcted myocardium than P40k-R. As to P20k-R, both its targeting efficacy and its level in infarcted myocardium at 3 hours postdose were comparable to P40k-R, but its level in blood and myocardium reduced obviously faster with time. As a whole, the results obtained here indicate that both loading in L-Lps and mono-PEGylation are effective in passively targeting drug to ischemic myocardium, but the former appears to induce stronger effects.
Acknowledgments
This work was supported by the Program for New Century Excellent Talents in University (NCET-13-0906), the National Natural Science Foundation of China (81073065), the funds from Shanghai Municipal Commission of Health and Family Planning (ZY3-CCCX-3-5001) and Science and Technology Commission of Shanghai Municipality (15DZ2292000), the Science and Technology Development Fund of Pudong New Area (PKF-2013-003), and the “085” Project (085ZY1219) of Shanghai University of TCM.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization. Health Statistics and Informatics Department, Causes of Death 2008 Summary Tables. Geneva: WHO; 2011. | ||
Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123(1):92–100. | ||
Ware JA, Simons M. Angiogenesis in ischemic heart disease. Nat Med. 1997;3(7):158–164. | ||
Paulis LE, Geelen T, Kuhlmann MT, et al. Distribution of lipid-based nanoparticles to infarcted myocardium with potential application for MRI-monitored drug delivery. J Control Release. 2012;162(2):276–285. | ||
Dvir T, Bauer M, Schroeder A, et al. Nanoparticles targeting the infarcted heart. Nano Lett. 2011;11(10):4411–4414. | ||
Takahama H, Minamino T, Asanuma H, et al. Prolonged targeting of ischemic/reperfused myocardium by liposomal adenosine augments cardioprotection in rats. J Am Coll Cardiol. 2009;53(8):709–717. | ||
Verma DD, Hartner WC, Levchenko TS, Bernstein EA, Torchilin VP. ATP-loaded liposomes effectively protect the myocardium in rabbits with an acute experimental myocardial infarction. Pharm Res. 2005;22(12):2115–2120. | ||
Verma DD, Hartner WC, Thakkar V, Levchenko TS, Torchilin VP. Protective effect of coenzyme Q10-loaded liposomes on the myocardium in rabbits with an acute experimental myocardial infarction. Pharm Res. 2007;24(11):2131–2137. | ||
Scott RC, Rosano JM, Ivanov Z, et al. Targeting VEGF-encapsulated immunoliposomes to MI heart improves vascularity and cardiac function. FASEB J. 2009;23(10):3361–3367. | ||
Binder A, Ali A, Chawla R, Aziz HA, Abbate A, Jovin IS. Myocardial protection from ischemia-reperfusion injury post coronary revascularization. Expert Rev Cardiovasc Ther. 2015;13(9):1045–1057. | ||
Nguyen J, Sievers R, Motion JP, Kivimae S, Fang Q, Lee RJ. Delivery of lipid micelles into infarcted myocardium using a lipid-linked matrix metalloproteinase targeting peptide. Mol Pharm. 2015;12(4):1150–1157. | ||
Xu DS, Feng Y, Zhou YH, Zhang X, Lin X, Deng HL. Active components of polysaccharide of Ophiopogon japonicus on acute myocardial ischemia. Chin Tradit Pat Med. 2004;26(10):832–837. | ||
Wang S, Zhang Z, Lin X, Xu DS, Feng Y, Ding K. A polysaccharide, MDG-1, induces S1P1 and bFGF expression and augments survival and angiogenesis in the ischemic heart. Glycobiology. 2010;20(4):473–484. | ||
Zheng Q, Feng Y, Xu DS, Lin X, Chen YZ. Influence of sulfation on anti-myocardial ischemic activity of Ophiopogon japonicus polysaccharide. J Asian Nat Prod Res. 2009;11(4):306–321. | ||
Lin X, Xu DS, Feng Y, Shen L. Determination of Ophiopogon japonicus polysaccharide in plasma by HPLC with modified postcolumn fluorescence derivatization. Anal Biochem. 2005;342(2):179–185. | ||
Lin X, Wang ZJ, Sun GL, Shen L, Xu DS, Feng Y. A sensitive and specific HPGPC-FD method for the study of pharmacokinetics and tissue distribution of radix ophiopogonis polysaccharide in rats. Biomed Chromatogr. 2010;24(8):820–825. | ||
Rodriguez M, Cai WJ, Kostin S, Lucchesi BR, Schaper J. Ischemia depletes dystrophin and inhibits protein synthesis in the canine heart: mechanism of myocardial ischemic injury. J Mol Cell Cardiol. 2005;38(5):723–733. | ||
Lukyanov AN, Hartner WC, Torchilin VP. Increased accumulation of PEG-PE micelles in the area of experimental myocardial infarction in rabbits. J Control Release. 2004;94(1):187–193. | ||
Sun GL, Lin X. Mechanisms and strategies for targeting drugs to myocardial ischemic regions. Acta Pharm Sin. 2010;45(7):827–832. | ||
Torchilin VP. Targeting of drugs and drug carriers within the cardiovascular system. Adv Drug Deliv Rev. 1995;17:75–101. | ||
Scott RC, Crabbe D, Krynska B, Ansari R, Kiani MF. Aiming for the heart: targeted delivery of drugs to diseased cardiac tissue. Expert Opin Drug Deliv. 2008;5(4):459–470. | ||
Galagudza M, Korolev D, Postnov V, et al. Passive targeting of ischemic-reperfused myocardium with adenosine-loaded silica nanoparticles. Int J Nanomedicine. 2012;7:1671–1678. | ||
Khaw BA, Torchilin VP, Vural I, Narula J. Plug and seal: prevention of hypoxic cardiocyte death by sealing membrane lesions with antimyosin-liposomes. Nat Med. 1995;1:1195–1198. | ||
Khaw BA, DaSilva J, Hartner WC. Cytoskeletal-antigen specific immunoliposome-targeted in vivo preservation of myocardial viability. J Control Release. 2007;120(1–2):35–40. | ||
Takahama H, Shigematsu H, Asai T, et al. Liposomal amiodarone augments anti-arrhythmic effects and reduces hemodynamic adverse effects in an ischemia/reperfusion rat model. Cardiovasc Drugs Ther. 2013;27(2):125–132. | ||
Levchenko TS, Hartner WC, Torchilin VP. Liposomes for cardiovascular targeting. Ther Deliv. 2012;3(4):501–514. | ||
Duncan R, Veronese FM. PEGylated protein conjugates: a new class of therapeutics for the 21st century. In: Veronese FM, editor. PEGylated Protein Drugs: Basic Science and Clinical Applications. Basel, Switzerland: Birkhäuser Verlag; 2009:1–9. | ||
Schlesinger N, Yasothan U, Kirkpatrick P. Pegloticase. Nat Rev Drug Discov. 2011;10:17–18. | ||
Sun GL, Lin X, Shen L, et al. Mono-PEGylated radix ophiopogonis polysaccharide for the treatment of myocardial ischemia. Eur J Pharm Sci. 2013;49(4):629–636. | ||
Xu DS, Feng Y, Lin X, Deng HL, Fang JN, Dong Q. Isolation, purification and structural analysis of a polysaccharide MDG-1 from Ophiopogon japonicus. Acta Pharm Sin. 2005;40(7):636–639. | ||
Lin X, Wang S, Jiang Y, et al. Poly(ethylene glycol)-radix ophiopogonis polysaccharide conjugates: preparation, characterization, pharmacokinetics and in vitro bioactivity. Eur J Pharm Biopharm. 2010;76(2):230–237. | ||
Ye J, Yang L, Sethi R, et al. A new technique of coronary artery ligation: experimental myocardial infarction in rats in vivo with reduced mortality. Mol Cell Biochem. 1997;176(1–2):227–233. | ||
Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63(3):131–135. | ||
Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2011;41:189–207. | ||
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. | ||
Torchilin VP. Drug targeting. Eur J Pharm Sci. 2000;11(suppl 2):S81–S91. | ||
Sun GL, Lin X, Hong YL, Feng Y, Ruan KF, Xu DS. PEGylation for drug delivery to ischemic myocardium: Pharmacokinetics and cardiac distribution of poly(ethylene glycol)s in mice with normal and ischemic myocardium. Eur J Pharm Sci. 2012;46(5):545–552. | ||
Lin X, Wang ZJ, Wang S, et al. Comparison of tissue distribution of a PEGylated radix ophiopogonis polysaccharide in mice with normal and ischemic myocardium. Eur J Pharm Biopharm. 2011;79(3):621–626. | ||
Horwitz LD, Kaufman D, Keller MW, Kong Y. Time course of coronary endothelial healing after injury due to ischemia and reperfusion. Circulation. 1994;90(5):2439–2447. | ||
Oerlemans MI, Koudstaal S, Chamuleau SA, de Kleijn DP, Doevendans PA, Sluijter JP. Targeting cell death in the reperfused heart: pharmacological approaches for cardioprotection. Int J Cardiol. 2013;165(3):410–422. | ||
Liu Z, Liu LL, Yang TT. Comparative study of myocardial infarction models in two kinds of rat. Prog Vet Med. 2010;31(4):19–25. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.