Back to Archived Journals » Gastrointestinal Cancer: Targets and Therapy » Volume 5
Targeting cancer stem cells in hepatocellular carcinoma
Authors He AR, Smith D, Mishra L
Received 26 February 2014
Accepted for publication 25 June 2014
Published 17 December 2014 Volume 2015:5 Pages 1—10
DOI https://doi.org/10.2147/GICTT.S44250
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Aiwu Ruth He,1 Daniel C Smith,1 Lopa Mishra2
1Lombardi Comprehensive Cancer Center, Georgetown University, Washington, DC, 2Department of Gastroenterology, Hepatology, and Nutrition, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Abstract: The poor outcome of patients with hepatocellular carcinoma (HCC) is attributed to recurrence of the disease after curative treatment and the resistance of HCC cells to conventional chemotherapy, which may be explained partly by the function of liver cancer stem cells (CSCs). Liver CSCs have emerged as an important therapeutic target against HCC. Numerous surface markers for liver CSCs have been identified, and include CD133, CD90, CD44, CD13, and epithelial cell adhesion molecules. These surface markers serve not only as tools for identifying and isolating liver CSCs but also as therapeutic targets for eradicating these cells. In studies of animal models and large-scale genomic analyses of human HCC samples, many signaling pathways observed in normal stem cells have been found to be altered in liver CSCs, which accounts for the stemness and aggressive behavior of these cells. Antibodies and small molecule inhibitors targeting the signaling pathways have been evaluated at different levels of preclinical and clinical development. Another strategy is to promote the differentiation of liver CSCs to less aggressive HCC that is sensitive to conventional chemotherapy. Disruption of the tumor niche essential for liver CSC homeostasis has become a novel strategy in cancer treatment. To overcome the challenges in developing treatment for liver CSCs, more research into the genetic makeup of patient tumors that respond to treatment may lead to more effective therapy. Standardization of HCC CSC tumor markers would be helpful for measuring the CSC response to these agents. Herein, we review the current strategies for developing treatment to eradicate liver CSCs and to improve the outcome for patients with HCC.
Keywords: cancer stem cell, therapeutic targets, surface maker, signaling pathways, transforming growth factor beta
Introduction
Hepatocellular carcinoma (HCC), a major cause of worldwide morbidity and mortality, is the third leading cause of cancer-related death.1 Age-adjusted HCC incidence rates in the USA tripled between 1975 and 2005.2 For early-stage disease, the curative treatment options include resection, radiofrequency ablation, and transplantation. For patients who have multifocal lesions in the liver without vascular invasion, transarterial chemoembolization has been shown to prolong survival. Treatment for patients with advanced HCC is limited. Sorafenib, a multikinase inhibitor, is the only systemic treatment approved by the US Food and Drug Administration that has conferred a modest survival benefit in this group by prolonging overall survival by 2 months in a randomized Phase III clinical trial.3 However, because of recent advances in the molecular pathogenesis of HCC, specifically in our understanding of cancer stem cell (CSC) biology, there is potential for the development of many novel pharmacologic targets and therapeutic strategies.
Liver cancer stem cells
A CSC concept was proposed several decades ago to explain two recurring observations, ie, that most cancers consist of phenotypically heterogeneous tumor cells resembling distinct stages of normal tissue development, and that only a fraction of cells from both hematologic and solid malignancies are tumorigenic.4–6
Accumulating evidence supports the origination of HCC from transformation of liver stem/progenitor cells. Approximately 40% of HCC cases have clonality and are thus believed to originate from progenitor/stem cells.7–10 The liver has high regenerative potential, and small oval hepatic progenitor cells around the peripheral branches of the bile ducts, the canals of Hering, can differentiate into biliary epithelial cells and hepatocytes.11 These oval liver progenitor cells share molecular markers with adult hepatocytes (albumin, cytokeratin 7, CK19, oval cell markers [OV6, A6, and OV1], chromogranin-A, and neural cell adhesion molecules) and fetal hepatocytes (α-fetoprotein).7,11 They are also positive for more common stem cell markers such as CD34+, Thy-1+, c-Kit+, and FMS-like tyrosine kinase 3.12
The role of liver progenitor cells in hepatocarcinogenesis was first suggested in the Solt-Farber and diethylnitrosamine models of experimental liver injury and hepatocarcinogenesis in the rat.13,14 Since then, more data have suggested that liver CSCs are responsible for cancer initiation, local recurrence, metastasis, and therapy resistance in subsets of HCC. In addition, microarray-generated molecular classifications of HCC suggest a potential stem cell origin in a subpopulation of human HCC and underline prognostic implications for the prospective analysis of putative CSCs.15 Taken together, liver CSCs have emerged as the therapeutic target for HCC, and there is increasing interest in identifying strategies eradicating CSCs.16,17
CSC markers and potential therapeutic targets against liver CSCs
Numerous surface markers for HCC stem cells have been identified, and include CD133, CD90, CD44, CD13, and epithelial cell adhesion molecules (EpCAMs).18–21 Although their roles in liver CSCs are not fully understood, studies have shown that targeting these markers can specifically inhibit liver CSCs with high efficacy. It is reported that the tumorigenicity and invasive capacity of liver CSCs can be impaired by targeting CSC surface markers, leading to reduction of the CSC pool.
A handful of new therapeutic agents have been developed to target CSC markers. VB4-845, an immune recombinant fusion protein targeting EpCAMs, has been shown to strongly suppress sphere formation and tumor formation in vivo by decreasing the cancer cell population expressing CD133 and CD13.22 Targeting of the CD44 isoforms in HCC was able to selectively deplete CD44-positive HCC cells.23 The anti-CD44 antibody-mediated liposomal nanoparticle has been developed to target CSCs and monitor cancer progression or regression in HCC cells.24 8-bromo-7-methoxychrysin, a synthetic analog of chrysin, has inhibited the proliferation, self-renewal, and invasion of liver CSCs in vitro and in vivo, downregulated the expression of liver CSC biomarkers CD133 and CD44, and induced epithelial-mesenchymal transition by downregulating the expression of Twist and β-catenin in liver CSCs.25 Therefore, direct targeting of liver CSC-specific markers may be a promising therapeutic strategy for eradicating liver CSCs.
Stemness signaling that serves as a therapeutic target for HCC CSCs
One alternative approach to targeting CSCs has been to examine the cellular pathways that are required for regulation of normal stem cells. Many signaling pathways observed in normal stem cells can also be detected in CSCs. These pathways include but are not limited to Wnt/β-catenin, Hedgehog, and Notch signaling pathways.26
Disturbing the signaling involved in normal stem cell fate reportedly decreased the self-renewal and proliferating capabilities of CSCs. The signaling pathways, which are proposed to regulate liver CSC function and to contribute to the aggressive behavior of HCC, are often found to have the characteristics summarized in Figure 1. By reviewing the literature, we found that seven pathways have four to five of the properties described in Figure 1 that provide promising targets for treating HCC against liver CSCs.
 | Figure 1 Properties of candidate signaling pathways that regulate liver cancer stem cell function. |
Wnt signaling
The Wnt signaling pathways are involved in determining the proliferation and fate of embryonic and adult stem cells. The cascade has also been shown to regulate cell axis patterning and cell migration in the developing embryo. Dysregulation of this pathway is found in the carcinogenesis of multiple cancer types, including HCC. Liver-targeted disruption of adenomatous polyposis coli (APC) in mice activates β-catenin signaling and leads to HCC tumorigenesis.27
The Wnt pathway shares a common initiating event, a Wnt family protein binding to a transmembrane Frizzled G-protein coupled receptor (along with other requisite coreceptors and ligands), thereby activating the intracellular protein Dishevelled. The activated Frizzled-Dishevelled complex leads to intracellular accumulation of β-catenin by deactivating the ubiquitinating β-catenin destruction complex. High amounts of β-catenin then translocate into the nucleus, where they induce phenotypic changes through transcription factor activation.28 Epithelial cell adhesion molecule (EpCAM), the downstream gene of Wnt signaling, forms a positive feedback loop in the promotion of liver CSC proliferation by activation of Wnt signaling. Upon proteolysis activation, EpCAM releases its intracellular domain, which translocates to the nucleus and associates with β-catenin and Lef-1 to transactivate genes and promote cell proliferation.29 EpCAM-positive HCCs have been shown to have a poor prognosis and more aggressive behavior after xenotransplantation in nonobese diabetic/severe combined immunodeficient mice, thereby rendering EpCAM a putative marker of HCC CSCs.29,30
The Wnt pathway can be inhibited by blocking β-catenin interaction with the TCF gene; when this pathway was inhibited in one study, CSC numbers and spheroid formation were reduced.31,32 These findings, showing the importance of the Wnt pathway in CSC viability, have led to a number of novel approaches to HCC treatment.32–37 However, no clinical trials are under way for Wnt-specific small molecule inhibitors in HCC.
TGF-β signaling
Transforming growth factor beta (TGF-β) signaling pathways can either promote or inhibit tumorigenesis, depending on the cell microenvironment. TGF-β is excreted in an autocrine fashion in varying amounts, depending on the cell type. Activated TGF-β surface receptors II and/or III (TGFBII/III) form a complex with TGF-R1, which then activates the protein kinase intracellular domain of TGF-RI. This protein kinase activates Smad transcription factors 2 and 3, either of which then forms a complex with Smad 4 proteins, translocating to the nucleus to regulate gene expression.
A functional role of TGF-β signaling in liver stem cell niches has been demonstrated through mouse genetics. Disruption of TGF-β signaling yields a phenotype similar to that of a human CSC disorder, Beckwith-Wiedemann syndrome, a disease characterized by stem cell alteration.38 Loss or reduced expression of the TGF-β receptor (TβRI/TβRII) or signaling molecules (eg, Smad 4) also enhances malignant progression in various human tumor types, cancer xenografts, and transgenic mice.39–45 This is at least partially due to the activation of mitogenic and oncogenic pathways involving CDK4, PRAJA, β-catenin, TERT, and c-MYC, that occurs when the TGF-β pathway is inactivated.46
Disruption of TGF-β signaling by genetically removing one copy of Sptbn1, a Smad3 adapter protein, results in spontaneous development of HCC; expression analysis of these tumors highlighted marked activation of genes involved in the interleukin (IL)-6 signaling pathway, including IL-6 and Stat3.10
Chronic inflammation is a major risk factor for cancer development. The interaction between liver CSCs and surrounding immune cells plays an important role in hepatocarcinogenesis. Suppressed TGF-β signaling activates Toll-like receptor 4/NANOG, a mediator of the immune and inflammatory response that increases pluripotency genes and tumorigenesis and promotes the chemoresistance of liver CSCs.47 In contrast, excessive TGF-β could increase expression of the CSC marker in HCC and render HCC highly tumorigenic after xenotransplantation.48 Furthermore, when liver CSCs were exposed to TGF-β for long periods of time (similar to what occurs in a cirrhotic liver), they gained CSC properties, including increased marker expression, tumorigenicity, chemoresistance, and self-renewal capacity. A number of drugs targeting aberrant TGF-β signaling are in development and have shown promising preclinical activity in arresting growth and metastasis of HCC.49–55 A TGF-β receptor tyrosine kinase inhibitor, Ly2157299, is currently being evaluated in HCC, with promising results.56
JAK-STAT
The Janus kinase (JAK)-STAT pathway is a unique intracellular signaling cascade that coactivates cytokine receptors after extracellular ligand binding, such as interferon, osteopontin, IL-6, and oncostatin. The binding of JAK protein to the intracellular domain of certain ligand-activated cytokine receptors (most notably interferon receptors) initiates tyrosine transphosphorylation between JAK proteins and the intracellular domain of the cytokine receptor. The resulting phosphorylated tyrosine binds the SH2 domain of STAT proteins, causing their dimerization/activation. Dimerized STAT proteins can interact with other proteins within the cytoplasm or translocate to the nucleus to act as transcription factors. The JAK-STAT pathway has been shown to play a key role in hematopoiesis, differentiation, and clonal expansion of immune cells, and immune cytokine transduction in nonimmune cells.57
Mice with disruption of TGF-β signaling develop HCC and are found to have activation of IL-6/JAK/STAT3 signaling. Formation of HCC can be prevented by inhibition of IL-6/JAK/STAT3, indicating that IL-6/JAK/STAT3 plays an important role in the transformation of liver CSCs.10,58 STAT3 inhibitors suppress the proliferation of HCC cells.58 Recently, He et al reported that the liver progenitor cells are premalignant cells, and become malignant if they are transplanted in the damaged liver because transformation of these cells requires the correct microenviroment. These progenitor cells depend on autocrine IL-6 production and activation of IL-6/JAK/STAT3 during transformation in order to form HCC.59 The IL-6/JAK/STAT3 signaling pathway may serve as a target for the prevention and treatment of HCC.
Notch
The Notch pathway plays an important role in cell-cell signaling, inducing proliferation, homeostasis, and/or differentiation, depending on cell lineage. The initiating cell expresses a variety of cell-bound proteins (termed DSL ligands) that bind to the extracellular domain of the target cell’s transmembrane Notch family protein, inducing proteolytic cleavage of the intracellular and extracellular Notch components by γ-secretase and tumor necrosis factor-α component enzyme, respectively. The Notch intracellular domain is activated by further cleaving, translocates to the nucleus, and associates with CSL family DNA transcription factors to induce phenotypic changes in the target cell.60
Notch overexpression has been shown to be oncogenic in lung, breast, colon, and brain CSCs, and pharmacologic targeting of this pathway is already being studied in clinical trials for these cancers.61–65 When the intracellular domain of Notch was conditionally expressed in liver progenitor cells, transgenic mice developed HCC within 12 months.66,67
Targeting the Notch pathway could allow inhibition of CSC self-renewal and a decrease in tumor growth. Inhibition of Notch signaling by γ-secretase inhibitors has decreased EpCAM
Hedgehog
The Hedgehog pathway is an important embryonic patterning cascade. Paracrine excretion of Hedgehog protein causes a variety of responses in the receiving cell, depending on the receiving cell type, concentration of Hedgehog protein, and concentration of the receiving cell’s transmembrane receptor patched protein. Without Hedgehog binding, transmembrane receptor patched protein inhibits the transmembrane G-coupled receptor Smoothened. Hedgehog therefore activates Smoothened, which in turn activates zinc transcription factors in the GLI family through an incompletely understood mechanism. GLI proteins accumulate in the nucleus, activating or inhibiting Hedgehog gene targets.68
Chronic fibrosing liver injury is a major risk factor for hepatocarcinogenesis in humans. Mice with targeted deletion of Mdr2 (the murine ortholog of MDR3) develop chronic fibrosing liver injury and eventually HCC. Mdr2(-/-) mice consistently expressed Hedgehog ligands and progressively accumulated Hedgehog-responsive liver myofibroblasts and progenitors with age. Treatment of aged Mdr2-deficient mice with the Hedgehog signaling antagonist GDC-0449 significantly inhibited hepatic Hedgehog activity, decreased liver myofibroblasts and progenitors, reduced liver fibrosis, promoted regression of intrahepatic HCCs, and decreased the number of metastatic HCCs without increasing mortality.69 Enhanced Hedgehog signaling activity was found to contribute to chemoresistance in HCC. A specific Hedgehog inhibitor, cyclopamine, not only significantly blocked Hedgehog signaling activity but also inhibited the proliferation of liver CSCs, suggesting that Hedgehog signaling is critical for the tumorigenicity of CSC subpopulations.
Many Hedgehog pathway inhibitors are being evaluated, and most agents focus on targeting the Smoothened. receptor.70 Currently, agents targeting this pathway, including vismodegib (the most studied Smoothened inhibitor), are in preclinical and Phase I trials for HCC.
PI3K/AKT/mTOR and IGF1
The phosphatidylinositol-4 5-bisphosphate 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway plays an important role in cell proliferation and survival. Activation of the PI3K/AKT/mTOR pathway has been shown to accelerate hepatocarcinogenesis driven by N-Ras or β-catenin.71,72 Mutational inactivation of phosphatase and tensin homologue is found in a variety of cancers, including HCC.73,74 Insulin growth-like factor/mTOR signaling has been shown to increase chemoresistance and tumor-imitating capacity in liver CSCs.75–78 Small molecule inhibitors of this pathway have shown promise in a variety of cancers, including HCC.79
c-MET (hepatocyte growth factor receptor)
Another transmembrane receptor affecting multiple proliferative signaling pathways is the hepatocyte growth factor receptor, encoded by the c-MET oncogene. Hepatocyte growth factor receptor binds to the paracrine scatter factor, dimerizes, and transphosphorylates itself to create two catalytic tyrosine residues on its cytoplasmic domain. c-MET-positive cells demonstrated CSC-like characteristics, such as chemoresistance, tumor sphere formation, and increased expression of CD44 and ABCG2; furthermore, PHA665752, a c-MET inhibitor, suppressed tumor sphere formation and inhibited CD44 expression.80 In addition, hepatocyte growth factor receptor released by stromal cells around liver CSCs were shown to be required for liver CSC homeostasis.81 A variety of small molecule hepatocyte growth factor receptor/c-MET inhibitors are currently in development.82
Differentiation of liver CSCs
The differentiation of cancer cells into less aggressive forms has been a successful treatment strategy, as demonstrated in the treatment of acute promyelocytic leukemia. In one study, the application of all-trans retinoic acid after normal chemotherapy resulted in a 90% remission rate and a 70% cure rate in acute promyelocytic leukemia.83 Therapeutic agents that promote the differentiation of liver CSCs have also been used to deplete these cells. Hepatocyte nuclear factor-1 alpha (HNF1α), one of the key transcription factors of the hepatocyte nuclear factor family, plays a critical role in hepatocyte differentiation. A recombinant adenovirus carrying the HNF1α gene inhibits HCC xenograft growth in mice by inducing the differentiation of hepatoma cells into mature hepatocytes and G(2)/M arrest.84
Oncostatin M, an IL-6-related cytokine known to induce the differentiation of hepatoblasts into hepatocytes, has been shown to induce the hepatocytic differentiation of EpCAM(+) HCC cells. EpCAM(+) HCC cells treated with oncostatin M have shown enhanced cell proliferation with expansion of the EpCAM-negative non-CSC population. A combination of oncostatin M and conventional chemotherapy with fluorouracil has efficiently eliminated HCC by targeting both CSCs and non-CSCs.20 The identification of valid differentiation pathways in CSCs may lead to new therapeutic strategies against liver CSCs.
Targeting microRNAs in liver CSC
MicroRNAs (miRNAs) are a class of small non-coding RNAs involved in the transcriptional and post-transcriptional regulation of gene expression. Increasing evidence has demonstrated that miRNAs also play a critical role in normal liver development and in the fine-tuning of fundamental biological liver processes.85 Studies of miRNAs as biomarkers and therapeutic targets in HCC have focused on the group of miRNAs maintaining the stemness of liver CSCs, and attribute the resistance of HCC to cytotoxic chemotherapy. The miRNA-181 family is highly expressed in EpCAM+ AFP+ cells. miRNA-181 cells exert their function by targeting Caudal type homeobox transcription factor 2, GATA binding protein 6, and Nemo-like kinase, which are essential for hepatic cell differentiation and the Wnt pathway.85
miRNA-130b is significantly elevated in CD133+ liver CSCs and is required for self-renewal, tumorigenicity, and chemoresistance.86 miRNA-221 is another tumor-promoting miRNA. Overexpression of miRNA-221 in the liver results in spontaneous HCC development.87 In contrast with miRNA-181 and miRNA-130b, miRNA-26a is a tumor suppressor miRNA. Expression of miR-26a was significantly downregulated in a MYC-induced hepatocarcinoma murine model (tet-o-MYC; LAP-tTA mice), and this result was further confirmed by detecting the expression profiling of miR-26a in human HCC and normal liver biopsy specimens.88 The tumor suppressive role of miR-26a may be related to it’s ability to regulate cell cycle progression by targeting cyclin D2 and cyclin E2, two influential players in G1/S cell phase transition. Ectopic expression of miRNA-26a results in blockage of cell proliferation and induction of tumor cell apoptosis. Furthermore, miRNAs are shown to regulate the drug sensitivity of HCC to chemotherapy. miRNA-21 is found to lower the sensitivity of HCC to chemotherapy by inhibiting phosphatase and tensin homologue or programmed cell death 4, but miRNA-101 sensitizes HCC cells to chemotherapy by inhibiting myeloid cell leukemia sequence 1, a well characterized antiapoptotic member of the Bcl-2 family.89,90 Understanding the function of miRNAs in liver CSCs will provide a novel strategy to develop treatments against liver CSCs.
Cancer stem cell niche
Disruption of the tumor niche that is essential for CSC homeostasis has become a novel strategy in cancer treatment.91 As the main source of extracellular matrix proteins in the tumor stroma, hepatic stellate cells decrease the sensitization of HCC cells to chemotherapeutic agents by promoting epithelial-mesenchymal transition and CSC-like features via hepatocyte growth factor/Met signaling.81,92 Apart from the molecular content surrounding liver CSCs, there is accumulating evidence that the physical environment is a critical mediator of HCC tumor behavior.93 The stiffness of the matrix is a strong predictor of HCC development. Increasing stiffness was found to promote HCC cell proliferation.93 In contrast, a soft environment induced reversible stem cell characteristics in HCC.94
Summary and future directions
Liver CSCs have emerged as a therapeutic target for HCC, resulting in a paradigm shift from traditional therapies to those aimed at eradicating CSCs. Strategies for eliminating liver CSCs have been developed to target CSC markers, signaling transduction pathways, and the CSC niche. Clinical trials using inhibitors of TGF-β signaling (LY2157299, PF-03446962), the Hedgehog pathway (GDC-0449, PF-04449913, BMS-833923, IPI-926, TAK-441), the Notch pathway (RO4929097, BMS-906024, MK0752), and Wnt pathways (PRI-724) have started to emerge, but their efficacy with regard to CSC function remains to be determined.54 Table 1 shows urrent clinical trial progress in targeting embryonic stem cell pathways shown to be overexpressed in HCC CSCs. Challenges remain, including the expressive heterogeneity of these pathways between patient tumors, which render agents targeting a single pathway less effective against HCC. More study of the genetic makeup of patient tumors that respond to these small molecule inhibitors may lead to more effective therapy and stronger clinical trial outcomes. Standardization of HCC CSC tumor markers would be helpful for measuring the CSC response to these agents. The CSC theory is an exciting new paradigm that has shown promise for future therapy ex vivo. The translational effectiveness of targeting CSCs remains to be determined.
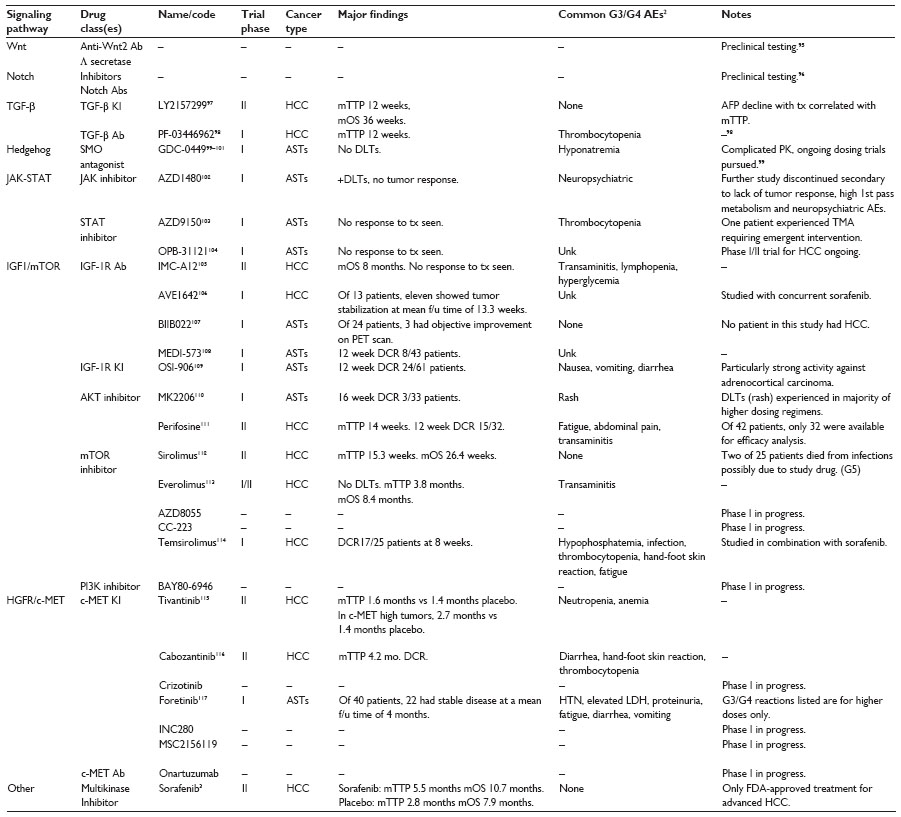 | Table 1 Clinical trials evaluating novel therapeutics targeting candidate signaling pathways that regulate liver cancer stem cell function |
Acknowledgments
This work was supported by American Cancer Society award number 118525-MRSG-10-068-01-TBE (ARH); R01 CA106614 (LM), R01 CA042857 (LM), R01 AA023146 (LM), P01 CA130821 (LM), Multidisciplinary Research Program Proposal (LM), and Science and Technology Acquisition and Retention Funding (LM), P30 CA016672 (RDP), and P30 DK56338 (ME).
Disclosure
The authors report no conflicts of interest in this work.
References
Parkin DM, Bray F, Ferlay J, et al. Estimating the world cancer burden: GLOBOCAN 2000. Int J Cancer. 2001;94:153–156. | |
Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009;27:1485–1491. | |
Genco C, Cabibbo G, Maida M, et al. Treatment of hepatocellular carcinoma: present and future. Expert Rev Anticancer Ther. 2013;13: 469–479. | |
Bruce WR, Van Der Gaag H. A quantitative assay for the number of murine lymphoma cells capable of proliferation in vivo. Nature. 1963;199:79–80. | |
Hamburger AW, Salmon SE. Primary bioassay of human tumor stem cells. Science. 1977;197:461–463. | |
Park CH, Bergsagel DE, McCulloch EA. Mouse myeloma tumor stem cells: a primary cell culture assay. J Natl Cancer Inst. 1971;46:411–422. | |
Roskams T. Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene. 2006;25:3818–3822. | |
Alison MR. Liver stem cells: implications for hepatocarcinogenesis. Stem Cell Rev. 2005;1:253–260. | |
Zender L, Spector MS, Xue W, et al. Identification and validation of oncogenes in liver cancer using an integrative onco genomic approach. Cell. 2006;125:1253–1267. | |
Tang Y, Kitisin K, Jogunoori W, et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc Natl Acad Sci U S A. 2008;105:2445–2450. | |
Roskams TA, Theise ND, Balabaud C, et al. Nomenclature of the finer branches of the biliary tree: canals, ductules, and ductular reactions in human livers. Hepatology. 2004;39:1739–1745. | |
Burke ZD, Thowfeequ S, Peran M, Tosh D. Stem cells in the adult pancreas and liver. Biochem J. 2007;404:169–178. | |
Sell S. Comparison of liver progenitor cells in human atypical ductular reactions with those seen in experimental models of liver injury. Hepatology. 1998;27:327–331. | |
Sell S, Leffert HL. Liver cancer stem cells. J Clin Oncol. 2008;26: 2800–2805. | |
Marquardt JU, Thorgeirsson SS. Stem cells in hepatocarcinogenesis: evidence from genomic data. Semin Liver Dis. 2010;30:26–34. | |
Yang W, Yan HX, Chen L, et al. Wnt/beta-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res. 2008;68:4287–4295. | |
Pang RW, Poon RT. Cancer stem cell as a potential therapeutic target in hepatocellular carcinoma. Curr Cancer Drug Targets. 2012;12: 1081–1094. | |
Mikhail S, He AR. Liver cancer stem cells. Int J Hepatol. 2011;2011: 486954. | |
Zhu Z, Hao X, Yan M, et al. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int J Cancer. 2010;126:2067–2078. | |
Yamashita T, Honda M, Nio K, et al. Oncostatin M renders epithelial cell adhesion molecule-positive liver cancer stem cells sensitive to 5-fluorouracil by inducing hepatocytic differentiation. Cancer Res. 2010;70:4687–4697. | |
Ma S, Chan KW, Hu L, et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132:2542–2556. | |
Ogawa K, Tanaka S, Matsumura S, et al. EpCAM-targeted therapy for human hepatocellular carcinoma. Ann Surg Oncol. 2014;21: 1314–1322. | |
Miletti-González KE, Chen S, Muthukumaran N, et al. The CD44 receptor interacts with P-glycoprotein to promote cell migration and invasion in cancer. Cancer Res. 2005;65:6660–6667. | |
Wang L, Su W, Liu Z, et al. CD44 antibody-targeted liposomal nanoparticles for molecular imaging and therapy of hepatocellular carcinoma. Biomaterials. 2012;33:5107–5114. | |
Ren KQ, Cao XZ, Liu ZH, et al. 8-bromo-5-hydroxy-7-methoxychrysin targeting for inhibition of the properties of liver cancer stem cells by modulation of Twist signaling. Int J Oncol. 2013;43:1719–1729. | |
Mishra L, Banker T, Murray J, et al. Liver stem cells and hepatocellular carcinoma. Hepatology. 2009;49:318–329. | |
Colnot S, Decaens T, Niwa-Kawakita M, et al. Liver-targeted disruption of APC in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proc Natl Acad Sci U S A. 2004;101:17216–17221. | |
Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis. 2008;4:68–75. | |
Maetzel D, Denzel S, Mack B, et al. Nuclear signalling by tumour-associated antigen EpCAM. Nat Cell Biol. 2009;11:162–171. | |
Yamashita T, Forgues M, Wang W, et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008;68:1451–1461. | |
Yamashita T, Ji J, Budhu A, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012–1024. | |
Lepourcelet M, Chen YN, France D, et al. Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer Cell. 2004;5:91–102. | |
Henrich CJ, Budhu A, Yu Z, et al. High-throughput screening for identification of inhibitors of epcam-dependent growth of hepatocellular carcinoma cells. Chem Biol Drug Des. 2013;82:131–139. | |
Chen Y, Yu D, Zhang C, et al. Lidamycin inhibits tumor initiating cells of hepatocellular carcinoma Huh7 through GSK3β/β-catenin pathway. Mol Carcinog. July 16, 2013. [Epub ahead of print.] | |
Wang F, He L, Dai WQ, et al. Salinomycin inhibits proliferation and induces apoptosis of human hepatocellular carcinoma cells in vitro and in vivo. PLoS One. 2012;7:e50638. | |
Wei W, Chua MS, Grepper S, So SK. Soluble Frizzled-7 receptor inhibits Wnt signaling and sensitizes hepatocellular carcinoma cells towards doxorubicin. Mol Cancer. 2011;10:16. | |
Wei Y, Shen N, Wang Z, et al. Sorafenib sensitizes hepatocellular carcinoma cell to cisplatin via suppression of Wnt/β-catenin signaling. Mol Cell Biochem. 2013;381:139–144. | |
Yao ZX, Jogunoori W, Choufani S, et al. Epigenetic silencing of beta-spectrin, a TGF-beta signaling/scaffolding protein in a human cancer stem cell disorder: Beckwith-Wiedemann syndrome. J Biol Chem. 2010;285:36112–36120. | |
Sun L, Wu G, Willson JK, et al. Expression of transforming growth factor beta type II receptor leads to reduced malignancy in human breast cancer MCF-7 cells. J Biol Chem. 1994;269:26449–26455. | |
Wang J, Han W, Zborowska E, et al. Reduced expression of transforming growth factor beta type I receptor contributes to the malignancy of human colon carcinoma cells. J Biol Chem. 1996;271:17366–17371. | |
Wang J, Sun L, Myeroff L, et al. Demonstration that mutation of the type II transforming growth factor beta receptor inactivates its tumor suppressor activity in replication error-positive colon carcinoma cells. J Biol Chem. 1995;270:22044–22049. | |
Markowitz S, Wang J, Myeroff L, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–1338. | |
Papageorgis P, Cheng K, Ozturk S, et al. Smad4 inactivation promotes malignancy and drug resistance of colon cancer. Cancer Res. 2011;71: 998–1008. | |
Borczuk AC, Kim HK, Yegen HA, Friedman RA, Powell CA. Lung adenocarcinoma global profiling identifies type II transforming growth factor-beta receptor as a repressor of invasiveness. Am J Respir Crit Care Med. 2005;172:729–737. | |
Gobbi H, Arteaga CL, Jensen RA, et al. Loss of expression of transforming growth factor beta type II receptor correlates with high tumour grade in human breast in-situ and invasive carcinomas. Histopathology. 2000;36:168–177. | |
Majumdar A, Curley SA, Wu X, et al. Hepatic stem cells and transforming growth factor β in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2012;9:530–538. | |
Chen CL, Tsukamoto H, Liu JC, et al. Reciprocal regulation by TLR4 and TGF-β in tumor-initiating stem-like cells. J Clin Invest. 2013;123:2832–2849. | |
You H, Ding W, Rountree CB. Epigenetic regulation of cancer stem cell marker CD133 by transforming growth factor-beta. Hepatology. 2010;51:1635–1644. | |
Vergaro V, Baldassarre F, De Santis F, Ciccarella G, Giannelli G, Leporatti S. TGF-beta inihibitor-loaded polyelectrolyte multilayers capsules for sustained targeting of hepatocarcinoma cells. Curr Pharm Des. 2012;18:4155–4164. | |
Mazzocca A, Antonaci S, Giannelli G. The TGF-β signaling pathway as a pharmacological target in a hepatocellular carcinoma. Curr Pharm Des. 2012;18:4148–4154. | |
Sun CK, Chua MS, He J, So SK. Suppression of glypican 3 inhibits growth of hepatocellular carcinoma cells through up-regulation of TGF-β2. Neoplasia. 2011;13:735–747. | |
Caja L, Sancho P, Bertran E, Fabregat I. Dissecting the effect of targeting the epidermal growth factor receptor on TGF-β-induced-apoptosis in human hepatocellular carcinoma cells. J Hepatol. 2011;55:351–358. | |
Zheng X, Gai X, Han S, et al. The human sulfatase 2 inhibitor 2,4-disulfonylphenyl-tert-butylnitrone (OKN-007) has an antitumor effect in hepatocellular carcinoma mediated via suppression of TGFB1/SMAD2 and Hedgehog/GLI1 signaling. Genes Chromosomes Cancer. 2013;52:225–236. | |
He C, Sun XP, Qiao H, et al. Downregulating hypoxia-inducible factor-2α improves the efficacy of doxorubicin in the treatment of hepatocellular carcinoma. Cancer Sci. 2012;103:528–534. | |
Baek HJ, Pishvaian MJ, Tang Y, et al. Transforming growth factor-β adaptor, β2-spectrin, modulates cyclin dependent kinase 4 to reduce development of hepatocellular cancer. Hepatology. 2011;53: 1676–1684. | |
ClinicalTrial.gov. A Study of LY2157299 in participants with hepatocellular carcinoma. Available from: http://clinicaltrials.gov/show/NCT01246986. Accessed July 28, 2014. | |
Bournazou E, Bromberg J. Targeting the tumor microenvironment: JAK-STAT3 signaling. JAKSTAT. 2013;2:e23828. | |
Lin L, Amin R, Gallicano GI, et al. The STAT3 inhibitor NSC 74859 is effective in hepatocellular cancers with disrupted TGF-beta signaling. Oncogene. 2009;28:961–972. | |
He G, Dhar D, Nakagawa H, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell. 2013;155:384–396. | |
Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–789. | |
Wang J, Sullenger BA, Rich JN. Notch signaling in cancer stem cells. Adv Exp Med Biol. 2012;727:174–185. | |
Hassan KA, Wang L, Korkaya H, et al. Notch pathway activity identifies cells with cancer stem cell-like properties and correlates with worse survival in lung adenocarcinoma. Clin Cancer Res. 2013;19: 1972–1980. | |
Won HY, Lee JY, Shin DH, et al. Loss of Mel-18 enhances breast cancer stem cell activity and tumorigenicity through activating Notch signaling mediated by the Wnt/TCF pathway. FASEB J. 2012;26:5002–5013. | |
Apostolou P, Toloudi M, Ioannou E, et al. Study of the interaction among Notch pathway receptors, correlation with stemness, as well as their interaction with CD44, dipeptidyl peptidase-IV, hepatocyte growth factor receptor and the SETMAR transferase, in colon cancer stem cells. J Recept Signal Transduct Res. 2013;33:353–358. | |
Fan X, Khaki L, Zhu TS, et al. Notch pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells. 2010;28:5–16. | |
Villanueva A, Alsinet C, Yanger K, et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology. 2012;143:1660–1669. e7. | |
Dill MT, Tornillo L, Fritzius T, et al. Constitutive Notch2 signaling induces hepatic tumors in mice. Hepatology. 2013;57:1607–1619. | |
Cohen DJ. Targeting the hedgehog pathway: role in cancer and clinical implications of its inhibition. Hematol Oncol Clin North Am. 2012;26:565–588. | |
Philips GM, Chan IS, Swiderska M, et al. Hedgehog signaling antagonist promotes regression of both liver fibrosis and hepatocellular carcinoma in a murine model of primary liver cancer. PLoS One. 2011;6:e23943. | |
Hadden MK. Hedgehog pathway inhibitors: a patent review (2009–present). Expert Opin Ther Pat. 2013;23:345–361. | |
Ho C, Wang C, Mattu S, et al. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology. 2012;55:833–845. | |
Stauffer JK, Scarzello AJ, Andersen JB, et al. Coactivation of AKT and β-catenin in mice rapidly induces formation of lipogenic liver tumors. Cancer Res. 2011;71:2718–2727. | |
Morgensztern D, McLeod HL. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer Drugs. 2005;16:797–803. | |
Hill R, Wu H. PTEN, stem cells, and cancer stem cells. J Biol Chem. 2009;284:11755–11759. | |
Alexia C, Fallot G, Lasfer M, Schweizer-Groyer G, Groyer A. An evaluation of the role of insulin-like growth factors (IGF) and of type-I IGF receptor signalling in hepatocarcinogenesis and in the resistance of hepatocarcinoma cells against drug-induced apoptosis. Biochem Pharmacol. 2004;68:1003–1015. | |
Lau CK, Yang ZF, Ho DW, et al. An Akt/hypoxia-inducible factor-1alpha/platelet-derived growth factor-BB autocrine loop mediates hypoxia-induced chemoresistance in liver cancer cells and tumorigenic hepatic progenitor cells. Clin Cancer Res. 2009;15:3462–3471. | |
Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene. 2008;27:1749–1758. | |
Wen W, Han T, Chen C, et al. Cyclin G1 expands liver tumor-initiating cells by Sox2 induction via Akt/mTOR signaling. Mol Cancer Ther. 2013;12:1796–1804. | |
Buitrago-Molina LE, Vogel A. mTor as a potential target for the prevention and treatment of hepatocellular carcinoma. Curr Cancer Drug Targets. 2012;12:1045–1061. | |
You H, Ding W, Dang H, Jiang Y, Rountree CB. c-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology. 2011;54:879–889. | |
Yu G, Jing Y, Kou X, et al. Hepatic stellate cells secreted hepatocyte growth factor contributes to the chemoresistance of hepatocellular carcinoma. PLoS One. 2013;8:e73312. | |
Cecchi F, Rabe DC, Bottaro DP. Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:553–572. | |
Ohno R, Asou N, Ohnishi K. Treatment of acute promyelocytic leukemia: strategy toward further increase of cure rate. Leukemia. 2003;17:1454–1463. | |
Chen Y, Verfaillie CM. MicroRNAs: the fine modulators of liver development and function. Liver Int. 2014;34:976–990. | |
Ji J, Yamashita T, Budhu A, et al. Identification ofMicroRNA-181 by genome-wide screening as a critical player in EpCAM-positive hepatic cancer stem cells. Hepatology. 2009;50:472–480. | |
Ma S, Tang KH, Chan YP, et al. miR-130b promotes CD133+ liver tumor-initiating cell growth and self-renewal via tumor protein 53-induced nuclear protein 1. Cell Stem Cell. 2010;7:694–707. | |
Callegari E, Elamin BK, Giannone F, et al. Liver tumorigenicity promoted by microRNA-221 in a mouse transgenic model. Hepatology. 2012;56:1025–1033. | |
Kota J, Chivukula RR, O’Donnell KA, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. | |
Tomimaru Y, Eguchi H, Nagano H, et al. MicroRNA-21 induces resistance to the anti-tumour effect of interferon-α/5-fluorouracil in hepatocellular carcinoma cells. Br J Cancer. 2010;103:1617–1626. | |
Su H, Yang JR, Xu T, et al. MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res. 2009;69:1135–1142. | |
Gilbertson RJ, Rich JN. Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat Rev Cancer. 2007;7:733–736. | |
Zeng X, Lin Y, Yin C, et al. Recombinant adenovirus carrying the hepatocyte nuclear factor-1alpha gene inhibits hepatocellular carcinoma xenograft growth in mice. Hepatology. 2011;54:2036–2047. | |
Schrader J, Gordon-Walker TT, Aucott RL, et al. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology. 2011;53:1192–1205. | |
Gokmen-Polar Y, Miller KD. Redefining the target again: chemotherapeutics as vascular disrupting agents? Cancer Cell. 2008; 14:195–196. | |
You L, He B, Xu Z, et al. An anti-Wnt-2 monoclonal antibody induces apoptosis in malignant melanoma cells and inhibits tumor growth. Cancer Res. 2004;64:5385–5389. | |
Aster JC, Blacklow SC. Targeting the Notch pathway: twists and turns on the road to rational therapeutics. J Clin Oncol. 2012;30: 2418–2420. | |
Faivre S, Santoro A, Kelley RK, et al. A Phase 2 study of novel transforming growth factor-beta (TGF-β) receptor 1 kinase inhibitor, LY2157299 monohydrate in patients with advanced hepatocellular carcinoma (HCC). Presented at the 2014 Gastrointestinal Cancers Symposium, January 16–18, 2014, San Francisco, CA, USA. | |
Simonelli M, Zucali PA, Thomas MB, et al. Phase I study of PF-03446962 (anti-ALK-1 mAb) in hepatocellular carcinoma (HCC). J Clin Oncol. 2013;31(Suppl 18):Abstr 4121. | |
Lum BL, Morrison GE, Chang I, et al. A phase I, open-label mass balance study of the hedgehog pathway inhibitor (HPI) GDC-0449 in healthy female subjects of non-childbearing potential. J Clin Oncol. 2010;28(Suppl 15):Abstr 2558. | |
LoRusso PM, Rudin CM, Reddy JC, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin Cancer Res. 2011;17:2502–2511. | |
Graham RA, Lum BL, Cheeti S, et al. Pharmacokinetics of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with locally advanced or metastatic solid tumors: the role of alpha-1-acid glycoprotein binding. Clin Cancer Res. 2011;17:2512–2520. | |
Plimack ER, Lorusso PM, McCoon P, et al. AZD1480: a phase I study of a novel JAK2 inhibitor in solid tumors. Oncologist. 2013;18: 819–820. | |
Hong DS, Younes A, Fayad L, et al. A phase I study of ISIS 481464 (AZD9150), a first-in-human, first-in-class, antisense oligonucleotide inhibitor of STAT3, in patients with advanced cancers. J Clin Oncol. 2013;31(Suppl 18):Abstr 8523. | |
Kurzrock R, Naing A, Falchook GS. A phase 1, open-label, dose escalation, nonrandomized study to assess the maximum tolerated dose, dose limiting toxicity, and pharmacokinetics of OPB-31121 in subjects with advanced solid tumors. Mol Cancer Ther. 2011;10 Suppl 1:B56a. | |
Abou-Alfa GK, Capanu M, O’Reilly EM, et al. A phase II study of cixutumumab (IMC-A12, NSC742460) in advanced hepatocellular carcinoma. J Hepatol. 2014;60:319–324. | |
Faivre S, Fartoux L, Bouattour F, et al. A phase I study of AVE1642, a human monoclonal antibody-blocking insulin-like growth factor-1 receptor (IGF-1R), given as a single agent and in combination with sorafenib as first-line therapy in patients with advanced hepatocellular carcinoma (HCC). J Clin Oncol. 2011;29(Suppl 4):Abstr 270. | |
Von Mehren M, Britten C, Lear K, et al. Phase I, dose-escalation study of BIIB022 (anti-IGF-1R antibody) in advanced solid tumors. J Clin Oncol. 2010;28(Suppl 15):Abstr 2612. | |
Haluska P, Menefee ME, Plimack ER, et al. Safety, pharmacokinetics, and antitumor activity of MEDI-573, an investigational monoclonal antibody that targets IGF-I and IGF-II, in adult patients with advanced solid tumors. J Clin Oncol. 2012;30 Suppl:Abstr TPS2618. | |
Carden CP, Kim ES, Jones RL, et al. Phase I study of intermittent dosing of OSI-906, a dual tyrosine kinase inhibitor of insulin-like growth factor-1 receptor (IGF-1R) and insulin receptor (IR) in patients with advanced solid tumors. J Clin Oncol. 2010;28(Suppl 15):Abstr A2530. | |
Tolcher AW, Yap TA, Fearen I, et al. A phase I study of MK-2206, an oral potent allosteric Akt inhibitor (Akti), in patients (pts) with advanced solid tumor (ST). J Clin Oncol. 2009;27(Suppl 15):Abstr 3503. | |
Campos LT, Nemunaitis J, Stephenson J, et al. Phase II study of single agent perifosine in patients with hepatocellular carcinoma (HCC). J Clin Oncol. 2009;27(Suppl 15):Abstr 15505. | |
Decaens T, Luciani A, Itti E, et al. Phase II study of sirolimus in treatment-naive patients with advanced hepatocellular carcinoma. Dig Liver Dis. 2012;44(7):610–616. | |
Zhu AX, Abrams TA, Miksad R, et al. Phase 1/2 study of everolimus in advanced hepatocellular carcinoma. Cancer. 2011;117(22): 5094–5102. | |
Kelley RK, Nimeiri HS, Munster PN, et al. Temsirolimus combined with sorafenib in hepatocellular carcinoma: a phase I dose-finding trial with pharmacokinetic and biomarker correlates. Ann Oncol. 2013;24(7):1900–1907. | |
Santoro A, Rimassa L, Borbath I, et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013;14(1):55–63. | |
Verslype C, Cohn A, Kelley R, et al. Activity of cabozantinib (XL184) in hepatocellular carcinoma: Results from a phase II randomized discontinuation trial (RDT). J Clin Oncol. 2012;30 Suppl:Abstr 4007. | |
Eder JP, Shapiro GI, Appleman LJ, et al. A phase I study of foretinib, a multi-targeted inhibitor of c-Met and vascular endothelial growth factor receptor 2. Clin Cancer Res. 2010;16(13):3507–3516. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.