Back to Journals » OncoTargets and Therapy » Volume 13
Targeting BRD9 for Cancer Treatment: A New Strategy
Authors Zhu X, Liao Y ![]() , Tang L
, Tang L ![]()
Received 15 October 2020
Accepted for publication 12 December 2020
Published 24 December 2020 Volume 2020:13 Pages 13191—13200
DOI https://doi.org/10.2147/OTT.S286867
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Takuya Aoki
Xiuzuo Zhu,1 Yi Liao,2 Liling Tang1
1Key Laboratory for Biorheological Science and Technology of Ministry of Education, College of Bioengineering, Chongqing University, Chongqing, People’s Republic of China; 2Department of Thoracic Surgery, Southwest Hospital, Army Medical University (Third Military Medical University), Chongqing, People’s Republic of China
Correspondence: Yi Liao Tel +86-23-68765333
Email [email protected]
Liling Tang Tel +86-23-65102507
Email [email protected]
Abstract: Bromodomain-containing protein 9 (BRD9) is a newly identified subunit of the non-canonical barrier-to-autointegration factor (ncBAF) complex and a member of the bromodomain family IV. Studies have confirmed that BRD9 plays an oncogenic role in multiple cancer types, by regulating tumor cell growth. The tumor biological functions of BRD9 are mainly due to epigenetic modification mediated by its bromodomain. The bromodomain recruits the ncBAF complex to the promoter to regulate gene transcription. This review summarizes the potential mechanisms of action of BRD9 in carcinogenesis and the emerging strategies for targeting BRD9 for cancer therapeutics. Although the therapeutic potential of BRD9 has been exploited to some extent, research on the detailed biological mechanisms of BRD9 is still in its infancy. Therefore, targeting BRD9 to study its biological roles will be an attractive tool for cancer diagnosis and treatment, but it remains a great challenge.
Keywords: BRD9, ncBAF, bromodomain, cancer, inhibitor
Introduction
The switch/sucrose non-fermentable (SWI/SNF) complex, a chromatin remodeling complex whose subunits are mutated in most malignant tumors, is one of the most common chromatin regulators in human malignancies.1 It is typically divided into two categories based on the subunit composition and function: the barrier-to-autointegration factor (cBAF) complex and polybromo-associated BAF (PBAF) complex. The core subunits of the cBAF complex are BAF250a (ARID1A) and BAF250b (ARID1B), and those of the PBAF complex are BAF180 (PBRM1) and BAF200 (ARID2).2,3 In addition to the core subunits, 7–15 auxiliary subunits such as BRM (SMARCA2), BRG1 (SMARCA4), BAF155 (SMARCC1), BAF170 (SMARCC2), and BAF47 (SMARCB1) participate in the assembly of the SWI/SNF complex.4 Numerous studies have established that the SWI/SNF complex is altered in many tumor types. For example, in malignant rhabdoid tumors (MRT),5–7 biallelic inactivation of SMARCB1 was first reported, and thereafter, in myoepithelial tumors8 and hepatoblastomas,9 repeated mutations were reported. In adenoid cystic carcinoma,10 SMARCA2, SMARCE1, and ARID1A genes were reported to be mutated. Several studies have revealed that SMARCA4 undergoes mutations at different frequencies in Burkitt’s lymphoma,11 lung adenocarcinoma,12 esophageal adenocarcinoma,13 and medulloblastoma.14–16 Moreover, researchers have found that the mutation frequency of ARID1A, which encodes the BAF250a subunit, is high in hepatocellular carcinoma,12,17 gastric cancer,18,19 bladder cancer,20,21 colorectal cancer,22 pancreatic cancer,23 Burkitt’s lymphoma,11 and cholangiocarcinoma.24 Overall, these studies have indicated that the SWI/SNF complex plays a critical role in the development and progression of human malignant tumors and could be a therapeutic target.
Notably, Wang et al25 defined a third category of the SWI/SNF complex in mouse stem cells: the non-canonical BAF (ncBAF) complex. BRD9 and glioma tumor suppressor candidate gene 1 (GLTSCR1) or GLTSCR1-like (GLTSCR1L) are unique members of the ncBAF complex. In addition, the ncBAF complex contains the following BAF subunits: BAF155, BAF60, SS18, BAF53a, and BRG1/BRM.26 Moreover, the ncBAF complex has not been reported to mutate repeatedly in cancer, unlike the cBAF and pBAF complexes. A study reported that prostate cancer cell line (PC3) proliferation and colony formation was dependent on GLTSCR1 expression, and knocking out GLTSCR1 could decreased PC3 cell proliferation and colony formation.26 Meanwhile, another study demonstrated that BRD9 was highly expressed and required for in SMARCB1-deficient MRT cells.25 In addition, researchers found that the protein level of BRD9 was significantly increased in acute myeloid leukemia (AML) cells than in CD34+ cells.27 Kang et al28 confirmed the presence of BRD9 amplification on chromosome 5p in patients with non-small cell lung cancer (NSCLC) through high-resolution array comparative genomic hybridization analysis. A previous study revealed that the gene copy number of BRD9 was significantly increased in the 5p15.33 region in 12.5% (2/16) of patients with papillary thyroid carcinoma.29 Similarly, based on single nucleotide polymorphism and fluorescence in situ hybridization analyses, Luigi Scotto et al30 demonstrated that BRD9 protein was overexpressed in cervical cancer. In addition, studies have demonstrated that BRD9 acted as a co-factor to stabilize the structure of the SS18-SSX fusion and to maintain its oncogenic transcription in synovial sarcoma (SS).31–33 These findings were in line with the finding that BRD9 and SS18-SSX were co-localized.34 Furthermore, BRD9 has been inextricably linked to inflammation and type 2 diabetes due to β-cell dysfunction.35–37 Wei et al38 confirmed that BRD9 inhibitors could restore β-cell function and reduce inflammation to a certain extent. Based on Kyoto Encyclopedia of Genes and Genomes analysis and Gene Set Enrichment Analysis, researchers found that genes involved in oxidative phosphorylation and the ribosomal pathway were significantly upregulated in cancers with BRD9 amplification, such as liver cancer and sarcoma.39
Interestingly, BRD9 appears to play a significant role in tumor suppression. SF3B1 is an RNA splicing factor that is frequently mutated in various cancer types, such as myelodysplasia,40,41 chronic lymphocytic leukemia,42 and melanoma.43 Inoue et al44 suggested that mutant SF3B1 recognized BRD9 introns and induced mis-splicing of BRD9, which ultimately led to the degradation of BRD9, and thus, promoted melanomagenesis. The whole exome sequencing revealed that BRD9 is one of the susceptibility genes for melanoma, and Gene Ontology analysis revealed that BRD9 is involved in cellular processes, such as DNA replication, DNA repair, and cell response to DNA damage stimuli.45 Furthermore, Park et al46 reported that the combination of BRD9 and lysine-specific histone demethylase 1 (LSD1) inhibitor may be a potential novel treatment for Merkel cell carcinoma (MCC), they claimed that LSD1 inhibition reduces the growth of MCC whereas inhibition reverses, at least partially, the anti-cancer benefit of LSD1 inhibition. Moreover, the AIPuFu database analysis revealed that BRD9 was differentially expressed in 23 malignancies (Figure 1); however, the function of BRD9 in these tumor types is yet to be determined, which will be interesting and meaningful.
Overall, these studies have indicated that BRD9 plays a critical role in tumor development. It is essential to explore the molecular mechanism of BRD9 in cancer progression, and targeting BRD9 will provide new directions for disease prevention and treatment.
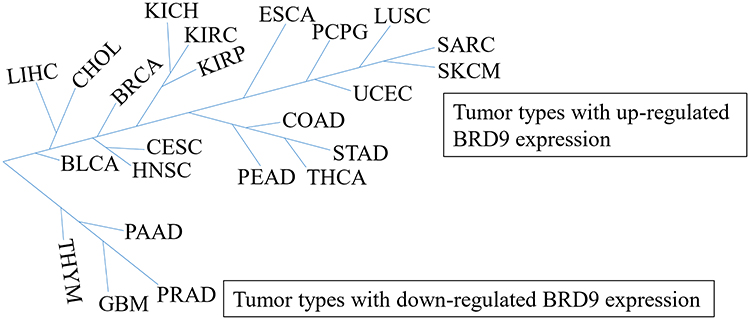 |
Figure 1 The tumor types with BRD9 differential expression. BDR9 is highly expressed in 19 cancer types including CHOL, LIHC, and BRCA, but is lowly expressed in four cancer types, such as THYM, PAAD, GBM and PRAD. According to the AIPuFu database, the URL: http://www.aipufu.com/index.html. |
BRD9 Structure
BRD9 contains a bromodomain and a DUF3512 domain.27 Although many researchers have focused on the bromodomain to determine the biological function of BRD9, few have studied the function of the DUF3512 domain. Till date, we only know that the DUF3512 domain is essential for the assembly of the ncBAF complex.25
“Epigenetics” first defined by Conrad Waddington, is a discipline that studies heritable changes in gene expression without involving changes in the nucleotide sequence of genes.47 These changes include DNA modifications (such as DNA methylation), covalent histone modifications (such as histone methylation, phosphorylation, acetylation, and ubiquitination), and RNA-mediated gene silencing.48,49 Functional bromodomains, usually approximately to 100 amino acids in length, can specifically recognize acetylated lysine residues on histone tails, and recruit different chromatin-modifying factors to specific sites to participate in transcription regulation.50,51 One common feature among all bromodomains: is that they have a hydrophobic pocket to which acetylated lysine residues bind. This hydrophobic region is formed between the helices αZ and αA (ZA loop) and between the helices αB and αC (BC loop).52 Histone acetylation is involved in protein stabilization,48 DNA repair,53 signal transduction,54 and post-translational modifications.55–57 Studies have indicated that an acetylated lysine is recognized and immobilized on a conserved asparagine residue in the hydrophobic pocket.58,59 The bromodomain family is divided into 8 subfamilies based on structural differences. BRD9, Bromodomain-containing protein 7 (BRD7), bromodomain and PHD finger containing 1 (BRPF1), bromodomain and PHD finger containing 2 (BRPF2), bromodomain and PHD finger containing 3 (BRPF3), ATPase family AAA domain containing 2 (ATAD2), and ATPase family AAA domain containing 2B (ATAD2B) belong to the bromodomain family IV.60 The BRPF1 protein is a component of the MOZ/HAT complex, and its bromodomain recognizes H2AK5ac, H4K12ac, H3K14ac, H4K8ac, and H4K5ac.61 BRPF2 and BRPF3 have high sequence similarity and are the subunits of the HBO1/HAT complex, which can acetylate histone H4.62 The HBO1/HAT complex is reportedly involved in DNA transcription and replication, and these processes are inseparable from the contribution of BRPF2/BRPF3.63 Although ATAD2 and ATAD2B are highly conserved, their functions appear to be quite different.64 ATAD2 is mainly located in reproductive tissues,65,66 and its bromodomain can recognize H4K5ac,67 H4K12ac, and H4K5acK12ac;68 ATAD2B is expressed in neural tissues,69 and its bromodomain and histone ligand have not been fully studied. BRD7 and BRD9 are the subunits of the PBAF and ncBAF complexes, respectively. Their bromodomains are highly homologous.70 However, their roles in tumor progression are quite different. BRD7 has been reported as a tumor suppressor,71 whereas BRD9 plays a role in cancer promotion.27 Although few biological ligands for the BRD9 bromodomain are known, BRD9 has been demonstrated to bind to diacetylated H4K5acK8ac and dipropionylated H4K5prK8pr.72
Potential Mechanisms of BRD9
BRD9-STAT5 Axis Participates in Tumor Progression
A recent study reported that the proliferation of AML cells depends on the function of BRD9.27 BRD9 is enriched in the downstream region of the MYC promoter and drives its transcription. Further analysis of this process revealed that the role of BRD9 in AML depends on the recognition of the acetyl lysine region by the bromodomain. Several studies have demonstrated that proteins containing bromodomains play a pivotal role in epigenetic regulation,73,74 because the bromodomain specifically recognizes acetylated lysines of histones and other proteins.51,74 BRD9 is recruited to chromatin binding sites. Another study demonstrated that BRD9 was overexpressed in the AML cell line,75 and acted as a key regulator of AML occurrence. The study revealed that the BRD9-STAT5 axis played an important role in the occurrence and maintenance of leukemia. In leukemia, BRD9 is overexpressed, and induces the activation of the signal transducer and activator of transcription 5 (STAT5) pathway. The activation of STAT5 is known to promote the proliferation and survival of AML cells and the occurrence of inflammation.76–79 Currently, no clinically effective STAT5 inhibitor is available for the treatment of leukemia.80 Therefore, knocking down BRD9 to decrease the activation of STAT5 and induce apoptosis through the Caspase8 signaling cascade may be a possible therapeutic strategy.
miR-140-3p-BRD9 Axis Participates in Tumor Progression
Lung cancer is one of the malignant tumors with high morbidity and mortality.81 It is divided into three subclasses: lung carcinoid, Squamous cell lung cancer (SCLC) and NSCLC.82 SCLC is a histological subtype of NSCLC. Patients with SCLC accounts for approximately one-third of those with NSCLC.83 However, no clinically effective, targeted therapeutic strategy for SCLC has been found. Many studies have demonstrated that the differential expression of miRNAs in lung cancer makes miRNAs carcinogenic or suppressive. For example, in lung cancer, miR-342-3p84 and miR-30d-5p85 are inhibited in lung cancer, whereas miR-29b,86 hsa-miR-3180, and miR-14,081 are highly expressed. Huang et al87 demonstrated that miR-140-3p was downregulated in NSCLC cells and tissues, and regulated the process of NSCLC by directly targeting BRD9. A negative correlation exists between the expression level of BRD9 and that of miR-140-3p. miR-140-3p directly targets BRD9 mRNA, inhibiting its protein translation and consequently downregulating the expression level of C-myc, and suppressing the proliferation of SCLC. Therefore, the miR-140-3p-BRD9 axis may be a promising therapeutic approach for the clinical treatment of SCLC.
Interaction Between the Phosphatidylinositol 3-Kinase Pathway and the Mitogen-Activated Protein Kinase Pathway Induces BRD9 Expression to Participate in Tumor Progression
Usually, tumor formation is closely related to a combination of multiple oncogenes or multiple oncogenic processes. A study reported that the activation of the phosphatidylinositol 3-kinase (PI3K) pathway was closely related to a variety of tumor biological processes, such as tumor cell proliferation, migration, metabolism, and other important processes.88 The PI3KCA gene catalyzes the activation of PI3K.89 Studies have demonstrated that the kirsten rat sarcoma (KRAS) and PIK3CA genes are significantly associated with colorectal cancer,90 lung cancer,91 and breast cancer,92–94 and their common mutations drive the malignant transformation. KRAS plays an indispensable role in the activation of the mitogen-activated protein kinase pathway.95 A study96 used double knock-in (DKI) breast epithelial (MCF-10A) cells harboring PIK3CA and KRAS to explore the interaction between PIK3CA and KRAS, and found that the carcinogenicity of KRAS-PIK3CA was because the KRAS-PIK3CA interaction induced the expression of BRD9. BRD9 regulated the proliferation and migration of DKI MCF-10A cells by combining with the MYC promoter.
Cancer Therapeutics for Targeting BRD9
Recently, the BET family has been extensively studied as a therapeutic target, and bromodomain inhibitors as antitumor agents have demonstrated remarkable clinical effect.97–101 Bromodomain inhibitors are divided into two categories: non-acetylated, and acetylated lysine mimetics. The former is a weak inhibitor, whereas the latter directly mimics the binding of an acetylated lysine to the bromodomain and competitively inhibits the binding of acetylated lysine residues to the hydrophobic binding pocket of the bromodomain.60 Gradually, as the biological function of BRD9 in tumorigenesis becomes clear, targeting the bromodomain of BRD9 will become a new and effective tumor treatment method. For example, small-molecule inhibitors of the BRD9 bromodomain selectively suppress tumor cell proliferation and survival and induce apoptosis.27,75,102,103 Indeed, scientists have researched and developed several effective BRD9 bromodomain inhibitors, such as BRD9 selective inhibitors (I-BRD9,104 BI-7273,105 and BI-9564106) and BRD7/9 inhibitors.107,108 LP99 is the first reported selective BRD7/9 inhibitor that effectively inhibits the binding of BRD7/9 to acetylated histones in vivo and in vitro; Moreover, LP99 inhibits the secretion of proinflammatory cytokine IL-6. These results suggest a role of BRD9 in inflammation.109 Notably, the probe compound of BRD9 designed by Crawford et al110 can inhibit the expression of the drug resistance gene aldehyde dehydrogenase 1 family member A1 (ALDH1A1). Interestingly, both ALDH1A1 and its promoter are involved in the regulation of acetylation,111,112 whereas the relationship between BRD9 and cancer resistance has not been described before. BRD9 inhibitors, BI-7273 and BI-9564, used to investigate the biological functions of BRD9 in vivo and in vitro were proven to be non-toxic by fragment-based screening.105,106 Based on the structural design, I-BRD9 has been identified as a selective cytochemical probe for BRD9.113 In addition, I-BRD9 downregulates cancer and immunology-related genes, such as SAMSN1,114 CLEC1,115 FES,116 and DUSP6.117 Studies on the selection mechanism of I-BRD9 for the BRD9 bromodomain have demonstrated that several residues in the ZA and ZB loops of the bromodomain, such as Asp144, Ile53, Lys91, Thr104, Pro82, Asn140, Asn100, and Phe44, can be used as important references for designing BRD9 inhibitors.104 The imidazo [1,5-a] pyrazin-8 (7H) -one derivative was designed and synthesized using the interaction of the inhibitor with the Asn and Tyr residues to inhibit BRD9 activity.118
In addition to the synthesis and design of protein inhibitors, targeting protein degradation by hijacking the ubiquitin-proteasome system can be another therapeutic strategy.119,120 Encouragingly, Remillard et al121 designed the heterobifunctional ligand dBRD9, a direct chemical degrader for BRD9, which linked the BRD9 bromodomain and the cereblon E3 ubiquitin ligase complex. Thereafter, they designed VZ185, a highly effective and fast degrader for BRD9.122 A previous study confirmed that BRD9 protein degraders are more effective than BRD9 bromodomain inhibitors.123 These data suggest that in addition to the bromodomain, other uncharacterized domains play vital roles in cancer.
Conclusion
Recently, the role of the SWI/SNF complex has been extensively studied in various malignant cancers. Repeated mutations in the subunits of the SWI/SNF complex in cancer make it a promising therapeutic target. Based on different subunit combinations, the SWI/SNF complex is classified into three categories: the cBAF, PBAF, and ncBAF complexes. The ncBAF complex lacks SNF5 and ARID subunits. BRD9 is one of the subunits that make up the ncBAF complex and is a member of the bromodomain family IV. BRD9 has been found to be overexpressed in malignant cancers, such as MRT, AML, SS, and SCLC. BRD9 is especially important for maintaining the growth and proliferation of MRT and AML cells. Meanwhile, BRD9 plays an anti-tumor role in melanoma and MCC. BRD9 is closely related to the biological processes of cells, such as cell proliferation and apoptosis (Figure 2). It consists of a bromodomain and a DUF3512 domain. The deletion of the DUF3512 domain affects the assembly of the ncBAF complex. The bromodomain is required for the biological functions of BRD9 during tumor formation. Bromodomain-containing proteins are involved in epigenetic regulation. The regulatory mechanism involves the binding of the bromodomain to acetylated lysine residues on histones and non-histones, and the recruitment of molecular chaperones to regulate gene transcription. Therefore, understanding the function of the bromodomain will aid in understanding the complex biological functions of BRD9. Currently, two treatment strategies targeting BRD9 are available. First, designing inhibitors to prevent the binding of bromodomains to acetylated lysine residues; and second, designing protein degraders to degrade proteins and inhibit their activity. Although the bromodomain inhibitors and degraders have demonstrated good therapeutic effect only to some extent, their great potential in cancer treatment has been demonstrated. However, the detailed mechanisms of the biological functions of BRD9 are unclear. We intend to focus on these mechanisms in our future research.
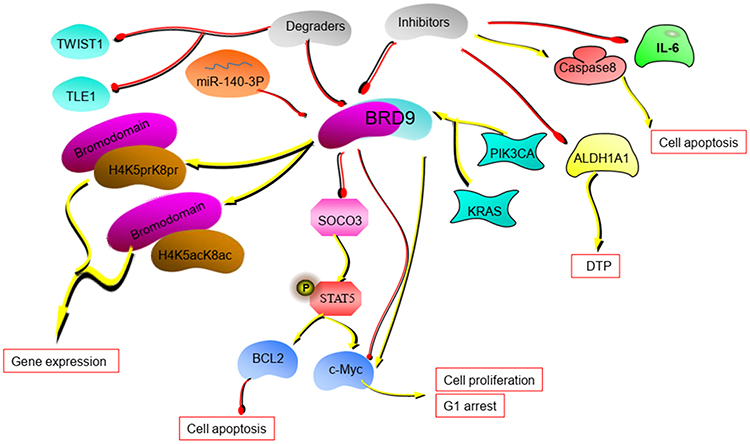 |
Figure 2 The potential mechanism of BRD9. BRD9 regulates tumor progression through the miR-140-3P-BRD9 axis and BRD9-STAT5 axis. BRD9 inhibitors and degraders can disrupt the tumor process. (The yellow lines in the figure represent the promotion between molecules, and the red lines represent the inhibition between molecules.). |
Abbreviations
BRD9, bromodomain-containing protein 9; MRT, malignant rhabdoid tumor; ncBAF, non-canonical barrier-to-autointegration factor; SWI/SNF, switch/sucrose non-fermentable; cBAF, canonical barrier-to-autointegration factor; PBAF, polybromo-associated BAF; GLTSCR1, glioma tumor suppressor candidate gene 1; AML, acute myeloid leukemia; SS, synovial sarcoma; LIHC, Liver hepatocellular carcinoma; BLCA, bladder urothelial carcinoma; CHOL, cholangiocarcinoma; BRCA, breast invasive carcinoma; HNSC, head and neck squamous cell carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; KIRP, kidney renal papillary cell carcinoma; KIRC, kidney renal clear cell carcinoma; KICH, kidney Chromophobe; READ, rectum adenocarcinoma; COAD, colon adenocarcinoma; THCA, thyroid carcinoma; STAD, stomach adenocarcinoma; ESCA, esophageal carcinoma; PCPG, pheochromocytoma and paraganglioma; UCEC, uterine corpus endometrial carcinoma; LUSC, lung squamous cell carcinoma; SKCM, skin cutaneous melanoma; SARC, sarcoma; THYM, thymoma; PAAD, pancreatic adenocarcinoma; GBM, glioblastoma multiforme; PRAD, prostate adenocarcinoma; BRD7, bromodomain-containing protein 7; BRPF1, bromodomain and PHD finger containing 1; BRPF2, bromodomain and PHD finger containing 2; BRPF3, bromodomain and PHD finger containing 3; ATAD2, ATPase family AAA domain containing 2; ATAD2B, ATPase family AAA domain containing 2B; STAT5, signal transducer and activator of transcription 5; SCLC, squamous cell lung cancer; PI3K, phosphatidylinositol 3-kinase; KRAS, kirsten rat sarcoma; DKI, double knock-in; ALDH1A1, aldehyde dehydrogenase 1 family member A1; LSD1, lysine-specific histone demethylase 1; MCC, Merkel cell carcinoma.
Author Contributions
All the authors contributed for the preparation of this manuscript. XZ and LT were responsible for confirming the topic. XZ were responsible for writing the first draft of this article. LT and YL contributed to furtherly editing and polishing the manuscript. All authors read and approved the final manuscript. All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This research was supported by the National Natural Sciences Foundation of China (No.31670952; No.81702921).
Disclosure
The authors declare that they have no competing interests.
References
1. Savas S, Skardasi G. The SWI/SNF complex subunit genes: their functions, variations, and links to risk and survival outcomes in human cancers. Crit Rev Oncol Hematol. 2018;123:114–131. doi:10.1016/j.critrevonc.2018.01.009
2. Masliah-Planchon J, Bieche I, Guinebretiere JM, Bourdeaut F, Delattre O. SWI/SNF chromatin remodeling and human malignancies. Annu Rev Pathol. 2015;10:145–171. doi:10.1146/annurev-pathol-012414-040445
3. Wu JI, Lessard J, Crabtree GR. Understanding the words of chromatin regulation. Cell. 2009;136(2):200–206. doi:10.1016/j.cell.2009.01.009
4. Kadoch C, Hargreaves DC, Hodges C, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45(6):592–601. doi:10.1038/ng.2628
5. Calderaro J, Masliah-Planchon J, Richer W, et al. Balanced translocations disrupting SMARCB1 are hallmark recurrent genetic alterations in renal medullary carcinomas. Eur Urol. 2016;69(6):1055–1061. doi:10.1016/j.eururo.2015.09.027
6. Bruggers CS, Bleyl SB, Pysher T, et al. Clinicopathologic comparison of familial versus sporadic atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system. Pediatr Blood Cancer. 2011;56(7):1026–1031. doi:10.1002/pbc.22757
7. Eaton KW, Tooke LS, Wainwright LM, Judkins AR, Biegel JA. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer. 2011;56(1):7–15. doi:10.1002/pbc.22831
8. Ammerlaan AC, Ararou A, Houben MP, et al. Long-term survival and transmission of INI1-mutation via nonpenetrant males in a family with rhabdoid tumour predisposition syndrome. Br J Cancer. 2008;98(2):474–479. doi:10.1038/sj.bjc.6604156
9. Trobaugh-Lotrario AD, Tomlinson GE, Finegold MJ, Gore L, Feusner JH. Small cell undifferentiated variant of hepatoblastoma: adverse clinical and molecular features similar to rhabdoid tumors. Pediatr Blood Cancer. 2009;52(3):328–334. doi:10.1002/pbc.21834
10. Ho AS, Kannan K, Roy DM, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet. 2013;45(7):791–798. doi:10.1038/ng.2643
11. Love C, Sun Z, Jima D, et al. The genetic landscape of mutations in burkitt lymphoma. Nat Genet. 2012;44(12):1321–1325.
12. Imielinski M, Berger AH, Hammerman PS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150(6):1107–1120. doi:10.1016/j.cell.2012.08.029
13. Dulak AM, Stojanov P, Peng S, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet. 2013;45(5):478–486. doi:10.1038/ng.2591
14. Parsons DW, Li M, Zhang X, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331(6016):435–439. doi:10.1126/science.1198056
15. Pugh TJ, Weeraratne SD, Archer TC, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488(7409):106–110. doi:10.1038/nature11329
16. Jones DT, Jager N, Kool M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488(7409):100–105. doi:10.1038/nature11284
17. Guichard C, Amaddeo G, Imbeaud S, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44(6):694–698. doi:10.1038/ng.2256
18. Wang K, Kan J, Yuen ST, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43(12):1219–1223. doi:10.1038/ng.982
19. Zang ZJ, Cutcutache I, Poon SL, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44(5):570–574. doi:10.1038/ng.2246
20. Gui Y, Guo G, Huang Y, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43(9):875–878. doi:10.1038/ng.907
21. Guo G, Sun X, Chen C, et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nature Genetics. 2013;45(12):1459–1463. doi:10.1038/ng.2798
22. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. doi:10.1038/nature11252
23. Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399–405. doi:10.1038/nature11547
24. Jiao Y, Pawlik TM, Anders RA, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet. 2013;45(12):1470–1473. doi:10.1038/ng.2813
25. Wang X, Wang S, Troisi EC, et al. BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat Commun. 2019;10(1):1881. doi:10.1038/s41467-019-09891-7
26. Alpsoy A, Dykhuizen EC. Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. J Biol Chem. 2018;293(11):3892–3903. doi:10.1074/jbc.RA117.001065
27. Hohmann AF, Martin LJ, Minder JL, et al. Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nat Chem Biol. 2016;12(9):672–679. doi:10.1038/nchembio.2115
28. Kang JU, Koo SH, Kwon KC, Park JW, Kim JM. Gain at chromosomal region 5p15.33, containing TERT, is the most frequent genetic event in early stages of non-small cell lung cancer. Cancer Genet Cytogenet. 2008;182(1):1–11. doi:10.1016/j.cancergencyto.2007.12.004
29. Yang C, Xu W, Gong J, Liu Z, Cui D. Novel somatic alterations underlie Chinese papillary thyroid carcinoma. Cancer Biomark. 2020;27(4):445–460. doi:10.3233/CBM-191200
30. Scotto L, Narayan G, Nandula SV, et al. Integrative genomics analysis of chromosome 5p gain in cervical cancer reveals target over-expressed genes, including Drosha. Mol Cancer. 2008;7:58. doi:10.1186/1476-4598-7-58
31. Banito A, Li X, Laporte AN, et al. The SS18-SSX oncoprotein hijacks KDM2B-PRC1.1 to drive synovial sarcoma. Cancer Cell. 2018;33(3):527–541.e528. doi:10.1016/j.ccell.2018.01.018
32. Brien GL, Remillard D, Shi J, et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. Elife. 2018:7. doi:10.7554/eLife.41305
33. McBride MJ, Pulice JL, Beird HC, et al. The SS18-SSX fusion oncoprotein hijacks BAF complex targeting and function to drive synovial sarcoma. Cancer Cell. 2018;33(6):1128–1141.e1127. doi:10.1016/j.ccell.2018.05.002
34. Middeljans E, Wan X, Jansen PW, Sharma V, Stunnenberg HG, Logie C. SS18 together with animal-specific factors defines human BAF-type SWI/SNF complexes. PLoS One. 2012;7(3):e33834. doi:10.1371/journal.pone.0033834
35. Ashcroft FM, Rorsman P. Diabetes mellitus and the beta cell: the last ten years. Cell. 2012;148(6):1160–1171. doi:10.1016/j.cell.2012.02.010
36. Donath MY, Dalmas E, Sauter NS, Boni-Schnetzler M. Inflammation in obesity and diabetes: islet dysfunction and therapeutic opportunity. Cell Metab. 2013;17(6):860–872. doi:10.1016/j.cmet.2013.05.001
37. Halban PA, Polonsky KS, Bowden DW, et al. beta-cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabetes Care. 2014;37(6):1751–1758. doi:10.2337/dc14-0396
38. Wei Z, Yoshihara E, He N, et al. Vitamin D switches BAF complexes to protect beta cells. Cell. 2018;173(5):1135–1149 e1115. doi:10.1016/j.cell.2018.04.013
39. Sima X, He J, Peng J, Xu Y, Zhang F, Deng L. The genetic alteration spectrum of the SWI/SNF complex: the oncogenic roles of BRD9 and ACTL6A. PLoS One. 2019;14(9):e0222305. doi:10.1371/journal.pone.0222305
40. Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–69. doi:10.1038/nature10496
41. Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384–1395. doi:10.1056/NEJMoa1103283
42. Wang L, Lawrence MS, Wan Y, et al. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med. 2011;365(26):2497–2506. doi:10.1056/NEJMoa1109016
43. Seiler M, Peng S, Agrawal AA, et al. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep. 2018;23(1):282–296.e284.
44. Inoue D, Chew GL, Liu B, et al. Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature. 2019;574(7778):432–436. doi:10.1038/s41586-019-1646-9
45. Campos C, Fragoso S, Luís R, et al. High-throughput sequencing identifies 3 novel susceptibility genes for hereditary melanoma. Genes (Basel). 2020;11(4):403.
46. Park DE, Cheng J, McGrath JP, et al. Merkel cell polyomavirus activates LSD1-mediated blockade of non-canonical BAF to regulate transformation and tumorigenesis. Nat Cell Biol. 2020;22(5):603–615. doi:10.1038/s41556-020-0503-2
47. Waddington CH. Preliminary notes on the development of the wings in normal and mutant strains of Drosophila. Proc Natl Acad Sci U S A. 1939;25(7):299–307. doi:10.1073/pnas.25.7.299
48. Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi:10.1016/j.cell.2007.02.005
49. Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science. 1999;286(5439):481–486. doi:10.1126/science.286.5439.481
50. Perez-Salvia M, Esteller M. Bromodomain inhibitors and cancer therapy: from structures to applications. Epigenetics. 2017;12(5):323–339. doi:10.1080/15592294.2016.1265710
51. Filippakopoulos P, Picaud S, Mangos M, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149(1):214–231. doi:10.1016/j.cell.2012.02.013
52. Sanchez R, Zhou -M-M. The role of human bromodomains in chromatin biology and gene transcription. Curr Opin Drug Discov Devel. 2009;12(5):659–665.
53. Celic I, Masumoto H, Griffith WP, et al. The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr Biol. 2006;16(13):1280–1289. doi:10.1016/j.cub.2006.06.023
54. Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 2000;19(6):1176–1179. doi:10.1093/emboj/19.6.1176
55. Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. doi:10.1126/science.1063127
56. Choudhary C, Kumar C, Gnad F, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325(5942):834–840. doi:10.1126/science.1175371
57. Zhao S, Xu W, Jiang W, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327(5968):1000–1004. doi:10.1126/science.1179689
58. Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399(6735):491–496. doi:10.1038/20974
59. Owen DJ, Ornaghi P, Yang JC, et al. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase gcn5p. EMBO J. 2000;19(22):6141–6149. doi:10.1093/emboj/19.22.6141
60. Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13(5):337–356. doi:10.1038/nrd4286
61. Poplawski A, Hu K, Lee W, et al. Molecular insights into the recognition of N-terminal histone modifications by the BRPF1 bromodomain. J Mol Biol. 2014;426(8):1661–1676. doi:10.1016/j.jmb.2013.12.007
62. Demont EH, Bamborough P, Chung CW, et al. 1,3-dimethyl benzimidazolones are potent, selective inhibitors of the BRPF1 bromodomain. ACS Med Chem Lett. 2014;5(11):1190–1195. doi:10.1021/ml5002932
63. Burke TW, Cook JG, Asano M, Nevins JR. Replication factors MCM2 and ORC1 interact with the histone acetyltransferase HBO1. J Biol Chem. 2001;276(18):15397–15408. doi:10.1074/jbc.M011556200
64. Leachman NT, Brellier F, Ferralli J, Chiquet-Ehrismann R, Tucker RP. ATAD2B is a phylogenetically conserved nuclear protein expressed during neuronal differentiation and tumorigenesis. Dev Growth Differ. 2010;52(9):747–755. doi:10.1111/j.1440-169X.2010.01211.x
65. Caron C, Lestrat C, Marsal S, et al. Functional characterization of ATAD2 as a new cancer/testis factor and a predictor of poor prognosis in breast and lung cancers. Oncogene. 2010;29(37):5171–5181. doi:10.1038/onc.2010.259
66. Ciro M, Prosperini E, Quarto M, et al. ATAD2 is a novel cofactor for MYC, overexpressed and amplified in aggressive tumors. Cancer Res. 2009;69(21):8491–8498. doi:10.1158/0008-5472.CAN-09-2131
67. Poncet-Montange G, Zhan Y, Bardenhagen JP, et al. Observed bromodomain flexibility reveals histone peptide- and small molecule ligand-compatible forms of ATAD2. Biochem J. 2015;466(2):337–346. doi:10.1042/BJ20140933
68. Koo SJ, Fernandez-Montalvan AE, Badock V, et al. ATAD2 is an epigenetic reader of newly synthesized histone marks during DNA replication. Oncotarget. 2016;7(43):70323–70335. doi:10.18632/oncotarget.11855
69. Zou JX, Guo L, Revenko AS, et al. Androgen-induced coactivator ANCCA mediates specific androgen receptor signaling in prostate cancer. Cancer Res. 2009;69(8):3339–3346. doi:10.1158/0008-5472.CAN-08-3440
70. Crawford TD, Tsui V, Flynn EM, et al. Diving into the water: inducible binding conformations for BRD4, TAF1(2), BRD9, and CECR2 bromodomains. J Med Chem. 2016;59(11):5391–5402. doi:10.1021/acs.jmedchem.6b00264
71. Harte MT, O’Brien GJ, Ryan NM, et al. BRD7, a subunit of SWI/SNF complexes, binds directly to BRCA1 and regulates BRCA1-dependent transcription. Cancer Res. 2010;70(6):2538–2547. doi:10.1158/0008-5472.CAN-09-2089
72. Flynn EM, Huang OW, Poy F, et al. A subset of human bromodomains recognizes butyryllysine and crotonyllysine histone peptide modifications. Structure. 2015;23(10):1801–1814. doi:10.1016/j.str.2015.08.004
73. Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502(7472):480–488. doi:10.1038/nature12751
74. Fujisawa T, Filippakopoulos P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Biol. 2017;18(4):246–262. doi:10.1038/nrm.2016.143
75. Del Gaudio N, Di Costanzo A, Liu NQ, et al. BRD9 binds cell type-specific chromatin regions regulating leukemic cell survival via STAT5 inhibition. Cell Death Dis. 2019;10(5):338. doi:10.1038/s41419-019-1570-9
76. Carow B, Rottenberg ME. SOCS3, a major regulator of infection and inflammation. Front Immunol. 2014;5:58. doi:10.3389/fimmu.2014.00058
77. Agger K, Miyagi S, Pedersen MT, Kooistra SM, Johansen JV, Helin K. Jmjd2/Kdm4 demethylases are required for expression of Il3ra and survival of acute myeloid leukemia cells. Genes Dev. 2016;30(11):1278–1288. doi:10.1101/gad.280495.116
78. Ferbeyre G, Moriggl R. The role of Stat5 transcription factors as tumor suppressors or oncogenes. Biochim Biophys Acta. 2011;1815(1):104–114. doi:10.1016/j.bbcan.2010.10.004
79. Lin JX, Leonard WJ. The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene. 2000;19(21):2566–2576. doi:10.1038/sj.onc.1203523
80. Furqan M, Akinleye A, Mukhi N, Mittal V, Chen Y, Liu D. STAT inhibitors for cancer therapy. J Hematol Oncol. 2013;6:90.
81. The L. Lung cancer: some progress, but still a lot more to do. Lancet. 2019;394(10212):1880.
82. Brinkmeyer JK, Moore DC. Necitumumab for the treatment of squamous cell non-small cell lung cancer. J Oncol Pharm Pract. 2018;24(1):37–41. doi:10.1177/1078155216682365
83. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10(9):1243–1260. doi:10.1097/JTO.0000000000000630
84. Xue X, Fei X, Hou W, Zhang Y, Liu L, Hu R. miR-342-3p suppresses cell proliferation and migration by targeting AGR2 in non-small cell lung cancer. Cancer Lett. 2018;412:170–178. doi:10.1016/j.canlet.2017.10.024
85. Chen D, Guo W, Qiu Z, et al. MicroRNA-30d-5p inhibits tumour cell proliferation and motility by directly targeting CCNE2 in non-small cell lung cancer. Cancer Lett. 2015;362(2):208–217. doi:10.1016/j.canlet.2015.03.041
86. Langsch S, Baumgartner U, Haemmig S, et al. miR-29b mediates NF-kappaB Signaling in KRAS-induced non-small cell lung cancers. Cancer Res. 2016;76(14):4160–4169. doi:10.1158/0008-5472.CAN-15-2580
87. Huang H, Wang Y, Li Q, Fei X, Ma H, Hu R. miR-140-3p functions as a tumor suppressor in squamous cell lung cancer by regulating BRD9. Cancer Lett. 2019;446:81–89. doi:10.1016/j.canlet.2019.01.007
88. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017;170(4):605–635.
89. Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554. doi:10.1126/science.1096502
90. Yaeger R, Chatila WK, Lipsyc MD, et al. Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell. 2018;33(1):125–136e123. doi:10.1016/j.ccell.2017.12.004
91. Campbell JD, Alexandrov A, Kim J, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016;48(6):607–616. doi:10.1038/ng.3564
92. Razavi P, Chang MT, Xu G, et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell. 2018;34(3):427–438e426.
93. Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi:10.1126/scisignal.2004088
94. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi:10.1158/2159-8290.CD-12-0095
95. Ramjaun AR, Downward J. Ras and phosphoinositide 3-kinase: partners in development and tumorigenesis. Cell Cycle. 2007;6(23):2902–2905. doi:10.4161/cc.6.23.4996
96. Bell CM, Raffeiner P, Hart JR, Vogt PK. PIK3CA cooperates with KRAS to promote MYC activity and tumorigenesis via the bromodomain protein BRD9. Cancers (Basel). 2019;11(11):1634. doi:10.3390/cancers11111634
97. Muller S, Filippakopoulos P, Knapp S. Bromodomains as therapeutic targets. Expert Rev Mol Med. 2011;13:e29. doi:10.1017/S1462399411001992
98. Hewings DS, Rooney TP, Jennings LE, et al. Progress in the development and application of small molecule inhibitors of bromodomain-acetyl-lysine interactions. J Med Chem. 2012;55(22):9393–9413. doi:10.1021/jm300915b
99. Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327):1067–1073. doi:10.1038/nature09504
100. Hewings DS, Wang M, Philpott M, et al. 3,5-dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. J Med Chem. 2011;54(19):6761–6770. doi:10.1021/jm200640v
101. Picaud S, Da Costa D, Thanasopoulou A, et al. PFI-1, a highly selective protein interaction inhibitor, targeting BET Bromodomains. Cancer Res. 2013;73(11):3336–3346. doi:10.1158/0008-5472.CAN-12-3292
102. Krämer KF, Moreno N, Frühwald MC, Kerl K. BRD9 inhibition, alone or in combination with cytostatic compounds as a therapeutic approach in rhabdoid tumors. Int J Mol Sci. 2017;18(7):1537. doi:10.3390/ijms18071537
103. Bevill SM, Olivares-Quintero JF, Sciaky N, et al. GSK2801, a BAZ2/BRD9 bromodomain inhibitor, synergizes with BET inhibitors to induce apoptosis in triple-negative breast cancer. Mol Cancer Res. 2019;17(7):1503–1518. doi:10.1158/1541-7786.MCR-18-1121
104. Su J, Liu X, Zhang S, Yan F, Zhang Q, Chen J. Insight into selective mechanism of class of I-BRD9 inhibitors toward BRD9 based on molecular dynamics simulations. Chem Biol Drug Des. 2019;93(2):163–176. doi:10.1111/cbdd.13398
105. Martin LJ, Koegl M, Bader G, et al. Structure-based design of an in vivo active selective BRD9 inhibitor. J Med Chem. 2016;59(10):4462–4475. doi:10.1021/acs.jmedchem.5b01865
106. Karim RM, Schonbrunn E. An advanced tool to interrogate BRD9. J Med Chem. 2016;59(10):4459–4461. doi:10.1021/acs.jmedchem.6b00550
107. Karim RM, Chan A, Zhu JY, Schönbrunn E. Structural basis of inhibitor selectivity in the BRD7/9 Subfamily of Bromodomains. J Med Chem. 2020;63(6):3227–3237. doi:10.1021/acs.jmedchem.9b01980
108. Clegg MA, Bamborough P, Chung CW, et al. Application of atypical acetyl-lysine methyl mimetics in the development of selective inhibitors of the Bromodomain-Containing Protein 7 (BRD7)/Bromodomain-Containing Protein 9 (BRD9) Bromodomains. J Med Chem. 2020;63(11):5816–5840. doi:10.1021/acs.jmedchem.0c00075
109. Clark PG, Vieira LC, Tallant C, et al. LP99: discovery and synthesis of the first selective BRD7/9 Bromodomain inhibitor. Angew Chem Int Ed Engl. 2015;54(21):6217–6221. doi:10.1002/anie.201501394
110. Crawford TD, Vartanian S, Cote A, et al. Inhibition of bromodomain-containing protein 9 for the prevention of epigenetically-defined drug resistance. Bioorg Med Chem Lett. 2017;27(15):3534–3541. doi:10.1016/j.bmcl.2017.05.063
111. Zhao D, Mo Y, Li MT, et al. NOTCH-induced aldehyde dehydrogenase 1A1 deacetylation promotes breast cancer stem cells. J Clin Invest. 2014;124(12):5453–5465. doi:10.1172/JCI76611
112. Rice KL, Izon DJ, Ford J, Boodhoo A, Kees UR, Greene WK. Overexpression of stem cell associated ALDH1A1, a target of the leukemogenic transcription factor TLX1/HOX11, inhibits lymphopoiesis and promotes myelopoiesis in murine hematopoietic progenitors. Leuk Res. 2008;32(6):873–883. doi:10.1016/j.leukres.2007.11.001
113. Theodoulou NH, Bamborough P, Bannister AJ, et al. Discovery of I-BRD9, a selective cell active chemical probe for Bromodomain containing protein 9 inhibition. J Med Chem. 2016;59(4):1425–1439. doi:10.1021/acs.jmedchem.5b00256
114. Yan Y, Zhang L, Xu T, et al. SAMSN1 is highly expressed and associated with a poor survival in glioblastoma multiforme. PLoS One. 2013;8(11):e81905. doi:10.1371/journal.pone.0081905
115. Sattler S, Reiche D, Sturtzel C, et al. The human C-type lectin-like receptor CLEC-1 is upregulated by TGF-beta and primarily localized in the endoplasmic membrane compartment. Scand J Immunol. 2012;75(3):282–292. doi:10.1111/j.1365-3083.2011.02665.x
116. Zhang S, Chitu V, Stanley ER, Elliott BE, Greer PA. Fes tyrosine kinase expression in the tumor niche correlates with enhanced tumor growth, angiogenesis, circulating tumor cells, metastasis, and infiltrating macrophages. Cancer Res. 2011;71(4):1465–1473. doi:10.1158/0008-5472.CAN-10-3757
117. Bagnyukova TV, Restifo D, Beeharry N, et al. DUSP6 regulates drug sensitivity by modulating DNA damage response. Br J Cancer. 2013;109(4):1063–1071. doi:10.1038/bjc.2013.353
118. Zheng P, Zhang J, Ma H, et al. Design, synthesis and biological evaluation of imidazo[1,5-a]pyrazin-8(7H)-one derivatives as BRD9 inhibitors. Bioorg Med Chem. 2019;27(7):1391–1404. doi:10.1016/j.bmc.2019.02.045
119. Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16(2):101–114. doi:10.1038/nrd.2016.211
120. Nicodeme E, Jeffrey KL, Schaefer U, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468(7327):1119–1123. doi:10.1038/nature09589
121. Remillard D, Buckley DL, Paulk J, et al. Degradation of the BAF complex factor BRD9 by heterobifunctional ligands. Angew Chem Int Ed Engl. 2017;56(21):5738–5743. doi:10.1002/anie.201611281
122. Zoppi V, Hughes SJ, Maniaci C, et al. Iterative design and optimization of initially inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a potent, fast, and selective von Hippel-Lindau (VHL) based dual degrader probe of BRD9 and BRD7. J Med Chem. 2019;62(2):699–726. doi:10.1021/acs.jmedchem.8b01413
123. Michel BC, D’Avino AR, Cassel SH, et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat Cell Biol. 2018;20(12):1410–1420. doi:10.1038/s41556-018-0221-1
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.