Back to Journals » ImmunoTargets and Therapy » Volume 14
Targeting B and T Lymphocyte Attenuator Regulates Lupus Disease Development in NZB/W Mice
Authors Gherardi L, Aubergeon L, Sayah M, Fauny JD, Dumortier H, Monneaux F ![]()
Received 30 August 2024
Accepted for publication 12 December 2024
Published 17 January 2025 Volume 2025:14 Pages 7—23
DOI https://doi.org/10.2147/ITT.S490573
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Léa Gherardi,* Lucie Aubergeon,* Mélanie Sayah, Jean-Daniel Fauny, Hélène Dumortier, Fanny Monneaux
CNRS UPR3572, Immunology, Immunopathology and Therapeutic Chemistry, Institute of Molecular and Cellular Biology, Strasbourg, 67084, France
*These authors contributed equally to this work
Correspondence: Fanny Monneaux, CNRS UPR3572, Institut de Biologie Moléculaire et Cellulaire, 2 Allée Konrad Roentgen, Strasbourg, 67084, France, Email [email protected]
Purpose: The co-inhibitory receptor B and T Lymphocyte Attenuator (BTLA) negatively regulates B and T cell activation. We have previously shown an altered BTLA expression by regulatory T cells and an impaired capacity of BTLA to inhibit CD4+ T cell activation in lupus patients. In this study, we analyzed BTLA expression and function in the NZB/W lupus-mouse model and examined the therapeutic potential of BTLA targeting.
Methods: BTLA expression and function were analyzed in young (10– 12-week-old) and old-diseased NZB/W mice (> 35-week-old with proteinuria) in comparison to age-related BALB/W control mice. 20– 22 weeks old NZB/W mice (n=10) were injected i.p with 3 mg/kg, twice a week for ten weeks, with the anti-BTLA antibody 6F7 or its isotype control.
Results: In old-diseased NZB/W mice, BTLA expression is slightly modified in B cell subsets whereas CD4+ T cells display impaired BTLA functionality. Administration of the 6F7 anti-BTLA antibody into 20– 22 week-old NZB/W mice resulted in a delayed onset of proteinuria (p< 0.01), limited kidney damages (p< 0.05) and an increased survival rate (p< 0.01) compared to isotype-treated mice. This beneficial effect was associated with a decrease in circulating B cell and spleen follicular B cell numbers. Regarding its mode of action, we demonstrated that the 6F7 antibody is not a depleting antibody and does not block HVEM binding to BTLA, but instead induces BTLA down modulation and exhibits in vivo agonist activity.
Conclusion: Overall, our data confirm the involvement of BTLA in lupus pathogenesis and provide the first evidence that BTLA is a potential therapeutic target for the treatment of lupus.
Keywords: systemic lupus erythematosus, BTLA, lupus mice, inhibitory receptors
Introduction
B and T lymphocyte attenuator (BTLA) is an inhibitory receptor of the CD28 superfamily that negatively regulates immune responses in synergy with CTLA-4/B7 and PD-1/PDL1 pathways.1 BTLA is expressed on CD4+ (among which Follicular Helper T cells; TFH) and CD8+ T cells, B cells, as well as on natural killer (NK) cells, NKT cells, macrophages and dendritic cells (DC).2,3 The ligand for BTLA is Herpes Virus-Entry Mediator (HVEM; TNFRS14),4 a TNF receptor family protein found on T, B, NK, DC and myeloid cells. The ligation of BTLA by HVEM attenuates T-cell activation, leading to decreased cell proliferation, cytokine production and cell cycle progression. The inhibitory role of BTLA in vivo was revealed by the analysis of BTLA-deficient mice characterized by a breakdown of self-tolerance, resulting in the development of an autoimmune hepatitis-like disease, lymphocytic infiltration in multiple organs5 and enhanced specific antibody responses and sensitivity to experimental autoimmune encephalomyelitis.1 On the contrary, BTLA engagement through the use of agonist antibodies leads to the induction of tolerance. Indeed, the administration of the agonist anti-BTLA antibody 3C10 was shown to prolong heart allograft survival via the induction of IL-10-producing regulatory T cells6 and to prevent atherosclerosis by shifting the balance between follicular and regulatory B cells.7 Moreover, the agonist BYK1 anti-BTLA antibody prevents the development of graft-versus-host disease, when administered at the time of transplantation.8 BTLA is thus considered as a critical receptor for maintaining peripheral tolerance.
Systemic lupus erythematosus (SLE) is a highly complex and heterogeneous autoimmune disease characterized by the presence of autoantibodies directed against a spectrum of self-antigens (including nuclear antigens) and by the subsequent formation of immune complexes.9 Hallmarks of SLE are immune system activation and inflammation in multiple organs, such as the skin, joints, kidneys, lungs and central nervous system. As autoantibody producers, B cells and their derivatives, plasma cells, are attractive targets to treat lupus.10 However, clinical trials aiming at depleting B cells revealed disappointing results, probably because plasma cells do not express typical B cell surface markers such as CD20 or CD22. Thus, modulation of plasma cell differentiation by targeting the T-B dialog may be an interesting alternative for lupus patients who are refractory to B cell depletion.11 Cross-talk between T and B cells, leading to B cell differentiation into autoantibody-producing cells, can be regulated by various cellular and molecular mechanisms such as regulatory T cells (Tregs) and co-inhibitory receptors. The latter therefore play a key role for the prevention of autoimmune diseases, and as such, any alteration of the expression and/or function of these inhibitory receptor, among them BTLA, could participate to the disturbed lymphocyte homeostasis observed in lupus.12
Data regarding BTLA involvement in lupus pathogenesis are still limited. In mouse-models of lupus, the only available study was performed with MLRlpr/lpr mice, in which genetic BTLA deletion leads to exacerbation of the disease13 suggesting a protective role of BTLA in lupus. In human SLE, relatively few studies have analyzed BTLA expression and function.12 Oster et al found a significant decrease of CD4+BTLA+ T cells in active lupus patients compared to healthy controls (HC) and Murphy et al reported a lower expression of BTLA on CD25hiCD127loCD4+ Tregs compared to Th1 and Th17 cells in lupus patients.14,15 Concerning B cells, BTLA was found to be lower expressed in IgD+CD27− naïve and CD27−IgD− memory B cells from SLE patients compared to HC,16 however, this result was not confirmed by a recent study in which BTLA expression was analyzed by mass cytometry.17 Our group previously described that the upregulation of BTLA expression upon activation is significantly lower in T cells from SLE patients compared to HC.18 In addition, we showed that there is a significant decrease in the capacity of BTLA to inhibit the proliferation and the activation of lupus CD4+ T cells compared to HC. This BTLA deficiency in lupus settings is due to poor BTLA recruitment to the immunological synapse following activation and can be corrected by restoring intracellular trafficking.18 Moreover, we recently observed that BTLA expression is significantly increased on activated Tregs (aTregs) from lupus patients.19 BTLA expression by lupus aTregs correlates with the diminution of the aTregs frequency, suggesting that the higher BTLA expression on the surface of lupus aTregs may account for the reduced frequency of this Treg subset in lupus patients. Altogether, these results highlight a potential involvement of a defective BTLA signaling pathway in lupus patients. As mouse models are crucial for designing and evaluating therapeutic protocols, we then decided to investigate BTLA expression and function in lupus mice.
In the present study, we characterized the phenotype and function of BTLA expressing cells in NZB/W mice, that spontaneously develop a syndrome resembling human SLE notably because of the development of severe immune complex-related glomerulonephritis.20 This often-used mouse model has largely contributed to our understanding of the lupus physiopathology and the recent comparison between human SLE and NZB/W mice by multi-omics analysis leads the authors to consider NZB/W mice as suitable model for deciphering the lupus pathogenesis as well as the response to newly developed therapeutic strategies.21 We found that diseased NZB/W mice display a similar pattern of BTLA expression and function to the one we previously observed in human SLE patients, highlighting the relevance of this mouse-model for evaluating the therapeutic potential of BTLA targeting. Very interestingly, we demonstrated that in vivo administration of an anti-BTLA antibody allowed to alleviate lupus nephritis in NZB/W mice.
Materials and Methods
Mice
Female BALB/W (BALB/c x NZW) F1 and NZB/W (NZB x NZW) F1 mice were bred in our animal facility (approved by French Veterinary Services, #I-67-482-2). All experiments were carried out in conformity with the 2010/63/UE European animal bioethics legislation and were approved by the Regional Ethics Committee of Strasbourg (CREMAS) and by the French Ministry of Higher Education and Research (APAFIS #2020041717124974).
Mouse Treatment and Disease Follow Up
NZB/W females (20–22 weeks of age) were randomly assigned to treatment groups and received either an i.p. administration of the anti-BTLA antibody (clone 6F7; 3 mg/kg/injection, BioXcell, Lebanon, NH, USA), or an appropriate mouse isotype antibody (IgG1κ; 3 mg/kg/injection, BioXcell,) twice a week for 10 weeks. Urine protein levels and mouse weight were determined twice a week and mice exhibiting high proteinuria (score >3) for at least two consecutive weeks were considered to be diseased. Proteinuria was evaluated in fresh urine samples using a colorimetric assay for albumin (Albustix, Siemens, Munich, Germany) and was semi-quantitatively estimated according to the scale recommended by the manufacturer. Blood samples were collected every 3 weeks to evaluate autoantibody levels in the serum and to perform a phenotypic analysis in peripheral blood. Mice were euthanized either i) at limit point achievements (high proteinuria and/or 20% weight loss and/or prostration) in order to evaluate clinical and biological signs of the disease (proteinuria, autoantibodies, survival) or ii) at 33 weeks of age to evaluate the therapeutic effect of anti-BTLA administration on kidney damages.
Depletion of NK Cells and Macrophages
NZB/W were treated with an anti-NK1.1 antibody (clone PK136, Biolegend; 200 µg i.p. on days −2, 0 and 3) to deplete NK cells. Mice were then administered with a high dose of the anti-BTLA 6F7 antibody (500 µg/mouse, i.p.) on day 0 and were sacrificed on day 5. For macrophages depletion, mice were treated with clodronate-loaded liposomes (Liposoma BV; 200 µL/mouse, i.v.; kind gift of Blandine Maître) one day before the 6F7 administration.
Cell Isolation and Cell Culture
Total or naive CD4+ T cells were negatively isolated from spleen using Mojosort Mouse CD4 T Cell Isolation Kit or Mojosort Mouse CD4 Naive T Cell Isolation Kit (Biolegend) according to the manufacturer’s instructions. In order to isolate leucocytes, kidneys were cut and digested for 30 min at 37°C with 0.5 mg/mL collagenase D (Roche) and 0.05 mg/mL DNase I (Roche) in complete medium. Leucocytes were isolated using a 72/36% discontinuous Percoll gradient (Sigma-Aldrich) and centrifugation (30 min, 400 g). The leucocyte-enriched cell suspension was harvested from the Percoll interface.
Analysis of BTLA Function in CD4+ T Cells
Purified CD4+ T cells or naive CD4+ T cells were stimulated with beads (Dynabeads M-450 Epoxy, ThermoFischer Scientific) coated with anti-CD3 (7.5%, clone 145–2C11, BD Biosciences)/anti-CD28 (7.5%, clone 37.51, BD Biosciences)/anti-BTLA (85%, clone 6F7, ThermoFischer Scientific) or IgG1κ antibodies (ratio bead/cell = 1:1). Flow cytometry analysis of CD69, CD25 and BTLA was performed after 24 hours and 48 hours of culture. In order to assess proliferation, cells were incubated for 10 min at 37°C with 5 µM CellTrace Violet (ThermoFisher Scientific). After washing, cells were activated as described above and cultured for 5 days.
BTLA Functional Analysis in B Cells
Splenocytes from mice that had received either the anti-BTLA 6F7 antibody or the isotype control (3 mg/kg/mouse, i.p. on day 0 and day 4) were collected on day 6 and stimulated with a polyclonal anti-IgM antibody (10 µg/mL, Jackson) and a polyclonal anti-IgG antibody (20 µg/mL, Jackson). Flow cytometry analysis of CD69 was performed after 6 hours of culture.
Flow Cytometry Analysis
Cells isolated from various tissues (spleen, blood, kidneys) were stained for 20 minutes at 4°C in staining buffer (2% FCS in PBS) with the following conjugated antibodies: CD4 (clone RM4-5), CD8 (clone 53–6.7), CD19 (clone 1D3), CD23 (clone B3B4), CD25 (clone PC61), CD62L (clone MEL-14), CD69 (clone H1.2F3), CD138 (clone 281–2) from BD Biosciences, CD3 (clone 145–2C11), CD44 (clone IM7), CD45 (clone 30F11), B220 (clone RA3-6B2), BTLA (clone 8F4) from Biolegend, BTLA (clone 6F7), CD21 (clone 8D9), Foxp3 (clone FJK-16s) from ThermoFischer Scientific. For mouse blood phenotyping, 50 µL of blood were first incubated with conjugated antibodies for 20 minutes at 4°C in staining buffer (2% FCS in PBS). Red blood cells were then lysed with lysis buffer (EasyLyse, Agilent Technologies, Santa Clara, USA) and cells were washed twice in staining buffer. Absolute cell numbers were determined in the blood using Precision Count Beads (Biolegend). Apoptosis was evaluated thanks to 4ʹ,6-diamidino-2-phenylindole (DAPI; Life Technologies, Carlsbad, USA) and Annexin-V-APC (BD Biosciences) staining. HVEM binding to BTLA was assessed by incubating cells with the HVEM-Fc recombinant protein (20 µg/mL; Biolegend) followed by a staining with an Alexa 488 conjugated-anti-IgG1 secondary Ab (clone HP6069, ThermoFischer Scientific). Cells were first incubated with rat anti-mouse CD16/32 monoclonal Ab (2.4G2, BioXcell) to block Fc receptors. For FoxP3 and BTLA intracellular staining, cells were fixed and successively permeabilized (FoxP3/staining buffer eBiosciences™ or BD Cytofix/Cytoperm™ and BD Cytoperm Permeabilization Buffer Plus™) according to recommended instructions. Dead cells were excluded using DAPI and single cells were discriminated from aggregates or doublets using SS-W versus SS-H and FS-W versus FS-H plots. Cell acquisition was performed using 10-color Flow Cytometer Gallios-Navios (Beckman Coulter, Brea, USA). Data were analyzed using FlowJo 10.4 software (Treestar, Oregon, USA).
Assessment of Kidney Disease
For histology, kidneys were fixed overnight at 4°C in paraformaldehyde 4% and embedded in paraffin (Leica). Sections of 5 µm were dewaxed, rehydrated and stained with hematoxylin and eosin, dehydrated and permanently mounted. Pathological changes in the kidneys were assessed by evaluating glomerular and tubular activity. Sections were scored using a 0–4 scale as follows: 0 = no lesion, 1 = lesions in <25% of glomeruli/tubules, 2 = lesions in 25–50% of glomeruli/tubules, 3 = lesions in 51–75% of glomeruli/tubules and 4 = lesions in >75% of glomeruli/tubules. Images were acquired with a 40x objective on a NanoZoomer S60 (Hamamatsu Photonics) and analyzed with the QuPath Software.22
For immunofluorescence, kidneys were frozen in Tissue-Tek® O.C.T™ Compound (CellPath, Mochdre) and 7 µm-sections were prepared. They were then mounted on SuperFrost™ Plus glass slides (ThermoFischer Scientific) fixed in cold acetone for 20 min, air dried, and washed in PBS. Sections were saturated with BSA 2% PBS and incubated overnight with an APC-conjugated anti-CD45 antibody (Biolegend) at 4°C. After fixation with paraformaldehyde 4%, slides were stained with DAPI (100 ng/mL) and mounted with Invitrogen Fluoromount-G Mounting Medium (ThermoFischer Scientific). Mosaic images were acquired with a 20x objective on a widefield fluorescent inverted microscope (Axiovert Z1 Zeiss) driven by Metamorph software (Molecular devices). Stitching of images was performed on Metamorph Software and quantification of infiltrate area was performed using both Fiji23 and Ilastik24 softwares.
Analysis of TCR Clustering by Immunofluorescence
Splenocytes (5x105) were first incubated in chambered cell culture slides (Falcon) with BSA 2% PBS and were then stained with anti-TCR-APC (clone H57-597; BD Biosciences) and anti-BTLA-PE (clone 6F7; ThermoFischer Scientific) in BSA 0.5% PBS for 2 hours at 4°C in the dark. After washing, cells were fixed thanks to IC fixation buffer (eBiosciences) for 30 minutes. Fixed cells were washed, stained with DAPI (100 ng/mL) and mounted with Invitrogen Fluoromount-G Mounting Medium (ThermoFischer Scientific). Mosaic images were acquired with a 100x objective on a widefield fluorescent inverted microscope (Axiovert Z1 Zeiss) driven by Metamorph software (Molecular devices). Images were treated on Fiji software.23 Cell segmentation and classification were done on Cellpose25 and Ilastik24 software respectively. Qupath22 software was used for counting different cell classes.
ELISA
Anti-dsDNA and anti-chromatin levels were determined using ELISA tests. Polystyrene plates were coated overnight at 37°C with dsDNA (Sigma, 100 ng/mL, in citrate buffer) or mouse chromatin (1 µg/mL expressed as dsDNA concentration in PBS prepared as described.26 Mouse sera (1/100 to 1/24,300 in PBS-Tween 0.05% BSA 0.5%) were added for 2 h, followed by goat anti-mouse IgG (1/20,000) supplemented with a goat anti-mouse IgG3 (1/10,000) conjugated to HRP for 30 min at 37°C in PBS containing 0.05% Tween and 0.5% BSA. Substrate solution (3,3ʹ,5,5ʹ-Tetramethylbenzidine, TMB) was added, the reaction was stopped with the addition of 1M HCl, and the final absorbance was read at 450 nm with the Varioskan Lux (ThermoFisher) lector.
Statistical Analyses
Data are shown as mean +/− SEM. Data were analyzed using a Mann–Whitney or Kruskal–Wallis tests. Data concerning survival or proteinuria were analyzed using a Log Rank test (Prism version 8.0; GraphPad Software). P-values <0.05 were considered significant.
Results
BTLA Expression in Lupus NZB/W Mice
To determine the role of BTLA in lupus pathogenesis in the NZB/W mouse model, we first examined BTLA expression on B and T lymphocyte subsets at various stages of the disease ie prior biological and clinical symptoms appearance (mice of 10–12 weeks of age) and after disease development (mice older than 35 weeks of age and displaying proteinuria), compared to haplotype-matched BALB/W mice. We found that BTLA is not differentially expressed on CD4+ T cells (or CD4+ T cell subsets) nor CD8+ T cells in lupus mice compared to control age-related BALB/W mice (Figure 1A and B). Regarding B cells, BTLA is similarly expressed by total CD19+ B cells from NZB/W mice and BALB/W mice (Figure 1A), however, we observed a significant increase of BTLA on CD23−CD21− B cells from lupus mice whatever the age and a higher BTLA expression on MZ B cells from old-diseased NZB/W mice compared to old BALB/W mice (Figure 1C). Interestingly, the frequency of CD23−CD21− B cells is significantly enhanced in NZB/W lupus mice compared to age-related BALB/c mice (Supplementary Figure 1). Finally, we confirmed that BTLA is expressed by NZB/W plasma cells (Figure 1D). Altogether, our results show that BTLA expression is not altered in NZB/W lupus mice compared to age-related control BALB/W mice, except slight modifications in some B cell subsets.
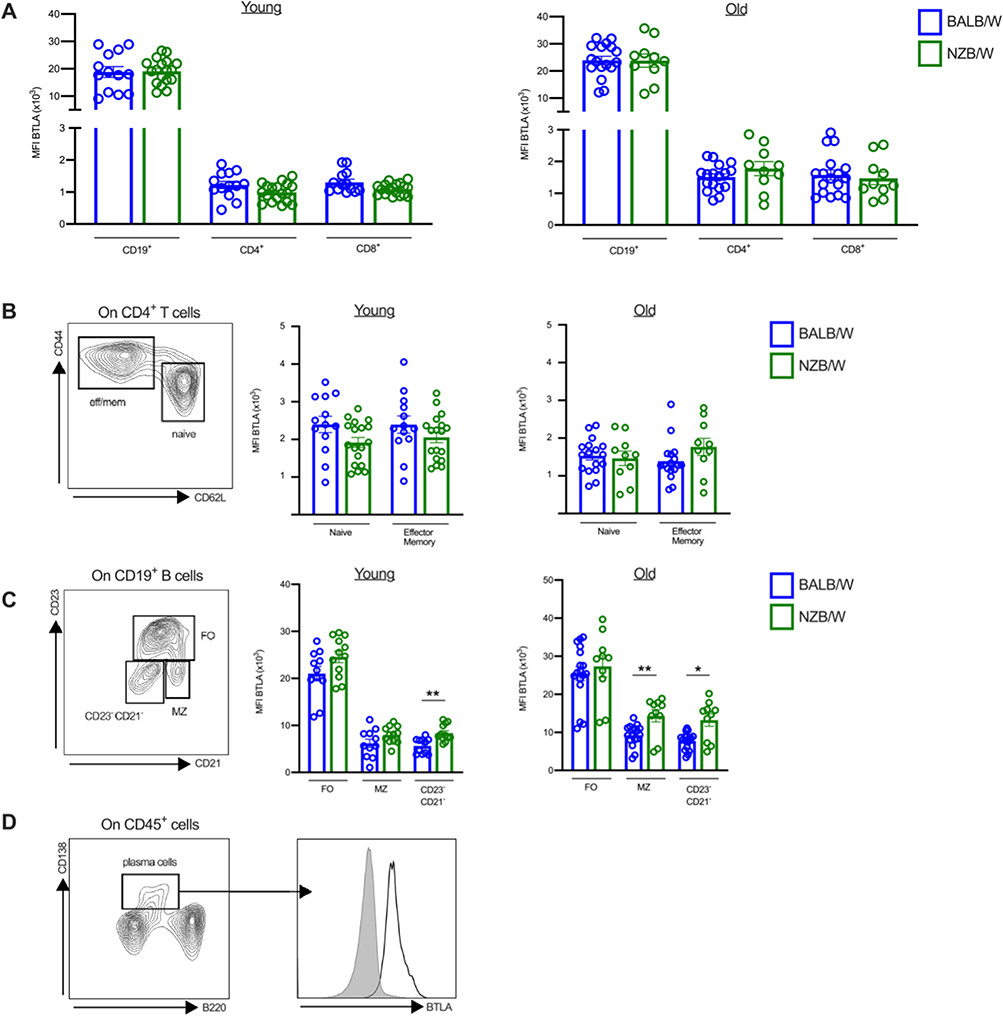 |
Figure 1 BTLA expression on T and B cell subsets. (A–C) Comparison of BTLA expression on CD19+ B cells, CD4+ and CD8+ T cells (A), CD4+ T cell subsets (B), and B cell subsets (C) in young (10–12-week-old, n=13) or old BALB/W (>35-week-old, n=17) and young (10–12-week-old, n=18) or old NZB/W (>35-week-old, n=10). (D) BTLA expression on plasma cells in old NZB/W mice. Results are expressed as mean ± SEM and each dot represents one mouse. *p<0.05; **p<0.01, Mann–Whitney test. |
BTLA Function in NZB/W Lupus Mice
We then wondered whether the engagement of BTLA with an agonist antibody (clone 6F7) could efficiently inhibit CD4+ T cell activation in lupus mice. We observed a decreased capacity of BTLA to inhibit CD69 and CD25 upregulation in old-diseased NZB/W mice (>35-week-old) compared to BALB/W mice of the same age (Figure 2A). Accordingly, proliferation inhibition is also defective in old-diseased lupus mice (Figure 2A). As there is a strong diminution of naive CD4+ T cells towards an enhancement of the effector/memory CD4+ T subsets in diseased-NZB/W mice (>35-week-old) compared to young (10–12-week-old) lupus mice (Figure 2B), we analyzed whether the defective BTLA signaling we noticed in old lupus mice, was linked to this shift toward an effector/memory phenotype. When naive CD4+ T cells from old-diseased NZB/W mice were used for function assessment, the defective BTLA functionality was not observed anymore (Figure 2C). Finally, BTLA upregulation upon activation, which is known to be induced following T cell activation, is significantly lower in total CD4+ T cells but not in naive CD4+ T cells from old-diseased NZB/W mice compared to control mice (Figure 2D). Unfortunately, we were not able to perform the reverse experiment as memory CD4+ T cells from old NZB/W mice are refractory to in vitro activation (Supplementary Figure 2). To exert its inhibitory function, BTLA has to be recruited to TCR clusters. In human SLE patients, we described that BTLA is excluded from pre-clustered TCR that are present in CD4+ T cells.18 As previously described by others,27 TCR pre-clusters can be visualized in the absence of stimulation in diseased-NZB/W mice (cell-23 compared to cell-5, Figure 2E, left panel) and interestingly, we observed that BTLA is not always properly recruited to them (Cell-7-37-38 and-6 are shown as examples, Figure 2E, right panel). Altogether, our results suggest that defective BTLA functionality in old diseased NZB/W mice is related to the high frequency of effector/memory CD4+ T cells, that are refractory to BTLA-mediated inhibition.
Figure 2 Continued. Figure 2 Impaired BTLA functionality in CD4+ T cells from old NZB/W mice. (A) CD4+ T cells from young (10–12-week-old, n=11) or old BALB/W (>35-week-old, n=13–16) and young NZB/W (10–12-week-old, n=11–16) or old NZB/W (>35-week-old, n=9–10) were stimulated with anti-CD3/CD28 antibody-coated beads in the presence of the anti-BTLA 6F7 antibody or its isotype control. The expression of CD69 at 24h, of CD25 at 48h, and CTV dilution at 5 days were analyzed by flow cytometry. Results are expressed as the percentages of inhibition of the activation and of the proliferation. (B) Frequencies of naive and effector/memory CD4+ T cell in young (n=13) and old (n=10) NZB/W mice are represented. (C) Naive CD4+ T cells from old BALB/W (n=14) and old NZB/W mice (n=10) were stimulated as in (A) and the percentages of inhibition of the activation and of the proliferation are represented. (D) Total CD4+ T cells (left) or naive CD4+ T cells (right) of old BALB/W (n=16) and old NZB/W mice (n=10) were stimulated with anti-CD3/CD28 antibody-coated beads and cultured for 24h. BTLA fold enhancement (ratio of BTLA MFI following activation/BTLA MFI without stimulation) is represented. (E) Unstimulated NZB/W splenocytes (>35-wk-old) were stained with anti-TCR-APC (in green) and anti-BTLA-PE (in red) and analyzed on a widefield fluorescent inverted microscope. Pre-clustered TCR are denoted by white arrows. Examples of non-clustered TCR (cell-5), pre-clustered TCR associated with BTLA (cell-23) and of pre-clustered TCR in which BTLA is excluded (cell-7, cell-37, cell-38 and cell-6) are represented. Scale bar: 3 µm. **p<0.01; ***p < 0.001; ****p<0.0001, Mann–Whitney test.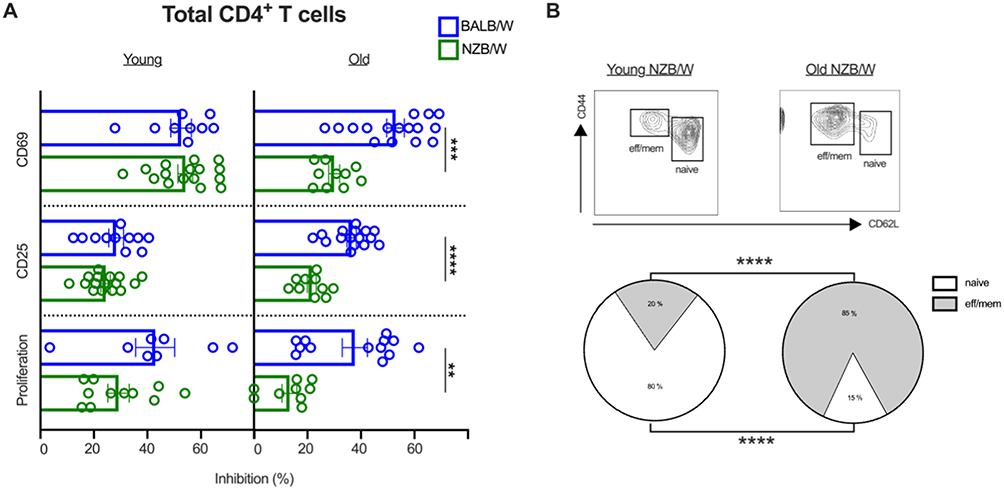
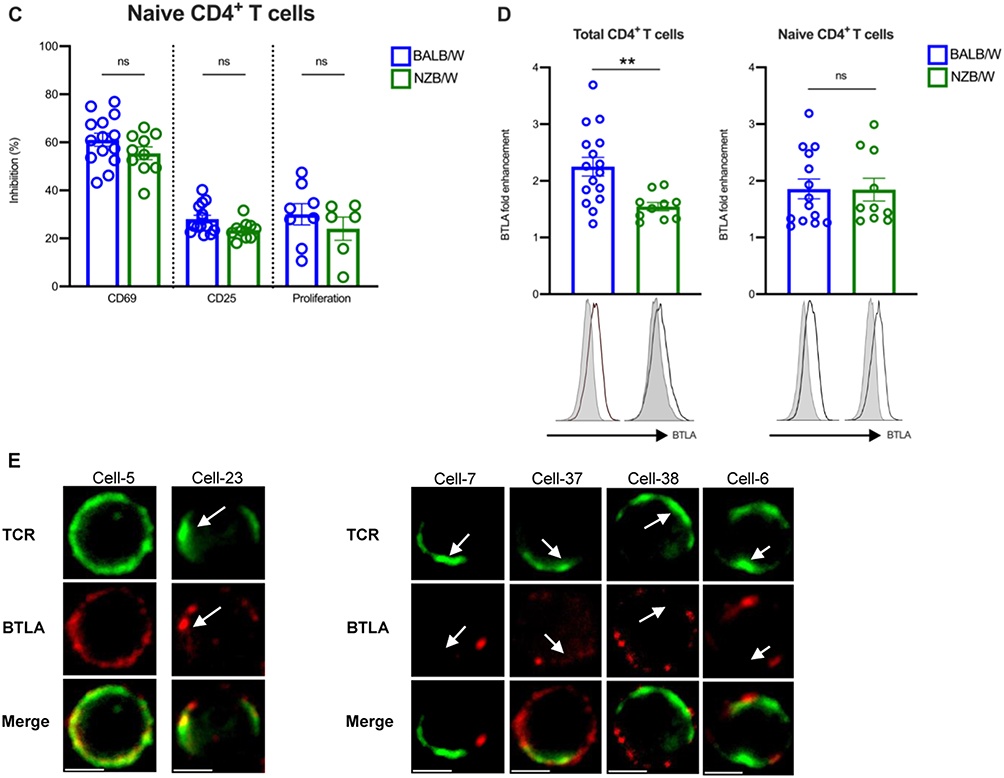
Administration of the 6F7 Anti-BTLA Antibody Delays Proteinuria Development and Extends Survival of NZB/W Mice
We administered the 6F7 antibody into 20–22-week-old NZB/W mice (Figure 3A), which can be considered as being in the preclinical phase of autoimmunity as these mice already exhibit mild to high levels of autoantibody titers without any overt pathological manifestations.28 The 6F7 antibody administration results in delayed proteinuria onset (Figure 3B) and limits the disease-related body weight loss of NZB/W lupus mice (Figure 3C). Consequently, 6F7-treated mice exhibited an extended lifespan (Figure 3D). The beneficial effect was accompanied by a significant decrease of peripheral CD19+ B cells. Indeed, circulating B cell frequencies were significantly reduced during anti-BTLA treatment (Figure 3E). Interestingly, the decrease in circulating B cell frequency perfectly corresponds to BTLA receptor occupancy (RO) which was evidenced by the inability of exogenous labeled 6F7 antibody to bind to CD19+ B cells and CD3+ T cells from 6F7-treated mice ex vivo (Figure 3F). The 6F7 administration does not lead to the diminution of T cell frequencies although the antibody efficiently binds BTLA expressed by T cells (Supplementary Figure 3). Unfortunately, we did not notice any significant differences regarding levels of circulating anti-dsDNA and anti-chromatin IgG antibodies (data not shown) in comparison with isotype control-treated mice.
 |
Figure 3 Administration of anti-BTLA 6F7 antibody to NZB/W mice delays the onset of proteinuria and prolongs survival. (A) NZB/W mice of 20–22 weeks of age received either i.p. administration of an anti-BTLA antibody (clone 6F7; 3 mg/kg/administration, n=10) or an appropriate mouse isotype antibody (IgG1κ; 3 mg/kg/administration, n=10) twice a week for 10 weeks. Mice were followed for various parameters and sacrificed at limit points. (B) Percentages of NZB/W mice that developed severe proteinuria in the group of anti-BTLA-treated mice and isotype-treated mice. (C) Body weight loss expressed as a ratio compared to the initial body weight in anti-BTLA-treated mice and isotype-treated mice. (D) Percentages of mice that had survived in anti-BTLA treated or isotype control treated mice. (E) Percentages of peripheral CD19+ B cells in the anti-BTLA-treated group and the isotype-treated group. 50 µL of blood were first incubated with conjugated antibodies, red blood cells were then lysed with lysis buffer, washed twice and analyzed by flow cytometry (F) Blood B were stained with a PE-conjugated 6F7 antibody to evaluate receptor occupancy (RO) in 6F7-treated mice during the course of the treatment. RO for each timepoint was calculated as follow: 1-[MFI 6F7/MFI 6F7at day 0]x100. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001, Log-rank (B and D) or Mann–Whitney test (C and E). |
Administration of the 6F7 Anti-BTLA Antibody Reduces Follicular B Cell Numbers in the Spleen and Leukocyte Infiltration in the Kidneys
To assess the consequences of BTLA targeting in the spleen and kidneys, we initiated a second therapeutic protocol in which mice were euthanized one week after the last antibody administration. We confirmed the decrease of B cell frequencies (and B cell numbers; Supplementary Figure 4) in the blood, except for one mouse (mouse NZB/W#1, highlighted with a green frame) (Figure 4A). This observation, along with our subsequent analyses (Supplementary Figure 5), suggest that the mouse NZB/W#1 did not respond to the anti-BTLA antibody administration, likely due to an experimental issue. Consequently, we decided to exclude this mouse from the following steps of the analysis. Splenomegaly is also reduced in 6F7-treated mice (Figure 4B) and accordingly, the total number of lymphocytes in the spleen seems to be decreased in 6F7-treated mice compared to isotype-treated mice (p = 0.056; Figure 4C). Interestingly, B lymphocytes are affected, particularly follicular (FO) B cells, which express the highest BTLA level (p < 0.001), but not T lymphocytes (Figure 4C and D). Contrary to what has been shown in the NOD diabetes mouse model,29 we did not notice any Treg expansion in the spleen of NZB/W lupus mice following 6F7 administrations (Figure 4E). Furthermore, we observed that immune cell proportions in the spleen of 6F7-treated mice, are more closely related to those of young, non-diseased NZB/W mice than those of old-diseased animals, suggesting that the 6F7 administration was able to revert immune cell alterations occurring during lupus development (Figure 4F).
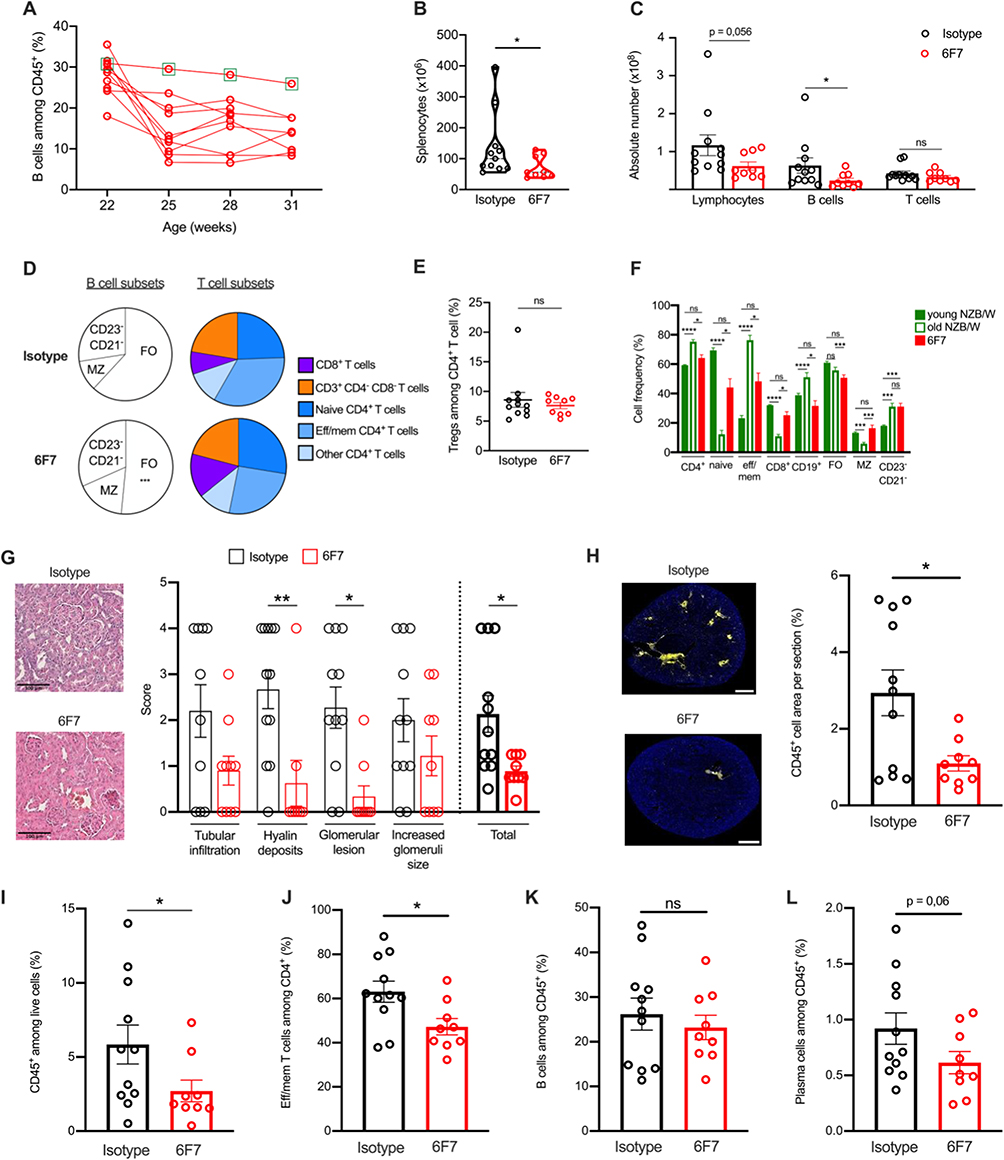 |
Figure 4 Anti-BTLA 6F7 antibody administration decreases spleen FO B cells and reduces kidney damages. NZB/W mice were treated as in Figure 3 and sacrificed one week after the end of the treatment. (A) Percentages of peripheral CD19+ B cells in the anti-BTLA-treated group. The NZB/W#1 mouse is highlighted with a green frame. (B) Comparison of splenocyte numbers between isotype and 6F7-treated mice. (C and D) Absolute numbers of lymphocytes, B and T cells (C), and frequencies of B and T cell subsets (D) in the spleen of isotype and 6F7-treated mice. (E) Frequency of spleen Tregs in isotype and 6F7-treated mice. (F) Lymphocyte frequencies in the spleen of young, old-diseased and 6F7-treated NZB/W mice. (G) Kidney sections from representative mice and comparison of pathological changes assessed by evaluating glomerular activity and tubulointerstitial activity between anti-BTLA and isotype treated mice. Sections were scored using a 0–4 scale as follows: 0 = no lesion, 1 = lesions in <25% of glomeruli/tubules, 2 = lesions in 25–50% of glomeruli/tubules, 3 = lesions in 51–75% of glomeruli/tubules and 4 = lesions in > 75% of glomeruli/tubules (H) Kidney sections were stained with anti-CD45 antibodies. Entire sections were reconstituted using Metamorph software and areas corresponding to tubulointerstitial infiltrates were quantified with Fiji software. Scale bars: 100 µm. (I–L) Frequencies of leukocytes (I), effector /memory CD4+ T cells (J), B cells (K) and plasma cells (L) in 6F7 or isotype-treated mice. *<0.05; **p<0.01; ***p<0.001; ****p<0.0001, Mann–Whitney or Kruskal–Wallis with post hoc analysis (Dunn’s test). |
Finally, anti-BTLA treated mice displayed limited kidney damages as shown by a reduction in glomerular size and cellularity, in mesangial expansion and deposits, and in glomerular damages, all being taken into consideration in the histopathological score (Figure 4G). Kidneys of anti-BTLA-treated mice were also less infiltrated by CD45+ immune cells (Figure 4H and I), with reduced infiltration of effector/memory CD4+ T cells (Figure 4J) but not B cells (Figure 4K), and a tendency to a decrease of plasma cell frequency (p = 0.06; Figure 4L).
6F7 Administration Down-Modulates BTLA Cell Surface Expression and Induces B Cell Hyporesponsiveness
Antibody targeting surface molecules can lead to different outcomes such as receptor down modulation, cell depletion, blockade of receptor/ligand interaction or agonist activities. To analyze whether the 6F7 antibody administration modifies BTLA expression, we used another anti-BTLA antibody (clone 8F4), which recognizes another epitope than the 6F7 clone (as confirmed by the double staining, Figure 5A, left panel) and is therefore still able to bind BTLA following 6F7 exposure (Figure 5A, right panel). We observed a reduced BTLA expression on spleen B cells (particularly FO B cells; Figure 5B) but only a slight diminution on T cells (Figure 5C) from 6F7-treated mice. The decrease of BTLA expression is similar when we compare both surface and intracellular staining, indicating that BTLA is not sequestered into the cells of mice that had received the 6F7 antibody (Figure 5D). Accordingly, we found that BTLA downmodulation is not due to its early internalization following 6F7 binding (Figure 5E). Subsequently, in order to address whether the 6F7 antibody harbors depleting properties in vivo, we administered mice with a high dose of the 6F7 antibody (500 µg; 15 mg/kg) and analyzed spleen lymphocyte numbers 5 days later. Moreover, in order to decipher whether antibody-dependent cellular cytotoxicity or phagocytosis (ADCC/ADCP) are involved, some mice were depleted for NK cells and macrophages, respectively, prior 6F7 administrations. No significant decrease in lymphocyte numbers was observed in the spleen 5 days upon 6F7 injection (Figure 6A), which correlates with the lack of depleting activity reported for the majority of mouse antibodies of the IgG1 isotype. Moreover, splenocytes incubated in the presence of the 6F7 antibody did not die from apoptosis (Figure 6B). We also explored whether 6F7 could act as a blocking antibody and found that the binding of a HVEM fusion protein to BTLA is preserved when splenocytes were incubated with saturating amounts of the 6F7 antibody (Figure 6C). Finally, we wondered whether the 6F7 administration influences B cell capacity to be further activated. We analyzed CD69 expression on B cells from mice that had received either the 6F7 antibody or the isotype control and evidenced that B cells from 6F7-treated mice are less responsive to BCR activation (Figure 6D), indicating an agonist activity of the 6F7 antibody in vivo. Altogether, our results show that the 6F7 antibody, is not a depleting antibody, induces a decrease of BTLA expression (particularly on cells expressing high levels of this molecule), does not block the HVEM binding to BTLA and reduces B cell activation.
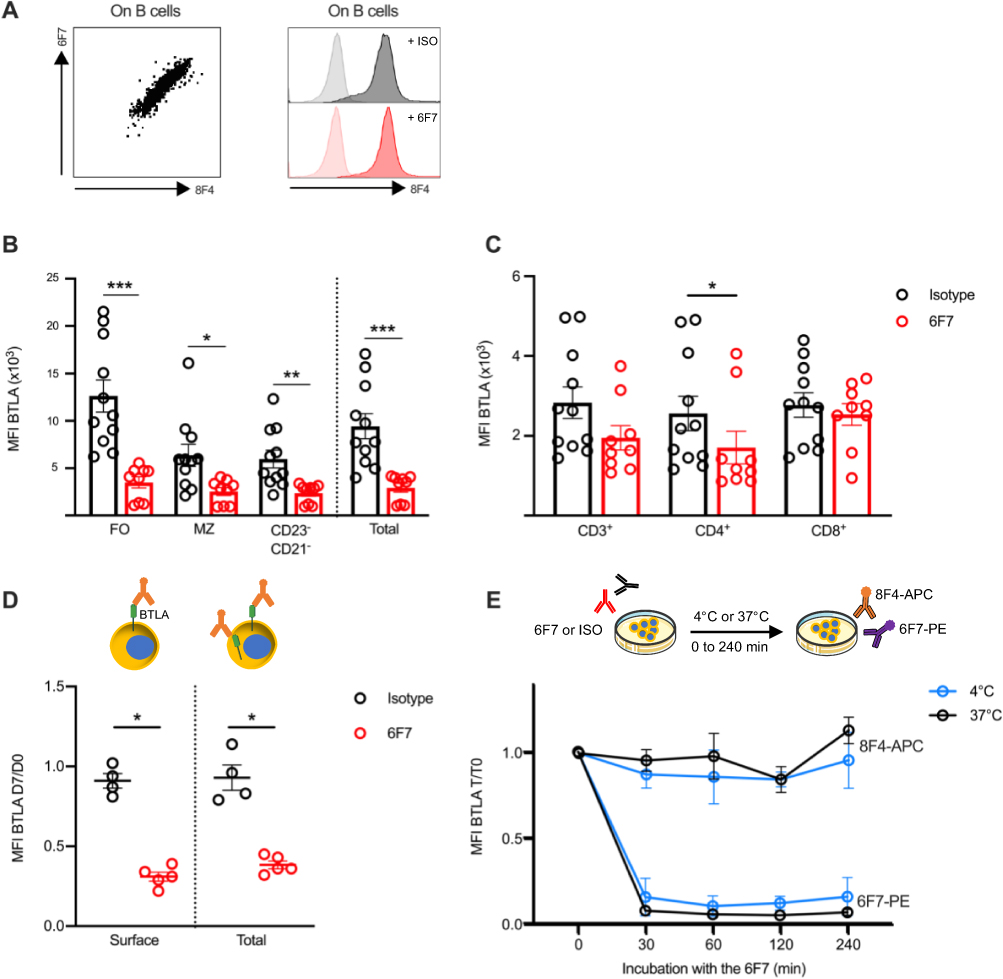 |
Figure 5 Administration of anti-BTLA 6F7 induces down modulation of BTLA expression. (A) The 8F4 anti-BTLA antibody can be used to detect BTLA expression following 6F7 exposure. Left: representative image of splenocytes co-stained with APC-conjugated anti-BTLA 8F4 and PE-conjugated anti-BTLA 6F7 antibodies. Right: splenocytes were incubated with saturating amounts of unlabeled 6F7 (10 µg/mL) or isotype control, and then stained with fluorescent-8F4 antibodies. (B and C) BTLA expression (detected by the 8F4 antibody) on spleen B cells (B) and T cells (C) of isotype or 6F7-treated mice sacrificed one week after the last administration. (D) Mice were administered with 3 mg/kg of 6F7 antibody (n=5) or isotype (n=4) at day 1 and day 4, and the blood was collected on day 7. Membrane (surface BTLA) and intracellular (total BTLA) staining with the 8F4 antibody were performed on B cells and BTLA expression is shown. Results are expressed as ratio of MFI between day 7 and day 0. (E) Splenocytes from BALB/W mice (n=3-4) were incubated with saturating amounts of 6F7 antibody (10 µg/mL) for 30 to 240 minutes at 4°C or 37°C and then stained with APC-conjugated 8F4 or PE-conjugated 6F7 antibodies. Results are expressed as ratio of MFI (T/ T0). *p<0.05; **p<0.01; ***p<0.001; Mann–Whitney test. |
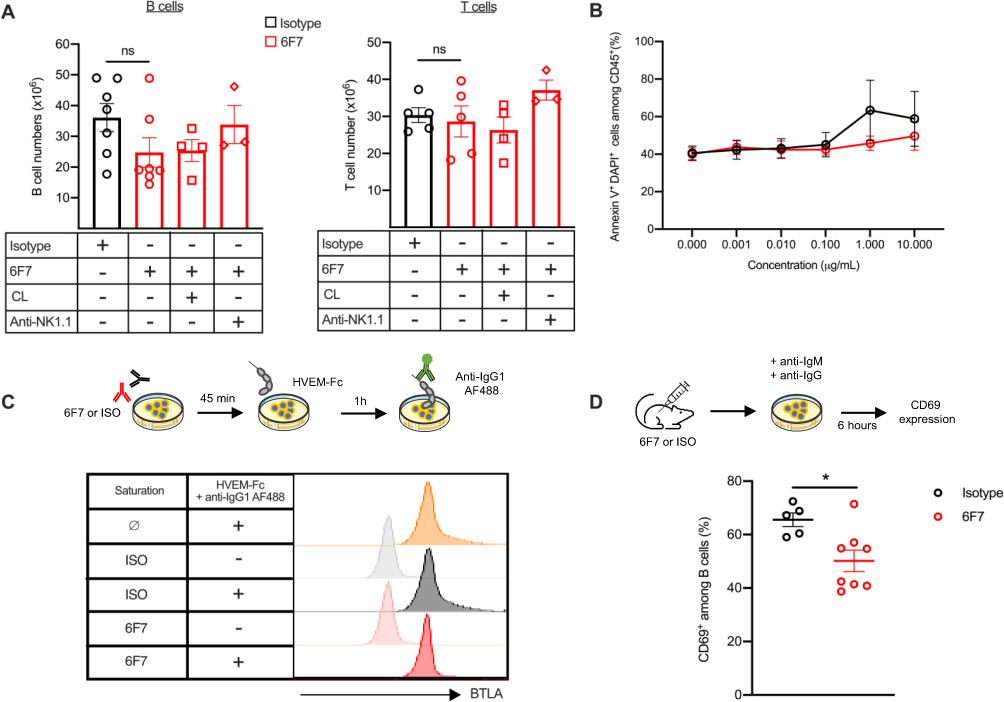 |
Figure 6 Activation of BTLA leads to B cell hyporesponsiveness. (A) Mice were administered with 500 µg of 6F7 antibody (15 mg/kg; n=7) or isotype (n=7) at day 0 and were sacrificed on day 5 to collect spleens and determined cell numbers. Some mice were firstly depleted in NK cells thanks to i.p. administrations of an anti-NK1.1 depleting antibody (200 µg on days −2, 0 and 3, n=3) or in macrophages thanks to i.v. administration of clodronate-loaded liposomes (200 µL on day −1, n=4). (B) Splenocytes were incubated with increasing concentrations of 6F7 (in red) or isotype (in black) antibodies for 24h. Cell death was measured with DAPI and Annexin-V staining on CD45+ cells. As control, apoptosis was analyzed in the presence of H2O2 (>90% of cell death). (C) Splenocytes were firstly incubated with saturating amounts of unlabeled 6F7 (10 µg/mL; 45 min at 4°C) or isotype antibodies. Cells were washed and incubated with HVEM-Fc mouse fusion protein (20 µg/mL; 1 h at 37°C) and binding of HVEM-Fc to BTLA was revealed with an AF488-conjugated anti-IgG1 antibody. One representative experiment of 4 with similar results is depicted. (D) Splenocytes from mice treated as in (A), were stimulated with anti-IgM (10 µg/mL) and anti-IgG antibodies (20 µg/mL) for 6 h. Cells were then incubated with anti-CD16/32 antibodies, stained with anti-CD19 and anti-CD69 conjugated antibodies and analyzed by flow cytometry. *p<0.05, Mann–Whitney. CL: clodronate-loaded liposomes. |
Discussion
Upon its discovery, research on the involvement of the BTLA pathway in autoimmune disorders has swiftly emerged. However, despite mounting evidence underscoring BTLA’s role in maintaining tolerance, its implication in lupus pathogenesis remains unclear. While recent studies have investigated BTLA expression and function in human patients’ cells,16,18,19 only one study in a mouse lupus model has shown that BTLA deficiency exacerbates lupus disease progression.13 In the present study, we conducted a comprehensive analysis of BTLA expression and function in the classic NZB/W lupus-mouse model. Our findings reveal no significant alteration in BTLA expression compared to control mice, except an enhancement on CD23−CD21− and MZ B cells particularly in aged diseased animals. CD23−CD21− B cells were firstly described by Hao et al as a novel subset of splenic B cells that accumulate with age in unimmunized mice and that are able to produce antibodies in response to TLR but not BCR stimulation.30 We also found that the frequency of CD23−CD21− B cells, that express higher BTLA levels, is significantly enhanced in diseased-NZB/W lupus mice compared to old-BALB/W. This higher BTLA expression could reflect an activated state of these cells. Indeed, activated cells typically exhibit increased BTLA levels and CD21−CD23− were shown to express several markers characteristics of memory activated cells including Fas, CD80, CD86 or MHC class II molecules.30
Additionally, we found that akin to human lupus T cells, the BTLA signaling pathway is impaired in CD4+ T cells from diseased-NZB/W lupus mice. Although we could not assess BTLA functionality in purified effector/memory CD4+ T cells from diseased NZB/W mice due to their poor proliferative response to antigenic stimulation in vitro31 and this work, our findings unequivocally show that the compromised BTLA functionality is not attributed to naive CD4+ T cells. In lupus patients, the impaired ability of BTLA to inhibit T cell activation is due to defective recruitment of BTLA to preexisting TCR clusters. Given that the latter have been observed even without stimulation in diseased NZB/W mice,27 and that BTLA is not properly recruited into part of them (present work), we propose that altered BTLA functionality in this mouse model is linked to the exclusion of BTLA from these pre-clustered TCR in effector/memory T cells.
Furthermore, we investigated the impact of administering an anti-BTLA antibody on disease features in NZB/W mice, revealing compelling evidence that targeting this pathway may be highly beneficial. Remarkably, in vivo treatment with the anti-BTLA 6F7 antibody significantly decreased the mortality rate among NZB/W mice and mitigated renal infiltration by lymphocytes, consequently delaying the onset of nephritis. Relapse occurred rapidly after the loss of BTLA receptor occupancy, suggesting that the antibody’s beneficial effect probably does not involve the induction of a tolerance mechanism but instead requires ongoing stimulation of the BTLA pathway. Consistent with this, we did not notice any modification of the Treg frequency in 6F7-treated animals. This result disagrees with data obtained by Truong et al, who showed that 6F7 administration to NOD diabetic mice led to the enhancement of CD4+FoxP3+ T cells.29 However, BTLA expression by DCs was found to be required for peripheral Treg induction.32 Although we did not analyze BTLA expression by DCs in our 6F7-treated lupus mice, we can speculate that as for B and CD4+T cells, DCs are affected by BTLA down-modulation. As a consequence, these BTLAlow DC may not be able to prone Treg differentiation.
Contrary to what we may expect, administration of the 6F7 did not result in reduced levels of anti-dsDNA or anti-nucleosome auto-antibodies. Some of these autoantibodies are produced by long-lived plasma cells (LLPCs), which are established very early in the spleen of NZB/W lupus mice.33 Spleen LLPCs can be detected as early as 6 weeks of age and their numbers continue to increase until they reach a plateau at 10 weeks of age. When we started the 6F7 administrations, at the age of 22 weeks, LLPCs were therefore well established in the spleen and may have produced large amounts of autoantibodies by long time. Accordingly, some mice already had anti-dsDNA autoantibodies prior the beginning of the treatment. However, it was demonstrated that pathogenic autoantibodies are mostly produced by LLPCs localized in inflamed-kidneys.34 Contrary to the spleen, LLPCs only begin to appear in the kidneys at 16 weeks of age, and their numbers constantly increase throughout the progression of the disease.33 Interestingly, we found that while frequency of CD138+ plasma cells decreases in the kidneys of 6F7-treated mice, it remains unchanged in the spleens (data not shown). This suggests that although 6F7 treatment does not significantly impact the well-established LLPCs in the spleen, it appears to limit their accumulation in kidneys. Finally, previous findings demonstrated that B cells alone (rather than autoantibodies) are sufficient for nephritis and cell infiltration.35 Accordingly, several studies have described efficient therapeutic strategies in lupus mice without concomitant decreases of anti-DNA antibody levels,36,37 while diminution of autoantibodies did not always translate to pathology improvements.38 In the Ldlr−/− model, administration of the agonist anti-BTLA 3C10 antibody7 attenuates atherosclerosis, without affecting total or antigen-specific antibody levels, aligning with data showing that allogenic humoral response remains unaffected following antibody-mediated blockade of the HVEM/BTLA pathway.39
Published data on the mode of action (MOA) of anti-BTLA antibodies in vivo vary depending on the clone that was used: the 6A6 antibody is a blocking but non-depleting antibody,8 while clones 3C10, BYK-1, 4G12b are classified as agonist antibodies.6,40,41 Contradictory data have been reported in the literature regarding the 6F7 antibody: Truong et al documented that the 6F7 antibody depletes B and T cells in the NOD diabetic model,29 whereas it exhibits in vivo an agonist activity in an induced-colitis model and in tuberculosis.42,43 Depleting antibodies can induce cell death through various mechanisms, including complement-dependent cytotoxicity (CDC), direct induction of apoptosis and ADCC or ADCP. The 6F7 antibody is an IgG1 isotype, which is unable to bind C1q and therefore does not efficiently activate the classical complement pathway. Furthermore, we evidenced that the 6F7 antibody inhibition of B cells does not involve induction of apoptosis. Lastly, it’s worth noting that mouse IgG1 does not bind the mouse FcRI and FcRIV which are the primary Fc receptors involved in ADCC or ADCP mediated by NK and myeloid cells, respectively.44 This limits the likelihood that the 6F7 antibody might have induced depletion through ADCC/ADCP mechanism, which was confirmed by our results. B cell depletion from the circulation could thus potentially reflect B cell migration into tissues rather than B cell killing. Our data showed that circulating B cells do not migrate into secondary lymphoid organs as their frequency is also decreased into the spleen (Figure 4C) and lymph nodes (data not shown), nor to inflamed kidneys (Figure 4K). Alternatively, circulating B cells could have been sequestered into the liver as sinusoidal endothelial cells express high levels of FcRIIb, which are able to bind mouse IgG1.
Our results demonstrate that administering the 6F7 antibody results in diminished BTLA expression in splenocytes. The 6F7 MOA thus resembles the one of the anti-BTLA clone PJ196, which did not deplete BTLA-expressing cells but caused its down regulation, promoting islet allograft acceptance.45 The mechanism leading to BTLA down-modulation does not rely on its internalization. Another hypothesis could be its direct cleavage from the membrane, as it was shown for the human anti-BTLA ANB032 developed by AnaptysBio and this assumption needs further investigations. BTLA downmodulation correlates with cell frequency declining: indeed, FO B cells exhibit the most substantial reduction of BTLA expression, and notably, they are the sole cell population wherein a significant decreased number was observed in the spleen. Accordingly, it was suggested that BTLA expression is necessary for the survival of FO B cells.39 In line with our findings, the administration of the agonist 3C10 anti-BTLA antibody also selectively reduced FO B cells in the Ldlr−/− mouse model of atherosclerosis.7 Interestingly, and not surprisingly given that BTLA is a negative regulator of Syk,46 the oral administration of a selective Syk inhibitor attenuates lupus nephritis and also reduces FO B cells without affecting the proportion of MZ B cells in the NZB/W lupus model.28
The scenario involving the NZB/W#1 mouse presents an intriguing puzzle: there’s no reduction in B cell frequencies in the blood, suggesting that the mouse does not react to the anti-BTLA treatment, as also evidenced by the absence of effect in all clinical parameters. Moreover, BTLA expression remains unchanged in spleen B cells of this mouse (contrary to other mice), hinting that the treatment’s efficacy may hinge on the antibody reaching the spleen. Altogether, our data provide evidence that BTLA downmodulation is part of the MOA of the 6F7 antibody.
Alternatively, the 6F7 antibody may affect survival of multiple cell type by modulating BTLA activity in a non-competitive manner with its ligand HVEM. Indeed, BTLA also serves as a ligand to deliver pro-survival cosignaling.39 Consequently, as BTLA expression diminishes following 6F7 exposure, fewer positive signals are engaged to HVEM-expressing cells. Additionally, the binding of HVEM is preserved in the presence of the 6F7 antibody thus allowing the endogenous inhibitory pathway to remain active. Besides reducing BTLA expression and FO B cell numbers, we found that 6F7 administration significantly reduces B cell responsiveness to BCR stimulation. This indicates that the 6F7 antibody acts in vivo through direct agonism of the BTLA pathway on B cells.
Current treatments for lupus do not adequately control disease activity and tissue damages and are associated with significant side effects. Therefore, the identification of new therapeutic targets is a major unmet clinical need. We suggest that utilizing a non-depleting anti-BTLA antibody may provide an improved safety profile by avoiding issues related to immune suppression. Unlike anti-CD20 antibodies, which deplete all B cell subsets, the 6F7 antibody specifically reduces but does not ablate FO B cells and attenuates without entirely halting B cell activation. This mechanism appears to address immune abnormalities that contribute to lupus symptoms. Therefore, activating the BTLA pathway emerges as a promising avenue for modulating lupus disease. Finally, although we did not perform a long term administration protocol into healthy BALB/W mice, we found that 6F7 administration also leads to a reduction of peripheral B cells (not shown), indicating a common MOA independently of the mouse strain, and that BTLA targeting may be efficient in the treatment of other B cell-related diseases. Even if the generation of agonist antibodies remains challenging, companies already underwent into this direction and several drugs are currently investigated in clinical trials in various autoimmune or inflammatory diseases.12 In 2023, Lilly completed the recruitment of 85 SLE patients for a Phase 2 clinical trial evaluating the safety and efficacy of LY3361237, an agonist anti-human BTLA antibody (22B3 antibody47). Unfortunately, the study was terminated recently due to a lack of efficacy. This disappointing result highlights the current need for depth understanding of BTLA involvement in lupus. Indeed, BTLA targeting to enhance its inhibitory function tightly depends on its surface expression and our group has described altered BTLA expression on Tregs19 and DN memory B cells48 from SLE patients. The use of agonist anti-BTLA antibodies may thus favor the inhibition of Tregs expressing higher BTLA in active lupus patients, whereas it will not target pathogenic DN memory B cells exhibiting very low BTLA levels. Moreover, altered BTLA functionality, in CD4+ T cells18 or B cells,16 may limit their inhibition through agonist antibodies. However, our results demonstrate that despite defective BTLA function in CD4+ T cells, the 6F7 administration was efficient to alleviate lupus symptoms in the NZB/W mouse model in vivo. Altogether, these data show that targeting the BTLA pathway is an interesting and promising avenue for the development of new therapeutic strategies in lupus, but also underline that a better understanding of BTLA biology is a prerequisite prior translation to the clinic.
Conclusion
In conclusion, our study highlights the pivotal role of BTLA signaling in lupus pathogenesis. To date, there are no reports exploring the potential of targeting this pathway in murine lupus. We demonstrated that administering the 6F7 antibody to pre-diseased NZB/W mice alleviates lupus development. The MOA of the non-depleting 6F7 antibody is multifaceted and includes i) downmodulation of BTLA expression, likely contributing to the decrease of FO B cell numbers and the reduction of positive signals to HVEM-expressing cells, ii) preservation of HVEM binding allowing the endogenous inhibitory pathway to remain active, iii) direct agonist activity (Figure 7).
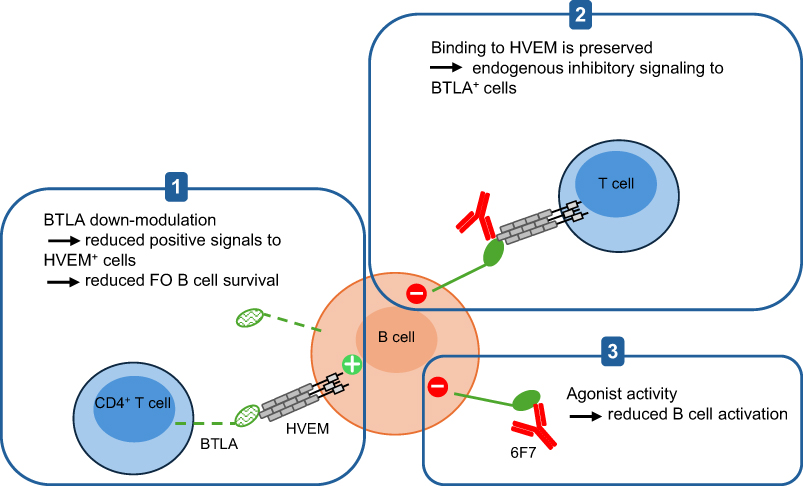 |
Figure 7 Proposed mechanism of action for the 6F7 anti-BTLA antibody. The 6F7 clone induces BTLA down-modulation and exhibits agonist properties. Moreover, the 6F7 anti-BTLA antibody does not block BTLA binding to its ligand HVEM. Altogether, these characteristics may reduce the survival of HVEM-positive cells and FO B cells (1), allow the endogenous inhibitory signaling through HVEM binding (2) and decrease B cell activation status (3). |
Acknowledgments
This work was supported by the French Centre National de la Recherche Scientifique (CNRS), the French Society of Rheumatology (grant to FM) and the French “Ministère de l’Enseignement et de la Recherche” (fellowship to LA and LG). We thank Delphine Lamon and Fabien Lhericel (CNRS UPR3572) for helping with mouse experiments and Hayet Dali for technical assistance. We thank Dr Blandine Maître (Etablissement Français du Sang, Strasbourg) for her generous gift of clodronate-loaded liposomes. This paper has been uploaded to BioRxiv as a preprint: https://www.biorxiv.org/content/10.1101/2024.05.28.596218v1
Disclosure
The authors report no conflicts of interest in this work.
References
1. Watanabe N, Gavrieli M, Sedy JR, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003;4:670–679. doi:10.1038/ni944
2. Han P, Goularte OD, Rufner K, et al. An inhibitory Ig superfamily protein expressed by lymphocytes and APCs is also an early marker of thymocyte positive selection. J Immunol. 2004;172:5931–5939. doi:10.4049/jimmunol.172.10.5931
3. M’Hidi H, Thibult M-L, Chetaille B, et al. High expression of the inhibitory receptor BTLA in T-follicular helper cells and in B-cell small lymphocytic lymphoma/chronic lymphocytic leukemia. Am J Clin Pathol. 2009;132:589–596. doi:10.1309/AJCPPHKGYYGGL39C
4. Sedy JR, Gavrieli M, Potter KG, et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol. 2005;6:90–98. doi:10.1038/ni1144
5. Oya Y, Watanabe N, Owada T, et al. Development of autoimmune hepatitis-like disease and production of autoantibodies to nuclear antigens in mice lacking B and T lymphocyte attenuator. Arthritis Rheum. 2008;58:2498–2510. doi:10.1002/art.23674
6. Uchiyama M, Jin X, Matsuda H, et al. An agonistic anti-BTLA mAb (3C10) induced generation of IL-10-dependent regulatory CD4+ T cells and prolongation of murine cardiac allograft. Transplantation. 2014;97:301–309. doi:10.1097/01.TP.0000438204.96723.8b
7. Douna H, Amersfoort J, Schaftenaar FH, et al. B- and T-lymphocyte attenuator stimulation protects against atherosclerosis by regulating follicular B cells. Eur Soc Cardiol. 2020;116:295–305.
8. Albring JC, Sandau MM, Rapaport AS, et al. Targeting of B and T lymphocyte associated (BTLA) prevents graft-versus-host disease without global immunosuppression. J Exp Med. 2010;207:2551–2559. doi:10.1084/jem.20102017
9. Kaul A, Gordon C, Crow MK, et al. Systemic lupus erythematosus. Nat Rev Dis Primers. 2016;2:16039. doi:10.1038/nrdp.2016.39
10. Sanz I, Lee FE. B cells as therapeutic targets in SLE. Nat Rev Rheumatol. 2010;6:326–337. doi:10.1038/nrrheum.2010.68
11. Sawaf M, Dumortier H, Monneaux F. Follicular helper T cells in systemic lupus erythematosus: why should they be considered as interesting therapeutic targets? J Immunol Res. 2016;2016:5767106. doi:10.1155/2016/5767106
12. Gherardi L, Dumortier H, Monneaux F. Altered BTLA expression and function in lymphocytes during lupus: consequences for therapeutic targeting. Curr Trends Immunol. 2023;24:59–69.
13. Oya Y, Watanabe N, Kobayashi Y, et al. Lack of B and T lymphocyte attenuator exacerbates autoimmune disorders and induces Fas-independent liver injury in MRL-lpr/lpr mice. Int Immunol. 2011;23:335–344. doi:10.1093/intimm/dxr017
14. Oster C, Wilde B, Specker C, et al. BTLA expression on Th1, Th2 and Th17 effector T-cells of patients with systemic lupus erythematosus is associated with active disease. Int J Mol Sci. 2019;20:4505. doi:10.3390/ijms20184505
15. Murphy KA, Bhamidipati K, Rubin SJS, et al. Immunomodulatory receptors are differentially expressed in B and T cell subsets relevant to autoimmune disease. Clin Immunol. 2019;209:108276. doi:10.1016/j.clim.2019.108276
16. Wiedemann A, Lettau M, Weißenberg SY, et al. BTLA expression and function are impaired on SLE B cells. Front Immunol. 2021;12:667991. doi:10.3389/fimmu.2021.667991
17. Szelinski F, Stefansi AL, Schrezenmeier E, et al. Plasmablast-like phenotype among antigen-experienced CXCR5-CD19low B cells in systemic lupus erythematosus. Arthritis Rheumatol. 2022;74:1556–1568. doi:10.1002/art.42157
18. Sawaf M, Fauny JD, Felten R, et al. Defective BTLA functionality is rescued by restoring lipid metabolism in lupus CD4+ T cells. JCI Insight. 2018;3:e99711.
19. Aubergeon L, Sawaf M, Felten R, et al. High BTLA expression likely contributes to contraction of the regulatory T cell subset in lupus disease. Front Immunol. 2021;12:767099. doi:10.3389/fimmu.2021.767099
20. Andrews BS, Eisenberg RA, Theofilopoulos AN, et al. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med. 1978;148:1198–1215. doi:10.1084/jem.148.5.1198
21. Okuma K, Oku T, Sasaki C, et al. Similarity and difference between systemic lupus erythematosus and NZB/W F1 mice by multi-omics analysis. Mod Rheumatol. 2024;34:359–368. doi:10.1093/mr/road024
22. Bankhead P, Loughrey MB, Fernandez JA, et al. QuPath: open source software for digital pathology image analysis. Sci Rep. 2017;4:16878. doi:10.1038/s41598-017-17204-5
23. Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi:10.1038/nmeth.2019
24. Berg S, Kutra D, Kroeger T, et al. Ilastik: interactive machine learning for (bio)image analysis. Nat Methods. 2019;16:1226–1232. doi:10.1038/s41592-019-0582-9
25. Stringer C, Wang T, Michaelos M, et al. Cellpose: a generalist algorithm for cellular segmentation. Nat Methods. 2021;18:100–106. doi:10.1038/s41592-020-01018-x
26. Lacotte S, Dumortier H, Décossas M, et al. Identification of new pathogenic players in lupus: autoantiboduy-secreting cells are present in nephritic kidneys of (NZBxNZW)F1 mice. J Immunol. 2010;184:3937–3945. doi:10.4049/jimmunol.0902595
27. Deng GM, Tsokos GC. Cholera toxin B accelerates disease progression in lupus-prone mice by promoting lipid raft aggregation. J Immunol. 2008;181:4019–4026. doi:10.4049/jimmunol.181.6.4019
28. Cho S, Jang E, Yoon T, et al. A novel selective spleen tyrosine kinase inhibitor SKI-O-703 (cevidophenib) ameliorates lupus nephritis and serum-induced arthritis in murine models. Clin Exp Immunol. 2023;211:31–45. doi:10.1093/cei/uxac096
29. Truong W, Hancock WW, Plester JC, et al. BTLA targeting modulates lymphocyte phenotype, function, and numbers and attenuates disease in nonobese diabetic mice. J Leukoc Biol. 2009;89:41–51. doi:10.1189/jlb.1107753
30. Hao Y, O’Neill P, Naradikian MS, et al. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood. 2011;118:1294–1304. doi:10.1182/blood-2011-01-330530
31. Gajewski TF, Schell SR, Nau G, et al. Regulation of T-cell activation: differences among T-cell subsets. Immunol Rev. 1989;111:79–110. doi:10.1111/j.1600-065X.1989.tb00543.x
32. Jones A, Bourque J, Kuehm L, et al. Immunomodulatory functions of BTLA and HVEM govern induction of extrathymic regulatory T cells and tolerance by dendritic cells. Immunity. 2016;15:1066–1077. doi:10.1016/j.immuni.2016.10.008
33. Taddeo A, Khodadadi L, Voigt C, et al. Long-lived plasma cells are early and constantly generated in New Zealand Black/New Zealand White F1 mice and their therapeutic depletion requires a combined targeting of autoreactive plasma cells and their precursors. Arthritis Res. 2015;17:39–50. doi:10.1186/s13075-015-0551-3
34. Espeli M, Bökers S, Giannico G, et al. Local renal autoantibody production in lupus nephritis. J Am Soc Nephrol. 2011;22:296–305. doi:10.1681/ASN.2010050515
35. Chan OTM, Hannum LG, Haberman AM, et al. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999;189:1639–1647. doi:10.1084/jem.189.10.1639
36. Jin X, Xu Q, Pu C, et al. Therapeutic efficacy of anti-CAR19 CAR-T cells in a mouse model of systemic lupus erythematosus. Cell Mol Immunol. 2021;18:1896–1903. doi:10.1038/s41423-020-0472-1
37. Wang H, Lu M, Zhai S, et al. ALW peptide ameliorates lupus nephritis in MRL/lpr mice. Arthritis Res Ther. 2019;21:261–273. doi:10.1186/s13075-019-2038-0
38. Tada Y, Koarada S, Tomiyoshi Y, et al. Role of inducible costimulator in the development of lupus in MRL/lpr mice. Clin Immunol. 2006;120:179–188. doi:10.1016/j.clim.2006.02.009
39. Rodriguez-Barbosa JI, Fernandez-Renedo C, Moral AMB, et al. T follicular helper expansion and humoral-mediated rejection are independent of the HVEM/BTLA pathway. Cell Mol Immunol. 2017;14:497–510. doi:10.1038/cmi.2015.101
40. Sakoda Y, Park JJ, Zhao Y, et al. Dichotomous regulation of GVHD through bidirectional functions of the BTLA-HVEM pathway. Blood. 2011;117:2506–2514. doi:10.1182/blood-2010-08-301325
41. Del Rio ML, Kaye J, Rodriguez-Barbosa JI. Detection of protein on BTLAlow cells and in vivo antibody-mediated down-modulation of BTLA on lymphoid and myeloid cells of C57BL/6 and BALB/c BTLA allelic variants. Immunobiol. 2010;215:570–578. doi:10.1016/j.imbio.2009.09.008
42. Steinberg MW, Turovskaya O, Shaikh R, et al. A crucial role for HVEM and BTLA in preventing intestinal inflammation. J Exp Med. 2008;205:1463–1476. doi:10.1084/jem.20071160
43. Liu J, Ming S, Song W, et al. B and T lymphocyte attenuator regulates autophagy in mycobacterial infection via the AKT/mTOR signal pathway. Int Immunopharmacol. 2021;91:107215. doi:10.1016/j.intimp.2020.107215
44. Stewart R, Hammond SA, Oberst M, et al. The role of Fc gamma receptors in the activity of immunomodulatory antibodies for cancer. J Immunother Cancer. 2014;2:29–38. doi:10.1186/s40425-014-0029-x
45. Truong W, Plester JC, Hancock WW, et al. Negative and positive co-signaling with anti-BTLA (PJ196) and CTLA4Ig prolongs islet allograft survival. Transplantation. 2007;84:1368–1372. doi:10.1097/01.tp.0000289995.70390.20
46. Vendel AC, Calemine-Fenaux J, Israel-Tomasevic A, et al. B and T lymphocyte attenuator regulates B cell receptor signaling by targeting Syk and BLNK. J Immunol. 2009;182:1509–1517. doi:10.4049/jimmunol.182.3.1509
47. Cheung TC, Atwell S, Bafeti L, et al. Epitope topography of agonist antibodies to the checkpoint inhibitory receptor BTLA. Structure. 2023;31:1–10. doi:10.1016/j.str.2023.05.011
48. Aubergeon L, Felten R, Gottenberg JE, et al. Subset of DN memory B cells expressing very low levels of inhibitory receptor BTLA is enriched in SLE patients. Cells. 2024 ;13:2063. doi:10.3390/cells13242063
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.