Back to Journals » Lung Cancer: Targets and Therapy » Volume 12
Targeting AXL in NSCLC
Received 8 June 2021
Accepted for publication 29 July 2021
Published 10 August 2021 Volume 2021:12 Pages 67—79
DOI https://doi.org/10.2147/LCTT.S305484
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sai-Hong Ou
Aubhishek Zaman,1,2 Trever G Bivona1,2
1Department of Medicine, University of California, San Francisco, CA, USA; 2UCSF Helen Diller Comprehensive Cancer Center, University of California, San Francisco, CA, USA
Correspondence: Trever G Bivona Email [email protected]
Abstract: State-of-the-art cancer precision medicine approaches involve targeted inactivation of chemically and immunologically addressable vulnerabilities that often yield impressive initial anti-tumor responses in patients. Nonetheless, these responses are overshadowed by therapy resistance that follows. AXL, a receptor tyrosine kinase with bona fide oncogenic capacity, has been associated with the emergence of resistance in an array of cancers with varying pathophysiology and cellular origins, including in non-small-cell lung cancers (NSCLCs). Here in this review, we summarize AXL biology during normal homeostasis, oncogenic development and therapy resistance with a focus on NSCLC. In the context of NSCLC therapy resistance, we delineate AXL’s role in mediating resistance to tyrosine kinase inhibitors (TKIs) deployed against epidermal growth factor receptor (EGFR) as well as other notable oncogenes and to chemotherapeutics. We also discuss the current understanding of AXL’s role in mediating cell-biological variables that function as important modifiers of therapy resistance such as epithelial to mesenchymal transition (EMT), the tumor microenvironment and tumor heterogeneity. We also catalog and discuss a set of effective pharmacologic tools that are emerging to strategically perturb AXL mediated resistance programs in NSCLC. Finally, we enumerate ongoing and future exciting precision medicine approaches targeting AXL as well as challenges in this regard. We highlight that a holistic understanding of AXL biology in NSCLC may allow us to predict and improve targeted therapeutic strategies, such as through polytherapy approaches, potentially against a broad spectrum of NSCLC sub-types to forestall tumor evolution and drug resistance.
Keywords: lung cancer, AXL, targeted therapy, drug resistance
Introduction
Non-small cell lung cancer (NSCLC) is a leading cause of cancer mortality worldwide.1 Recent improvements in biomarker-driven therapeutics have resulted in the replacement of cytotoxic chemotherapy with more specific and personalized precision medicines or targeted therapy treatments in many NSCLC patients.2,3 Unfortunately, targeted therapy response in tumors with oncogenic alterations, such as epidermal growth factor receptor (EGFR) mutations, inevitably results in therapy resistance and lethal tumor progression in patients.4,5 AXL is a receptor tyrosine kinase that is positioned upstream of a wide array of biological processes, some of which include cellular proliferation and differentiation, cytoskeletal rearrangement and cellular migration and cellular survival signaling.6,7 AXL activation can also play an important role in lineage plasticity programs such as the epithelial-to-mesenchymal transition (EMT) and the emergence of resistance to targeted inhibition of oncogenic receptor tyrosine kinases (RTKs) including EGFR in NSCLC.6,7 Several studies support an expanded role of AXL in NSCLC pathogenesis and drug resistance. In this review, we discuss AXL biology, the current understanding of the role of AXL in cancer, lineage plasticity and therapy resistance in NSCLC.4,8 We also discuss current AXL kinase inhibitors and their clinical development and comment on potential future directions.
AXL Molecular Signaling
AXL is one of the three RTKs belonging to the TAM family of receptors – TYRO3, AXL, METRK. Protein ligands for TAM family receptors, protein S (PROS1) and growth-arrest-specific 6 (GAS6), are both secreted and share both structural and functional homology.7,9–11 For example, during phagocytosis, phosphatidyl serine present in lipid membranes, which is often a hallmark for phagocytosed cells, can bind to the ligands and then present itself to TAM family receptors located on macrophages thereby modulating the receptor’s dimerization and kinase activation (Figure 1).11 Structurally, TAM family receptors have three common domains, two of which are extracellular, the Immunoglobulin (Ig)-like extracellular domain and fibronectin type III domain and another that is an intracellular kinase domain. Thus, in response to various biological processes such as inflammatory responses, TAM family receptors integrate extracellular cues through ligand binding to induce productive intracellular signaling (Figure 1).10
 |
Figure 1 Schematic representation of AXL’s structure and cell-biological functionality during canonical cellular signaling based on current literature. Ig: immunoglobulin-like domain; FNIII: fibronectin type III domain. |
According to The Human Protein Atlas, AXL expression is mostly limited to smooth muscle, lung, kidney, testis and blood in normal cells under physiological conditions.11 During normal homeostatic conditions, AXL requires a protein ligand and a lipid-based moiety for optimal RTK activation.9 However, depending on the cellular contexts, a varying degree of mechanistic flexibility is permitted during AXL activation (Figure 1).10 For example, Gas6 binding causes dimerization of AXL, which in turn forms a quadruple protein complex consisting of two GAS6 ligands and two AXL receptors (Figure 1). This event is followed by trans-autophosphorylation of AXL in its intracellular kinase domain activation loop. The hinge region of the ATP active site in the kinase domain consists of a DFG (aspartate–phenylalanine–glycine) patch that is often structurally important for pharmacological inhibitor design. Some of the major phosphorylation sites include Y698, Y702, Y779, Y821, and Y866.12 However, the exact sequence of phosphorylation events is yet to be determined. Like other RTK signaling mechanisms, downstream effector and adaptor molecules such as PLCγ, PI3K, and growth factor receptor-bound protein 2 (GRB2) then dock onto the phospho-sites present within the AXL cytoplasmic tails (Figure 1).12–15 In a cell type-dependent context, AXL activation promotes the recruitment of GRB2 and then mediates cancer cell proliferation by activating the MAPK and PI3K signaling cascades.13,16,17 AXL can also activate cellular immune and inflammatory signaling pathways (such as NF-κB or SOCS-1 cytokine signaling) and cytoskeletal rearrangement pathways (such as Src/FAK signaling) to promote cellular migration, invasion and often epithelial to mesenchymal transition (Figure 1).17–22 Src can be phosphorylated by AXL to activate FAK, subsequently inducing the migration and invasion of cancer cells. In many cell-biological contexts, crosstalk between other RTKs, including other TAM family members, EGFR, VEGFR, MET has also been reported.23 Several variations of this prototypical ligand-mediated and dimerization-dependent activation hierarchy have been observed. For example, ligand-independent activation via overexpression of AXL or cell-to-cell binding with other receptors and both ligand dependent and independent heterophilic dimerization of AXL with other RTKs has been observed.24 These non-canonical activation processes, while less operative in normal physiological conditions, can often be aberrantly active during oncogenic progression to support cellular survival and tumor immune suppression (Figure 1).25,26
AXL as an Oncogene in NSCLC
Recent literature has uncovered a growing catalog of oncogenes including EGFR, BRAF, KRAS and ALK that shape the genomic landscape of NSCLC.2,3 AXL too has been shown to have oncogenic potential in NSCLC. High AXL expression was noted in a subset of lung adenocarcinoma (LAC) and high AXL was associated with increasing tumor grade as well as metastasis and predicted poorer survival in patients.25,27–30 Furthermore, treatment of patient-derived NSCLC tumors with high AXL in mice with a monoclonal antibody against active AXL demonstrated antitumor activity, indicating a potential causal role of AXL in NSCLC tumorigenesis.31 Yang et al evaluated the therapeutic potential of the AXL/MET selective inhibitors, BGB324 and cabozantinib, in cell and mouse xenograft models of lung squamous cell carcinoma (SCC).32 In preclinical settings, these AXL targeting compounds alone were significantly efficacious. AXL has also been reported as an oncogene in thyroid cancers, pancreatic ductal carcinoma (PDAC), gastric cancer, metastatic prostate cancer and melanoma.33–38
Interestingly, AXL mutations (single nucleotide mutations, deep deletions and amplifications) are not very common in NSCLC. Querying of The Cancer Genome Atlas (TCGA) datasets revealed that less than 4% of lung squamous carcinomas and less than 2% of the lung adenocarcinomas demonstrate detectable AXL mutation (Figure 2).39 Recently, an AXL fusion with – MAP3K12 Binding Inhibitory Protein (MBIP) has been reported as a potential mechanism of oncogenesis.40 AXL-MBIP fusions can occur when the tyrosine kinase domain of AXL is chromosomally rearranged into a chimera with the leucine zipper (LZ) domain of MBIP, resulting in a gene fusion that possesses both AXL kinase activity and the dimerization property conferred by the MBIP LZ domain. However, the clinical and functional significance of the AXL fusion as well as AXL mutations are yet to be determined.
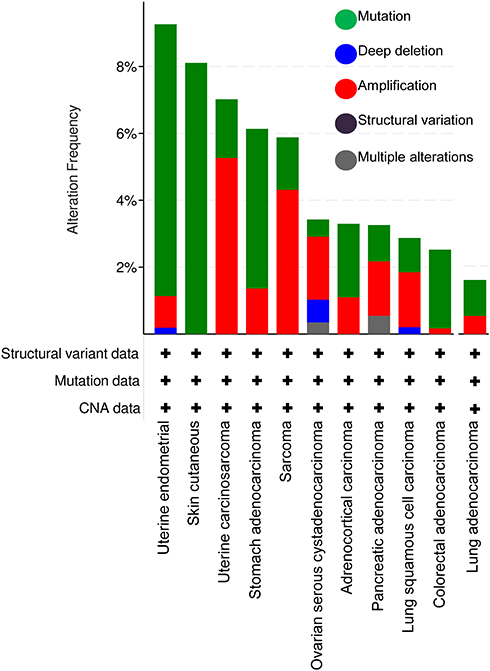 |
Figure 2 AXL genetic alterations in TCGA cohort by cBioPortal analysis. |
The mechanistic basis of AXL mediated oncogenesis, although context specific, appears to share some commonalities. Gas6 and AXL upregulation has been shown to establish a feedback amplification loop by engaging MAPK, PI3K/AKT and WNT1 cell signaling and thereby facilitating a form of lineage plasticity: an epithelial to mesenchymal transition (EMT) in cancer cells (Figure 1).9,17,37 AXL upregulation was also shown to abrogate innate immune signaling in the tumor cells yet engage interferon β innate immune signaling through a TBK1- NF-κB axis.18,41 AXL mediated mechanism of rewiring tumor microenvironment towards a more immunosuppressive and oncogenic state has been attributed to modulation of cancer associated fibroblasts (CAFs) via the regulation of AXL-regulated downstream cytokine expression and secretion.41–43
AXL and NSCLC Therapy Resistance
Driven by recent technological advancements in molecular profiling of chemically and immunologically addressable vulnerabilities in NSCLC, an increasing application of precision medicine by targeted inactivation of tumor oncogenes and by immunogenic activation of host anti-tumor surveillance has become standard treatment in many patients.2,3,44–46 However, impressive responses to these molecular therapies are typically followed by relapse due to therapy-resistant disease emergence.4,5 In NSCLC, AXL has been shown to play an important role in mediating resistance to therapy. We summarize below key aspects of AXL mediated therapy resistance.
Role of AXL in EGFR Mutant NSCLC
NSCLC tumors harboring activating mutations in EGFR that affect the kinase domain often initially respond to treatment with an EGFR tyrosine kinase inhibitor (TKI) such as osimertinib or erlotinib. However, acquired resistance to EGFR TKI treatment almost invariably occurs. In one of the earliest studies, AXL was shown to mediate resistance to EGFR TKI treatment.47 This study indicated AXL in association with an EMT was hallmark of EGFR TKI therapy resistance. Using human NSCLC HCC827 patient-derived cell line with deletion of exon 19 of EGFR (p.Glu746_Ala750del) Zhang et al showed that HCC827 cells and tumors are initially sensitive to erlotinib treatment and eventually develop acquired resistance to EGFR TKIs, both in vitro and in vivo. These models of resistance showed strong upregulation of AXL and GAS6. Furthermore, knockdown or pharmacological inhibition of AXL in these tumor models sensitized the tumors to EGFR TKI treatment and AXL upregulation was observed in a significant fraction of EGFR mutant NSCLC human tumors obtained from patients with acquired EGFR TKI resistance. These combined observations placed AXL as a causative genetic component that drives EGFR TKI therapy resistance. A range of different subsequent observations in lung cancer established AXL as an important gene that regulates resistance to several EGFR TKIs including afatinib, gefitinib, erlotinib and osimertinib in EGFR mutant NSCLC.48,49
Osimertinib, a third-generation EGFR TKI, inhibits EGFR with oncogenic-activating mutations and/or the EGFR p.Thr790Met (T790M) mutation that can cause resistance to first-generation EGFR TKIs such as erlotinib. Notably, osimertinib does not substantially inhibit wild-type EGFR or other kinases, such as AXL, AKT1, or HER3. Osimertinib resistance can arise by mechanisms that include acquisition of the EGFR-C797S mutation, loss of the T790M mutation, activation of a bypass survival pathway, and histological transformations including small cell or squamous cell transformation. A Phase III clinical trial demonstrated that in patients with EGFR-mutated NSCLC, progression-free survival (PFS) was longer in those patients treated with first-line osimertinib than for those treated with gefitinib or erlotinib.50 A comprehensive analysis of 29 patient tumor samples indicated that AXL expression negatively correlated with osimertinib response.51 Interestingly, AXL upregulation resulted in acquired resistance in absence of the EGFR T790M mutation. Wang et al indicated that blockade of AXL activation was sufficient to overcome acquired resistance to EGFR TKI treatment in NSCLC.52 Taniguchi et al used biochemical analyses of EGFR mutated patient-derived cell lines to show that AXL is upregulated upon osimertinib treatment.53 Similar to the observations of Zhang et al, knockdown of AXL was sufficient to confer sensitivity to osimertinib, indicating a causative role of AXL in osimertinib resistance. In these settings, active AXL cooperated with EGFR and HER3 to coordinate cellular tolerance to osimertinib. AXL overexpression caused increased cellular growth and resistance to therapy both in vivo and in vitro. AXL overexpression correlated with low response to EGFR TKI in EGFR mutant cell lines and cell line-derived xenografts.53 Altogether, these data place AXL not only as an important mediator of EGFR TKI resistance, but also as a critical component of EGFR RTK cellular survival network. In a separate study, co-expression of CUB Domain Containing Protein 1 (CDCP1) and AXL was observed in EGFR mutation-positive tumors, which limited the efficacy of EGFR TKIs via an intrinsic resistance mechanism.54 Moreover, additional studies indicated that autophagy-dependent mechanisms are located downstream of AXL. Interestingly, AXL upregulation was also present during the drug tolerant persister (DTP) state in response to EGFR TKI treatment in this context. These observations are consistent with the association of AXL and autophagy in regulating drug tolerance and resistance in NSCLC.55
Role of AXL in Other Oncogene Driven NSCLC
Accumulating evidence indicates pathway crosstalk between other oncogenes and AXL. For example, crosstalk between AXL and mTOR has been observed.56 In one study, dual inhibition of EGFR and AXL using gelatin nanoparticles covalently conjugated with EGFR targeting antibody and AXL siRNA (GAbsiAXL), downregulated both EMT and mTOR signaling in vitro.57 GAbsiAXL sensitized cells through induction of p53. In this context, AXL directly upregulated the PI3K downstream effector mTOR through activation of the phospholipase Cγ (PLCγ)-protein kinase C (PKC) signaling axis.
Additionally, in lung squamous carcinoma, head and neck cancer (H&N) and esophageal squamous cell carcinoma (ESCC), AXL was shown not only to be a biomarker but also to play an active role in emergence of resistance to PI3K pathway perturbation.58,59 Elkabets et al demonstrated a mTOR activation in tumors with acquired resistance to a PI3K inhibitor. Interestingly, they report that EGFR dimerizes with AXL in the acquired resistance settings and the heterophilic complex induced productive downstream PKC/mTOR activation that was sufficient to promote PI3K inhibitor resistance.60 In accordance with this, PKC inhibition via PKC412 blocked the emergence of resistance by synergizing with the PI3K inhibitor treatment. Based on these findings, the authors suggested that other cancer types characterized by EGFR dependency, such as lung cancer, may adopt a similar mechanism to escape PI3Kα or other oncogene inhibition. Interestingly, crosstalk between AXL and KRAS has been reported in metastatic colon cancer and ovarian cancer via modulation of AXL downstream transcription factors such as connective tissue growth factor (CTGF), yes associated protein (YAP1) and tafazzin.53 Interestingly, AXL has also been shown to be a target of YAP1, which may help cancer cells establish an AXL-YAP/TAZ feedback amplification loop.53 Despite some indications, existence of similar pathway crosstalk in NSCLC is yet to be fully explored.61
Role of AXL in EMT
In response to precision medicine therapeutics, upregulation of AXL is often coupled with an EMT transition.47 These observations, initially reported by Zhang et al, have further been verified and elaborated in a wide array of subsequent studies. In NSCLC, a robust EMT signature that predicted resistance to EGFR and PI3K/AKT inhibitors identified AXL as the driving force for the phenotype.62 Studies indicated ~20% of TKI resistant NSCLC patient tumors overexpress AXL with an increase in EMT and a decrease in p53 expression. Using comprehensive-OMICS-based analyses of 54 NSCLC cell lines, Byers et al reported that in NSCLC an EMT gene signature predicted intrinsic resistance to EGFR and PI3K inhibitors in vitro. AXL was present in cells with mesenchymal signature and AXL and EGFR dual perturbation using the pharmacological inhibitor SGI-7079 and erlotinib showed strong anti-tumor activity in vitro and in vivo. Furthermore, an AXL associated mesenchymal gene expression signature was able to predict patient with poorer prognosis due to relapse or metastatic development.62 Taken together, these observations indicated that AXL and perhaps its downstream EMT program play a causal role in drug resistance in NSCLC. Melchionna et al also showed that AXL is upregulated by a dynamic and paracrine interplay between cancer cells and CAFs. CAFs upregulate hMENA, a member of the actin regulatory protein of Ena/VASP family.41 This, in turn, results in elevated secretion of the AXL ligand Gas6 by CAFs, causing emergence of an EMT phenotype and drug resistance. Given that EMT activation is a recurrent and persistent phenotype during drug resistance events in a range of different cancer types, it will be interesting to determine if AXL activation is a convergent signaling mechanism operating in these diverse settings.
Role of AXL in Chemo-Resistance
AXL plays a crucial role in mediating resistance to chemotherapy and radiotherapy. In one of the earliest studies on AXL dependent chemoresistance, Wilson et al demonstrated that AXL was strongly overexpressed in a panel of 643 human cancer cells of multiple origin, including NSCLC. Using an unbiased small molecule screen, the authors reported a selective synergy between AXL and chemotherapeutic reagents that are anti-mitotic in nature.63 The mechanism of AXL mediated chemoresistance was attributed, in this context, to a TGFβ-induced EMT. Ramkumar et al, on the other hand, demonstrated that high AXL expression and EMT were frequently expressed in subsets of treatment-naive and treatment-relapsed NSCLC.64 Mechanistically, the authors attributed this resistance to AXL’s capacity to regulate the DNA damage response (DDR). The authors showed that in response to DNA damaging agents, such as WEE1 inhibitors, AXL governs a resistance program via activating mTOR, which consequently activated DNA repair pathway protein CHK1. mTOR inactivation caused suppression of this CHK1 pathway and thereby restored DNA damage mediated sensitivity. The authors also used BGB324, a specific, selective and nanomolar inhibitor of AXL, to perturb this AXL-mediated CHK1 pathway response and introduce replication stress in NSCLC, SCLC and large cell neuroendocrine carcinoma (LCNEC) patient-derived cell lines.64 Hence, the AXL mediated mechanism of chemoresistance in NSCLC appears to be multi-faceted. Furthermore, in a range of other observations, AXL mediated chemoresistance has been reported in renal cell carcinoma (RCC) and PDAC.33,36 Given the similarity of observations between NSCLC and other cancers in terms of AXL’s role in chemoresistance, it would be interesting to determine if the mechanistic pathways that actively contribute to resistance in other cancers (such as AXL/TBK1 signaling axis) are also active in NSCLC. Nevertheless, accumulating evidence suggests a potentially pervasive role of AXL in chemoresistance in tumors of different lineages.
Role of AXL in Inflammation and Tumor Microenvironment
Like other TAM family members, AXL contributes to resolution of inflammation by dampening the innate immune response.9,18 In normal physiologic conditions, AXL is also involved in restoring tissue function through promotion of tissue repair and clearance of apoptotic cells. However, in cancer development, this biological function of AXL is thought to be coopted by tumors to escape anti-tumor immunosurveillance. Accumulating evidence indicated that TAM receptors are promising therapeutic targets present on tumor-associated macrophages. TAM receptors have been shown to reshape macrophages to a pro-tumor M2-like type.65,66 Moreover, apoptotic tumor cell clearance (also known as efferocytosis) is supported by TAMs, causing tumor immune tolerance by hiding the so-called “eat me” signals on tumor from immune cells.42,66,67 Hence, the GAS6/AXL signaling axis creates a microenvironment that is suitable for tumor growth. This is consistent with reports from Aguilera et al that AXL knockout resulted in slower growth and increased radiosensitivity in a breast cancer model.68 Detailed mechanistic analyses revealed that the CD8+ T-cell response was decreased in AXL upregulated tumors, indicating a novel role for AXL in suppressing antigen presentation through MHC class I and enhancing cytokine release, which promotes a suppressive myeloid microenvironment. Moreover, in lung adenocarcinoma, AXL expression was shown to significantly correlate with expression of programmed death-ligand 1 (PD-L1), an immune checkpoint molecule and CXC chemokine receptor 6 (CXCR6) a chemokine that modulates immunosurveillance. Pharmacological perturbation of AXL yielded a more productive tumor clearance and greater response to EGFR targeted therapies. Recent literature indicates additional evidence for AXL mediated suppression of cellular anti-tumor immunity.68 Recently, Melchionna et al demonstrated that secretory GAS6 from CAFs that are pro‐tumor in nature express higher levels of hMENA/hMENADv6 compared to normal fibroblasts and activate AXL in the tumor.41 The authors demonstrated that AXL overexpression was associated with enrichment of pro-tumor CAFs, which express hMENA, a member of the actin regulatory protein of Ena/VASP family, as discussed above. In their model, a feed-forward crosstalk between CAFs and tumor cells maintained immunosuppression in AXL high cells and this was mediated by secretion of GAS6 from CAFs and activation of AXL on tumor cells. Clinically, a high hMENA/GAS6/AXL gene expression signature was associated with worse prognosis in NSCLC. In accordance with this, Terry et al demonstrated that lung cancer clones with a mesenchymal signature that had high AXL expression displayed intrinsic resistance to both natural killer cells and cytotoxic T lymphocyte (CTL)-mediated killing.69 Furthermore, this phenotype can be reverted using a pharmacological inhibitor of AXL. The authors further investigated the cellular signaling events associated with AXL upregulation and found AXL inhibition upregulates ICAM1 and ULBP1, two biomarkers associated with improved survival in NSCLC patients, as well as downregulate MAPK signaling and thereby causes tumor cytotoxicity. Hence, AXL was nominated as a biomarker for immune escape from NK and CTL immunity in NSCLC. Consistently, Du et al indicated that in refractory cancer models AXL inhibitors can alter the tumor microenvironment and convert so-called “cold” immune checkpoint inhibitor unresponsive tumors to responsive.70 The authors used sitravatinib, a selective TAM inhibitor and showed potent antitumor activity via activation of innate and adaptive immune cells. Sitravatinib not only reduced tumor burden and angiogenesis, but also enriched immunosuppressive (M2) to immunostimulatory (M1) macrophage as well as enhanced the efficacy of PD-1 blockade. These data suggest that AXL inhibitors can be a potent strategy for altering tumor microenvironments.
Role of AXL in Tumor Heterogeneity
Intratumor heterogeneity is one of the crucial confounders of therapy response. Boshuizen et al reported that AXL-high cells are resistant to MAPK pathway inhibitors, whereas AXL-low cells are sensitive to these inhibitors, rationalizing a differential therapeutic approach.71 The authors uncovered this heterogeneity initially in melanoma, and this interesting phenomenon appeared to be conserved in patient-derived xenografts (PDXs) of melanoma, lung, pancreas and cervical cancer origin. The group developed an antibody–drug conjugate, AXL-107-MMAE, comprising a human AXL antibody linked to the microtubule-disrupting agent monomethyl auristatin E. In their analyses, the authors reported a robust reduction of tumor burden in the PDXs. Interestingly, they found that MAPK inhibitors (ie, BRAF/MEK inhibitors) adaptively activated expression of AXL and hence they observed a synergy between AXL-107-MMAE and MAPK inhibitors. Since this study was largely focused on melanoma, extending this analogous investigation in NSCLC would be of tremendous interest. Another study reported low AXL level as a predictive biomarker for therapy-induced necroptosis and programmed cell death (PCD) in cancers of diverse origin.72 The authors found that the dual inhibition blocked both MAPK and PI3K/AKT cell signaling pathways and in result upregulated Bim, a potent molecule that initiates cellular apoptosis. It remains to be better determined what role AXL plays in necroptosis and PCD in NSCLC.72 This is consistent with the observation by Zhang et al that the AXL immunohistochemistry pattern was heterogenous even in the acquired resistance settings, consistent with similar findings for other bypass track RTKs, such as the kinase MET.47 In response to targeted therapeutics, how crosstalk between AXL and other activated RTKs gives rise to tumor heterogeneity is yet to be fully understood.
Strategies to Overcome AXL Mediated Therapy Resistance
An overarching goal for the rapidly evolving field of cancer genomics is to map the dynamic evolution of tumors, including emergence of resistance. Conventional single intervention therapies, in this context, are increasingly becoming inadequate to address the issue of therapy resistance, highlighting the need for a paradigm shift toward combinatorial intervention strategies or rational “polytherapy”.4,5 A conserved phenotype of AXL upregulation in a wide range of oncogene mediated cancers presents an opportunity for AXL inhibition as a conserved intervention strategy.42 For instance, a confluence of preclinical and translational data suggests that EGFR mutant patients receiving EGFR TKI may benefit from a combination with an AXL inhibitor.73,74
Several inhibitors of AXL have been developed and tested in preclinical and clinical settings. Based on selectivity of the inhibitors, they can be classified into two types – AXL selective and multi-targeted inhibitors. Based on AXL-inhibitor binding modes, these agents can further be classified into either type I or type II. Like most RTK TKIs, AXL inhibitors structurally mimic ATP.56,75 Type I inhibitors bind to the active conformation of the AXL DFG motif oriented toward the active site, which is often called the DFG-in conformation, whereas type II inhibitors bind DFG residues in an inactive AXL and are oriented away from the active site towards an allosteric region.56 Here, we briefly review a set of AXL inhibitors, their mechanisms of action, and clinical trials (Table 1) in NSCLC.
 |
Table 1 AXL Inhibitors in Clinical Trials |
BGB324/R428/Bemcentinib: A Selective (Type I)
BGB324 is a selective and potent nanomolar inhibitor of AXL. It competes with ATP in the AXL binding pocket and significantly perturbs AXL downstream signaling marked by a treatment mediated decrease in EMT markers, PI3K/AKT/mTOR pathway and inflammatory responses. BGB324 is actively being pursued in clinical trials as a therapeutic strategy in metastatic and therapy resistant NSCLC in the clinical settings.76
ONO-7475 (Type I)
ONO-7475 is nanomolar inhibitor selective for AXL and MER. ONO-7475 suppresses the emergence of EGFR TKI resistant cells in EGFR mutant cancers.73 The preclinical efficacy of this compound is yet to be extended into clinical settings.
TP-0903 (Type I)
TP-0903, a selective inhibitor of AXL that causes apoptosis of NSCLC, leukemia and breast cancer cells with AXL upregulation.77 However, due to some off target effects on ALK and Aurora kinases the compound has so far been used only in preclinical settings.
BMS-777607/ASLAN002 (Type II)
BMS-777607 is a small molecule inhibitor of MET and AXL with modest activity against tumor cells. It perturbs AXL downstream biological processes, such as growth factor-dependent cell migration and invasion and signaling pathways such as MAPK and PI3K/AKT pathways.78,79 Due to higher preclinical potency of this compound against cancer cell migration and invasion, the use of this agent was theorized as specifically more promising for blocking tumor metastasis.
CB469 (Type I)
CB469, developed by CMG Pharmaceutical, is a dual inhibitor of AXL and MET. Recently, this compound was shown to synergize with EGFR TKI treatment to block emergence of resistance to EGFR TKIs including osimertinib and erlotinib in EGFR mutant NSCLC tumors in vivo and in vitro.74
NPS-1034 (Type II)
NPS-1034 targets both AXL and MET. It inhibits AXL and downstream MAPK and PI3K signaling and causes a reduction in AXL mediated resistance in EGFR mutant NSCLC cancer cells by lowering cell proliferation and EMT. Interestingly, NPS-1034 also modulated efficacy of gefitinib in chemo-resistant cell lines.80
Cabozantinib/BMS-907351/XL184 (Type II)
Cabozantinib, a potent RTK inhibitor and an FDA approved therapy in advanced stage RCC, inhibits a wide variety of kinases implicated in tumor initiation and development, including VEGFR2, FLT3, MET, KIT, RET, and AXL.81,82 Cabozantinib can markedly improve progression-free survival (PFS), overall response rate,33 and overall survival,6 effects that are partially attributed to AXL inhibition.
RXDX-106/CEP-40783 (Type II)
RXDX-106 selectively inhibits TAMs (including AXL) and MET RTKs. It favors a “hot” tumor microenvironment causing tumor-clearance by immune cells.83 RXDX-106 also inhibits MAPK and PI3K signaling and has displayed potency against TAM-driven tumors.84,85
Crizotinib/PF-02341066 (Type I)
Crizotinib, an FDA approved therapeutic against ALK driven metastatic NSCLC, inhibits MET, ALK, and AXL.86,87 In NSCLC, Crizotinib lowers tumor cell proliferation and invasion in ALK rearranged and mutant cancer as well as in AXL upregulated cancers. However, the efficacy of crizotinib is limited by resistance and the relatively lower potency against AXL compared to other oncogenic targets such as MET has dampened enthusiasm for targeting AXL-driven tumor processes such as drug resistance.88
Sitravatinib/MGCD516 (Type II)
Sitravatinib, a newly developed multi-targeted RTK inhibitor, potently blocks activity of an array of RTKs, including EPHA3, Ret, VEGFR, PDGFR, KIT, the TRK family, DDR2, MET, and AXL.70,89 This agent shows potency against cancers with AXL upregulation and amplification in NSCLC as well as in other cancers in preclinical models.89
AXL Targeted Antibodies
A growing body of literature also highlights the potential for antitumor efficacy of AXL-targeted monoclonal antibodies (mAbs) to perturb AXL signaling and downstream pathways, supporting this distinct class of agents as a viable therapeutical alternative to AXL kinase inhibitors.71,90–93 Koopman et al demonstrated that AXL-targeting antibody–drug conjugate Enapotamab vedotin (EnaV) displayed impressive efficacy as a monotherapeutic agent in 90% of AXL expressing NSCLC. The authors also showed the efficacy of EnaV in EGFR mutant PDX tumors that had acquired resistance to EGFR TKI treatment.91 Consistent with this, Marrocco et al showed that polytherapeutic and synergistic combinations of mAbs and EGFR-specific TKIs were able to stall the emergence of resistance in EGFR mutant lung cancer models by downregulating the MAPK and PI3K/AKT pathways.90
In one of the earliest studies reporting on AXL-targeted mAbs, Kariolis et al engineered and developed a “decoy AXL receptor” that antagonized cellular AXL dimerization and activation by sequestering the AXL ligand Gas6.93 The authors also demonstrated in AXL dependent metastatic cancer models that the decoy molecules successfully inhibited metastasis and disease progression.93 However, since these decoy therapeutics could potentially also stabilize Gas6 from cellular degradation machinery, use of this approach warrants vigilance and is yet to be tested widely in other preclinical models.
Current Challenges and Prospects
The role of AXL in NSCLC is a rapidly evolving field. Intratumor heterogeneity is a key factor contributing to therapeutic failure and, hence, cancer lethality. Heterogeneous tumors show partial therapy responses, allowing for the emergence of drug-resistant clones that often express high levels of AXL.71 This is particularly relevant for AXL since AXL appears to be involved in both intrinsic and adaptive resistance mechanisms in NSCLC.61 Moreover, dynamic state-transitions between heterogeneous populations, which may show different levels of AXL expression, are a confounder. Furthermore, the crosstalk between AXL and other oncogenes in both treatment naive and treated tumors appears to be complex and heterogeneous. This interplay between AXL and other oncogenes (eg, MET, BRAF, EGFR, ALK) needs to be understood better to help shape the development of AXL inhibitors in the clinic. Furthermore, although the role of AXL in EGFR mutant tumors with EGFR TKI resistance has been demonstrated by several studies, variability in terms of downstream mechanisms is apparent. From a mode-of-action point of view, this poses an additional challenge. Moreover, accumulating evidence also indicates AXL can condition of the tumor cellular milieu. Detailed investigation of this phenomenon is warranted.
There remains an immense untapped opportunity in the field targeted inhibition of AXL and AXL downstream pathways and biological processes. First, strategies for targeting AXL have been mostly focused on developing small molecule inhibitors against the kinase activation domain. Although monoclonal antibodies against AXL have mostly been used as tool compounds, therapeutic potential for such molecules remains to be tested clinically.94 An ongoing clinical trial testing an antibody–drug conjugate targeting AXL high cells has been encouraging in this regard.95 Furthermore, analogous to EGFR, stapled peptides against AXL have been a field less explored.96,97 The majority of AXL inhibitors have so far been mostly broad spectrum and displayed off target effects. Further pharmaco-chemical investigations to identify more specific, potent and efficacious AXL inhibitors are of tremendous importance.
AXL expression levels could be one of the potential biomarkers for poorer response to targeted therapeutics. However, there remain some challenges in this regard. Specifying a biomarker requires careful estimation of biomarker levels and treatment response criteria. Unlike mutational biomarkers with discrete variables, transcriptomic (as well as proteomic) biomarkers have higher degrees of freedom due to the continuous nature of their variables. Hence, defining “high” or “low” biomarker states is critically important yet challenging for transcriptomic biomarkers such as AXL. For example, in a panel of 643 cell lines, a median cutoff was initially set by Wilson et al to define “AXL high” and “AXL low” cohorts.63 Omics-guided accurate biomarker prediction algorithms, which require control of false-positive rates without affecting enrichment of predictive signals, are often based on correlation between gene signatures (eg, expression, mutation, etc.) and response vectors (eg, viability, tumor response, tumor progression). Establishing AXL as a biomarker is by nature susceptible to all of the challenges associated with this process and requires further investigation in human NSCLC samples with associated molecular and treatment response metadata.
In terms of perturbation of downstream biological processes, the possibility of reconditioning tumor microenvironment using AXL inhibition presents an exciting therapeutic potential. Over the course of the evolving precision medicine era, the prospect of immunotherapy as a consolidation therapy following targeted inhibitor induction therapy has been theorized as a promising approach in NSCLC. However, many NSCLCs are generally poorly immunogenic. AXL inhibitors, by virtue of their ability to condition the tumor microenvironment to favor tumor clearance, may present a unique opportunity in this regard. In this context, AXL inhibition in combination with immunotherapies is yet to be tested in NSCLC. AXL, as a modulator of cellular EMT, is positioned as a central signaling node controlling cellular lineage plasticity and protecting the so-called “stem-like” cell niche population.98,99 It would be interesting to determine the role of AXL throughout different stages in the course of therapy-driven tumor evolution, including during the emergence of “drug tolerant persister” (reversible) and acquired resistant (irreversible) cells.53,100–104 This could set the stage for inhibition of AXL signaling as a mechanism by which to block or reverse lineage plasticity programs such as EMT or stem-like cellular states that contribute to cellular persistence and resistance during therapy in NSCLC and other cancers.
In summary, we have attempted to summarize recent developments in AXL’s role in the underlying cancer biology and treatment of NSCLC. We also discussed strategies, both currently available and to-be-explored, for perturbing AXL driven biological programs. We hope that a better mechanistic understanding of AXL and its role in NSCLC evolutionary trajectories, both in the presence or absence of precision medicine intervention, will eventually allow us to predict and derive upfront polytherapy approaches for stalling tumor growth and improve patient outcomes.
Acknowledgments
The authors acknowledge funding support from the National Institutes of Health (R01CA231300, U54CA224081, R01CA204302, R01CA211052 and R01CA169338) to T.G.B.
Disclosure
T.G.B. is an advisor to Array Biopharma, Revolution Medicines, Novartis, AstraZeneca, Takeda, Springworks, Jazz Pharmaceuticals, Relay Therapeutics, Rain Therapeutics, Engine Biosciences, and receives research funding from Novartis, Strategia, Kinnate, and Revolution Medicines. The authors report no other conflicts of interest in this work.
References
1. Howlader N, Forjaz G, Mooradian MJ, et al. The effect of advances in lung-cancer treatment on population mortality. N Engl J Med. 2020;383(7):640–649. doi:10.1056/NEJMoa1916623
2. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489(7417):519–525. doi:10.1038/nature11404
3. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. doi:10.1038/nature13385
4. Okimoto RA, Bivona TG. AXL receptor tyrosine kinase as a therapeutic target in NSCLC. Lung Cancer. 2015;6:27–34.
5. Zaman A, Bivona TG. Emerging application of genomics-guided therapeutics in personalized lung cancer treatment. Ann Transl Med. 2018;6(9):160. doi:10.21037/atm.2018.05.02
6. Janssen JW, Schulz AS, Steenvoorden AC, et al. A novel putative tyrosine kinase receptor with oncogenic potential. Oncogene. 1991;6(11):2113–2120.
7. Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131(6):1124–1136. doi:10.1016/j.cell.2007.10.034
8. O’Bryan JP, Frye RA, Cogswell PC, et al. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell Biol. 1991;11(10):5016–5031.
9. Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol. 2013;5(11):a009076. doi:10.1101/cshperspect.a009076
10. Graham DK, DeRyckere D, Davies KD, Earp HS. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer. 2014;14(12):769–785. doi:10.1038/nrc3847
11. Uhlen M, Fagerberg L, Hallstrom BM, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. doi:10.1126/science.1260419
12. Fridell YW, Jin Y, Quilliam LA, et al. Differential activation of the Ras/extracellular-signal-regulated protein kinase pathway is responsible for the biological consequences induced by the Axl receptor tyrosine kinase. Mol Cell Biol. 1996;16(1):135–145. doi:10.1128/MCB.16.1.135
13. Stenhoff J, Dahlback B, Hafizi S. Vitamin K-dependent Gas6 activates ERK kinase and stimulates growth of cardiac fibroblasts. Biochem Biophys Res Commun. 2004;319(3):871–878. doi:10.1016/j.bbrc.2004.05.070
14. Braunger J, Schleithoff L, Schulz AS, et al. Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene. 1997;14(22):2619–2631. doi:10.1038/sj.onc.1201123
15. Bellosta P, Zhang Q, Goff SP, Basilico C. Signaling through the ARK tyrosine kinase receptor protects from apoptosis in the absence of growth stimulation. Oncogene. 1997;15(20):2387–2397. doi:10.1038/sj.onc.1201419
16. Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res. 2008;100:35–83.
17. Lee WP, Wen Y, Varnum B, Hung MC. Akt is required for Axl-Gas6 signaling to protect cells from E1A-mediated apoptosis. Oncogene. 2002;21(3):329–336. doi:10.1038/sj.onc.1205066
18. Demarchi F, Verardo R, Varnum B, Brancolini C, Schneider C. Gas6 anti-apoptotic signaling requires NF-kappa B activation. J Biol Chem. 2001;276(34):31738–31744. doi:10.1074/jbc.M104457200
19. Hasanbasic I, Cuerquis J, Varnum B, Blostein MD. Intracellular signaling pathways involved in Gas6-Axl-mediated survival of endothelial cells. Am J Physiol Heart Circ Physiol. 2004;287(3):H1207–H1213. doi:10.1152/ajpheart.00020.2004
20. Nielsen-Preiss SM, Allen MP, Xu M, et al. Adhesion-related kinase induction of migration requires phosphatidylinositol-3-kinase and ras stimulation of rac activity in immortalized gonadotropin-releasing hormone neuronal cells. Endocrinology. 2007;148(6):2806–2814. doi:10.1210/en.2007-0039
21. Allen MP, Xu M, Linseman DA, et al. Adhesion-related kinase repression of gonadotropin-releasing hormone gene expression requires Rac activation of the extracellular signal-regulated kinase pathway. J Biol Chem. 2002;277(41):38133–38140. doi:10.1074/jbc.M200826200
22. Nakagawa R, Naka T, Tsutsui H, et al. SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002;17(5):677–687. doi:10.1016/S1074-7613(02)00449-1
23. Asiedu MK, Beauchamp-Perez FD, Ingle JN, Behrens MD, Radisky DC, Knutson KL. AXL induces epithelial-to-mesenchymal transition and regulates the function of breast cancer stem cells. Oncogene. 2014;33(10):1316–1324. doi:10.1038/onc.2013.57
24. Vouri M, Croucher DR, Kennedy SP, An Q, Pilkington GJ, Hafizi S. Axl-EGFR receptor tyrosine kinase hetero-interaction provides EGFR with access to pro-invasive signalling in cancer cells. Oncogenesis. 2016;5(10):e266. doi:10.1038/oncsis.2016.66
25. Rikova K, Guo A, Zeng Q, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–1203. doi:10.1016/j.cell.2007.11.025
26. Guo A, Villen J, Kornhauser J, et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A. 2008;105(2):692–697. doi:10.1073/pnas.0707270105
27. Shieh YS, Lai CY, Kao YR, et al. Expression of axl in lung adenocarcinoma and correlation with tumor progression. Neoplasia. 2005;7(12):1058–1064. doi:10.1593/neo.05640
28. Ishikawa M, Sonobe M, Nakayama E, et al. Higher expression of receptor tyrosine kinase Axl, and differential expression of its ligand, Gas6, predict poor survival in lung adenocarcinoma patients. Ann Surg Oncol. 2013;20(Suppl 3):S467–S476. doi:10.1245/s10434-012-2795-3
29. Wu Z, Bai F, Fan L, et al. Coexpression of receptor tyrosine kinase AXL and EGFR in human primary lung adenocarcinomas. Hum Pathol. 2015;46(12):1935–1944. doi:10.1016/j.humpath.2015.08.014
30. Qu XH, Liu JL, Zhong XW, Li XI, Zhang QG. Insights into the roles of hnRNP A2/B1 and AXL in non-small cell lung cancer. Oncol Lett. 2015;10(3):1677–1685. doi:10.3892/ol.2015.3457
31. Iida S, Miki Y, Suzuki T, et al. Activation of AXL and antitumor effects of a monoclonal antibody to AXL in lung adenocarcinoma. Anticancer Res. 2014;34(4):1821–1827.
32. Yang PW, Liu YC, Chang YH, et al. Cabozantinib (XL184) and R428 (BGB324) inhibit the growth of esophageal squamous cell carcinoma (ESCC). Front Oncol. 2019;9:1138. doi:10.3389/fonc.2019.01138
33. Ludwig KF, Du W, Sorrelle NB, et al. Small-molecule inhibition of axl targets tumor immune suppression and enhances chemotherapy in pancreatic cancer. Cancer Res. 2018;78(1):246–255. doi:10.1158/0008-5472.CAN-17-1973
34. Axelrod HD, Valkenburg KC, Amend SR, et al. AXL is a putative tumor suppressor and dormancy regulator in prostate cancer. Mol Cancer Res. 2019;17(2):356–369. doi:10.1158/1541-7786.MCR-18-0718
35. Zhou L, Liu XD, Sun M, et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene. 2016;35(21):2687–2697. doi:10.1038/onc.2015.343
36. Xiao Y, Zhao H, Tian L, et al. S100A10 is a critical mediator of gas6/axl-induced angiogenesis in renal cell carcinoma. Cancer Res. 2019;79(22):5758–5768. doi:10.1158/0008-5472.CAN-19-1366
37. Engelsen AST, Wnuk-Lipinska K, Bougnaud S, et al. AXL is a driver of stemness in normal mammary gland and breast cancer. iScience. 2020;23(11):101649. doi:10.1016/j.isci.2020.101649
38. Bae CA, Ham IH, Oh HJ, et al. Inhibiting the GAS6/AXL axis suppresses tumor progression by blocking the interaction between cancer-associated fibroblasts and cancer cells in gastric carcinoma. Gastric Cancer. 2020;23(5):824–836. doi:10.1007/s10120-020-01066-4
39. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi:10.1158/2159-8290.CD-12-0095
40. Seo JS, Ju YS, Lee WC, et al. The transcriptional landscape and mutational profile of lung adenocarcinoma. Genome Res. 2012;22(11):2109–2119. doi:10.1101/gr.145144.112
41. Melchionna R, Spada S, Di Modugno F, et al. The actin modulator hMENA regulates GAS6-AXL axis and pro-tumor cancer/stromal cell cooperation. EMBO Rep. 2020;21(11):e50078. doi:10.15252/embr.202050078
42. Myers KV, Amend SR, Pienta KJ. Targeting Tyro3, Axl and MerTK (TAM receptors): implications for macrophages in the tumor microenvironment. Mol Cancer. 2019;18(1):94. doi:10.1186/s12943-019-1022-2
43. Wu G, Ma Z, Cheng Y, et al. Targeting Gas6/TAM in cancer cells and tumor microenvironment. Mol Cancer. 2018;17(1):20. doi:10.1186/s12943-018-0769-1
44. Wei SC, Levine JH, Cogdill AP, et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell. 2017;170(6):1120–1133. doi:10.1016/j.cell.2017.07.024
45. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. doi:10.1126/science.aaa8172
46. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205–214. doi:10.1016/j.cell.2015.03.030
47. Zhang Z, Lee JC, Lin L, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44(8):852–860. doi:10.1038/ng.2330
48. Bae SY, Hong JY, Lee HJ, Park HJ, Lee SK. Targeting the degradation of AXL receptor tyrosine kinase to overcome resistance in gefitinib-resistant non-small cell lung cancer. Oncotarget. 2015;6(12):10146–10160. doi:10.18632/oncotarget.3380
49. Yoshida T, Zhang G, Smith MA, et al. Tyrosine phosphoproteomics identifies both codrivers and cotargeting strategies for T790M-related EGFR-TKI resistance in non-small cell lung cancer. Clin Cancer Res. 2014;20(15):4059–4074. doi:10.1158/1078-0432.CCR-13-1559
50. Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378(2):113–125. doi:10.1056/NEJMoa1713137
51. Wang R, Yamada T, Kita K, et al. Transient IGF-1R inhibition combined with osimertinib eradicates AXL-low expressing EGFR mutated lung cancer. Nat Commun. 2020;11(1):4607. doi:10.1038/s41467-020-18442-4
52. Wang FLX, Bartholdy BA, Cheng H, Halmos B. Blockade of AXL activation overcomes acquired resistance to EGFR tyrosine kinase inhibition in non-small cell lung cancer. Transl Cancer Res. 2019;8(6):2425–2438. doi:10.21037/tcr.2019.09.61
53. Taniguchi H, Yamada T, Wang R, et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat Commun. 2019;10(1):259. doi:10.1038/s41467-018-08074-0
54. Karachaliou N, Chaib I, Cardona AF, et al. Common co-activation of AXL and CDCP1 in EGFR-mutation-positive non-smallcell lung cancer associated with poor prognosis. EBioMedicine. 2018;29:112–127. doi:10.1016/j.ebiom.2018.02.001
55. Lotsberg ML, Wnuk-Lipinska K, Terry S, et al. AXL targeting abrogates autophagic flux and induces immunogenic cell death in drug-resistant cancer cells. J Thorac Oncol. 2020;15(6):973–999. doi:10.1016/j.jtho.2020.01.015
56. Sen T, Tong P, Diao L, et al. Targeting AXL and mTOR pathway overcomes primary and acquired resistance to WEE1 inhibition in small-cell lung cancer. Clin Cancer Res. 2017;23(20):6239–6253. doi:10.1158/1078-0432.CCR-17-1284
57. Suresh D, Zambre A, Mukherjee S, et al. Silencing AXL by covalent siRNA-gelatin-antibody nanoconjugate inactivates mTOR/EMT pathway and stimulates p53 for TKI sensitization in NSCLC. Nanomedicine. 2019;20:102007. doi:10.1016/j.nano.2019.04.010
58. Giles KM, Kalinowski FC, Candy PA, et al. Axl mediates acquired resistance of head and neck cancer cells to the epidermal growth factor receptor inhibitor erlotinib. Mol Cancer Ther. 2013;12(11):2541–2558. doi:10.1158/1535-7163.MCT-13-0170
59. Liu L, Greger J, Shi H, et al. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 2009;69(17):6871–6878. doi:10.1158/0008-5472.CAN-08-4490
60. Elkabets M, Pazarentzos E, Juric D, et al. AXL mediates resistance to PI3Kα inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell. 2015;27(4):533–546. doi:10.1016/j.ccell.2015.03.010
61. Konen JM, Rodriguez BL, Padhye A, et al. Dual inhibition of MEK and AXL targets tumor cell heterogeneity and prevents resistant outgrowth mediated by the epithelial-to-mesenchymal transition in NSCLC. Cancer Res. 2021;81(5):1398–1412. doi:10.1158/0008-5472.CAN-20-1895
62. Byers LA, Diao L, Wang J, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19(1):279–290. doi:10.1158/1078-0432.CCR-12-1558
63. Wilson C, Ye X, Pham T, et al. AXL inhibition sensitizes mesenchymal cancer cells to antimitotic drugs. Cancer Res. 2014;74(20):5878–5890. doi:10.1158/0008-5472.CAN-14-1009
64. Ramkumar K, Stewart CA, Cargill KR, et al. AXL inhibition induces DNA damage and replication stress in non-small cell lung cancer cells and promotes sensitivity to ATR inhibitors. Mol Cancer Res. 2021;19(3):485–497. doi:10.1158/1541-7786.MCR-20-0414
65. Mahajan NP, Earp HS. An SH2 domain-dependent, phosphotyrosine-independent interaction between Vav1 and the Mer receptor tyrosine kinase: a mechanism for localizing guanine nucleotide-exchange factor action. J Biol Chem. 2003;278(43):42596–42603. doi:10.1074/jbc.M305817200
66. Choi JY, Seo JY, Yoon YS, Lee YJ, Kim HS, Kang JL. Mer signaling increases the abundance of the transcription factor LXR to promote the resolution of acute sterile inflammation. Sci Signal. 2015;8(365):ra21. doi:10.1126/scisignal.2005864
67. Lu Q, Gore M, Zhang Q, et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature. 1999;398(6729):723–728. doi:10.1038/19554
68. Aguilera TA, Rafat M, Castellini L, et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat Commun. 2016;7:13898. doi:10.1038/ncomms13898
69. Terry S, Abdou A, Engelsen AST, et al. AXL targeting overcomes human lung cancer cell resistance to NK- and CTL-mediated cytotoxicity. Cancer Immunol Res. 2019;7(11):1789–1802. doi:10.1158/2326-6066.CIR-18-0903
70. Du W, Huang H, Sorrelle N, Brekken RA. Sitravatinib potentiates immune checkpoint blockade in refractory cancer models. JCI Insight. 2018;3(21). doi:10.1172/jci.insight.124184
71. Boshuizen J, Koopman LA, Krijgsman O, et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat Med. 2018;24(2):203–212. doi:10.1038/nm.4472
72. Umemura S, Sowa Y, Iizumi Y, Kitawaki J, Sakai T. Synergistic effect of the inhibitors of RAF/MEK and AXL on KRAS-mutated ovarian cancer cells with high AXL expression. Cancer Sci. 2020;111(6):2052–2061. doi:10.1111/cas.14414
73. Okura N, Nishioka N, Yamada T, et al. ONO-7475, a novel AXL inhibitor, suppresses the adaptive resistance to initial EGFR-TKI treatment in EGFR-mutated non-small cell lung cancer. Clin Cancer Res. 2020;26(9):2244–2256. doi:10.1158/1078-0432.CCR-19-2321
74. Yang YM, Jang Y, Lee SH, Kang B, Lim SM. AXL/MET dual inhibitor, CB469, has activity in non-small cell lung cancer with acquired resistance to EGFR TKI with AXL or MET activation. Lung Cancer. 2020;146:70–77. doi:10.1016/j.lungcan.2020.05.031
75. Messoussi A, Peyronnet L, Feneyrolles C, Cheve G, Bougrin K, Yasri A. Structural elucidation of the DFG-Asp in and DFG-Asp out states of TAM kinases and insight into the selectivity of their inhibitors. Molecules. 2014;19(10):16223–16239. doi:10.3390/molecules191016223
76. Sheridan C. First Axl inhibitor enters clinical trials. Nat Biotechnol. 2013;31(9):775–776. doi:10.1038/nbt0913-775a
77. Sinha S, Boysen J, Nelson M, et al. Targeted Axl inhibition primes chronic lymphocytic leukemia B cells to apoptosis and shows synergistic/additive effects in combination with BTK inhibitors. Clin Cancer Res. 2015;21(9):2115–2126. doi:10.1158/1078-0432.CCR-14-1892
78. Dai Y, Siemann DW. BMS-777607, a small-molecule met kinase inhibitor, suppresses hepatocyte growth factor-stimulated prostate cancer metastatic phenotype in vitro. Mol Cancer Ther. 2010;9(6):1554–1561. doi:10.1158/1535-7163.MCT-10-0359
79. Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7(6):504–516. doi:10.1038/nrd2530
80. Shin JS, Hong SW, Moon JH, et al. NPS-1034, a novel MET inhibitor, inhibits the activated MET receptor and its constitutively active mutants. Invest New Drugs. 2014;32(3):389–399. doi:10.1007/s10637-013-0039-4
81. Grullich C. Cabozantinib: multi-kinase Inhibitor of MET, AXL, RET, and VEGFR2. Recent Results Cancer Res. 2018;211:67–75.
82. Timar J, Dome B. Antiangiogenic Drugs and Tyrosine Kinases. Anticancer Agents Med Chem. 2008;8(5):462–469.
83. Lew ED, Oh J, Walsh C, et al. RXDX-106, a novel, selective and potent small molecule TAM (TYRO3, AXL, MER) inhibitor, demonstrates efficacy in TAM-driven tumors. AACR; 2017.
84. Yokoyama Y, Lew ED, Seelige R, et al. Immuno-oncological efficacy of RXDX-106, a novel TAM (TYRO3, AXL, MER) family small-molecule kinase inhibitor. Cancer Res. 2019;79(8):1996–2008. doi:10.1158/0008-5472.CAN-18-2022
85. Kim JE, Kim Y, Li G, et al. MerTK inhibition by RXDX-106 in MerTK activated gastric cancer cell lines. Oncotarget. 2017;8(62):105727–105734. doi:10.18632/oncotarget.22394
86. Zou HY, Li Q, Lee JH, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67(9):4408–4417. doi:10.1158/0008-5472.CAN-06-4443
87. Christensen JG, Zou HY, Arango ME, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6(12 Pt 1):3314–3322. doi:10.1158/1535-7163.MCT-07-0365
88. Cui JJ, Tran-Dube M, Shen H, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med Chem. 2011;54(18):6342–6363. doi:10.1021/jm2007613
89. Patwardhan PP, Ivy KS, Musi E, de Stanchina E, Schwartz GK. Significant blockade of multiple receptor tyrosine kinases by MGCD516 (Sitravatinib), a novel small molecule inhibitor, shows potent anti-tumor activity in preclinical models of sarcoma. Oncotarget. 2016;7(4):4093–4109. doi:10.18632/oncotarget.6547
90. Marrocco I, Romaniello D, Vaknin I, et al. Upfront admixing antibodies and EGFR inhibitors preempts sequential treatments in lung cancer models. EMBO Mol Med. 2021;13(4):e13144. doi:10.15252/emmm.202013144
91. Koopman LA, Terp MG, Zom GG, et al. Enapotamab vedotin, an AXL-specific antibody-drug conjugate, shows preclinical antitumor activity in non-small cell lung cancer. JCI Insight. 2019;4(21). doi:10.1172/jci.insight.128199
92. Romaniello D, Mazzeo L, Mancini M, et al. A combination of approved antibodies overcomes resistance of lung cancer to osimertinib by blocking bypass pathways. Clin Cancer Res. 2018;24(22):5610–5621. doi:10.1158/1078-0432.CCR-18-0450
93. Kariolis MS, Miao YR, Jones DS, et al. An engineered Axl ‘decoy receptor’ effectively silences the Gas6-Axl signaling axis. Nat Chem Biol. 2014;10(11):977–983. doi:10.1038/nchembio.1636
94. Codony-Servat J, Garcia-Roman S, Molina-Vila MA, et al. Anti-epidermal growth factor vaccine antibodies enhance the efficacy of tyrosine kinase inhibitors and delay the emergence of resistance in EGFR mutant lung cancer cells. J Thorac Oncol. 2018;13(9):1324–1337. doi:10.1016/j.jtho.2018.04.030
95. Tap WD, O’Reilly EM. A phase I/II study of BA3011 in patients with advanced cancer. ClinicalTrialsgov. 2018.
96. Maisel SA, Broka D, Atwell B, et al. Stapled EGFR peptide reduces inflammatory breast cancer and inhibits additional HER-driven models of cancer. J Transl Med. 2019;17(1):201. doi:10.1186/s12967-019-1939-7
97. Sinclair JK, Schepartz A. Influence of macrocyclization on allosteric, juxtamembrane-derived, stapled peptide inhibitors of the epidermal growth factor receptor (EGFR). Org Lett. 2014;16(18):4916–4919. doi:10.1021/ol502426b
98. Eskiocak B, McMillan EA, Mendiratta S, et al. Biomarker accessible and chemically addressable mechanistic subtypes of BRAF melanoma. Cancer Discov. 2017;7(8):832–851. doi:10.1158/2159-8290.CD-16-0955
99. Falletta P, Sanchez-Del-Campo L, Chauhan J, et al. Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes Dev. 2017;31(1):18–33. doi:10.1101/gad.290940.116
100. Hangauer MJ, Viswanathan VS, Ryan MJ, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551(7679):247–250. doi:10.1038/nature24297
101. Ramirez M, Rajaram S, Steininger RJ, et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat Commun. 2016;7:10690. doi:10.1038/ncomms10690
102. Swayden M, Chhouri H, Anouar Y, Grumolato L. Tolerant/persister cancer cells and the path to resistance to targeted therapy. Cells. 2020;9(12):2601. doi:10.3390/cells9122601
103. Sharma SV, Lee DY, Li B, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141(1):69–80. doi:10.1016/j.cell.2010.02.027
104. Nakamichi S, Seike M, Miyanaga A, et al. Overcoming drug-tolerant cancer cell subpopulations showing AXL activation and epithelial-mesenchymal transition is critical in conquering ALK-positive lung cancer. Oncotarget. 2018;9(43):27242–27255. doi:10.18632/oncotarget.25531
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.