Back to Journals » Biologics: Targets and Therapy » Volume 8
Targeted therapy for sarcomas
Authors Forscher C, Mita M, Figlin R
Received 11 October 2012
Accepted for publication 9 July 2013
Published 17 March 2014 Volume 2014:8 Pages 91—105
DOI https://doi.org/10.2147/BTT.S26555
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Charles Forscher,1 Monica Mita,2 Robert Figlin3
1Sarcoma Program, Samuel Oschin Comprehensive Cancer Institute, Cedars-Sinai Medical Center, Los Angeles, CA, USA; 2Experimental Therapeutics Program, Samuel Oschin Comprehensive Cancer Institute, Cedars-Sinai Medical Center, Los Angeles, CA, USA; 3Academic Development Program, Samuel Oschin Comprehensive Cancer Institute, and Division of Hematology/Oncology, Cedars-Sinai Medical Center, Los Angeles, CA, USA
Abstract: Sarcomas are tumors of mesenchymal origin that make up approximately 1% of human cancers. They may arise as primary tumors in either bone or soft tissue, with approximately 11,280 soft tissue tumors and 2,650 bone tumors diagnosed each year in the United States. There are at least 50 different subtypes of soft tissue sarcoma, with new ones described with ever-increasing frequency. One way to look at sarcomas is to divide them into categories on the basis of their genetic make-up. One group of sarcomas has an identifiable, relatively simple genetic signature, such as the X:18 translocation seen in synovial sarcoma or the 11:22 translocation seen in Ewing's sarcoma. These specific abnormalities often lead to the presence of fusion proteins, such as EWS-FLI1 in Ewing's sarcoma, which are helpful as diagnostic tools and may become therapeutic targets in the future. Another group of sarcomas is characterized by complex genetic abnormalities as seen in leiomyosarcoma, osteosarcoma, and undifferentiated sarcoma. It is important to keep these distinctions in mind when contemplating the development of targeted agents for sarcomas. Different abnormalities in sarcoma could be divided by tumor subtype or by the molecular or pathway abnormality. However, some existing drugs or drugs in development may interfere with or alter more than one of the presented pathways.
Keywords: sarcoma, targeted agents, tyrosine kinase inhibitors, mTor inhibition
Angiogenesis
c-kit, PDGFRA, and gastrointestinal stromal tumor
Gastrointestinal stromal tumor (GIST) is a malignant sarcoma felt to arise from the interstitial cell of Cajal and is characterized most commonly by activating mutations in c-kit, and less commonly in platelet-derived growth factor receptor alpha (PDGFRA). Approximately 5,000 cases are diagnosed in the United States each year, with the stomach being the most common location. Imatinib, a tyrosine kinase inhibitor initially developed as an agent for chronic myelogenous leukemia and designed to target the product of its fusion protein Bcr-Abl, was also found to inhibit c-kit. The initial report of activity in 20011,2 was followed by trials in both the United States and Europe that demonstrated objective response rates in the range of 50% to 70%.3,4 Interestingly, patients with complete responses, partial responses, and stable disease have similar rates of progression-free survival (PFS) compared with patients with stable disease using imatinib when using conventional response criteria such as Response Evaluation Criteria in Solid Tumours (RECIST) with traditional computed tomography scanning.5 Approximately 50% of patients with responsive disease are still alive at 5 years compared with less than 10% survival at 2 years for nonresponders. This observation has prompted a search for alternative assessment schemas and has led some to argue that “we should desist from using RECIST in GIST.”6 The most common mutation site is at exon 11 and is usually sensitive to treatment with imatinib at 400 mg. Other mutations, such as exon 9, may benefit from higher doses of imatinib.7,8 A smaller percentage of GISTs have mutations in the platelet-derived growth factor receptor alpha, and these tumors may also be sensitive to imatinib. Wild-type GISTs, without a mutation, are less sensitive to imatinib.
Patients with advanced disease should continue to receive imatinib until disease progression. This issue was addressed in a French discontinuation study in which patients with responsive GIST were randomly assigned to continue or stop imatinib at 1 year.9 The majority of patients developed recurrence of disease within 6 months on discontinuation of imatinib, but disease control could often be achieved on resumption of the drug. This finding was also observed with further randomization at 3 years.10 The study did suggest that brief “honeymoon” periods off imatinib could be possible without undue patient risk.
For those whose GISTs progress while receiving imatinib, sunitinib is approved for second-line therapy. Of note, after initial progression on standard-dose imatinib, the next recommended treatment is increasing the dose to 800 mg. The dose of sunitinib is 50 mg/day for 4 weeks with a 2-week rest period. Although the objective response rate to sunitinib is 7%, the time to progression on this agent is prolonged significantly compared with placebo, at 27.3 weeks with sunitinib versus 6.4 weeks for placebo.11
Other agents are in the process of evaluation for patients whose tumors progress on imatinib. Regorafenib, an oral multikinase inhibitor, has shown activity in both Phase II and Phase III trials in patients progressing on both imatinib and sunitinib, with a clinical benefit rate of 54% at 16 weeks (two partial response, 10 stable disease) in the Phase II trial12 and a median PFS of 4.8 months compared with 0.9 months for placebo in the Phase III trial.13 This agent blocks vascular endothelial growth factor receptor 2–3 (VEGFR2-3), c-kit, TIE2, PDGFR beta (PDGFRB), fibroblast growth factor receptor 1, RET (rearranged during transfection), RAF, and p38 mitogen-activated protein kinase. It is now approved for use in advanced GISTs after progression on imatinib and sunitinib. Masitinib, a newer tyrosine kinase inhibitor, has been compared with sunitinib in patients with imatinib-resistant GIST. Masitinib was administered at a dose of 12 mg/day versus 50 mg/day sunitinib. Although median PFS rates were similar for the two agents, at 3.9 months for masitinib and 3.8 months for sunitinib, the adverse effect profile was more favorable for the masitinib group.14 A smaller study of 30 patients has evaluated masitinib in imatinib-naïve patients with a 20% response rate (RR) by RECIST and an 86% RR by fluorodeoxyglucose positron emission scanning. Disease control rate in this study (complete response, partial response, stable disease) was 96.7%.15
Dasatinib, another tyrosine kinase inhibitor with activity against KIT (CD117), PDGFR, ABL, and SRC, was evaluated in a Phase II study. Although the drug demonstrated activity by overall response rate with a partial response of 32% (15/47) by Choi criteria, it did not meet the target progression-free rate of more than 30% at 6 months.16
Sorafenib, which targets KIT, VEGFR, PDGFRB, and BRAF, has also been studied in refractory GIST. In 38 patients refractory to either imatinib alone or both imatinib and sunitinib, partial response was observed in 13% (1 imatinib resistant, 4 imatinib and sutent resistant), and stable disease was seen in 55% (3 IM-RES, 18 IM-SU RES). Median progression-free survival was 5.2 months and median overall survival was 11.6 months with 1-year survival of 50% and 2-year survival of 26%. With an overall disease control rate of 68%, sorafenib was felt to have definite activity in IM-SU-resistant GIST.
Another approach under evaluation for imatinib-resistant GIST is to combine other agents with imatinib. Everolimus (RAD001), an m-Tor inhibitor, has been combined with imatinib. Patients were stratified as to whether they had progressed on imatinib alone (stratum 1) or on imatinib and sunitinib (stratum 2). Everolimus was administered at 2.5 mg/day, and imatinib was given at 600 mg/day. Four of 23 patients in stratum 1 were progression free at 4 months, as were 13 (37%) of 35 patients in stratum 2.17 The authors concluded that the combination of everolimus and imatinib was worthy of further investigation in GIST.
After the dramatic effect of imatinib in advanced GIST, several studies were initiated to evaluate the use of imatinib in the adjuvant setting for resected GIST. The first published study, performed by the American College of Surgeons Oncology Group, randomly assigned patients with KIT-positive GIST of at least 3 cm in size who underwent complete resection to receive imatinib at 400 mg daily (n = 359) versus placebo (n = 354) for 1 year in a double-blind manner. With a primary endpoint of recurrence-free survival (RFS), at 19.7 months, 30 patients (8%) in the imatinib group and 70 (20%) in the placebo group had tumor recurrence. The RFS rate was 98% in the imatinib group and 83% in the placebo group at 1 year, with a hazard ratio of 0.35 (one-sided P < 0.0001).18 As patients receiving the placebo were eligible to crossover to imatinib on progression, an overall survival advantage for imatinib could not be evaluated.
A subsequent study19 conducted by the Working Group for Medical Oncology and the Scandinavian Sarcoma Group compared 12 versus 36 months of imatinib in the adjuvant setting for high-risk GIST. The primary endpoint of RFS for the 36-month group was 65.6% versus 47.9% for the 12-month group with a hazard ratio of 0.46 (P < 0.0001). In addition, overall survival was superior in the 36-month imatinib group compared with the 12-month group (92% versus 81.7%; hazard ratio, 0.45; P = 0.019).19 On the basis of this study, the use of adjuvant imatinib for 36 months is now considered the standard of care for high-risk gastrointestinal stromal tumors.
Neoadjuvant imatinib administration is being employed with the goal of downsizing tumors and increasing the potential resectability of advanced GIST. This indication is not a standard practice in GIST, but it is used in some cases with locally advanced tumors. In the Radiation Therapy Oncology Group/American College of Radiology Imaging Network trial,20 neoadjuvant imatinib was given at 600 mg/day. Thirty patients with localized GIST had RECIST responses of 7% partial response, 83% stable disease, and 10% unknown. Twenty-two patients with metastatic GIST had RECIST responses of 4.5% partial response, 91% stable disease, and 4.5% progression. Two-year PFS was 83% for those with localized disease and 77% for those with metastatic disease.20 Other trials have included both preoperative and postoperative imatinib demonstrating radiographic and pathologic responses with 3–7 days of preoperative imatinib.21
PDGFRB and dermatofibrosarcoma protuberans
Dermatofibrosarcoma protuberans (DFSP) is a skin sarcoma that is typically a localized problem but in rare cases may metastasize and can undergo fibrosarcomatous transformation. It is associated with a translocation at t(17;22), which fuses the COL1A1 gene with the PDGF-B gene, leading to overexpression of PDGF-B. Two Phase II trials were conducted, one by the European Organisation for Research and Treatment of Cancer (EORTC) and the other by the Southwest Oncology Group, and the data from these trials were combined for analysis.22 The conclusion was that imatinib was active in DFSP with an objective response rate approaching 50%. Responses were observed in both standard DFSP and in DFSP undergoing fibrosarcomatous transformation. Doses in these studies were from 400–800 mg. Additional studies have explored the use of imatinib in DFSP as preoperative therapy.23 Imatinib was administered at a dose of 600 mg/day for 2 months before reevaluation. Overall response rate was 36%. Imatinib has been approved for use in DFSP at a dose of 800 mg.
PDGFRB and chordoma
Chordoma is a tumor that arises from primitive notochordal remnants, that can behave in an aggressive local fashion, and that in rare cases may metastasize. These tumors are felt to be chemotherapy-insensitive. After tumor evaluations demonstrating either expression or phosphorylation of PDGFRB, imatinib was instituted in six patients with advanced chordoma at a dose of 800 mg daily.24 This initial study, which suggested a clinical benefit for imatinib, has been confirmed by a larger Phase II study of 50 patients. The larger study using overall response rate by RECIST showed a single partial response (2%) and 35 patients with stable disease (70%), and a clinical benefit rate of 64%.25 Median PFS was 9 months. Responses can be associated with tumor liquefaction and/or hemorrhage.26
ASPS/transcription factor E3 fusion, VEGF, and ASPS
Alveolar soft part sarcoma (ASPS) is a chemotherapy-insensitive tumor associated with a specific genetic translocation at t(17-X), which results in the ASPS/transcription factor E3 fusion protein. It often grows slowly but inexorably and is frequently associated with lung metastases. Given its long natural history, brain metastases are not infrequent. Recently, several targeted agents have demonstrated responses in this disease. Both sunitinib and cediranib have elicited responses. In one series, two of five patients with ASPS had partial response by RECIST criteria, with an additional patient showing stable disease with sunitinib at 37.5 mg/day.27 Analysis of tumor samples showed activation of PDGFR, epidermal growth factor receptor, MET, and RET. An additional study from this group confirmed activity for sunitinib mediated by PDGFRB, VEGFR2, and RET.28 Promising activity for cediranib, a selective VEGF signaling inhibitor, has also been reported.29 Seven patients were treated with cediranib at an initial dose of 45 mg daily. Four patients had partial responses, two patients had minor responses, and one had stable disease.29 Additional confirmative studies with this agent in this disease are in progress.30
VEGF inhibition, insulin-like growth factor 1 receptor inhibition, and solitary fibrous tumor
Hemangiopericytoma/solitary fibrous tumors are tumors of uncertain histogenesis that are often localized and low grade but that may be multifocal and/or metastatic. Their behavior is often unpredictable. Recent reports have suggested activity for the use of VEGF inhibitors in conjunction with chemotherapy for this entity. At the University of Texas MD Anderson Cancer Center, 14 patients with hemangiopericytoma/solitary fibrous tumor received chemotherapy with temozolomide at 150 mg/m2 orally on days 1–7 and 15–21 with bevacizumab at 5 mg/kg on days 8 and 22.31 Eleven (79%) of 14 patients had partial response by Choi criteria, two patients had stable disease, and one patient had progressive disease. Median PFS was estimated to be 9.7 months.
Another study of targeted therapy in solitary fibrous tumors evaluated sunitinib in eleven patients at a dose of 37.5 mg/day and in one patient who received figitumumab, an insulin-like growth factor receptor inhibitor.32 Ten patients were evaluable for response, with six showing partial response with Choi criteria (all with stable disease by RECIST), one showing stable disease, and three showing progressive disease. Response duration was more than 6 months in five patients. PDGFR, RET, VEGFRi/2, and insulin-like growth factor 1 receptor (IGF1R) were activated in these tumors. The single patient who progressed on sunitinib appears to have experienced disease stabilization on figitumumab.
VEGF inhibition and angiosarcoma
Given the vascular nature of angiosarcoma, it is tempting to assume that these tumors should be the ideal targets for VEGF inhibitors. Several recent studies have been performed evaluating VEGF inhibition in this disease. A study from the French Sarcoma Group evaluated sorafenib in advanced sarcoma using a primary endpoint of progression-free rate at 9 months according to RECIST. Patients received sorafenib at 400 mg twice a day. Two strata were evaluated: one for superficial disease (26 patients) and one for visceral angiosarcoma (15 patients). The progression-free rate at 9 months was 3.8% in the superficial angiosarcoma group and 0% in the visceral disease group. Median PFS rates were 1.8 months and 3.8 months. Thirty patients (78%) had received prior chemotherapy. No responses were observed in the chemotherapy-naïve group, but a 40% tumor control rate and a 23% response rate were seen in pretreated patients. The researchers’ conclusion was that sorafenib showed limited antitumor activity in angiosarcoma.33 Another study of sorafenib that included 40 patients with vascular sarcomas showed a response rate of 13%, a 3-month PFS rate of 64%, and a median PFS time of 4 months.34 A study of bevacizumab in angiosarcoma and epithelioid hemangioendothelioma enrolled 32 patients receiving bevacizumab at 15 mg/kg every 21 days. Thirty patients were evaluable with four having a partial response (two angiosarcoma and two hemangioendothelioma). Fifteen patients had stable disease (50%), with a mean time to progression of 26 weeks.35 These studies taken together suggest that further progress is needed in these highly aggressive sarcomas and that VEGF inhibition with currently available agents alone is inadequate.
VEGF inhibition and pazopanib
Pazopanib is an oral angiogenesis inhibitor that targets VEGF receptors, PDGF receptor, and c-kit. It has been tested in Phase II and Phase III trials in sarcomas. In the Phase II trial, 142 patients were enrolled in four different strata: adipocytic soft tissue sarcoma, leiomyosarcoma, synovial sarcoma, and other sarcomas. The primary endpoint was progression-free rate at 12 weeks. Pazopanib was administered at 800 mg/day. The adipocytic group was closed after the first stage because of insufficient activity; the progression-free rate was 26%. The progression-free rates in the other groups were 44% for leiomyosarcoma, 49% for synovial sarcoma, and 39% for other sarcoma subtypes.36
This study was followed by a Phase III trial evaluating pazopanib in metastatic nonadipocytic soft tissue sarcomas after failure of standard chemotherapy. Patients were randomly assigned to receive pazopanib at 800 mg/day or placebo in a 2:1 ratio. The study was double-blinded without crossover. The primary endpoint was PFS. Of the patients, 369 were evaluable. Median PFS was 4.6 months with pazopanib and 1.6 months for placebo. The hazard ratio was 0.31 (P < 0.0001). Overall survival was 12.5 months for the pazopanib group and 10.7 months for the placebo group.37 On the basis of the results of this trial, pazopanib has been approved to use in refractory, metastatic sarcomas in the US.
M-CSF, COL6A3, and pigmented villonodular synovitis
Pigmented villonodular synovitis is a benign process that can be locally destructive and that is associated with frequent local recurrences. It is characterized by a t(1;2) translocation, which results in the fusion of COL6A3 and M-CSF genes. Both imatinib and nilotinib have demonstrated activity in this disease. A complete response has been observed with imatinib,38 and nilotinib demonstrated a 12-week progression-free rate of 85.7% in 33 patients at a dose of 800 mg/day, although no objective responses were seen.39
Anaplastic lymphoma kinase and inflammatory myofibroblastic tumor
Inflammatory myofibroblastic tumor combines a spindle cell proliferation with an inflammatory infiltrate. These tumors may have aberrant expression of ALK, the anaplastic lymphoma kinase. Recently, an ALK inhibitor, crizotinib, was shown to be active in non-small-cell lung cancer with ALK gene rearrangements and was approved for use in that entity. Two patients with inflammatory myofibroblastic tumor have been treated with crizotinib, with a partial response of 6 months’ duration in a single patient with ALK-rearranged disease.40
IGF1R belongs to the family of receptors that are known to activate cellular proliferation, differentiation, and growth, as well as prevention from apoptosis through interaction with the ligands insulin, and IGF1 and IGF2. IGF1R has been implicated in the pathogenesis of both soft and bone sarcomas.41
IGF1R and Ewing’s sarcoma
Ewing’s sarcoma is the second most common bone tumor in children and young adults. It is associated with t(11;22), which produces the EWS-FLI1 fusion protein. This fusion protein binds to IGF binding protein 3, which upregulates IGF1.42 Several agents have been studied in attempting to target this pathway. R1507 is a monoclonal antibody directed at IGF1R. One hundred eleven patients with Ewing’s sarcoma were treated with R1507 weekly at 9 mg/kg. A response rate of 14.4% with single complete response and nine partial responses was seen with a median survival of 6.9 months. A small subset of patients treated with this agent has experienced durable remissions.43 Unfortunately, clinical development of this agent has been discontinued.
Ganitumab is another monoclonal antibody directed against IGF1R. It has been evaluated in a Phase II trial of patients with metastatic Ewing’s sarcoma or desmoplastic small cell tumors, which also contain fusions of the EWS gene. Thirty-eight patients were treated at 12 mg/kg every 2 weeks, with 35 patients evaluable for response. There were two partial responses (6%), and 17 patients with stable disease (49%). Four patients had stable disease longer than 24 weeks, yielding a clinical benefit response of 17%. No apparent relationship was observed between tumor response and IGF1 levels of EWS gene translocation.44
Figitumumab is a fully human IgG2 monoclonal antibody that targets IGF1R. A Phase I study that included 16 patients with Ewing’s sarcoma had two patients with objective responses, one complete response, and one partial response, and an additional six patients had stable disease lasting at least 4 months.45 A Phase 2 study of figitumumab has also been conducted in which 106 evaluable patients were treated with figitumumab at 30 mg/kg every 4 weeks. Fifteen patients (14.2%) had partial response and 25 had stable disease. Median overall survival was 8.9 months. High pretreatment levels of IGF1 were associated with a survival benefit.46
Despite the initial enthusiasm for this group of agents, their activity in Ewing’s sarcoma appears to be modest, with relatively low response rates and responses often of brief duration. One explanation for this observation may be the development of alternate pathways for growth in these tumors. One recent paper studying two patients with initial responses to IGF1R antibody followed by progression, revealed upregulation of p-Akt and p-m-Tor, with a subsequent response to combined IGF1R inhibition with m-Tor inhibition. A second patient was found to have activation of the ERK pathway at emergence of resistant tumor.47 The implication of this study may be that multiple pathways may need to be inactivated for more durable responses.
RANKL and giant cell tumor of bone
Giant cell tumor of bone is a benign condition that presents as an intraosseous lytic lesion that may be associated with a soft tissue component. It can recur locally and can rarely metastasize. Surgery is the usual treatment of choice. Receptor activator of nuclear factor kappa-B ligand (RANKL) is the ligand for tumor necrosis factor receptor, RANK. The stromal cells of giant cell tumor express RANKL, and denosumab is a monoclonal antibody directed against RANKL. A recent study of denosumab in 37 patients with recurrent or unresectable giant cell tumor of bone demonstrated responses in 30 of 35 evaluable patients. Response was defined as elimination of at least 90% of giant cells or no radiological progression of the target lesion up to week 25. Denosumab was administered at 120 mg subcutaneously every 28 days, with a loading dose given on days 8 and 15 of cycle 1. Adverse events were reported in 33 of 37 patients, most commonly pain or headache.48 Histologic evaluation of tumor samples demonstrated a decrease of 90% or more in 20/20 samples evaluated, as well as a reduction in tumor stromal cells.49
MDM2, CDK4, and liposarcoma
CDK4 and MDM2 are amplified in most well-differentiated/dedifferentiated liposarcomas. Their presence can help confirm the diagnosis of liposarcoma and serve to distinguish benign lipomas (which do not express CDK4 or MDM2) from true sarcomas. PD0332991 is a CDK4 inhibitor. A recent Phase II trial using oral PD0332991 at 200 mg/day for 14 days in a 21-day cycle enrolled 29 patients, of whom 27 were evaluable for the primary endpoint of PFS higher than 40% at 12 weeks. PFS was 70% at 12 weeks, and median PFS was 18 weeks.50 On the basis of these results, a Phase III trial is planned.
Hedgehog signaling and chondrosarcoma
Hedgehog signaling plays a role in cartilage tumorigenesis and is activated in chondrosarcoma. GDC-0449 is an inhibitor of the hedgehog pathway. A Phase II trial51 has been performed with GDC-0449 in advanced chondrosarcoma, using a dose of 150 mg daily. The primary endpoint of the trial is 6-month nonprogression rate according to RECIST, with a 40% nonprogressive disease rate. At the planned interim analysis, four of 17 patients had stable disease. Accrual is now at 40 patients. The drug appears to be well-tolerated.51 Additional trials with this agent in combination with a Notch inhibitor, RO4929097, in advanced sarcomas are in progress.52
Because of the importance of the mammalian target of rapamycin (mTOR) pathway, the abnormalities and overexpression found in different types of sarcomas, as well as the numerous studies of mTOR inhibitors as single agents or in combination in sarcomas, the mTOR pathway, interactions with other pathways, and ongoing clinical studies are presented separately.
mTOR inhibitors
mTOR was identified in 1995 as the principal protein kinase targeted by RAPAMYCIN. The natural product RAP (sirolimus; Rapamune®; Wyeth-Ayerst, Philadelphia, PA, USA) is a lipophilic macrolide isolated more than 20 years ago from a strain of Streptomyces hygroscopicus found in the soil of Easter Island (Rapa Nui). Initially,53–55 the antiproliferative effect of RAP was principally attributed to its ability to modulate the synthesis of critical proteins required for ribosome biosynthesis, protein translation, and G1 to S cell cycle phase traverse, with the final result being cell cycle arrest in G1 phase.56
RAP and other therapeutics targeting mTOR are particularly active against malignancies driven by activated PI3K, Akt, or both, as well as by PTEN (phosphatase and tensin homologue deleted from chromosome 10) gene mutations as the tumor becomes dependent on this pathway for growth. PTEN protein, the product of the PTEN tumor suppressor gene, opposes the activation of PI3K.57 Studies in PTEN knockout mice have demonstrated that PTEN-deficient tumor cells are very sensitive to the growth inhibitory effects of RAP and its analogs.58
RAP binds intracellularly to FK-BP12, a low-molecular-weight cytosolic protein, and forms a complex with a high affinity for mTOR.59,60 The inhibition of the mTOR kinase activity blocks the activation of two downstream signaling elements: the 40S ribosomal protein S6 kinase (p70s6k) and the eukaryotic initiation factor 4E binding protein1 (4E-BP1).61,62 By inhibiting mTOR, and therefore the translation of proteins that are essential for cell cycle traverse, cell growth, survival, and cell division, RAP and RAP analogs have antiproliferative and immunosuppressive effects.
mTOR exists in two different complexes, mTORC1 and mTORC2.63,64 TORC1 is considered sensitive to rapamycin, whereas TORC2 was until recently believed to be totally rapamycin-insensitive. Recent studies show that prolonged exposure to RAP analogs can lead to inhibition of mTORC2.63,65 mTORC1 is a trimeric protein kinase formed of the mTOR catalytic subunit, the regulatory-associated protein of mTOR (raptor), and a mammalian LST8/G-protein β-subunit-like protein (mLST8/GβL).63,66–68 The functions of the mTORC1 components are only partly understood. Studies suggest that raptor presents downstream target substrates to the mTOR kinase domain for phosphorylation in the presence of enough nutrients and also stabilizes mTOR; it is also a scaffolding protein, as it binds to downstream targets of mTOR, eukaryotic initiation factor 4E binding protein 1 and protein S6 kinase 1, via TOR signaling motif.69,70 The full catalytic activity of TOR requires interaction with LST8, with downregulation of mLST8 resulting in a decrease in mTOR kinase activity.71–73 FKBP12-rapamycin when bound to mTORC1 can abrogate mTORC1 kinase activity both in vitro and in vivo.
TORC2 consists of mTOR, rictor (rapamycin-insensitive companion of mTOR), GβL, and mammalian stress-activated protein kinase interacting protein 1.74,75 TORC2 can form multimeric supercomplexes, which have been shown to have a more active kinase activity than monomeric TORC2.76 Recently, novel components such as Protor 1 (protein observed with Rictor-1) and Protor 2 have been shown to interact with Rictor.77 mTORC2 is not bound by FKBP12-rapamycin, and it has been suggested that TORC2 mediates the spatial control of cell growth.74,75,78 Knockdown of components of mTORC2 such as rictor or mTOR results in loss of both actin polymerization and cell spreading.64,75
The mTOR pathway, as well as the mechanism of action of mTOR inhibitors, their clinical characteristics, and suggested mechanism of resistance, have been extensively explored during the last few years, and a large amount of information is now available.71,79,80
There is preclinical evidence supporting the use of mTOR inhibitors in sarcomas. Studies have revealed that PTEN is involved in the pathogenesis of sarcomas including GIST, leiomyosarcomas, and rhabdomyosarcomas.81–83 Analysis of 51 cases of soft tissue sarcomas revealed mutations of the PTEN/MMAC1 gene in two cases (3.9%), both being leiomyosarcoma arising from the intraabdominal cavity.81 Another study suggests that Akt Ser473 may be a key target residue for PTEN to modulate the effects of IGF2 on activating the PI3K/Akt pathway in rhabdomyosarcoma cells.82 Further preclinical studies are warranted to allow a better understanding of the biology of these heterogeneous tumors, thus leading to novel therapeutic options.
Rapamycin analogs in clinical development
RAP’s poor aqueous solubility and chemical stability precluded its use at doses susceptible to producing an effect as anticancer agent, and therefore new RAP analogs were developed. The RAP analogs currently used as anticancer agents include temsirolimus (CCI-779; Wyeth, Cambridge, MA, USA), everolimus (RAD 001, Novartis Pharmaceuticals AG, Basel, Switzerland), and ridaforolimus (AP23573, Merck & Co, Inc, Whitehouse Station, NJ, USA). These agents have demonstrated antiproliferative activity against a diverse range of malignancies in preclinical studies, and clinical evaluations have been very successful in a range of malignancies.
Temsirolimus
Temsirolimus (CCI-779) is a water-soluble RAP ester and was the first rapamycin analog selected for clinical development. In preclinical studies, CCI-779 and RAP demonstrated similar growth inhibitory profiles. Both agents elicited tumor growth inhibition rather than tumor regression in a wide variety of human tumor xenografts.
Two intermittent schedules were selected for initial development of CCI-779: a weekly 30-minute intravenous (IV) infusion and a 30-minute IV infusion daily for 5 days every 2 weeks. Main toxicities were mucositis, hypercholesterolemia, thrombocytopenia, rash, and fatigue. The weekly temsirolimus was approved as first-line treatment for poor-prognosis metastatic renal carcinoma.84–91
Everolimus is an orally bioavailable hydroxyethyl ether of RAP. The agent has demonstrated impressive antiproliferative activity against several human tumor cell lines as well as a broad range of human tumor xenografts.27 Moreover, additional studies have revealed that RAD 001 may have antiangiogenic properties.28 Early clinical trials demonstrated tolerability with a daily dose of 10 mg. Adverse events are similar with what was described with other mTOR inhibitors including mucositis, rash, hyperglycemia and hypercholesterolemia, anemia, thrombocytopenia, fatigue, and rarely, pneumonitis. Everolimus is currently approved for treatment of metastatic renal cell carcinoma (MRCCA) and pancreatic neuroendocrine tumors. Several other studies revealed promising results in other tumor types, including a combination with aromatase inhibitors in breast cancer (Everolimus in Combination With Exemestane in the Treatment of Postmenopausal Women With Estrogen Receptor Positive Locally Advanced or Metastatic Breast Cancer Who Are Refractory to Letrozole or Anastrozole; BOLERO2).92–99
Ridaforolimus (AP23573) is a nonprodrug of RAP and has demonstrated prominent antiproliferative activity against several cancers in vitro and in vivo. AP23573 has favorable pharmaceutical and pharmacological characteristics, and early studies in human xenograft models have shown a potent inhibition of tumor growth with a five times a day oral administration schedule of AP23573.100 Two formulations (IV and oral) were subject to clinical testing. For the IV formulation, two schedules of administration were explored: five times a day every other week and weekly.101,102 Toxicities included rash, mucositis, hypercholesterolemia, fatigue, and thrombocytopenia. Dose limiting toxicity in both schedules was determined to be mucositis. The recommended dose for Phase II studies is 15 mg IV daily for 5 days every other week and 100 mg IV on the weekly schedules, respectively. Interestingly, at the lowest dose level of 3 mg in the five times a day schedule, one patient with metastatic Müllerian sarcoma experienced a confirmed and durable partial response for more than 36 months. Antitumor activity was seen over the entire dose range on both schedules in several tumor types, including Müllerian sarcoma, GIST, Ewing’s sarcoma, renal cell carcinoma, lymphoma and non-small-cell lung cancer. Other hematological studies or combination studies were also performed.103,104
mTOR inhibitors in sarcomas
Ridaforolimus phase II sarcoma study
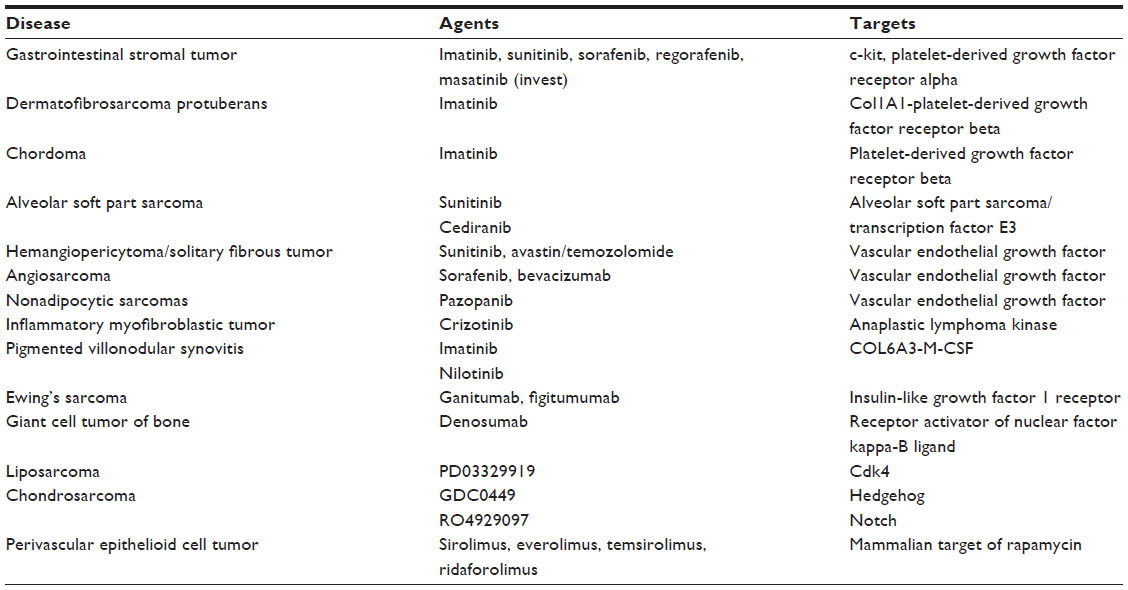
One the basis of the results seen in the Phase I study, ridaforolimus was tested in a Phase II clinical study in patients with previously treated advanced bone or soft tissue sarcoma.105 Patients with advanced sarcomas were stratified into four cohorts on the basis of histologic type: bone sarcoma, leiomyosarcoma, liposarcoma, and other soft tissue sarcomas excluding GIST. Patients were eligible regardless of the number of previous therapies. Ridaforolimus was administered at 12.5 mg IV daily for 5 days every 2 weeks on the basis of the results from Phase I studies. The study used a Simon two-stage design. For each cohort, treatment was defined as active if the proportion of patients with clinical benefit response that included partial response, complete response, or stable disease for at least 16 weeks was more than 25%. The use of stable disease and clinical benefit as a study endpoint was decided on the basis of studies revealing that for certain cancers such as sarcomas, sustained, stable disease is increasingly being recognized as a relevant response to treatment.106–108 In addition, studies by EORTC have demonstrated that a 6-month PFS of 30% or more can be considered as a reference value to suggest drug activity for first-line therapy.109 For second-line therapy, a 3-month PFS of 40% or greater would suggest drug activity, and 20% and lower would suggest inactivity.109 In ridaforolimus, all cohorts met the criteria to enter stage 2 of the trial. A total of 213 patients were treated, with 193 patients evaluable for response. In all four categories of sarcoma, clinical benefit was seen in approximately 25% of patients. The most frequent adverse effects were mucositis, rash, hyperlipidemia, fatigue, and thrombocytopenia. Overall, 54 (28%) patients had clinical benefit, including 5 with partial response. The antitumor activity of AP23573 in this study is summarized in Table 1.
 | Table 1 Targets and targeted agents in sarcoma |
A subgroup of 76 patients underwent positron emission tomography (PET) with 18F-fluorodeoxyglucose, and the metabolic response was analyzed on days 3 to 5 and 48 to 56, using the EORTC PET study group criteria.110 Partial metabolic responses were seen in 26% of the patients on first evaluation and 46% on second evaluation; stable metabolic response was seen in 69% of patients. Although preliminary, these results are encouraging in determining the role of PET in the evaluation of early response in patients with sarcoma treated with mTOR inhibitors.
The encouraging results of the Phase II study led to a Phase III study in patients with advanced sarcomas the SUCCEED (Sarcoma Multi-Center Clinical Evaluation of the Efficacy of Ridaforolimus) study. The purpose of the study was to evaluate whether maintenance therapy with oral ridaforolimus, by preventing and controlling tumor growth for a prolonged period of time in patients with metastatic soft tissue or bone sarcomas responding to chemotherapy, will result in clinically significant improvement in PFS compared with oral placebo. The study was completed and results were presented in 2011. This was an international, double-blind study randomized 1:1 between ridaforolimus (40 mg orally for 5 days/week) and placebo as maintenance therapy in patients with metastatic sarcoma after stable disease or objective response to prior chemotherapy treatment scan. Patients were stratified by sarcoma type and line of previous treatment. The primary endpoint was PFS based on independent radiological review; secondary endpoints include overall survival best target lesion response, cancer-related symptoms, and safety and tolerability.
In less than 3 years, 711 patients were randomized and analyzed for efficacy, which constitutes a relatively large study for this indication. The baseline characteristics were well-balanced between the study groups. Compared with placebo, ridaforolimus met the prespecified study endpoint with a statistically significant improvement in PFS (hazard ratio, 0.72; P = 0.0001, stratified log-rank), and median PFS improved by 21%. Ridaforolimus also demonstrated both improvement (hazard ratio, 0.69; P < 0.0001) and a 52% gain (22.4 weeks for ridaforolimus versus 14.7 weeks for placebo) in median PFS; PFS benefit was noted across all prespecified strata.
Follow-up for overall survival is ongoing, but the results seen at data cutoff indicate a trend favoring ridaforolimus. Although incidence of stomatitis (52%) and other adverse events was higher in patients receiving ridaforolimus, the overall safety profile was consistent with other mTOR inhibitors. This trial met the primary endpoint of improved PFS in patients with metastatic soft tissue or bone sarcomas, maintaining benefit from prior standard chemotherapy treatments.111 However, despite positive results on the predefined primary endpoints, the study did not reveal a significant improvement of the overall survival. Therefore, the US Food and Drug Administration did not grant approval for ridaforolimus as maintenance therapy for patients with sarcoma. Further studies are planned or ongoing with ridaforolimus.
Temsirolimus Phase II sarcoma study
Forty-one patients with advanced soft tissue sarcomas were enrolled in a first-line Phase II study of temsirolimus (CCI-779) at 25 mg weekly for 3 weeks every 4 weeks.112 Histologic types were diverse and included malignant fibrous histiocytoma, sarcoma not otherwise specified, fibrosarcoma, liposarcoma, leiomyosarcoma, endometrial sarcoma, synovial, hemangio/angiosarcoma, hemangiopericytoma, and neurofibrosarcoma. One patient with fibrosarcoma had a partial response for 36 weeks. Twenty-eight patients have progressed, and the estimated median time to progression was 2 months. The toxicity profile was considered acceptable, with few grade 3 events, including anemia, neutropenia, dyspnea, nausea, stomatitis, fatigue, and hyperglycemia. The authors concluded that CCI-779 failed to demonstrate sufficient activity as first-line therapy in patients with advanced soft tissue sarcoma to justify further development.112
Sirolimus case reports
Four cases of patients with sarcoma failing two to six lines of chemotherapy who were treated with sirolimus were presented at the American Society of Clinical Oncology 2006 Annual Meeting.113 Patients with malignant fibrous histiocytoma, leiomyosarcoma, angiosarcoma, and osteosarcoma were treated with 4–8 mg sirolimus daily for a median of 16 weeks with or without daily oral cyclophosphamide 200 mg every other week. Three patients had improvement in tumor-related symptoms, and two had improvement in performance status. Three patients had radiographic improvement and continued receiving treatment at the time of the presentation. The researchers concluded that sirolimus treatment was associated with improvement in tumor-related symptoms, performance status, and biochemical markers of disease activity, and that it inhibited tumor growth in patients with advanced sarcoma failing prior therapies. A formal study of sirolimus in advanced sarcoma is considered.
Combinations studies with mTOR inhibitors
Combination everolimus–imatinib in GIST
GISTs historically represent a distinct category of soft tissue sarcoma, with a dramatic evolution and a poor prognosis until the approval of imatinib. This new targeted therapy inhibiting c-kit has impressive efficacy, including prolonged responses and remissions for patients with GIST. Primary resistance to imatinib in advanced GIST is rare; however, ultimately, most patients develop secondary resistance. The Akt-mTOR pathway was incriminated as a potential molecular pathway mediating resistance to imatinib.114 In addition, synergism in vitro between imatinib and everolimus has been seen in human GIST resistant to imatinib. As a result, a Phase I/II trial was performed in patients with GIST refractory to imatininb.115 Thirty-one patients received a continuous dose of imatinib 600 mg/day in combination with everolimus either weekly or daily. Dose limiting toxicity occurred in 3 patients at 25 mg/day everolimus and were stomatitis, thrombocytopenia, and gastritis. The recommended dose of everolimus was 2.5 mg/day in combination with imatinib 600 mg/day. Two patients achieved partial response, and an additional eight patients had stable disease for more than 4 months. Molecular analyses were not available at the time of the presentation, and mature data are expected.
Combination of IGFR inhibitors and mTOR inhibitors in sarcomas
There is growing preclinical evidence that inhibiting IGFR and mTOR pathways together could result in synergistic activity. Two studies of combinations of mTOR inhibitors and IGFR inhibitors were recently presented. One Phase II study evaluated the combination of cixutumumab (IMC-A12) and temsirolimus in patients with IGFR1-positive and IGFR1-negative bone and soft tissue sarcoma.116 The study design included three cohorts: IGF1R-positive soft tissue sarcomas (group A), IGF1R-positive bone sarcomas (group B), and IGF1R-negative bone and soft tissue sarcomas (group C). An optimal Simon two-stage design was used for each group. A 40% PFS rate was considered promising, and a 20% PFS rate was considered nonpromising. Beginning in February 2010, 383 patients were tested for IGFR and 171 were treated. Cixutumumab was administered at 6 mg/kg and temsirolimus at 25 mg IV weekly. Pre- and posttreatment biopsies and plasma measurements for IGF1R and IGFB3 were obtained. By intent to treat, each group achieved the primary 12 week PFS (32% group A, 38% group B, and 43% group C). The effect was independent of IGF1R status by immunohistochemistry. Plasma biomarkers did not correlate with PFS. Although the authors conclude that this combination should be further explored, the extensive progressive disease markers and target screening proved once again that a better understanding of the target and the identification of effective predictive biomarkers for clinical benefit will be key in the rational development of mTOR inhibitors and other targeted therapies.
The second study presented a combination of everolimus and CP-751,871 in patients with advanced sarcomas and other solid tumors.117 This was a Phase I single-center study to determine the tolerability and recommended Phase II dose for the combination. The study demonstrated that a full-dose combination of CP-751,871 and everolimus appears safe and well-tolerated. Sixteen sarcoma patients were enrolled, and the majority experienced stable disease. Further follow-up is needed.
Combination of mTOR inhibitors with chemotherapy in sarcomas
A dose-finding study of temsirolimus and liposomal doxorubicin for patients with advanced sarcomas was reported in 2011.118 Fifteen patients were enrolled and treated at 3 dose levels. The recommended Phase II dose was 30 mg/m2 liposomal doxorubicin and 20 mg temsirolimus. The combination was considered tolerable at the indicated dose, and a Phase II study to determine efficacy in advanced sarcomas is ongoing.
Another Phase I study in patients with refractory sarcomas combined irinotecan and temsirolimus.119 Fourteen patients were enrolled. The recommended Phase II dose of this weekly combination was 80 mg/m2 irinotecan and 20 mg temsirolimus. The study is being expanded to a Phase II study, and results are pending.
Activity of mTOR inhibitors in specific sarcoma subtypes
mTOR inhibitors activity in PEComa
Perivascular epithelioid cell tumors (PEComas) are extremely rare tumors only recently recognized as a distinct entity by the World Health Organization. PEComas are part of a family of mesenchymal neoplasm with myelomelanocytic differentiation and share a specific cell type, “the perivascular epithelioid cell,” or PEC. PEComas are a group of neoplasms sharing morphological and immunohistochemical features and include angiomyolipomas, clear-cell “sugar” tumors of the lung, lymphangioleiomyomatosis, and others, some of them strongly associated with tuberous sclerosis complex. Disorders caused by mutation of the TSC1 or TSC2 genes usually result in overactivation of the mTOR pathway, as their gene products regulate negatively mTORC. It is also believed that PEComas share the activation of the mTOR pathway with lymphangioleiomyomatosis and angiomyolipomas, and therefore all PEComas could be sensitive to mTOR inhibition.
Recently, two publications revealed not only significant radiographic responses and confirmed target inhibition in three and two cases, respectively, but also emphasized the major clinical improvement experienced by the patients.120,121 Therefore, based on the preclinical and clinical evidence of efficacy of mTOR inhibitors in PEComas, larger studies are ongoing.
Discussion
The number of active treatments for patients with sarcoma is limited. Therefore, there is a need for more active and less toxic drugs to be included in the panoply of available treatment for sarcomas. The broad spectrum of histopathological and molecular sarcoma subtypes as well as the clinical behavior of these tumors was the reason the clinical studies and preclinical discoveries have been delayed.
Targeted drugs including antiangiogenic agents, mTOR inhibitors, IGFR-inhibitors, and other drugs are currently tested as single agents or in combination for the treatment of sarcomas.
Although there are several ways of altering angiogenesis, and access to small molecules or monoclonal antibodies targeting one or several elements involved in angiogenesis is increasing, the results are not yet practice-altering with the exception of the recent approval of pazopanib. New studies are ongoing, and results are awaited specifically in sarcoma subtypes in which angiogenesis was proven to represent an essential step for malignant transformation and progression.
Inhibition of signaling received from activation of growth factors such as EGFR and IGFR has been linked to sarcomatous transformation, and many preclinical studies support the use of IGFR inhibitors in sarcomas. Several clinical studies with monoclonal antibodies and small molecules established the tolerability of most of these agents, but their role in sarcoma treatment is still under question, although in selected sarcoma subtypes (Ewing’s), the clinical benefit is significant.
The mTOR pathway is one of the most important pathways for tumor transformation and growth and was shown to be altered in sarcomas. Significant clinical evidence is also available. Of the three rapamycin analogs available, ridaforolimus administered IV showed promising results in a Phase II study in patients with advanced sarcomas with a clinical benefit of approximately 25%. The Phase III SUCCEED trial confirmed the role of ridaforolimus as maintenance therapy for patients with sarcoma responding to chemotherapy with a significant increase in PFS compared with in the placebo group. However, the difference in survival did not reach statistical significance; therefore, ridaforolimus did not receive US FDA approval for this setting. Anecdotal responses in patients with sarcoma were also seen with sirolimus either alone or in combination with cyclophosphamide. Therefore, mTOR inhibitors may represent an effective and tolerable therapeutic option for patients with sarcoma. However, there are still some challenges and questions to be addressed in future studies, such as dose, schedule, and appropriate design for Phase III studies as well as identification of biomarkers that would allow prospective selection of patients likely to benefit from treatment with mTOR inhibitors.
Sarcomas are different diseases in terms of pathology, clinical presentation, molecular characteristics, and prognosis. However, because of their rarity, most clinical studies usually include various histological types. Therefore, it is very difficult to analyze subtle differences in response and/or clinical benefit by sarcoma type. Future large randomized studies should take into consideration these issues for study design. The dose and schedule to be used in Phase II and III studies are also of critical importance. An optimal assessment of the treatment effect on disease progression should remain the main focus in clinical trials, as these agents are more likely to behave as cytostatic drugs, and therefore may produce a significant difference in PFS or overall survival in Phase III studies, even if the response rate in Phase II studies is relatively low. Also, as noted previously for several targeted drugs, even low doses demonstrated improvement in overall survival in selective patients. Therefore, for targeted “cytostatic” drugs, it is crucial to define in Phase I and II studies the optimal dose that produces biological activity with minimal adverse effects, thus offering the option of prolonged drug administration without detrimental influence on patient’s quality of life. It is also essential that the schedule of administration for most of the orally administered targeted drugs be defined in early clinical trials, as protracted, uninterrupted administration may result in less common adverse effects such as lung toxicity. Intermittent schedules of administration may be therefore more tolerable and more compatible with a prolonged treatment. When deciding the use of different tyrosine-kinase inhibitors for different sarcoma subtypes, it is critical to assess the intracellular targets and pathways to better evaluate possible efficacy. The most recent promising results in patients with PEComas support a better understanding of molecular biology in these rare tumors in finding new cures.
Furthermore, mainly for targeted therapies, determining the select patient population that could significantly benefit from treatment is the next critical step for the development of these drugs. It is clear from several studies that these agents are effective and probably have more than cytostatic effects in selected patients. Identifying the predictive biomarkers of response and tolerability will allow not only a better patient selection but also improvement of the therapeutic index, as well as of the use and indication of the mTOR inhibitors.
Conclusion
Significant advances in molecular biology and genetic research have allowed better identification of molecular differences between the types of sarcomas. These molecular signatures are important now not only for appropriate diagnosis but also for identification and use of targeted therapies. mTOR inhibitors, IGFR inhibitors, tyrosine kinase inhibitors, and antiangiogenic agents represent promising drugs for the treatment of sarcomas. However, several questions still remain to be answered, particularly regarding optimal dose and schedule, as well as the optimal design for future Phase III trials. Despite a significant number of Phase II studies (either completed or ongoing), a limited number of agents are moving into Phase III testing in sarcoma. The next step for targeted agents is to identify biomarkers and validate screening to fully validate the targets and determine the optimal treatment strategy.
Disclosure
The authors report no conflicts of interest in this work.
References
Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med. 2001;344(14):1052–1056. | |
van Oosterom AT, Judson I, Verweij J, et al; European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group. Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet. 27 2001;358(9291):1421–1423. | |
Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–480. | |
Blanke CD, Demetri GD, von Mehren M, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26(4):620–625. | |
Benjamin RS, Choi H, Macapinlac HA, et al. We should desist using RECIST, at least in GIST. J Clin Oncol. 2007;25(13):1760–1764. | |
Heinrich MC, Owzar K, Corless CL, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26(33):5360–5367. | |
Debiec-Rychter M, Sciot R, Le Cesne A, et al; EORTC Soft Tissue and Bone Sarcoma Group; Italian Sarcoma Group; Australasian GastroIntestinal Trials Group. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42(8):1093–1103. | |
Blay JY, Le Cesne A, Ray-Coquard I, et al. Prospective multicentric randomized phase III study of imatinib in patients with advanced gastrointestinal stromal tumors comparing interruption versus continuation of treatment beyond 1 year: the French Sarcoma Group. J Clin Oncol. 2007;25(9):1107–1113. | |
Le Cesne A, Ray-Coquard I, Bui BN, et al; French Sarcoma Group. Discontinuation of imatinib in patients with advanced gastrointestinal stromal tumours after 3 years of treatment: an open-label multicentre randomised phase 3 trial. Lancet Oncol. 2010;11(10):942–949. | |
Demetri GD, van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–1338. | |
George S, von Mehren M, Heinrich MC, et al. A multicenter phase II study of regorafenib in patients (pts) with advanced gastrointestinal stromal tumor (GIST), after therapy with imatinib (IM) and sunitinib (SU). J Clin Oncol. 2011;29(15 May 20 Supplement):10007. | |
Demetri GD, Reichardt P, Kang Y-K, et al. Randomized phase III trial of regorafenib in patients (pts) with metastatic and/or unresectable gastrointestinal stromal tumor (GIST) progressing despite prior treatment with at least imatinib (IM) and sunitinib (SU): GRID trial. J Clin Oncol. 2012;30:Abstract LBA10008. | |
Adenis A, Le Cesne A, Nguyen BB, et al. Masitinib mesylate in imatinib-resistant advanced GIST: a randomized phase II trial. J Clin Oncol. 2012;30:Abstract 10007. | |
Le Cesne A, Blay JY, Bui BN, et al. Phase II study of oral masitinib mesilate in imatinib-naïve patients with locally advanced or metastatic gastro-intestinal stromal tumour (GIST). Eur J Cancer. 2010;46(8):1344–1351. | |
Trent JC, Walthen K, von Mehren M, et al. A phase II study of dasatinib for patients with imatinib-resistant gastrointestinal stromal tumor (GIST). J Clin Oncol. 2011;29:Abstract 10006. | |
Schöffski P, Reichardt P, Blay JY, et al. A phase I-II study of everolimus (RAD001) in combination with imatinib in patients with imatinib-resistant gastrointestinal stromal tumors. Ann Oncol. 2010;21(10):1990–1998. | |
Dematteo RP, Ballman KV, Antonescu CR, et al; American College of Surgeons Oncology Group (ACOSOG) Intergroup Adjuvant GIST Study Team. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. 2009 28;373(9669):1097–1104. | |
Joensuu H, Eriksson M, Hatrmann K, et al. Twelve versus 36 months of adjuvant imatinib (IM) as treatment of operable GIST with a high risk of recurrence: Final results of a randomized trial (SSGXVIII/AIO). Proc Am Soc Clin Oncol. 2011;Abstract LBA1. | |
Eisenberg BL, Harris J, Blanke CD, et al. Phase II trial of neoadjuvant/adjuvant imatinib mesylate (IM) for advanced primary and metastatic/recurrent operable gastrointestinal stromal tumor (GIST): early results of RTOG 0132/ACRIN 6665. J Surg Oncol. 2009;99(1):42–47. | |
McAuliffe JC, Hunt KK, Lazar AJ, et al. A randomized, phase II study of preoperative plus postoperative imatinib in GIST: evidence of rapid radiographic response and temporal induction of tumor cell apoptosis. Ann Surg Oncol. 2009;16(4):910–919. | |
Rutkowski P, Van Glabbeke M, Rankin CJ, et al; European Organisation for Research and Treatment of Cancer Soft Tissue/Bone Sarcoma Group; Southwest Oncology Group. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: pooled analysis of two phase II clinical trials. J Clin Oncol. 2010;28(10):1772–1779. | |
Kérob D, Porcher R, Vérola O, et al. Imatinib mesylate as a preoperative therapy in dermatofibrosarcoma: results of a multicenter phase II study on 25 patients. Clin Cancer Res. 2010;16(12):3288–3295. | |
Casali PG, Messina A, Stacchiotti S, et al. Imatinib mesylate in chordoma. Cancer. 2004;101(9):2086–2097. | |
Stacchiotti S, Longhi A, Ferraresi V, et al. Phase II study of imatinib in advanced chordoma. J Clin Oncol. 2012;30(9):914–920. | |
Mercier F, Guiot MC, Bojanowski MW. Treatment of chordoma with imatinib complicated by intracranial hemorrhage: a case showing dissociation between biological effect and therapeutic outcome. J Neurooncol. 2012;107(2):435–437. | |
Stacchiotti S, Tamborini E, Marrari A, et al. Response to sunitinib malate in advanced alveolar soft part sarcoma. Clin Cancer Res. 2009;15(3):1096–1104. | |
Stacchiotti S, Negri T, Zaffaroni N, et al. Sunitinib in advanced alveolar soft part sarcoma: evidence of a direct antitumor effect. Ann Oncol. 2011;22(7):1682–1690. | |
Gardner K, Judson I, Leahy M, et al. Activity of cediranib, a highly potent and selective VEGF signaling inhibitor, in alveolar soft part sarcoma. J Clin Oncol. 2009;27:15s, Abstract 10523. | |
National Cancer Institute. Phase II study of cediranib (AZD2171) in patients with alveolar soft part sarcoma. Available from: http://clinicaltrials.gov/show/NCT00942877. NLM identifier: NCT00942877. Accessed July 25, 2013. | |
Park MS, Patel SR, Ludwig JA, et al. Activity of temozolomide and bevacizumab in the treatment of locally advanced, recurrent, and metastatic hemangiopericytoma and malignant solitary fibrous tumor. Cancer. 2011;117(21):4939–4947. | |
Stacchiotti S, Negri T, Palassini E, et al. Sunitinib malate and figitumumab in solitary fibrous tumor: patterns and molecular bases of tumor response. Mol Cancer Ther. 2010;9(5):1286–1297. | |
Ray-Coquard I, Italiano A, Bompas E, et al; French Sarcoma Group (GSF/GETO). Sorafenib for patients with advanced angiosarcoma: a phase II Trial from the French Sarcoma Group (GSF/GETO). Oncologist. 2011;7(2):260–266. | |
Maki RG, D’Adamo DR, Keohan ML, et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J Clin Oncol. 2009;27(19):3133–3140. | |
Agulnik M, Yarber JL, Okuno SH, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24(1):257–263. | |
Sleijfer S, Ray-Coquard I, Papai Z, et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: a phase II study from the European organisation for research and treatment of cancer-soft tissue and bone sarcoma group (EORTC study 62043). J Clin Oncol. 2009;27(19):3126–3132. | |
van der Graaf WT, Blay JY, Chawla SP, et al; EORTC Soft Tissue and Bone Sarcoma Group; PALETTE study group. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879–1886. | |
Blay JY, El Sayadi H, Thiesse P, Garret J, Ray-Coquard I. Complete response to imatinib in relapsing pigmented villonodular synovitis/tenosynovial giant cell tumor (PVNS/TGCT). Ann Oncol. 2008;19(4):821–822. | |
Gelderblom H, Pérol D, Chevreau C, et al. An open-label international multicentric phase II study of nilotinib in progressive pigmented villo-nodular synovitis (PVNS) not amenable to a conservative surgical treatment. J Clin Oncol. 2013;31:Abstract 10516. | |
Butrynski JE, D’Adamo DR, Hornick JL, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363(18):1727–1733. | |
Scotlandi K, Picci P. Targeting insulin-like growth factor 1 receptor in sarcomas. Curr Opin Oncol. 2008;20(4):419–427. | |
Prieur A, Tirode F, Cohen P, Delattre O. EWS/FLI-1 silencing and gene profiling of Ewing cells reveal downstream oncogenic pathways and a crucial role for repression of insulin-like growth factor binding protein 3. Mol Cell Biol. 2004;24(16):7275–7283. | |
Pappo AS, Patel S, Crowley J, et al. Activity of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF1R), in patients (pts) with recurrent or refractory Ewing’s sarcoma family of tumors (ESFT): Results of a phase II SARC study. J Clin Oncol. 2010;28:15s, Abstract 10000. | |
Tap WD, Demetri G, Barnette P, et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J Clin Oncol. 2012;30(15):1849–1856. | |
Olmos D, Postel-Vinay S, Molife LR, et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: a phase 1 expansion cohort study. Lancet Oncol. 2010;11(2):129–135. | |
Juergens H, Daw NC, Geoerger B, et al. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J Clin Oncol. 2011;29(34):4534–4540. | |
Subbiah V, Naing A, Brown RE, et al. Targeted morphoproteomic profiling of Ewing’s sarcoma treated with insulin-like growth factor 1 receptor (IGF1R) inhibitors: response/resistance signatures. PLoS One. 6 2011;6(4):e18424. | |
Thomas D, Henshaw R, Skubitz K, et al. Denosumab in patients with giant-cell tumour of bone: an open-label, phase 2 study. Lancet Oncol. 2010;11(3):275–280. | |
Branstetter DG, Nelson SD, Manivel JC, et al. Denosumab induces tumor reduction and bone formation in patients with giant-cell tumor of bone. Clin Cancer Res. 2012;18(16):4415–4424. | |
Dickson MA, Tap WD, Keohan ML, et al. Phase II Trial of the CDK4 Inhibitor PD0332991 in Patients With Advanced CDK4-Amplified Well-Differentiated or Dedifferentiated Liposarcoma. J Clin Oncol. 2013;31(16):2024–2028. | |
Italiano A, Le Cesne A, Cassier PA, et al. GDC-0449 in patients with advanced chondrosarcomas: A French Sarcoma Group (FSG)/French and US. National Cancer Institutes phase II collaborative study. J Clin Oncol. 2013;30:Abstract 10005. | |
Gounder MM, Dickson MA, Wu N, et al. A first-in-human, phase Ib combination study to assess safety, pharmacokinetics (PK), and pharmacodynamics (PD) of a hedgehog inhibitor, GDC-0449, with a Notch inhibitor, RO4929097, in patients with advanced sarcoma. J Clin Oncol. 2013;30:Abstract 10004. | |
Vézina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo). 1975;28(10):721–726. | |
Baker H, Sidorowicz A, Sehgal SN, Vézina C. Rapamycin (AY-22,989), a new antifungal antibiotic. III. In vitro and in vivo evaluation. J Antibiot (Tokyo). 1978;31(6):539–545. | |
Eng CP, Sehgal SN, Vézina C. Activity of rapamycin (AY-22,989) against transplanted tumors. J Antibiot (Tokyo). 1984;37(10):1231–1237. | |
Wiederrecht GJ, Sabers CJ, Brunn GJ, Martin MM, Dumont FJ, Abraham RT. Mechanism of action of rapamycin: new insights into the regulation of G1-phase progression in eukaryotic cells. Prog Cell Cycle Res. 1995;1:53–71. | |
Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15(4):356–362. | |
Neshat MS, Mellinghoff IK, Tran C, et al. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc Natl Acad Sci U S A. 2001;98(18):10314–10319. | |
Fruman DA, Wood MA, Gjertson CK, Katz HR, Burakoff SJ, Bierer BE. FK506 binding protein 12 mediates sensitivity to both FK506 and rapamycin in murine mast cells. Eur J Immunol. 1995;25(2):563–571. | |
Sekulić A, Hudson CC, Homme JL, et al. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000;60(13):3504–3513. | |
Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, Thomas G. Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70s6k. EMBO J. 1997;16(12):3693–3704. | |
Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–963. | |
Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6(9):729–734. | |
Loewith R, Jacinto E, Wullschleger S, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10(3):457–468. | |
Zeng Z, Sarbassov dos D, Samudio IJ, et al. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood. 2007;109(8):3509–3512. | |
Kim DH, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110(2):163–175. | |
Hara K, Maruki Y, Long X, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110(2):177–189. | |
Yonezawa K, Tokunaga C, Oshiro N, Yoshino K. Raptor, a binding partner of target of rapamycin. Biochem Biophys Res Commun. 2004;313(2):437–441. | |
Harris TE, Lawrence JC Jr. TOR signaling. Sci STKE. 2003; 2003(212):re15. | |
Nojima H, Tokunaga C, Eguchi S, et al. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003;278(18):15461–15464. | |
Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–484. | |
Jiang BH, Liu LZ. Role of mTOR in anticancer drug resistance: perspectives for improved drug treatment. Drug Resist Updat. 2008;11(3):63–76. | |
Kim DH, Sarbassov DD, Ali SM, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003;11(4):895–904. | |
Frias MA, Thoreen CC, Jaffe JD, et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol. 2006;16(18):1865–1870. | |
Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–1302. | |
Wullschleger S, Loewith R, Oppliger W, Hall MN. Molecular organization of target of rapamycin complex 2. J Biol Chem. 2005;280(35):30697–30704. | |
Pearce LR, Huang X, Boudeau J, et al. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem J. 2007;405(3):513–522. | |
Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6(11):1122–1118. | |
Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5(8):671–688. | |
Kurmasheva RT, Huang S, Houghton PJ. Predicted mechanisms of resistance to mTOR inhibitors. Br J Cancer. 2006;95(8):955–960. | |
Ricci R, Maggiano N, Castri F, et al. Role of PTEN in gastrointestinal stromal tumor progression. Arch Pathol Lab Med. 2004;128(4):421–425. | |
Saito T, Oda Y, Kawaguchi K, et al. PTEN/MMAC1 gene mutation is a rare event in soft tissue sarcomas without specific balanced translocations. Int J Cancer. 2003;104(2):175–178. | |
Wan X, Helman LJ. Levels of PTEN protein modulate Akt phosphorylation on serine 473, but not on threonine 308, in IGF-II-overexpressing rhabdomyosarcomas cells. Oncogene. 2003;22(50):8205–8211. | |
Gibbons JJ, Discafani C, Peterson R, et al. The effect of CCI-779, a novel macrolide anti-tumor agent, on the growth of human tumor cells in vitro and in nude mouse in vivo. Proc Am Assoc Cancer Res. 1999;40:301, Abstract 2000. | |
Hidalgo M, Buckner JC, Erlichman C, et al. A phase I and pharmacokinetic study of temsirolimus (CCI-779) administered intravenously daily for 5 days every 2 weeks to patients with advanced cancer. Clin Cancer Res. 2006;12(19):5755–5763. | |
Raymond E, Alexandre J, Faivre S, et al. Safety and pharmacokinetics of escalated doses of weekly intravenous infusion of CCI-779, a novel mTOR inhibitor, in patients with cancer. J Clin Oncol. 2004;22(12):2336–2347. | |
Atkins MB, Hidalgo M, Stadler WM, et al. Randomized phase II study of multiple dose levels of CCI-779, a novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory renal cell carcinoma. J Clin Oncol. 2004;22(5):909–918. | |
Hudes G, Carducci M, Tomczack P, et al. A phase 3, randomized, 3-arm study of temsirolimus (TEMSR) or interferon-alpha (IFN) or the combination of TEMSR + IFN in the treatment of first-line, poor-risk patients with advanced renal cell carcinoma (adv RCC). J Clin Oncol. 2006;24(18S June 20 Supplement):LBA4. | |
Chan S, Scheulen ME, Johnston S, et al. Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol. 2005;23(23):5314–5322. | |
Carpenter JT, Roché H, Campone M, et al. Randomized 3-arm, phase 2 study of temsirolimus (CCI-779) in combination with letrozole in postmenopausal women with locally advanced or metastatic breast cancer. J Clin Oncol. 2005;23(16S June 1 Supplement):564. | |
Galanis E, Buckner JC, Maurer MJ, et al; North Central Cancer Treatment Group. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol. 2005;23(23):5294–5304. | |
Fourouzesh B, Buckner J, Adjei A, et al. Phase I, bioavailability and pharmacokinetic study of oral dosage of CCI-779 administered to patients with advanced solid malignancies. Proceedings of the 13th EORTC-NCI-AACr symposium. Eur J Cancer. 2002;38(Suppl 7); S54, Abstract 168. | |
O’Reilly T, Vaxelaire J, Muller M. In vivo activity of RAD001, an orally active rapamycin derivative, in experimental tumor models. Proc Am Assoc Cancer Res. 2002:43:Abstract 359. | |
Lane HA, Schnell C, Theuer A, et al. Antiangiogenic activity of RAD001, an orally active anticancer agent. Proc Amer Assoc Cancer Res. 2002;43:184. | |
O’Donnell A, Faivre S, Judson I, et al. A phase I study of the oral mTOR inhibitor RAD001 as monotherapy to identify the optimal biologically effective dose using toxicity, pharmacokinetic (PK) and pharmacodynamic (PD) endpoints in patients with solid tumors. Proc Am Soc Clin Oncol. 2003;22:200:Abstract 803. | |
Amato RJ, Misellati A, Khan M, Chiang S. A phase II trial of RAD001 in patients (Pts) with metastatic renal cell carcinoma (MRCC). J Clin Oncol. 2006;24(18S June 20 Supplement):4530. | |
Lerut E, Roskams T, Goossens E, et al. Molecular pharmacodynamic (MPD) evaluation of dose and schedule of RAD001 (everolimus) in patients with operable prostate carcinoma (PC). J Clin Oncol. 2005;23(16S June 1 Supplement):3071. | |
Reardon D, Quinn JA, Rich JN, et al. A phase I trial of imatinib, hydroxyurea and RAD001 for patients with recurrent malignant glioma. J Clin Oncol. 2006;24(18S June 20 Supplement):1580. | |
Yao JC, Phan A, Chang DZ, et al. Phase II study of RAD001 (everolimus) and depot octreotide (sandostatin LAR) in advanced low grade neuroendocrine carcinoma (LGNET). J Clin Oncol. 2007;25(18S June 20 Supplement):4503. | |
Milton DT, Kris MG, Azzoli CG, et al. Phase I/II trial of gefitinib and RAD001 (everolimus) in patients (pts) with advanced non-small cell lung cancer (NSCLC). J Clin Oncol. 2005;23(16S June 1 Supplement):7104. | |
Clackson T, Metcalf CA, Rivera VM, et al. Broad anti-tumor activity of ap23573, an mTOR inhibitor in clinical development. Proc Am Soc Clin Oncol. 2003;22(220):Abstract 882. | |
Mita M, Rowinsky EK, Goldston ML, et al. Phase I, pharmacokinetic (PK) and pharmacodynamic (PD) study of AP23573, an mTOR inhibitor administered IV daily X5 every other week in patients with refractory or advanced malignancies. J Clin Oncol. 2004;22(14S):Abstract 3076. | |
Desai AA, Janish L, Berk LR, et al. A phase I trial of a novel mTOR inhibitor AP23573 administered weekly (wkly) in patients (pts) with refractory or advanced malignancies: A pharmacokinetic (PK) and pharmacodynamic (PD) analysis. J Clin Oncol. 2004;22(14S July 15 Supplement):3150. | |
Perotti A, Maur M, Viganò L, et al. Phase Ib pharmacokinetic (PK) and pharmacodynamic (PD) study to define the optimal dose for combining the mTOR inhibitor AP23573 with capecitabine (CAPE). J Clin Oncol. 2006;24(No 18S June 20 Supplement):3065. | |
Feldman E, Giles F, Roboz G, et al. A phase 2 clinical trial of AP23573, an mTOR inhibitor, in patients with relapsed or refractory hematologic malignancies. J Clin Oncol. 2005;23(16S June 1 Supplement):6631. | |
Chawla SP, Tolcher AW, Staddon AP, et al. Updated results of a phase II trial of AP23573, a novel mTOR inhibitor, in patients (pts) with advanced soft tissue or bone sarcomas. J Clin Oncol. 2006;24(18S June 20 Supplement):9505. | |
Blay JY, van Glabbeke M, Verweij J, et al. Advanced soft-tissue sarcoma: a disease that is potentially curable for a subset of patients treated with chemotherapy. Eur J Cancer. 2003;39(1):64–69. | |
Le Cesne A, Blay JY, Judson I, et al. Phase II study of ET-743 in advanced soft tissue sarcomas: a European Organisation for the Research and Treatment of Cancer (EORTC) soft tissue and bone sarcoma group trial. J Clin Oncol. 2005;23(3):576–584. | |
Le Cesne A, Judson I, Crowther D, et al. Randomized phase III study comparing conventional-dose doxorubicin plus ifosfamide versus high-dose doxorubicin plus ifosfamide plus recombinant human granulocyte-macrophage colony-stimulating factor in advanced soft tissue sarcomas: A trial of the European Organization for Research and Treatment of Cancer/Soft Tissue and Bone Sarcoma Group. J Clin Oncol. 2000;18(14):2676–2684. | |
Van Glabbeke M, Verweij J, Judson I, Nielsen OS; EORTC Soft Tissue and Bone Sarcoma Group. Progression-free rate as the principal end-point for phase II trials in soft-tissue sarcomas. Eur J Cancer. 2002;38(4):543–549. | |
Sankhala KK, Chawla SP, Iagaru A, et al. Early response evaluation of therapy with AP23573 (an mTOR inhibitor) in sarcoma using [18F]2-fluoro-2-deoxy-D-glucose (FDG) positron emission tomography (PET) scan. J Clin Oncol. 2005;23(16S June 1 Supplement):9028. | |
Chawla SP, Blay J, Ray-Coquard IL, et al. Results of the phase III, placebo-controlled trial (SUCCEED) evaluating the mTOR inhibitor ridaforolimus (R) as maintenance therapy in advanced sarcoma patients (pts) following clinical benefit from prior standard cytotoxic chemotherapy (CT). J Clin Oncol. 2011;29:Abstract 10005. | |
Okuno SH, Mahoney MR, Bailey HH, et al. A multicenter phase 2 consortium (P2C) study of the mTOR inhibitor CCI-779 in advanced soft tissue sarcomas (STS). J Clin Oncol. 2006;24(18S June 20 Supplement):9504. | |
Schuetze SM, Baker LH, Maki RG. Sirolimus reduced tumor-related morbidity and resulted in biochemical and radiographic response in patients with progressive sarcoma. J Clin Oncol. 2006;24(18S June 20 Supplement):9503. | |
Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24(29):4764–4774. | |
van Oosterom A, Reichardt P, Blay J-Y, et al. A phase I/II trial of the oral mTOR-inhibitor everolimus (E) and imatinib mesylate (IM) in patients (pts) with gastrointestinal stromal tumor (GIST) refractory to IM: study update. J Clin Oncol. 2005;23(16S June 1 Supplement):9033. | |
Schwartz GK, Tap WD, Qin LX, et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, phase 2 trial. Lancet Oncol. 2013;14(4):371–382. | |
Quek R, Wang Q, Morgan JA, et al. Combination mTOR and IGF-1R inhibition: phase I trial of everolimus and figitumumab in patients with advanced sarcomas and other solid tumors. Clin Cancer Res. 2011;17(4):871–879. | |
Leob DM, DeLorenzo KA, Chen ARS, et al. A dose-finding study of temsirolimus and liposomal doxorubicin for patients with advanced soft tissue or bone sarcoma. J Clin Oncol. 2011;29:Abstract 10026. | |
Movva S, Bocklage T, Schmit B, et al. Phase I study of irinotecan and temsirolimus in patients with refractory sarcomas. J Clin Oncol. 2011;29:Abstract 10027. | |
Wagner AJ, Malinowska-Kolodziej I, Morgan JA, et al. Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol. 2010;28(5):835–840. | |
Italiano A, Delcambre C, Hostein I, et al. Treatment with the mTOR inhibitor temsirolimus in patients with malignant PEComa. Ann Oncol. 2010;21(5):1135–1137. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.