Back to Journals » OncoTargets and Therapy » Volume 14
Targeted Therapy for Advanced or Metastatic Cholangiocarcinoma: Focus on the Clinical Potential of Infigratinib
Authors Yu J ![]() , Mahipal A
, Mahipal A ![]() , Kim R
, Kim R ![]()
Received 30 July 2021
Accepted for publication 6 October 2021
Published 23 October 2021 Volume 2021:14 Pages 5145—5160
DOI https://doi.org/10.2147/OTT.S272208
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Arseniy Yuzhalin
James Yu,1 Amit Mahipal,2 Richard Kim3
1Department of Internal Medicine, Adventhealth Orlando, Orlando, FL, USA; 2Department of Medical Oncology, Mayo Clinic, Rochester, MN, USA; 3Department of Gastrointestinal Oncology, H. Lee Moffitt Cancer Center, Tampa, FL, USA
Correspondence: Richard Kim
Department of Gastrointestinal Oncology, H. Lee Moffitt Cancer Center, 12902 Magnolia Drive, Tampa, FL, 33612, USA
Tel +1 813-745-1277
Fax +1 813-449-8553
Email [email protected]
Abstract: Cholangiocarcinoma is one of the most aggressive cancers, with a 5-year survival rate of 11– 44% after surgical resection. However, there is no established systemic therapy after failure of the gemcitabine plus cisplatin first-line therapy with exception of FOLFOX. Fibroblast growth factor receptor (FGFR) genomic aberrations have been detected in cholangiocarcinoma, and targeting these genomic aberrations with FGFR inhibitors has shown remarkable clinical benefits in advanced cholangiocarcinoma. In this article, we provide up-to-date information on the clinical development of selective FGFR inhibitors in advanced cholangiocarcinoma, focusing on infigratinib. In a Phase 1 trial, infigratinib showed a safe profile. In a following Phase 2 trial, infigratinib showed remarkable efficacy in advanced cholangiocarcinoma with FGFR2 fusions or rearrangements, and the Food and Drug Administration (FDA) approved infigratinib for cholangiocarcinoma in May 2021 largely based on tumor response and duration of response. Currently infigratinib is on a Phase 3 trial (PROOF301) as a first-line setting compared to the GEMCIS therapy in advanced cholangiocarcinoma. Given that the FGFR genomic aberrations including FGFR2 fusions are rarely accompanied with other targetable mutations, infigratinib and other FGFR inhibitors are continuously expected to be the novel targeted agents in cholangiocarcinoma harboring these aberrations. Acquired resistance to infigratinib was reported in several recent studies which could potentially be a barrier to overcome. Active clinical trials including PROOF301 are expected to elucidate the clinical benefits of infigratinib in this disease. Infigratinib combined with immunotherapy is also a potential future direction of investigation in cholangiocarcinoma.
Keywords: infigratinib, cholangiocarcinoma, fibroblast growth factor receptor, FGFR inhibitors, targeted therapy
Introduction
Cholangiocarcinoma (CCA) is the second-most common primary liver tumor after hepatocellular carcinoma and constitutes 10–15% of hepatobiliary malignancies and approximately 3% of all gastrointestinal malignancies.1 CCA is classically categorized anatomically into intrahepatic (iCCA) and extrahepatic cholangiocarcinoma (eCCA). eCCA is further subdivided into perihilar and distal CCA.2 Surgery is considered to be the only potential curative management, but less than one third of CCA patients present with resectable stage at the time of diagnosis, mainly due to late onset, nonspecific clinical symptoms and lack of a proper screening system.3 Five-year survival rates after surgical resection is 22–44% in iCCA, 27–37% in distal CCA and 11–41% in perihilar CCA, respectively.3–5 Unfortunately, even after complete resection, the majority of patients develop recurrent disease. For patients with advanced or metastatic disease, systemic therapy remains the primary treatment option. Gemcitabine and cisplatin combination therapy (GEMCIS) is considered the standard first-line treatment for patients with advanced CCA based on promising efficacy in the Phase III ABC-02 trial with median overall survival (OS) of 11.7 months.6 At this time, there is no established second-line therapy for advanced CCA per current guidelines even though FOLFOX is the most commonly used regimen. (NCCN Guidelines, Version 2.2021-April 16, 2021).
In the past decade, several genomic studies expanded our knowledge of molecular pathogenesis and revealed the distinct genomic aberrations (GAs) in intrahepatic, perihilar, and distal CCA. This genomic profiling led to the discovery of targetable gene mutations and began the era of novel targeted therapy for CCA. Among GAs in advanced or metastatic CCA, fibroblast growth factor receptor 2 (FGFR2) fusions, Isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2), and BRAF have been considered promising targets. FGFR inhibitors including infigratinib, futibatinib and pemigatinib, have demonstrated promising efficacy in refractory CCA and are currently undergoing phase III trials, comparing them to first line GEMCIS therapy. In this article, we review current novel targeted therapy options for advanced or metastatic CCA, focusing on the clinical potential of infigratinib.
Genetic Profiling of CCA and Targetable Mutations
Comprehensive transcriptomic and genomic analyses of CCA have established the distinct molecular heterogenicity between iCCA and eCCA. Lowery et al explored genomic profile of 195 CCA patients with 78% iCCA and 22% eCCA by using a targeted next-generation sequencing (NGS) of 410 cancer-associated genes.7 The most common mutated genes were IDH1 (25%), TP53 (24%), ARID1A (21%), BAP1 (15%), KRAS (13%), PBRM1 (12%), SMAD4 (9%), and ATM (8%). The most commonly altered genes in iCCA were IDH1 (30%), ARID1A (23%), BAP1 (20%), TP53 (20%), and FGFR2 gene fusions (14%). BAP1 mutations and FGFR2 gene fusions were identified exclusively in iCCA. KRAS, SMAD4, and STK11 alterations were more commonly seen in eCCA. In another study by Weinberg et al with 1,502 biliary tract cancers (BTCs) including 825 iCCA, 249 eCCA, the most prevalent mutations in all BTCs were TP53 (42.7%), ARID1A (21.7%), KRAS (15.7%), IDH1 (8.7%), CDKN2A (7.8%), BAP1 (6.7%), SMAD4 (6.5%), and PIK3CA (6.0%). iCCA had higher rates of IDH1 (14.5%), BAP1 (9.5%), and PBRM1 (7.5%) mutations and FGFR2 fusions (% not reported) while eCCA had higher rates of KRAS (37.3%), CDKN2A (22.0%), and BRCA1 (3.9%) mutations.8 A study by Nakamura et al of a 260-case biliary tract cancer (BTC) cohort, including 145 iCCA, 86 eCCA with whole-exome, and transcriptome sequencing, suggested the IDH1/2, FGFR2 fusion, FGFR3, EPHA2, and BAP1 genes preferably contribute tumorigenesis in iCCA, while protein kinase cAMP-activated catalytic subunit alpha (PRKACA), beta (PRKACB), ELF3, and AT-rich interactive domain-containing protein 1B (ARID1B) are preferably involved in eCCA.9
Multiple genomic studies including the above demonstrated FGFR2 fusions exclusively occur in iCCA at 13–20% and rarely reported in eCCA.7–9 Currently, there are no available selective target agents for KRAS (except KRAS G12C), TP53 or ARID1A.10 The main identified targetable aberrations were FGFR2 fusions,11,12 IDH1 mutations,13 BRAF,14 Her2/neu,15 and rarely NTRK fusions and BRCA.16
The Role of the FGFR Genomic Aberrations in CCA
The fibroblast growth factor receptors (FGFRs) are a family of transmembrane tyrosine kinase receptors composed of 4 subtypes (FGFR1-4).17,18 FRGRs have homologous structures consisting of an extracellular ligand-binding domain, a transmembrane helix and an intracellular tyrosine kinase domain.17,19 The extracellular domain of FGFRs can bind 22 different fibroblast growth factors (FGFs) depending on the cellular context.20 This FGF-FGFR binding leads to dimerization of FGFR and activates the intracellular kinase domains, which subsequently activates multiple downstream intracellular signaling pathways. Key downstream signaling pathways activated by FGF-FGFR signaling are the Ras-Raf-MAPK pathway, the PI3-AKT-mTOR pathway, the JAK-STAT and PLCγ-PKC pathway21 which is involved in cellular proliferation, differentiation, survival, migration, and angiogenesis.22,23 Dysregulation of FGFRs has been implicated in oncogenesis, tumor progression and resistance to anticancer therapy, and is detected in various types of malignancies including breast, ovarian, prostate, hepatocellular, and non-small cell lung cancer.24,25
FGFR signaling is known to cause oncogenesis through multiple mechanisms, including ligand independent manners such as: (1) FGFR gene amplification, which leads to receptor accumulation and activates the downstream signaling pathways; (2) activating mutations of FGFR resulting in increased dimerization of the receptors without ligand-binding; (3) oncogenic FGFR gene fusions with other protein partners originated by chromosomal rearrangements, resulting in either increasing dimerization of the receptors or falling to the promoter regions of a different protein, which leads to receptor hyperactivation or ligand-dependent manners such as (4) autocrine fashion.24,26 Among those FGFR GAs, amplifications is the most common in human malignancies, at 66%, followed by FGFR mutations at 26%.27 FGFR fusions only account for 8% of FGFR GAs in human cancer.27
However, FGFR2 fusions are particularly common in CCA, with approximately 15% in iCCA and rare in eCCA.12,28 FGFR2–BICC1, FGFR2–PPHLN1, FGFR2-AHCYL1 fusions have been frequently observed in patients with iCCA, although over 100 different FGFR2 partners have been reported (Table 1).7,12,26,28,29 In an analysis of 377 CCA cases, 95 cases had FGFR GAs, among which FGFR2 fusion (n=63, 66.3%) was most common.29 Javle et al studied 3,634 CCA patients and reported that nearly 20% of CCA patients harbor targetable kinase GAs, half of which were FGFR2 fusions.30 In iCCA, FGFR GAs were reported to be mutually exclusive with other genes including IDH, KRAS, BRAF, ERBB2 and co-occurred with BAP1.7,12,30,31 While FGFR2 fusions have been well identified as oncogenic drivers, the oncogenic potential of FGFR2 amplifications and mutations in CCA has been less identified.32 FGFR GAs, especially FGFR2 fusions, have been considered as novel targets for CCA managements.
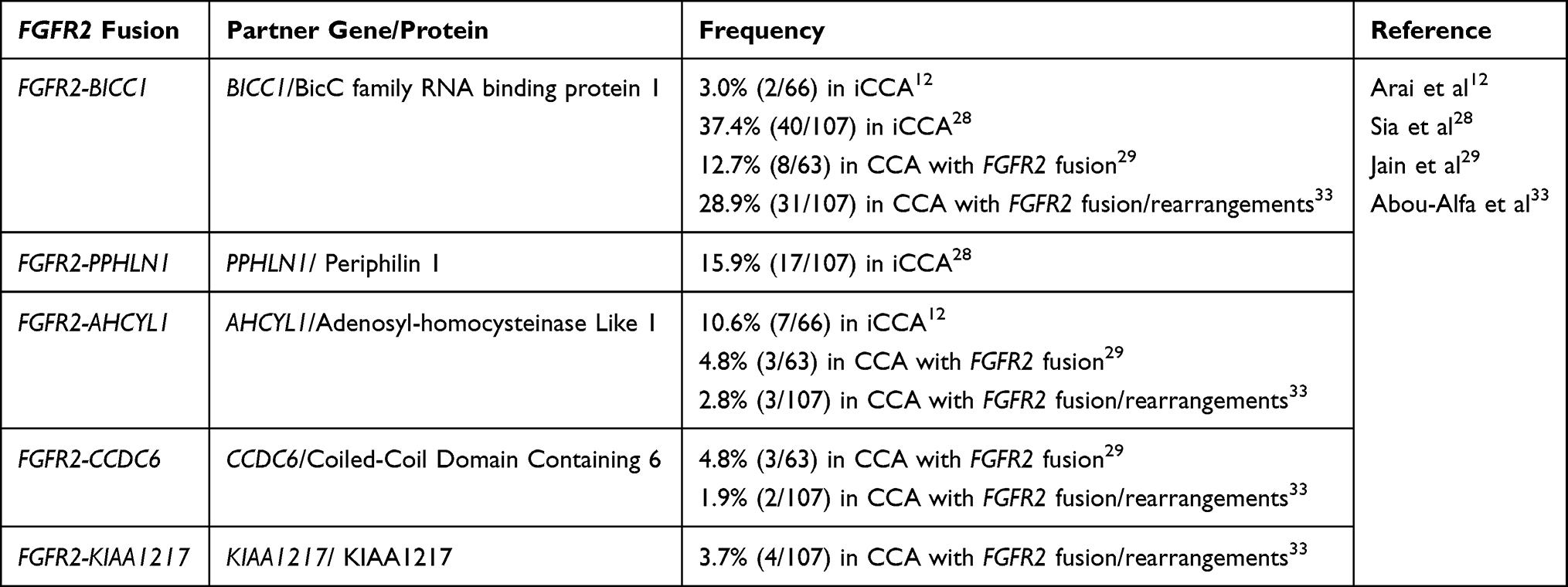 |
Table 1 Commonly Reported FGFR2 Fusion Partner Proteins in CCA |
Mechanisms of FGFR Inhibitors
Based on the spectrum of targeting pathways and molecular structure, targeting agents for FGF-FGFR pathway could be classified into selective tyrosine kinase inhibitors (TKIs), non-selective TKIs, FGFR monoclonal antibodies (mAb), and heat shock protein inhibitors.34,35 Non-selective TKIs, including pazopanib and ponatinib, which have blocking activity against FGFR, have been investigated in CCA.36,37 However, pazopanib in combination with gemcitabine demonstrated a low response rate in BTCs, which precluded further testing.38 In addition, the off-target side effects due to the nature of non-selective TKI are considered a significant weakness compared to selective FGFR TKIs.39,40 Bemarituzumab is a monoclonal antibody therapies against FGFR2b and showed its safety profile in a Phase I trial with multiple solid tumors and are currently being evaluated in a phase III trial as front-line therapy for patients with FGFR2b-overexpressing gastroesophageal cancer.41,42 However, its clinical efficacy in CCA has not been studied. Therefore, selective FGFR TKIs are considered a reasonable option for CCA with FGFR GAs at this time.
Multiple selective FGFR inhibitors including infigratinib, pemigatinib and futibatinib, are in the advanced stage of drug development in CCA with FGFR GAs. Infigratinib is a selective FGFR inhibitor that demonstrated its efficacy in clinical data in CCA populations and was approved by the USFDA in May 2021 for CCA with FGFR2 fusions or rearrangements,43 while in April 2020 pemigatinib became the first FGFR-targeted agent to gain regulatory approval from the USFDA for use in previously treated CCA patients with FGFR2 fusions or rearrangements. Many FGFR TKIs are being developed and have demonstrated their clinical benefits in multiple malignancies.44 However, challenges including resistance and adverse effects (AEs) have led to continuous development of novel FGFR inhibitors. There are more than 40 inhibitors targeting FGF-FGFRs pathways in clinical development.45 The main differences among the selective FGFR inhibitors include binding affinity and half-maximal inhibitory concentration (IC50) to FGFR 1–4 and being reversible or irreversible inhibitors (Table 2).
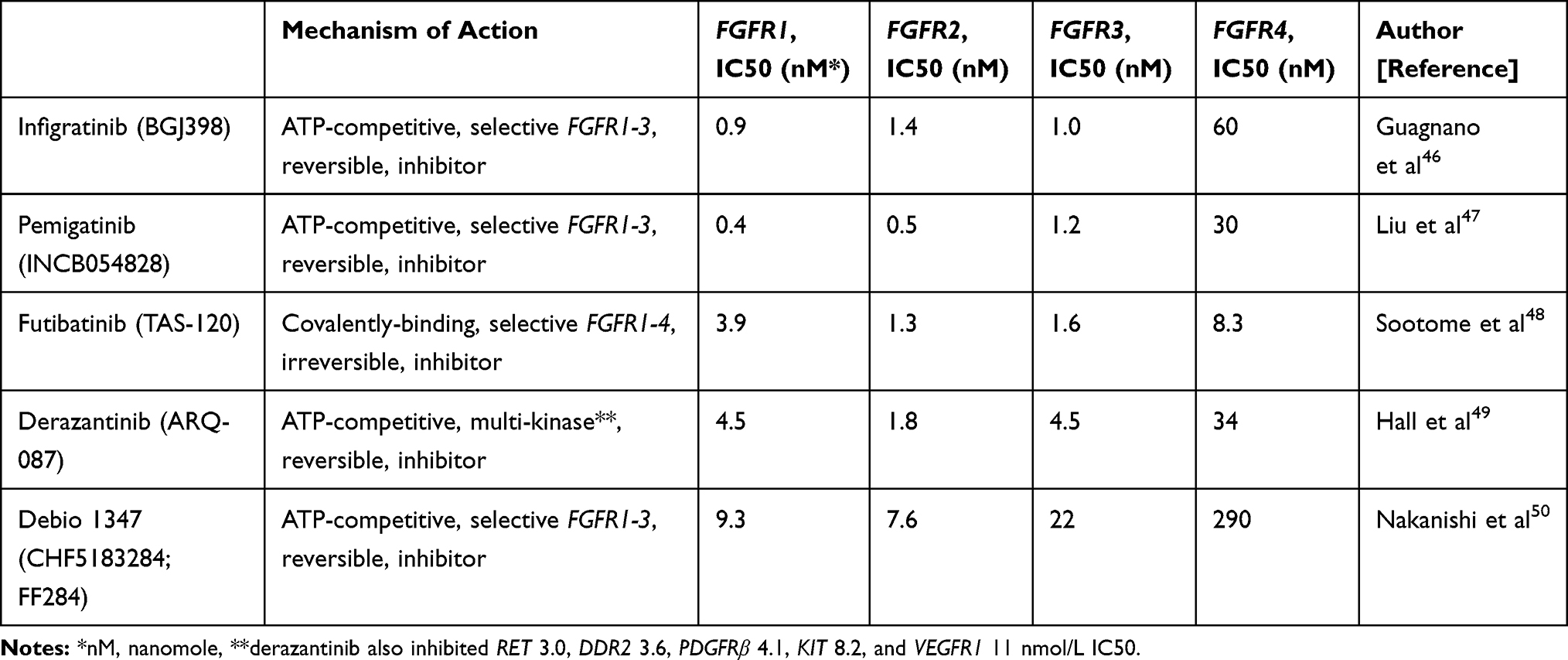 |
Table 2 Mechanism of Action and Target Half- Maximal Inhibitory Concentration (IC50) of FGFR Inhibitors |
Clinical Experience with Infigratinib (BGJ398) in CCA
Infigratinib is a reversible ATP-competitive FGFR 1–3 inhibitor. In preclinical analyses, infigratinib inhibited FGFR1, FGFR2, and FGFR3 with 0.9, 1.4, 1.0 nmol/L IC50 in biochemical (Table 1) and 6.5, 5.8, and 5.8 nmol/L IC50 in cellular auto-phosphorylation assays respectively, while FGFR4 showed 38- to 60-fold lower-potency biochemical and cellular assays compared to FGFR1–3. Other tyrosine kinase including VEGFR, BRAF, and EGFR displayed lower than 70-fold potency compared with FGFR1–3. Based on these results, infigratinib is determined as a selective, pan-FGFR predominant FGFR1-3 inhibitor.46 Infigratinib showed it can inhibit tumor growth and proliferation in preclinical cancer cell line models with FGFR1-3 GAs.46,51
Based on these preclinical data, a phase I basket trial involving 132 patients with multiple solid malignancies harboring FGFR2 GAs (including amplification, mutation, and fusion), most commonly, lung (36.4%), breast (32.6%), bladder/urothelial (9.1%), colon (3.8%), was performed to determine the maximum tolerated dose (MTD) and recommended Phase II dose (RP2D).52 The dose-escalation cohort received 5, 10, 20, 40, 60, 100, 125, and 150 mg infigratinib once daily in a continuous 28-day cycle. Dose-limited toxicities (DLTs) occurred in 4 out of 34 patients in the dose-determining cohort, including grade 3 increases in ALT/AST (n=2), hyperphosphatemia (n=1), and grade 1 corneal toxicity (n=1), which were reversible after drug interruption. MTD was determined as 125mg once daily. Among 43 patients treated at 125 mg once daily dosing, median time to first dose interruption was 22 days with a median duration of interruption of 7 days due to AEs, majority of which were due to hyperphosphatemia. Thus, an intermittent 3-weeks-on/1-week-off schedule of 125 mg once daily was introduced in the dose-expansion arm. Less patients experienced AEs required dose adjustment on the intermittent schedule, though the rate of AEs-related discontinuation remained the same. RP2D was determined to be 125 mg once daily administered in cycles of 21-days on and 7-days off schedule. Infigratinib demonstrated a tolerable safety profile, with most patients (95.5%) experiencing at least one AE. The most common treatment-emergent AEs (TEAEs) across all doses were hyperphosphatemia (74.2%), constipation (40.2%), and decreased appetite (40.2%). Common AEs in patients treated at the MTD (n=57) included hyperphosphatemia (82.5%), constipation (50.9%), decreased appetite (45.6%), and stomatitis (45.6%). The FGFR pathway is established as playing an important role in FGF23-mediated phosphate homeostasis.53,54 Thus, this high frequency of hyperphosphatemia was considered as an indirect indicator of FGFR inhibition.45 Three CCA patients (2 with FGFR2 fusions and 1 with FGFR2 genetic mutation) were included in the basket trial, all of which had a remarkable reduction in the tumor size.52
Following up on phase 1 basket trial, a phase II single-arm clinical trial (NCT02150967) was initiated investigating infigratinib in advanced CCA.43,52 Sixty-one patients with CCA refractory to gemcitabine harboring FGFR GAs (48 FGFR2 fusions, 8 FGFR2 mutations, 3 FGFR2 amplifications, 4 FGFR3 amplifications and 1 FGFR1 amplification) were enrolled. With a median treatment duration of 4.7 months, 9 subjects achieved a partial response (PR), all of which were FGFR2 fusions, and 37 patients qualified for stable disease (SD). The overall response rate (ORR) was 14.8% and the disease control rate (DCR) was 75.4% among patients with all FGFR GAs, and a higher ORR of 18.5% and DCR of 83.3% were demonstrated in FGFR2-fusion patients. In contrast, none of the four patients with FGFR3 amplification responded to infigratinib. However, there was no statistically significant association between antitumor activity and types of FGFR GAs. The median progression-free survival (PFS) was 5.8 months and median OS was not reached at the time of publication. A total of 50 patients discontinued the therapy, most commonly due to progressive disease (60.7%). A similar safety profile was observed in the infigratinib phase 1 basket trial.43,52 Five patients died on the study; four as a result of disease progression and one as a result of an ischemic bowel. Updated data specific to FGFR2 gene fusions or rearrangements of 108 patients including 83 (77%) cases of FGFR2 fusions was presented on Jan. 2021.55 With the median follow-up of 10.6 months, ORR was 23.1% and PFS was 7.3 months. A higher ORR of 34% was seen in the second-line setting, compared to 13.8% in the third or later-line setting. Most common TEAEs (any grade) were hyperphosphatemia (76.9%), eye disorders (67.6%), stomatitis (54.6%), and fatigue (39.8%). Common grade 3/4 TEAEs were stomatitis (14.8%; all G3), hyponatremia (13.0%; all G3), and hypophosphatemia (13.0%; 13 G3, 1 G4). Central serous retinopathy/retinal pigment epithelium detachment occurred in 16.7% of patients, one case of which was grade 3.55 Largely based on infigratinib’s promising efficacy shown in the updated phase 2 trial data, the FDA approved infigratinib in May 2021 for previously treated, unresectable locally advanced or metastatic CCA with a FGFR2 fusion or other rearrangement. This is the second approval of FGFR inhibitors in CCA. Given the promising ORR and DCR in the phase II trial,43 a phase III multicenter, open-label, randomized trial comparing infigratinib as the first line therapy to the standard GEMCIS regimen in advanced/metastatic CCA with FGFR2 fusions or translocation is under investigation (PROOF 301, NCT03773302).56 With naive to systemic anti-cancer therapy in the metastatic/unresectable setting as an inclusion criterion, the study is eligible to prior neoadjuvant or adjuvant therapy if completed prior to 6 months ago. Gallbladder cancer (GBC) or ampulla of Vater carcinoma are not eligible. The target enrollment is approximately 384 patients and randomized in a 2:1 ratio, with groups receiving either the above RP2D dose of infigratinib or intravenous GEMCIS on days 1 and 8 of a 21-day treatment cycle until disease progression, or other discontinuation criteria is met.56 Participants who experience disease progression while receiving GEMCIS will be allowed to cross over to the infigratinib arm. The primary end point is PFS confirmed by blinded independent reviewers with secondary end points that include OS, PFS as determined by the investigator, ORR, DCR, best overall response (BOR), duration of response and safety.56
Other FGFR Inhibitors in CCA
Pemigatinib (INCB054828), another ATP-competitive and selective FGFR 1–3 inhibitor, has been investigated the most, along with infigratinib.47 In a phase II study (FIGHT-202) with 146 advanced/metastatic CCA patients refractory to prior systemic therapy, including 107 cases of FGFR2 fusions/rearrangements, pemigatinib showed a promising efficacy of ORR 35.8%, DCR of 82.2%, and 6.9 months of PFS in patients harboring FGFR2 fusions or rearrangements, with a tolerable safety profile.33 On the contrary, no remarkable clinical benefits were seen in the other FGFR GAs groups. Given these encouraging results, the FDA approved pemigatinib in refractory advanced or metastatic CCA with FGFR2 fusions or other rearrangement, which was the first FDA approval of FGFR inhibitors for CCA. Currently, a Phase III, open-label, randomized trial is comparing pemigatinib versus GEMCIS as the first-line treatment of patients with unresectable/metastatic CCA and naive to prior systemic with FGFR2 rearrangements (NCT03656536).57 The target enrollments are approximately 432 patients randomized in a 1:1 ratio for each treatment arm with PFS as the primary endpoint.57
Futibatinib is a highly selective and irreversible FGFR1-4 inhibitor.48 In a phase I dose-escalation study (FOENIX-101; NCT02052778) of 86 patients with advanced solid tumors, futibatinib showed a tolerable safety profile.58 In the dose-expansion cohort with 45 CCA patients harboring FGFR GAs, including 28 cases of FGFR2 fusion, futibatinib showed a promising ORR of 25% and DCR of 79% among FGFR2 fusion.59 It is notable that the study included 13 cases priorly treated to reversible FGFR inhibitors, and among the 13 patients, four (three with FGFR2 gene fusions and one with FGFR2 amplification) had PR on futibatinib.59 This finding suggested futibatinib could overcome resistance to ATP-competitive FGFR inhibitors in selective cases.60 An open-label, single-arm, phase II (FOENIX-CCA2) trial with 103 advanced/metastatic iCCA cases refractory to prior systemic therapy and naive to FGFR inhibitors with harboring FGFR2 fusions or other rearrangements is ongoing.61,62 Interim results with 67 patients demonstrated the ORR of 37.3%, DCR of 82.1%, and the median PFS of 7.2 months with the median DOR of 8.3 months.62 A phase III trial (FOENIX-CCA3, NCT04093362) with metastatic/unresectable iCCA naive to systemic therapy with harboring FGFR2 rearrangements is recruiting patients to compare futibatinib versus GEMCIS.63 The target enrollments are approximately 216 patients randomized in a 1:1 ratio for each treatment arm with PFS as the primary endpoint.63
Derazantinib (ARQ-087) is an ATP-competitive and selective FGFR1–3 inhibitor with inhibiting of several other kinases, including RET, DDR2, PDGFRβ, KIT, and VEGFR1.49 In a phase I study (NCT01752920) with 80 multiple advanced solid tumors, derazantinib showed a similar safety profile to infigratinib.64 A following phase I/II, open-label study (NCT01752920) enrolled 29 patients with unresectable iCCA refractory, intolerant or not eligible for first-line chemotherapy with harboring FGFR2 fusion. Derazantinib demonstrated an ORR of 20.7% and a DCR of 82.8% with 5.7 months of PFS.65 Based on this promising efficacy, a pivotal, open-label, single-arm, phase II trial (FIDES-01, NCT03230318) is ongoing with pretreated iCCA patients with FGFR2 gene fusions, mutations or amplifications (M/A).32 Enrolment into the FGFR2 fusions cohort of 100 patients has been completed and the target enrollments for FGFR2 M/A is 43.32,39 Debio 1347 (CHF5183284; FF284) is another ATP-competitive and selective FGFR 1–3 inhibitor.50 In a phase I trial with multiple advanced solid tumors harboring FGFR1–3 Gas, Debio 1347 showed remarkable response in 4 cases CCA harboring FGFR2 fusions (2 cases of SD and 2 cases of PR).66,67 Currently, a phase II Basket trial (FUZE) with 63 advanced/metastatic multiple solid tumors with FGFR1-3 gene fusion/rearrangement is under investigation with BTC as cohort arm 1.68 Tables 3 and 4 summarize clinical trials that evaluated or are evaluating the clinical benefits of the FGFR inhibitors mentioned above in patients with advanced and/or metastatic CCA.
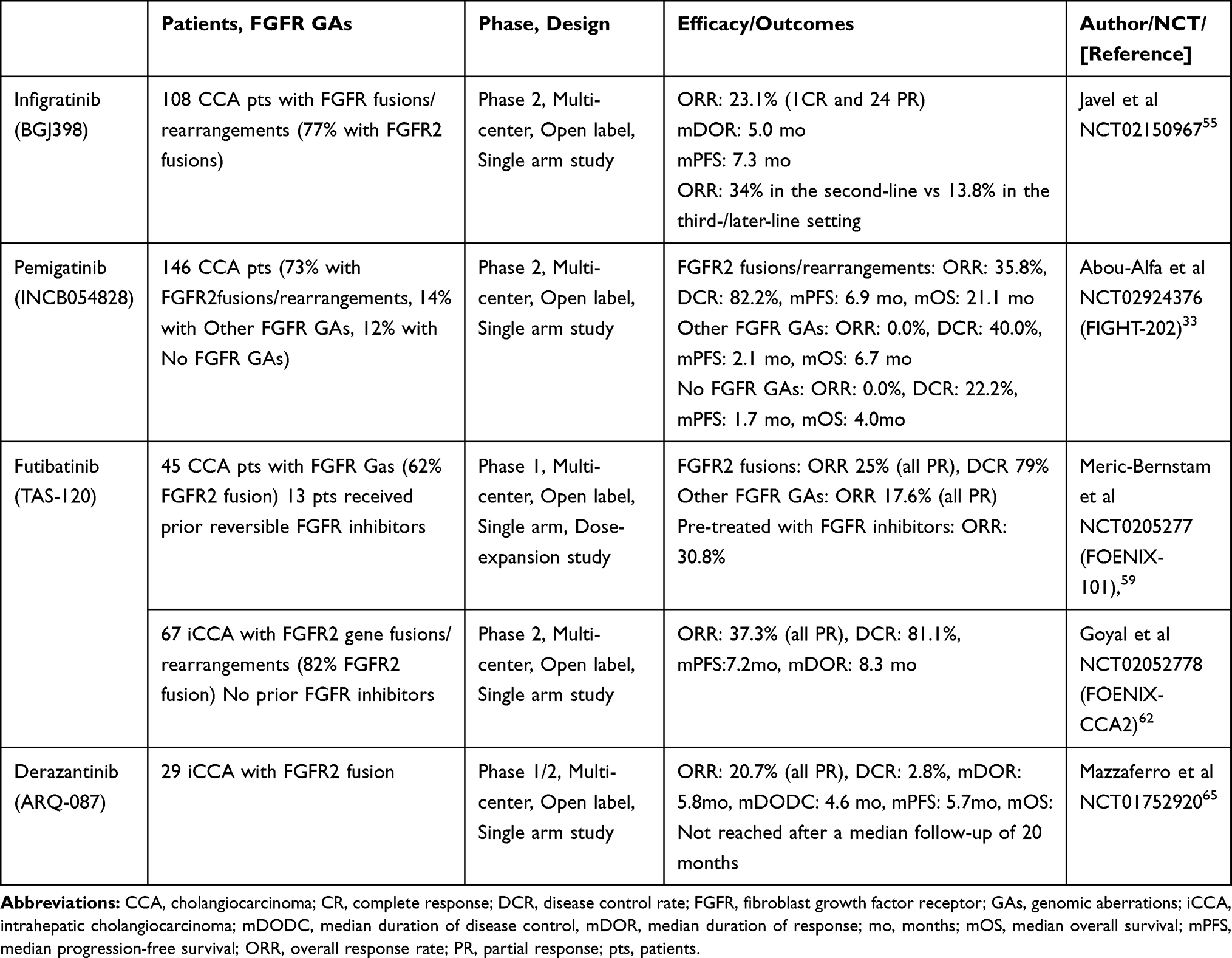 |
Table 3 Key Phase I and II Clinical Trials of FGFR Inhibitors in CCA |
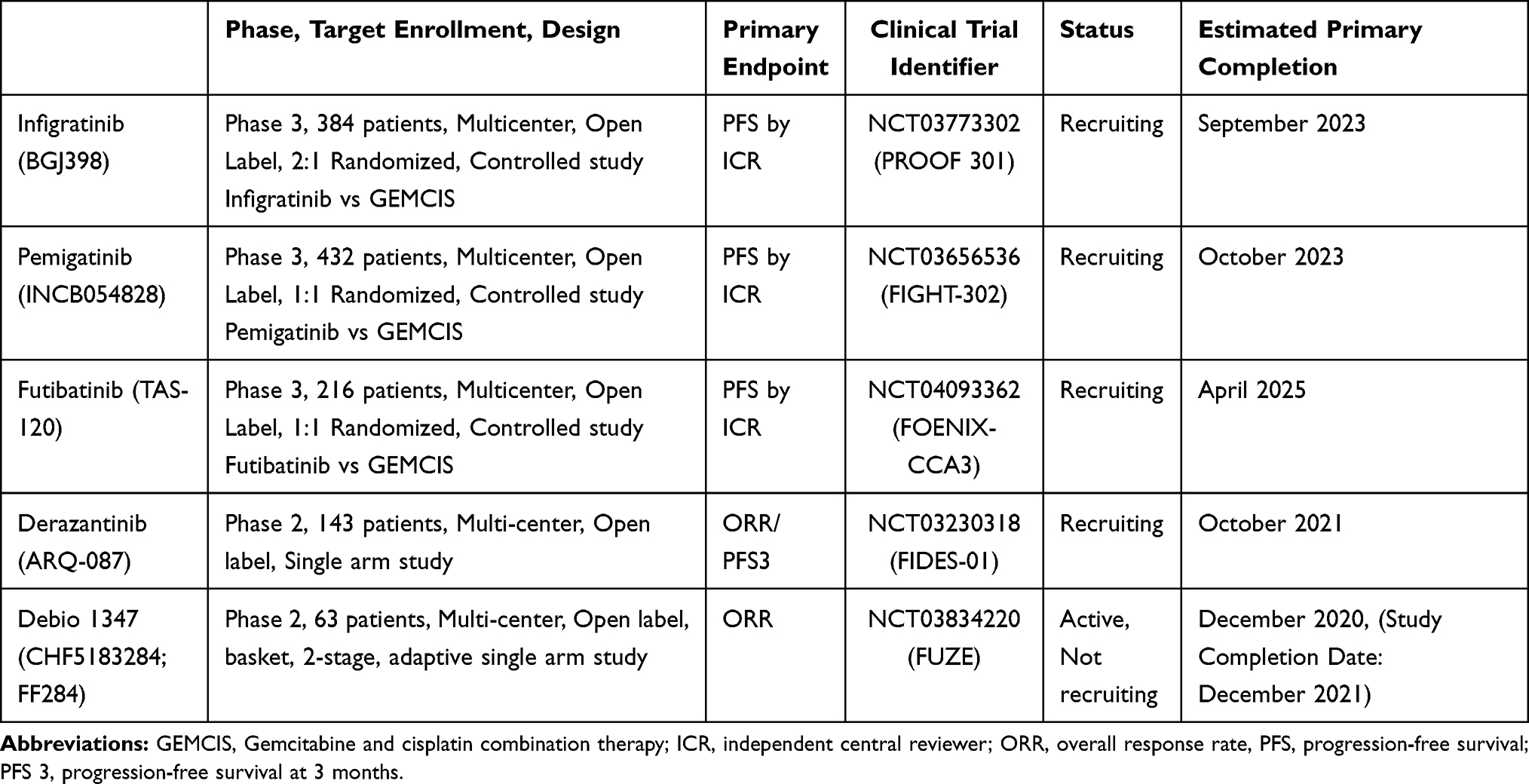 |
Table 4 Active Ongoing Phase II or III Clinical Trials of FGFR Inhibitors in CCA |
Toxicity of Infigratinib
Like other FGFR inhibitors, the most common AE of infigratinib is hyperphosphatemia, which was reported in 72–82.5% of patients but was rarely grade 4 or 5.43,52 Hyperphosphatemia is identified as an on-target side effect of FGFR inhibitors caused by FGF23-FGFR signaling blockade in the kidneys and a following increase in renal phosphate reabsorption.69 Hyperphosphatemia can be managed by initiation of phosphate lowering therapy such as magnesium hydroxide, calcium carbonate, calcium acetate, and, sevelamer along with low phosphorus diet.70 Dose reduction can be considered when serum phosphate level is greater than 7 mg/dL and cannot be lowered by above agents.70 Other common AEs included fatigue (36%), stomatitis (29.5%) and alopecia (26%) which were also commonly demonstrated in other FGFR inhibitors.43 Ocular toxicities are another well-documented FGFR inhibitor side effect. Dry eye (21.3%) was commonly reported which can be managed or prevented most cases with ocular lubricants.70 Blurred vision (14.8%) was also reported. Although they were not significantly reported in the Phase 1 and 2, severe ocular toxicities such as central serous retinopathy or retinal pigment epithelium detachment reported in 16.7% of patients in updated interim data of the phase2.43,52,55 There is no consensus for the management of ocular toxicity of FGFR inhibitors, regular ophthalmologic evaluation including optical coherence tomography and fundus exam is recommended. In grade 1–3 ocular toxicities which resolve in a month, low dose FGFR inhibitor could be restarted with a thorough follow up with ophthalmology. With grade 4 ocular toxicity, FGFR inhibitors including infigratinib should be permanently stopped.70
One of the key differences among FGFR inhibitors is their selectivity of each FGFR subtype. Compared to other FGFR inhibitors including futibatinib and pemigatinib, infigratinib binds relatively less strongly to FGFR4, as shown in Table 2. Theoretically this weak FGFR4 inhibition effect can cause less gastrointestinal toxicity such as severe diarrhea, low food consumption, decreased body weight, or liver toxicity.71,72 Based on our literature review, in RP2D dosage, infigratinib showed fewer incidences of AEs compared to other agents (including derazantinib, pemigatinib or futibatinib) in diarrhea (15% vs 21–37%), nausea (15% vs 25%-45%) or decreased appetite (12% vs 17–24%) (Table 5). Table 5 summarized toxicity profiles of each FGFR inhibitor demonstrated in clinical trials.
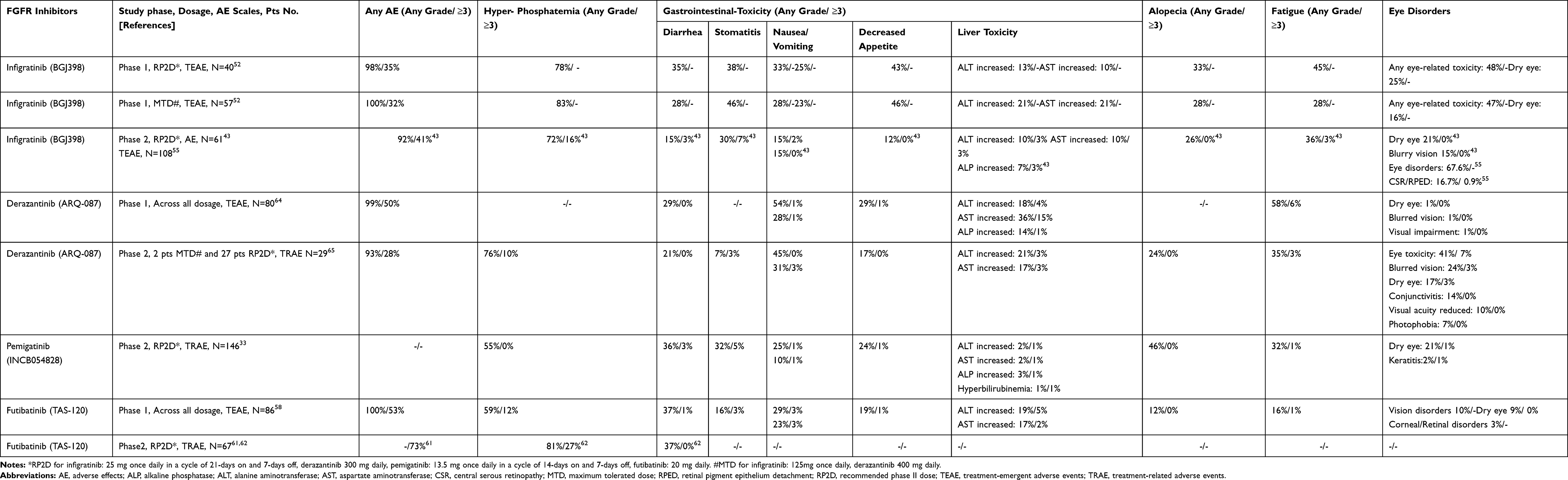 |
Table 5 Toxicity Profile of FGFR Inhibitors in Cholangiocarcinoma Clinical Trials |
Resistance of CCA to Infigratinib
Primary and acquired resistances to FGFR inhibitors including infigratinib have been reported in CCA. It has been suggested that co-occurring mutations with FGFR fusion could be a mechanism of primary resistance to FGFR inhibitors.39,73 Notably, Silverman et al reported shorter PFS with no significant difference in the ORR in CCA patients treated with pemigatinib when FGFR GAs accompanied with tumor-suppressor gene loss mutations including BAP1, CDKN2A/B, TP53, PBRM1, ARID1A, or PTEN.73 However, given the no significant response, the decreased PFS could be due to the adverse survival effects of those tumor-suppressor genes mutations but not because of resistance to FGFR inhibitors.
One of the remarkable acquired resistances to FGFR inhibitors is FGFR mutations that change the ATP binding pocket of FGFR proteins. In clinical setting, this resistance was first reported from three cases during the follow up of the infigratinib phase II trial (NCT02160041).74 All three cases were iCCA with FGFR fusion initially responded to infigratinib followed by short interval disease progression. Following cell-free DNA (cfDNA) showed FGFR2 V565F gatekeeper mutation in all three cases with two cases had 4 other point mutations in the FGFR2 kinase domain. The p.V564F gatekeeper mutation confers resistance by inducing a structural change in the ATP-binding pocket where infigratinib competitively bind. The five other point mutations spanning five amino acid residues were also predicted to induce less favorable infigratinib-binding condition by destabilize the inactive conformation of the kinase where infigratinib binds. It is also remarkable, those FGFR-mutations induced resistance was rapidly acquired in 4–8 months after the first exposure to infigratinib.74 Similar mechanism of resistance was observed in pemigatinib from 8 cases of FIGHT-202 trial with initial tumor shrinkage followed by progressive disease with six unique FGFR2 mutations spanning five amino acid residues.73 Futibatinib which is a third-generation, irreversible FGFR inhibitor, covalently binds to the ATP pocket of FGFR75 and expected to overcome this acquired FGFR2 kinase domain mutations induced resistance to ATP-competitive FGFR inhibitors. Goyal et al reported 4 iCCA patients with FGFR2-fusion who developed resistance to infigratinib (n=3) or Debio 1347 (n=1) followed by futibatinib treatment.60 All four patients demonstrated clinical benefit (2 SD and 2 PR). In vitro analyses, futibatinib showed only minimal or modest changes in activity against each of the nine acquired FGFR2 mutations in the study (2- to 7-fold IC50 increase) with the exception of V565F (103-fold).60 This finding also indicates that next-generation FGFR inhibitors need to be focused on inhibiting gatekeeper mutations. Notably, the authors also reported that clonal outgrowth of multiple mutations occurred more readily at lower concentrations of infigratinib in the in vitro analysis, and suggested that reduced drug exposure may play an important role in acquisition of resistance.60 While further studies are warranted to confirm this finding, it also highlights the importance of minimizing unnecessary dose reduction or interruption while aggressively managing hyperphosphatemia, which is the most common reason for dose reduction or interruption.
Another remarkable acquired resistance to infigratinib is the Akt activation mediated mechanism. The PI3-AKT-mTOR is one of the FGFR downstream pathways which plays a crucial role in cell growth and survival. An improper activation of this pathway is known to cause a competitive growth advantage, metastatic competence, angiogenesis, and therapy resistance.76 In a preclinical analysis, Datta et al analyzed small cell lung cancer harboring FGFR1 amplification and urothelial carcinoma harboring FGFR3 fusion/amplification cell lines which acquired resistance after chronic infigratinib exposure.77 By performing proteomic analysis, they found the Akt pathway was activated in both the resistant cell lines, despite ongoing FGFR blockade in the presence of infigratinib. Treatment of the resistant cells with Akt siRNA or GSK2141795, an oral, competitive, pan-isoform Akt inhibitor which showed safety profile in an recent phase 1,78 was able to restore sensitivity of resistant cell lines.77 Recently, Krook et al also showed upregulation of the PI3-AKT-mTOR signaling pathway in cells harboring the FGFR2 p.E565A mutation which was found in infigratinib resistant iCCA tumor.79 This PI3-AKT-mTOR pathway upregulated resistance could potentially be overcome by combination of infigratinib with one of PI3K, AKT or mTOR inhibitors. Further clinical research is warranted.
Future Direction of Infigratinib and Other FGFR Inhibitors in CCA
Infigratinib (BGJ398) is an oral, ATP-competitive pan-FGFR inhibitor. Along with pemigatinib, infigratinib is one of the two most clinically advanced and investigated FGFR inhibitors in cholangiocarcinoma managements. FGFR2 fusions are common in iCCA with occurring in approximately 15% of cases and have been well identified as oncogenic drivers in CCA, while roles of other FGFR genomic aberrations have been relatively less identified. Infigratinib demonstrated promising efficacy in a phase II trial of patients with advanced cholangiocarcinoma harboring FGFR genomic aberrations and refractory to the first line therapy. Based on these promising results, the FDA recently approved infigratinib for CCA, and an ongoing phase III trial (PROOF301, NCT03773302) is designed to compare infigratinib versus the first line therapy of GEMCIS in CCA harboring FGFR2 fusions or translocation. In iCCA, FGFR GAs were reported to be mutually exclusive with other targetable genes including IDH and BRAF. Given that the FGFR GAs including fusions are rarely accompanied with other targetable mutations, infigratinib and other FGFR inhibitors are expected to continue to be the novel targeted agents in CCA harboring FGFR GAs.
Competition of FGFR Inhibitors in CCA Harboring FGFR GAs
FGFR GAs are one of few targetable mutations in CCA. With active attention and investigation of these GAs, numerous selective FGFR inhibitions have been developed, and currently some of them including infigratinib, pemigatinib and futibatinib are at the same stage of investigation in Phase 3 clinical trials as a potential first-line therapy. The other two FGFR inhibitors, derazantinib and debio 1347, are in phase 2 trials.
PROOF 301 and FIGHT-302 trials are expected to conclude in September and October 2023 respectively and results will tell us whether each agent is superior to GEMCIS, which is the current first-line therapy. Additionally, the long-term follow-up of PROOF 301 and FIGHT 302 cohorts will give us a better understanding regarding the achieved resistance to those ATP-competitive FGFR inhibitors. The FOENIX-CCA3 is estimated to conclude in April 2025. The results will tell us the direct comparison between futibatinib to GEMCIS and allow indirect comparison with infigratinib and pemigatinib. The result of FOENIX-CCA3 and the long-term follow-up of the cohort will also tell us whether futibatinib as a covalently binding FGFR inhibitor has the potential to overcome the achieved resistance to ATP-competitive agents.
Ultimately, the direct drug-to-drug comparison will be necessary to thoroughly elucidate the benefits and superiority of each FGFR inhibitor. However those studies most likely will not be conducted.
Based on further investigation of resistances by long-term follow up of clinical trial cohorts of each FGFR inhibitors and learning to sequence these inhibitors will be the key for future trials. As described above, based on current data, development of new point mutations is one of the plausible causes of developing resistance to FGFR inhibitors.80 Certain FGFR inhibitor such as futibatinib have distinctive capabilities to target the newly developed resistant FGFR point mutations.81 Therefore better understanding of mechanisms of primary and secondary resistance will be crucial to improve patient stratification and to sequence these FGFR inhibitors.
Potential Role of Liquid Biopsy in CCA Treated with FGFR Inhibitors
The circulating tumor DNA (ctDNA) analysis has strong potential in the future direction of FGFR inhibitors in CCA. In advanced/metastatic CCA, the sampling tissue for biopsy is limited due to its invasiveness, anatomical difficulty to approach, and difficulty in obtaining sufficient sample for cancer genome profiling.82,83 The application of ctDNA analysis can assist disease response, monitoring, and early detection of recurrence.83,84 In addition, as demonstrated by Goyal et al with their recent suggestion of p.V564F gatekeeper mutation as possible acquired resistance of reversible FGFR inhibitors by monitoring ctDNA during the follow up of the infigratinib phase II trial,74 ctDNA analysis can assist the identification of resistance mechanisms to FGFR inhibitors and guide the further treatment development and selection.83,85 Also, in the real world, ctDNA analysis can potentially help clinicians for quick actionable application of the next agents by detecting resistance in the meaningful time. As suggested close ctDNA monitoring can be integrated with clinical trials, or can become the standard clinical management for CCA in the near future.81 However, sensitive of fusion detection must improve with ctDNA before we can adapt liquid biopsy as standard of care.
Combination with Immunotherapy
Immunotherapy has been investigated in the management of CCA. Pembrolizumab showed promising efficacy of 41% (9/22) ORR in advanced cholangiocarcinoma with mismatch repair deficiency (dMMR).86 Also, in KEYNOTE-158 trial with multiple solid tumors, pembrolizumab showed a higher response rate in high-tumor mutational burden (TMB) compared to non-TMB-high group (29% vs 6%).87 Although CCA was not included in this study, the FDA approved pembrolizumab for patients with any solid tumor, including CCA, that has high TMB. There is much pre-clinical evidence demonstrating potential benefits of combining FGFR inhibitors with immune checkpoint inhibitors (ICIs). As PD-1 and CTLA-4 signaling are involved in functional suppression of cytotoxic T cells through regulatory T cells, ICIs which block such signaling can lead to sustainable remission.88 On the contrary, FGFR inhibitors are shown to target other immune cells, such as myeloid-derived suppressor cells or tumor-associating macrophages of M2 type, and are expected to indirectly repress PD-L1 expression on tumor cells and stromal/immune cells through normalization of the tumor microenvironment.88 In vivo data with lung cancer mouse model showed the combination of FGFR inhibitors and anti–PD-1 leads to expansion of T-cell clones, and this remodeling of the immune microenvironment can enhance antitumor immunity and survival.89 Therefore, combination therapy using FGFR inhibitors and ICIs may be a promising choice for cancer managements.
The potential clinical benefit of FGFR inhibitor and ICI combination could be investigated, particularly for advanced CCA patients harboring FGFR GAs with high-TMB or dMMR/high levels of MSI (MSI-H), or programmed cell death 1 ligand 1 (PD-L1) overexpression. This FGFR inhibitors and ICI combination is actively evaluated in urothelial cancer. Vofatamab, which is a FGFR3 inhibitor with pembrolizumab, showed a safety profile with an encouraging efficacy in urothelial cancer in a phase 1b/2 study.90 Also, FGFR inhibitors and ICI combination is actively investigated in multiple phase 1 or 2 trials in urothelial cancer having FGFR2 genetic alterations (derazantinib plus atezolizumab, phase I/II[FIDES02], NCT04045613; futibatinib plus pembrolizumab, phase II, NCT04601857; and pemigatinib plus pembrolizumab, phase 2 [FIGHT-205], NCT04003610; rogaratinib plus atezolizumab, phase 1b/2 [FORT-2], NCT 03473756). Such trials will provide indirect insights on this combination versus FGFR inhibitor monotherapy.
Conclusion
Tumor resistance to reversible FGFR inhibitors including infigratinib could be a barrier for the clinical benefits of infigratinib in CCA. In select cases, futibatinib, which covalently binds to FGFR, has been suggested as an agent to overcome resistance induced by ATP-binding pocket structural changes with FGFR acquired mutation. Hopefully, second generation FGFR inhibitors will be developed that have more durable responses and are able to overcome resistance. Active clinical trials including PROOF301 in iCCA are expected to elucidate the clinical benefits of infigratinib, including clinical efficacy and safety compared to current first line GEMCIS in CCA with harboring FGFR2, as well as the impact of resistance with the long-term follow-up of the cohort. The result of this trial is expected to affect potential usage of infigratinib in CCA in potentially in first line setting. However, with many FGFR 2 inhibitors being available we will need to better understand how to incorporate all of these agents in treatment of these patients. Furthermore, although it has been less investigated, infigratinib and immune checkpoint inhibitor combination therapy is also a potential future direction of investigation of infigratinib in the management of advanced cholangiocarcinoma.
Abbreviations
AEs, adverse effects; BOR, best overall response; BTC, biliary tract cancer; CCA, Cholangiocarcinoma; CR, complete response; DCR, disease control rate; DLTs, dose-limited toxicities; dMMR, mismatch repair deficiency; DOR, duration of response; eCCA, extrahepatic cholangiocarcinoma; FGFR, fibroblast growth factor receptor; FGFs, fibroblast growth factors; GAs, genomic aberrations; GBC, gallbladder cancer; GEMCIS, gemcitabine and cisplatin combination therapy; iCCA, intrahepatic cholangiocarcinoma; ICI, immune checkpoint inhibitor; ICR, independent central reviewer; M/A, mutations or amplifications; MTD, maximum tolerated dose; NGS, next-generation sequencing; ORR, overall response rate; OS, overall survival; PFS, progression-free survival; PR, partial response; RP2D, recommended phase II dose; SD, stable disease; TEAEs, treatment-emergent adverse effects; TKIs, tyrosine kinase inhibitors; TMB, tumor mutational burden.
Acknowledgments
This research received no administrative and technical support, or donations.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research received no external funding.
Disclosure
Dr. Richard Kim received honorarium from, Incyte, QED, Lilly, BMS and Bayer, outside of the submitted work. The other authors report no conflicts of interest for this work.
References
1. Bergquist A, von Seth E. Epidemiology of cholangiocarcinoma. Best Pract Res Clin Gastroenterol. 2015;29(2):221–232. doi:10.1016/j.bpg.2015.02.003
2. de Groen PC, Gores GJ, LaRusso NF, Gunderson LL, Nagorney DM. Biliary tract cancers. N Engl J Med. 1999;341(18):1368–1378. doi:10.1056/NEJM199910283411807
3. Khan SA, Davidson BR, Goldin RD, et al. Guidelines for the diagnosis and treatment of cholangiocarcinoma: an update. Gut. 2012;61(12):1657–1669. doi:10.1136/gutjnl-2011-301748
4. Sano T, Shimada K, Sakamoto Y, Yamamoto J, Yamasaki S, Kosuge T. One hundred two consecutive hepatobiliary resections for perihilar cholangiocarcinoma with zero mortality. Ann Surg. 2006;244(2):240–247. doi:10.1097/01.sla.0000217605.66519.38
5. Ustundag Y, Bayraktar Y. Cholangiocarcinoma: a compact review of the literature. World J Gastroenterol WJG. 2008;14(42):6458–6466. doi:10.3748/wjg.14.6458
6. Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362(14):1273–1281. doi:10.1056/NEJMoa0908721
7. Lowery MA, Ptashkin R, Jordan E, et al. Comprehensive molecular profiling of intrahepatic and extrahepatic cholangiocarcinomas: potential targets for intervention. Clin Cancer Res off J Am Assoc Cancer Res. 2018;24(17):4154–4161. doi:10.1158/1078-0432.CCR-18-0078
8. Weinberg BA, Xiu J, Lindberg MR, et al. Molecular profiling of biliary cancers reveals distinct molecular alterations and potential therapeutic targets. J Gastrointest Oncol. 2019;10(4):652–662. doi:10.21037/jgo.2018.08.18
9. Nakamura H, Arai Y, Totoki Y, et al. Genomic spectra of biliary tract cancer. Nat Genet. 2015;47(9):1003–1010. doi:10.1038/ng.3375
10. Nagasaka M, Li Y, Sukari A, Ou S-HI, Al-Hallak Mn, Azmi AS. KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat Rev. 2020;84:101974. doi:10.1016/j.ctrv.2020.101974
11. Wu Y-M, Su F, Kalyana-Sundaram S, et al. Identification of Targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3(6):636–647. doi:10.1158/2159-8290.CD-13-0050
12. Arai Y, Totoki Y, Hosoda F, et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology. 2014;59(4):1427–1434. doi:10.1002/hep.26890
13. Borger DR, Tanabe KK, Fan KC, et al. Frequent Mutation of Isocitrate Dehydrogenase (IDH)1 and IDH2 in Cholangiocarcinoma Identified Through Broad-Based Tumor Genotyping. The Oncologist. 2012;17(1):72–79. doi:10.1634/theoncologist.2011-0386
14. Long GV, Stroyakovskiy D, Gogas H, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386(9992):444–451. doi:10.1016/S0140-6736(15)60898-4
15. Javle M, Churi C, Kang HC, et al. HER2/neu-directed therapy for biliary tract cancer. J Hematol OncolJ Hematol Oncol. 2015;8(1):58. doi:10.1186/s13045-015-0155-z
16. Drilon A, Laetsch TW, Kummar S, et al. Efficacy of Larotrectinib in TRK Fusion–positive cancers in adults and children. N Engl J Med. 2018;378(8):731–739. doi:10.1056/NEJMoa1714448
17. Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. 2004;20(11):563–569. doi:10.1016/j.tig.2004.08.007
18. Touat M, Ileana E, Postel-Vinay S, André F, Soria J-C. Targeting FGFR Signaling in Cancer. Clin Cancer Res. 2015;21(12):2684–2694. doi:10.1158/1078-0432.CCR-14-2329
19. Mohammadi M, Olsen SK, Ibrahimi OA. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005;16(2):107–137. doi:10.1016/j.cytogfr.2005.01.008
20. Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2(3):
21. Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16(2):139–149. doi:10.1016/j.cytogfr.2005.01.001
22. Sarabipour S, Hristova K. Mechanism of FGF receptor dimerization and activation. Nat Commun. 2016;7(1):10262. doi:10.1038/ncomms10262
23. Katoh M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat Rev Clin Oncol. 2019;16(2):105–122. doi:10.1038/s41571-018-0115-y
24. Babina IS, Turner NC. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer. 2017;17(5):318–332. doi:10.1038/nrc.2017.8
25. Dienstmann R, Rodon J, Prat A, et al. Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol. 2014;25(3):552–563. doi:10.1093/annonc/mdt419
26. De Luca A, Esposito Abate R, Rachiglio AM, et al. FGFR fusions in cancer: from diagnostic approaches to therapeutic intervention. Int J Mol Sci. 2020;21:18. doi:10.3390/ijms21186856
27. Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res off J Am Assoc Cancer Res. 2016;22(1):259–267. doi:10.1158/1078-0432.CCR-14-3212
28. Sia D, Losic B, Moeini A, et al. Massive parallel sequencing uncovers actionable FGFR2 – PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun. 2015;6(1):6087. doi:10.1038/ncomms7087
29. Jain A, Borad MJ, Kelley RK, et al. Cholangiocarcinoma With FGFR genetic aberrations: a unique clinical phenotype. JCO Precis Oncol. 2018;1(2):1–12. doi:10.1200/PO.17.00080
30. Javle MM, Murugesan K, Shroff RT, et al. Profiling of 3,634 cholangiocarcinomas (CCA) to identify genomic alterations (GA), tumor mutational burden (TMB), and genomic loss of heterozygosity (gLOH). J Clin Oncol. 2019;37(15_suppl):4087. doi:10.1200/JCO.2019.37.15_suppl.4087
31. Kongpetch S, Jusakul A, Lim JQ, et al. Lack of Targetable FGFR2 fusions in endemic fluke-associated cholangiocarcinoma. JCO Glob Oncol. 2020;2(6):628–638. doi:10.1200/GO.20.00030
32. Javle MM, Shaib WL, Braun S, et al. FIDES-01, a phase II study of derazantinib in patients with unresectable intrahepatic cholangiocarcinoma (iCCA) and FGFR2 fusions and mutations or amplifications (M/A). J Clin Oncol. 2020;38(4_suppl):TPS597–TPS597. doi:10.1200/JCO.2020.38.4_suppl.TPS597
33. Abou-Alfa GK, Sahai V, Hollebecque A, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 2020;21(5):671–684. doi:10.1016/S1470-2045(20)30109-1
34. Mahipal A, Tella SH, Kommalapati A, Anaya D, Kim R. FGFR2 genomic aberrations: achilles heel in the management of advanced cholangiocarcinoma. Cancer Treat Rev. 2019;78:1–7. doi:10.1016/j.ctrv.2019.06.003
35. Ghedini GC, Ronca R, Presta M, Giacomini A. Future applications of FGF/FGFR inhibitors in cancer. Expert Rev Anticancer Ther. 2018;18(9):861–872. doi:10.1080/14737140.2018.1491795
36. Harris PA, Boloor A, Cheung M, et al. Discovery of 5-[[4-[(2,3-Dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (Pazopanib), a Novel and Potent Vascular Endothelial Growth Factor Receptor Inhibitor. J Med Chem. 2008;51(15):4632–4640. doi:10.1021/jm800566m
37. O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a Pan-BCR-ABL Inhibitor for Chronic Myeloid Leukemia, Potently Inhibits the T315I Mutant and Overcomes Mutation-Based Resistance. Cancer Cell. 2009;16(5):401–412. doi:10.1016/j.ccr.2009.09.028
38. Sgouros J, Aravantinos G, Koliou G-A, et al. First Line Gemcitabine/Pazopanib in Locally Advanced and/or Metastatic Biliary Tract Carcinoma. A Hellenic Cooperative Oncology Group Phase II Study. Anticancer Res. 2020;40(2):929–938. doi:10.21873/anticanres.14026
39. Goyal L, Kongpetch S, Crolley VE, Bridgewater J. Targeting FGFR inhibition in cholangiocarcinoma. Cancer Treat Rev. 2021;95. doi:10.1016/j.ctrv.2021.102170
40. Yang W, Sun Y. Promising Molecular Targets for the Targeted Therapy of Biliary Tract Cancers: an Overview. OncoTargets Ther. 2021;14:1341–1366. doi:10.2147/OTT.S297643
41. Catenacci DVT, Rasco D, Lee J, et al. Phase I Escalation and Expansion Study of Bemarituzumab (FPA144) in Patients With Advanced Solid Tumors and FGFR2b-Selected Gastroesophageal Adenocarcinoma. J Clin Oncol off J Am Soc Clin Oncol. 2020;38(21):2418–2426. doi:10.1200/JCO.19.01834
42. Catenacci DV, Tesfaye A, Tejani M, et al. Bemarituzumab with modified FOLFOX6 for advanced FGFR2-positive gastroesophageal cancer: FIGHT Phase III study design. Future Oncol. 2019;15(18):2073–2082. doi:10.2217/fon-2019-0141
43. Javle M, Lowery M, Shroff RT, et al. Phase II Study of BGJ398 in Patients With FGFR-Altered Advanced Cholangiocarcinoma. J Clin Oncol. 2017;36(3):276–282. doi:10.1200/JCO.2017.75.5009
44. Porta R, Borea R, Coelho A, et al. FGFR a promising druggable target in cancer: molecular biology and new drugs. Crit Rev Oncol Hematol. 2017;113:256–267. doi:10.1016/j.critrevonc.2017.02.018
45. Botrus G, Raman P, Oliver T, Bekaii-Saab T. Infigratinib (BGJ398): an investigational agent for the treatment of FGFR-altered intrahepatic cholangiocarcinoma. Expert Opin Investig Drugs. 2021;1–8. doi:10.1080/13543784.2021.1864320
46. Guagnano V, Kauffmann A, Wöhrle S, et al. FGFR Genetic Alterations Predict for Sensitivity to NVP-BGJ398, a Selective Pan-FGFR Inhibitor. Cancer Discov. 2012;2(12):1118–1133. doi:10.1158/2159-8290.CD-12-0210
47. Liu PCC, Koblish H, Wu L, et al. INCB054828 (pemigatinib), a potent and selective inhibitor of fibroblast growth factor receptors 1, 2, and 3, displays activity against genetically defined tumor models. PLoS One. 2020;15(4):e0231877. doi:10.1371/journal.pone.0231877
48. Sootome H, Fujita H, Ito K, et al. Futibatinib Is a Novel Irreversible FGFR 1–4 Inhibitor That Shows Selective Antitumor Activity against FGFR-Deregulated Tumors. Cancer Res. 2020;80(22):4986–4997. doi:10.1158/0008-5472.CAN-19-2568
49. Hall TG, Yu Y, Eathiraj S, et al. Preclinical Activity of ARQ 087, a novel inhibitor targeting FGFR dysregulation. PLoS One. 2016;11:9. doi:10.1371/journal.pone.0162594
50. Nakanishi Y, Akiyama N, Tsukaguchi T, et al. The fibroblast growth factor receptor genetic status as a potential predictor of the sensitivity to CH5183284/Debio 1347, a novel selective FGFR inhibitor. Mol Cancer Ther. 2014;13(11):2547–2558. doi:10.1158/1535-7163.MCT-14-0248
51. Guagnano V, Furet P, Spanka C, et al. Discovery of 3-(2,6-Dichloro-3,5-dimethoxy-phenyl)-1-{6-[4-(4-ethyl-piperazin-1-yl)-phenylamino]-pyrimidin-4-yl}-1-methyl-urea (NVP-BGJ398), A Potent and Selective Inhibitor of the Fibroblast Growth Factor Receptor Family of Receptor Tyrosine Kinase. J Med Chem. 2011;54(20):7066–7083. doi:10.1021/jm2006222
52. Nogova L, Sequist LV, Perez Garcia JM, et al. Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Kinase Inhibitor, in Patients With Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: results of a Global Phase I, Dose-Escalation and Dose-Expansion Study. J Clin Oncol. 2017;35(2):157–165. doi:10.1200/JCO.2016.67.2048
53. Perwad F, Zhang MYH, Tenenhouse HS, Portale AA. Fibroblast growth factor 23 impairs phosphorus and vitamin D metabolism in vivo and suppresses 25-hydroxyvitamin D-1α-hydroxylase expression in vitro. Am J Physiol-Ren Physiol. 2007;293(5):F1577–F1583. doi:10.1152/ajprenal.00463.2006
54. Gattineni J, Bates C, Twombley K, et al. FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol-Ren Physiol. 2009;297(2):F282–F291. doi:10.1152/ajprenal.90742.2008
55. Javle MM, Roychowdhury S, Kelley RK, et al. Final results from a phase II study of infigratinib (BGJ398), an FGFR-selective tyrosine kinase inhibitor, in patients with previously treated advanced cholangiocarcinoma harboring an FGFR2 gene fusion or rearrangement. J Clin Oncol. 2021;39(3_suppl):265. doi:10.1200/JCO.2021.39.3_suppl.265
56. Makawita S, Abou-Alfa G, Roychowdhury S, et al. Infigratinib in patients with advanced cholangiocarcinoma with FGFR2 gene fusions/translocations: the PROOF 301 trial. Future Oncol. 2020;16(30):2375–2384. doi:10.2217/fon-2020-0299
57. Bekaii-Saab TS, Valle JW, Van Cutsem E, et al. FIGHT-302: first-line pemigatinib vs gemcitabine plus cisplatin for advanced cholangiocarcinoma with FGFR2 rearrangements. Future Oncol. 2020;16(30):2385–2399. doi:10.2217/fon-2020-0429
58. Bahleda R, Meric-Bernstam F, Goyal L, et al. Phase I, first-in-human study of futibatinib, a highly selective, irreversible FGFR1–4 inhibitor in patients with advanced solid tumors. Ann Oncol. 2020;31(10):1405–1412. doi:10.1016/j.annonc.2020.06.018
59. Meric-Bernstam F, Arkenau H, Tran B, et al. Efficacy of TAS-120, an irreversible fibroblast growth factor receptor (FGFR) inhibitor, in cholangiocarcinoma patients with FGFR pathway alterations who were previously treated with chemotherapy and other FGFR inhibitors. Ann Oncol. 2018;29:v100. doi:10.1093/annonc/mdy149
60. Goyal L, Shi L, Liu LY, et al. TAS-120 overcomes resistance to ATP-competitive FGFR inhibitors in patients with FGFR2 fusion-positive intrahepatic cholangiocarcinoma. Cancer Discov. 2019;9(8):1064–1079. doi:10.1158/2159-8290.CD-19-0182
61. Goyal L, Meric-Bernstam F, Hollebecque A, et al. FOENIX-CCA2: a phase II, open-label, multicenter study of futibatinib in patients (pts) with intrahepatic cholangiocarcinoma (iCCA) harboring FGFR2 gene fusions or other rearrangements. J Clin Oncol. 2020;38(15_suppl):108. doi:10.1200/JCO.2020.38.15_suppl.108
62. Furuse I, Goyal L, Meric-Bernstam F, et al. Efficacy, safety, and quality of life (QoL) with futibatinib in patients (pts) with intrahepatic cholangiocarcinoma (iCCA) harbouring FGFR2 fusions/rearrangements: FOENIX-CCA2.
63. Borad MJ, Bridgewater JA, Morizane C, et al. A phase III study of futibatinib (TAS-120) versus gemcitabine-cisplatin (gem-cis) chemotherapy as first-line (1L) treatment for patients (pts) with advanced (adv) cholangiocarcinoma (CCA) harboring fibroblast growth factor receptor 2 (FGFR2) gene rearrangements (FOENIX-CCA3). J Clin Oncol. 2020;38(4_suppl):TPS600. doi:10.1200/JCO.2020.38.4_suppl.TPS600
64. Papadopoulos KP, El-Rayes BF, Tolcher AW, et al. A Phase 1 study of ARQ 087, an oral pan-FGFR inhibitor in patients with advanced solid tumours. Br J Cancer. 2017;117(11):1592–1599. doi:10.1038/bjc.2017.330
65. Mazzaferro V, El-Rayes BF, Droz Dit Busset M, et al. Derazantinib (ARQ 087) in advanced or inoperable FGFR2 gene fusion-positive intrahepatic cholangiocarcinoma. Br J Cancer. 2019;120(2):165–171. doi:10.1038/s41416-018-0334-0
66. Voss MH, Hierro C, Heist RS, et al. A Phase I, Open-Label, Multicenter, Dose-escalation Study of the Oral Selective FGFR Inhibitor Debio 1347 in Patients with Advanced Solid Tumors Harboring FGFR Gene Alterations. Clin Cancer Res. 2019;25(9):2699–2707. doi:10.1158/1078-0432.CCR-18-1959
67. Cleary JM, Iyer G, Oh D-Y, et al. Final results from the phase I study expansion cohort of the selective FGFR inhibitor Debio 1,347 in patients with solid tumors harboring an FGFR gene fusion. J Clin Oncol. 2020;38(15_suppl):3603. doi:10.1200/JCO.2020.38.15_suppl.3603
68. Hyman DM, Goyal L, Grivas P, et al. FUZE clinical trial: a phase 2 study of Debio 1347 in FGFR fusion-positive advanced solid tumors irrespectively of tumor histology. J Clin Oncol. 2019;37(15_suppl):TPS3157. doi:10.1200/JCO.2019.37.15_suppl.TPS3157
69. Wöhrle S, Bonny O, Beluch N, et al. FGF receptors control vitamin D and phosphate homeostasis by mediating renal FGF-23 signaling and regulating FGF-23 expression in bone. J Bone Miner Res. 2011;26(10):2486–2497. doi:10.1002/jbmr.478
70. Mahipal A, Tella SH, Kommalapati A, Yu J, Kim R. Prevention and treatment of FGFR inhibitor-associated toxicities. Crit Rev Oncol Hematol. 2020;155:103091. doi:10.1016/j.critrevonc.2020.103091
71. Packer LM, Pollock PM. Paralog-Specific Kinase Inhibition of FGFR4: adding to the Arsenal of Anti-FGFR Agents. Cancer Discov. 2015;5(4):355–357. doi:10.1158/2159-8290.CD-15-0246
72. Pai R, French D, Ma N, et al. Antibody-Mediated Inhibition of Fibroblast Growth Factor 19 Results in Increased Bile Acids Synthesis and Ileal Malabsorption of Bile Acids in Cynomolgus Monkeys. Toxicol Sci. 2012;126(2):446–456. doi:10.1093/toxsci/kfs011
73. Silverman IM, Hollebecque A, Friboulet L, et al. Clinicogenomic Analysis of FGFR2-Rearranged Cholangiocarcinoma Identifies Correlates of Response and Mechanisms of Resistance to Pemigatinib. Cancer Discov. 2021;11(2):326–339. doi:10.1158/2159-8290.CD-20-0766
74. Goyal L, Saha SK, Liu LY, et al. Polyclonal secondary FGFR2 mutations drive acquired resistance to FGFR inhibition in patients with FGFR2 fusion-positive cholangiocarcinoma. Cancer Discov. 2017;7(3):252–263. doi:10.1158/2159-8290.CD-16-1000
75. Kalyukina M, Yosaatmadja Y, Middleditch MJ, Patterson AV, Smaill JB, Squire CJ. TAS-120 Cancer Target Binding: defining Reactivity and Revealing the First Fibroblast Growth Factor Receptor 1 (FGFR1) Irreversible Structure. ChemMedChem. 2019;14(4):494–500. doi:10.1002/cmdc.201800719
76. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol. 2014;4. doi:10.3389/fonc.2014.00064
77. Datta J, Damodaran S, Parks H, et al. Akt Activation Mediates Acquired Resistance to Fibroblast Growth Factor Receptor Inhibitor BGJ398. Mol Cancer Ther. 2017;16(4):614–624. doi:10.1158/1535-7163.MCT-15-1010
78. Burris HA, Siu LL, Infante JR, et al. Safety, pharmacokinetics (PK), pharmacodynamics (PD), and clinical activity of the oral AKT inhibitor GSK2141795 (GSK795) in a phase I first-in-human study. J Clin Oncol. 2011;29(15_suppl):3003. doi:10.1200/jco.2011.29.15_suppl.3003
79. Krook MA, Lenyo A, Wilberding M, et al. Efficacy of FGFR Inhibitors and Combination Therapies for Acquired Resistance in FGFR2-Fusion Cholangiocarcinoma. Mol Cancer Ther. 2020;19(3):847–857. doi:10.1158/1535-7163.MCT-19-0631
80. Aitcheson G, Mahipal A, John BV. Targeting FGFR in intrahepatic cholangiocarcinoma [iCCA]: leading the way for precision medicine in biliary tract cancer [BTC]? Expert Opin Investig Drugs. 2021;30(4):463–477. doi:10.1080/13543784.2021.1900821
81. Rizzo A, Ricci AD, Brandi G. Futibatinib, an investigational agent for the treatment of intrahepatic cholangiocarcinoma: evidence to date and future perspectives. Expert Opin Investig Drugs. 2021;30(4):317–324. doi:10.1080/13543784.2021.1837774
82. Pantel K, Alix-Panabières C. Real-time liquid biopsy in cancer patients: fact or fiction? Cancer Res. 2013;73(21):6384–6388. doi:10.1158/0008-5472.CAN-13-2030
83. Rizzo A, Ricci AD, Tavolari S, Brandi G. Circulating Tumor DNA in Biliary Tract Cancer: current Evidence and Future Perspectives. Cancer Genomics Proteomics. 2020;17(5):441–452. doi:10.21873/cgp.20203
84. Ettrich TJ, Schwerdel D, Dolnik A, et al. Genotyping of circulating tumor DNA in cholangiocarcinoma reveals diagnostic and prognostic information. Sci Rep. 2019;9(1):13261. doi:10.1038/s41598-019-49860-0
85. Rizzo A, Ricci AD, Brandi G. Pemigatinib: hot topics behind the first approval of a targeted therapy in cholangiocarcinoma. Cancer Treat Res Commun. 2021;27:100337. doi:10.1016/j.ctarc.2021.100337
86. Marabelle A, Le DT, Ascierto PA, et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: results From the Phase II KEYNOTE-158 Study. J Clin Oncol. 2019;38(1):1–10. doi:10.1200/JCO.19.02105
87. Marabelle A, Fakih M, Lopez J, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21(10):1353–1365. doi:10.1016/S1470-2045(20)30445-9
88. Katoh M. FGFR inhibitors: effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int J Mol Med. 2016;38(1):3–15. doi:10.3892/ijmm.2016.2620
89. Palakurthi S, Kuraguchi M, Zacharek SJ, et al. The Combined Effect of FGFR Inhibition and PD-1 Blockade Promotes Tumor-Intrinsic Induction of Antitumor Immunity. Cancer Immunol Res. 2019;7(9):1457–1471. doi:10.1158/2326-6066.CIR-18-0595
90. Siefker-Radtke AO, Currie G, Abella E, et al. FIERCE-22: clinical activity of vofatamab (V) a FGFR3 selective inhibitor in combination with pembrolizumab (P) in WT metastatic urothelial carcinoma, preliminary analysis. J Clin Oncol. 2019;37(15_suppl):4511. doi:10.1200/JCO.2019.37.15_suppl.4511
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.