Back to Journals » Journal of Hepatocellular Carcinoma » Volume 4
Targeted therapy and personalized medicine in hepatocellular carcinoma: drug resistance, mechanisms, and treatment strategies
Authors Galun D, Srdic-Rajic T, Bogdanovic A ![]() , Loncar Z, Zuvela M
, Loncar Z, Zuvela M
Received 24 February 2017
Accepted for publication 20 May 2017
Published 11 July 2017 Volume 2017:4 Pages 93—103
DOI https://doi.org/10.2147/JHC.S106529
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmed Kaseb
Danijel Galun,1,2 Tatjana Srdic-Rajic,3 Aleksandar Bogdanovic,1 Zlatibor Loncar,2,4 Marinko Zuvela1,2
1Hepato-Pancreato-Biliary Unit, University Clinic for Digestive Surgery, Clinical Center of Serbia, 2Medical School, University of Belgrade, 3Institute for Oncology and Radiology of Serbia/Unit for Experimental Oncology, 4Emergency Center, Clinical Center of Serbia, Belgrade, Serbia
Abstract: Hepatocellular carcinoma (HCC) is characterized by a growing number of new cases diagnosed each year that is nearly equal to the number of deaths from this cancer. In a majority of the cases, HCC is associated with the underlying chronic liver disease, and it is diagnosed in advanced stage of disease when curative treatment options are not applicable. Sorafenib is a treatment of choice for patients with performance status 1 or 2 and/or macrovascular invasion or extrahepatic spread, and regorafenib is the only systemic treatment found to provide survival benefit in HCC patients progressing on sorafenib treatment. Other drugs tested in different trials failed to demonstrate any benefit. Disappointing results of numerous trials testing the efficacy of various drugs indicate that HCC has low sensitivity to chemotherapy that is in great part caused by multidrug resistance. Immunotherapy for HCC is a new challenging treatment option and involves immune checkpoint inhibitors/antibody-based therapy and peptide-based vaccines. Another challenging approach is microRNA-based therapy that involves two strategies. The first aims to inhibit oncogenic miRNAs by using miRNA antagonists and the second strategy is miRNA replacement, which involves the reintroduction of a tumor-suppressor miRNA mimetic to restore a loss of function.
Keywords: hepatocellular carcinoma, drug resistance, multimodal treatment, chemotherapy
Introduction
Hepatocellular carcinoma (HCC) is characterized by a growing incidence worldwide.1,2 It is a leading cause of cancer-related deaths, as it is the fifth most common cancer and the third most important cause of cancer-related death in men.2–5 The growing number of new cases diagnosed each year is nearly equal to the number of deaths from this cancer, indicating that health care systems worldwide have no efficient answer to this deadly disease.
In some regions, HCC is the fastest rising cause of cancer-related death.6–10 In spite of increased research and new treatment modalities, the overall prognosis for HCC patients remains poor.11
In a majority of the cases, HCC is associated with underlying chronic liver disease. Among different risk factors for the development of HCC, the two factors prevail: one is chronic hepatitis C virus infection and the other one is obesity and the metabolic syndrome.12–14 These two factors are responsible for the growing incidence of HCC.12,14 The epidemic of obesity is not only relevant for the USA but for Asia as well with a tendency to become the leading cause of the rise in HCC incidence.10,12 Another important risk factor is chronic hepatitis B virus infection responsible for ~50% of all worldwide HCC cases.2,3,5 Among 400 million people with hepatitis B virus (HBV) infection, ~25% will develop HCC.3,5,8,12
A majority of HCC cases are diagnosed in advanced stage of disease when curative treatment options (liver transplantation, liver resection, ablative procedures) cannot be applied. In principle, treatment allocation should be decided by a multidisciplinary tumor board for two reasons: the first one is to balance between different treatment modalities and the second is due to complexity of the disease (chronic liver disease associated with cancer). Appropriate patient selection and tailored treatment modality guided by personalized-based medicine should offer every HCC patient the best possible treatment outcome.15
In recent years, different staging systems for HCC have been developed, although none is universally accepted.16–20 However, Barcelona Clinic Liver Cancer (BCLC) staging system endorsed by the American Association for the Study of the Liver (AASL) and European Association for the Study of the Liver (EASL), despite its limitations, has emerged as the most useful to guide HCC treatment.21,22
According to the BCLC staging system, chemotherapy is indicated for HCC patients with Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 1 or 2 and/or macrovascular invasion or extrahepatic spread (BCLC stage C).21 Based on the opinions of Asian experts, this category could be further stratified into a locally advanced stage without extrahepatic spread and an extrahepatic advanced stage.23,24 This stratification has an impact on selecting the most optimal treatment approach.23 Further stratification of patients in BCLC stage C according to liver function, or other prognostic variables, can be also explored.25,26
In the past, various conventional chemotherapy regimens were tested in numerous trials demonstrating disappointing results in the management of HCC.27 Finally, the two milestone studies have positioned sorafenib, the first drug that demonstrated benefit in HCC patients, as a treatment of choice for BCLC stage C patients according to the EASL– European Organization for Research and Treatment of Cancer (EORTC) guidelines.21,28,29
However, the management of HCC patients in the BCLC C group should not be limited to sorafenib treatment only. The multimodal treatment of this patient population is feasible according to the treatment algorithm used in Asian countries.30–34 The possible treatment modalities are transcatheter arterial chemoembolization (TACE),35 hepatic arterial infusion chemotherapy (HAIC),36 and radiotherapy (RT),37–40 or combinations of locoregional therapies and sorafenib.41–43 Drug resistance and toxicity limits the use of sorafenib or other drugs in the management of HCC patients.44
Drug resistance
Disappointing results of numerous trials testing the efficacy of various drugs indicate that HCC has low sensitivity to chemotherapy. This is in great part caused by multidrug resistance (MDR).45 Drug efflux pump, epithelial–mesenchymal transition (EMT), hypoxia-inducible factor1-α (Hif1-α), and DNA damage repair are known mechanisms responsible for MDR.46–49
Several authors found that respective MDR mechanisms are closely related to intracellular calcium.50–53 Wen et al explored the roles of intracellular calcium on various MDR-relevant mechanisms in HCC cells.54 In their study, HCC cells were treated by stimulation with doxorubicin, hypoxia, and ionizing radiation, representing three models of MDR.54 The authors found that the MDR-related mechanisms, EMT, Hif1-α-signaling, and DNA damage repair are all calcium dependent in HCC cells.54
Other authors studied the mechanisms of sorafenib resistance and found the important role of fibroblast growth factor 19 (FGF19).55–57 FGF19 is a metabolic regulator gene belonging to the hormone-like FGF family of signal molecules, and acts as an oncogenic driver in HCC.58–60
Gao et al found that FGF19 is essential for sorafenib efficacy and resistance in the treatment of HCC.61 The authors have demonstrated that elevated FGF19 expression or hyperactivation of FGF19/FGFR4 signaling in HCC cells is one of the main mechanisms of sorafenib resistance.61 In the same study, it was shown that blocking FGF19/FGFR4 axis by ponatinib, the third-generation tyrosine–kinase inhibitor, can overcome the resistance of HCC cells to sorafenib by enhancing reactive oxygen species-associated apoptosis.61
These and similar studies may provide the basis for developing treatment strategies to prevent single-drug resistance. Inhibition of FGF19/FGFR4 signaling is one of the possible strategies for overcoming sorafenib resistance in HCC.
Molecular targeted therapy
Sorafenib
Sorafenib is a molecular multikinase inhibitor of several tyrosine protein kinases (VEGFR and PDGFR); Raf kinases (C-Raf and B-Raf); and intracellular serine/threonine kinases (C-Raf, wild-type B-Raf, and mutant B-Raf)62–64 (Table 1). This is the first molecular targeted agent that demonstrated survival benefit in nonresectable HCC patients.28,29 Sorafenib induces autophagy which in turn suppresses tumor growth.65
 | Table 1 Summary of sorafenib, tivantinib, and regorafenib mechanism of action, effect, and side effects |
The two milestone studies have established sorafenib, as a treatment of choice for HCC patients with ECOG PS of 1 or 2 and/or macrovascular invasion or extrahepatic spread according to the EASL–EORTC guidelines.21,28,29
The findings of SHARP/Phase III trial conducted in the Western world have demonstrated prolonged median survival from 7.9 months (placebo group) to 10.7 months (sorafenib group) (hazard rate [HR]=0.69; 95% CI: 0.55–0.87; p=0.00058).28 Sorafenib also improved the time to radiological progression (from 2.8 months to 5.5 months).28 The results of another Phase III trial, Asia-Pacific trial, have demonstrated a median overall survival of 6.5 months for a treatment group compared to 4.2 months for a placebo group (HR =0.68; 95% CI: 0.50–0.93; p=0.014)29 (Table 2).
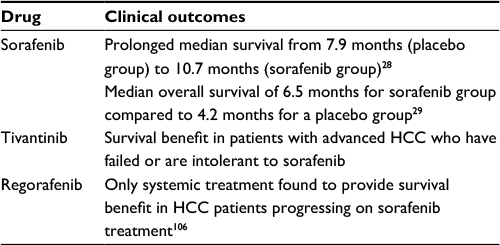 | Table 2 Summary of sorafenib, tivantinib, and regorafenib clinical outcomes Abbreviation: HCC, hepatocellular carcinoma. |
At the beginning, sorafenib was introduced as a well-tolerated drug. However, a subanalysis of the SHARP and Asia-Pacific trials and results of other studies have shown suboptimal tolerability of sorafenib; it was down-dosed in >50% patients and interrupted in 45% of patients due to severe adverse events (AEs) or compromised liver function.28,29,66–68
According to the results of several studies, the most important side effects are gastrointestinal (diarrhea 43%, increased lipase 41%, increased amylase 30%, nausea 23%, anorexia 16%, vomiting 16%, and constipation 15%), dermatologic (rash/desquamation 40%, hand–foot skin reaction 30%, alopecia 27%, pruritus 19%, and dry skin 11%), cardiovascular (hypertension 17%, angioedema, and congestive heart failure), hematologic (hypoalbuminemia 49%, hemorrhage 15%, anemia and thrombocytopenia), and nervous system side effects (neuropathy 13% and headache 10%).28,29,69–72
The safety of sorafenib treatment in real-life conditions was evaluated in Phase IV, GIDEON trial; the second interim analysis from 2011 showed a median survival of 10.3 months for Child A patients and 4.8 months for Child B patients and AE rate comparable to the two leading studies.73
Further studies on sorafenib and other drugs demonstrated disappointing results.74 Phase III trials based on head-to-head comparison between sorafenib and other drugs (sunitinib, brivanib, erlotinib) failed to demonstrate any benefit. 74 The same results were found in Phase III trials on second-line therapies comparing new biological agents (everolimus, brivanib, and ramucirumab) versus placebo.74
The combination regimen of sorafenib and chemotherapeutic agent (“sorafenib plus concept”) was assessed in several Phase II/III trials; however, the combination failed to show superiority in clinical trials.75,76
The impact of viral status on survival in HCC patients receiving sorafenib was analyzed by Jackson et al.77 They conducted a meta-analysis of three large prospective randomized trials (total of 3,256 patients) in which sorafenib was the control arm.77 They found an improved overall survival in patients who were hepatitis C virus (HCV) positive and HBV negative and no evidence of any improvement in overall survival attributable to sorafenib in patients who were HBV positive and HCV negative.77 The same results were found in subgroup analysis of the SHARP trial demonstrating the superior median overall survival in HCV-positive patients.78
In the same study, the subgroup of alcohol-related HCC patients was analyzed.78 The sorafenib group was associated with a longer median overall survival (10.3 vs 8.0 months) and time to progression (5.5 vs 3.9 months) and a higher disease control rate (54.4% vs 38.8%) than placebo.78 Although in alcoholics the surveillance for HCC could be less effective because of the socioeconomic context, the stage at the diagnosis and the prognosis are not different compared to viral HCCs.79
Nonalcoholic steatohepatitis (NASH)-associated HCC patients are another important subset of patients because of the increasing incidence of NASH worldwide.8,10,12 Weinmann et al have analyzed a large cohort of HCC patients (1,119) treated in an 11-year period and compared the outcome of NASH–HCC and non-NASH–HCC patients managed by sorafenib.80 Systemic therapy with sorafenib as a first-line treatment was significantly more frequently performed in NASH–HCC (17.8% vs 3.5%, p<0.001). The overall survival in NASH–HCC was 4.22 months shorter compared to non-NASH–HCC (median 11.28 vs 15.5 months).80 However, in the absence of cirrhosis, NASH–HCC patients demonstrated a trend to an increased overall survival compared to non-NASH–HCC patients (43.4 vs 25 months, p=0.748).80
The safety and efficacy of sorafenib in HCC patients with diabetes was confirmed by Di Constanzo et al.81 They found a comparable frequency of main AEs in both diabetes and nondiabetes groups, with the exception of rash, which was more frequent among diabetes patients (27.5% vs 17.6%, p=0.047). The median overall survival was 9 months in nondiabetes and 10 months in the diabetes group (p=0.535).81 Median time to progression was longer in diabetes than in the nondiabetes group (p=0.038), suggesting a better anticancer effect of sorafenib in patients with diabetes.81 However, the concomitant use of sorafenib and metformin was associated with shorter median progression-free survival compared to patients receiving sorafenib alone (2.6 vs 5.0 months, p=0.029).82 The median overall survival of patients treated with the combination was 10.4 months compared to 15.1 months for patients who were not given metformin therapy (p=0.014). These results suggest possible resistance to sorafenib in metformin-treated patients.82
The possible use of sorafenib in adjuvant settings was assessed in the STORM trial. The trial enrolled 1,114 patients randomized to take either sorafenib or placebo.83 However, the study did not met its primary and secondary endpoints since no differences were observed regarding recurrence-free survival (33.4 vs 33.8 months; HR =0.94; 95% CI: 0.78–1.13, p=0.26), time to recurrence (38.6 vs 35.8 months; HR =0.89, 95% CI: 0.73–1.08), and overall survival (not reached vs not reached, HR =0.99, 95% CI: 0.76–1.30, p=0.48).83
The combination of sorafenib and transarterial chemoembolization for HCC patients in intermediate stage (BCLC B group) was evaluated in the SPACE trial; however, the primary end point was not reached.84 Among 307 patients randomized, 154 received sorafenib and 153 received placebo. Median time to progression for patients receiving sorafenib plus drug-eluting bead (DEB)-TACE or placebo plus DEB-TACE was similar (169 vs 166 days, respectively; HR =0.797, p=0.072).84
Tivantinib
Tivantinib (ARQ 197) is a small, oral, highly selective inhibitor of c-MET receptor tyrosine kinase85 (Table 1). The MET encodes for cellular mesenchymal–epithelial transition factor (c-MET).86 Tivantinib inhibits in a non-ATP-competitive manner MET autoactivation by selectively targeting the inactive nonphosphorylated form of the kinase.87,88 The inhibition of c-MET by tivantinib promotes apoptosis and cell growth arrest.89–91 Recent studies have shown that tivantinib possesses cytotoxic activity, even in cells that lack c-MET.90–94 Also, some studies hypothesize that tivantinib can activate cyclin B195 and inhibit microtubule.90,94 Several preclinical and clinical studies showed that tivantinib can act as a single agent85,96 and in combination with sorafenib.97–99
In Phase I and II trials, tivantinib has shown a survival benefit in patients with advanced HCC who have failed or are intolerant to sorafenib.100–102 Twenty-one patients were enrolled to a 28-day treatment protocol. The tumor response was evaluated only in 16 patients, but tumor regression was not recorded. The median disease progression interval was 3.3 months (range 1.47–5.3 months)97 (Table 2).
Those patients who have tumors with high expression of MET (MET-high) appear to derive the greatest benefit from tivantinib therapy. Currently, two large randomized double-blind placebo-controlled Phase III trials METIV-HCC trail [NCT01755767] and JET-HCC trail [NCT02029157]) are evaluating tivantinib in patients with MET-high advanced HCC.103,104
Regorafenib
Regorafenib is a multikinase inhibitor of VEGFR1-3, c-KIT, TIE-2, PDGFR-β, FGFR-1, RET, c-RAF, BRAF, and p38 MAP kinase105 (Table 1). Its structure is similar to sorafenib; however, higher potency is expected due to the addition of a fluorine atom in the central phenyl ring.106
In the multicenter analysis of treatment pattern and outcomes in Hong Kong, 45 consecutive patients with metastatic colorectal cancer treated with regorafenib after failure of all standard systemic options were analyzed.107 Initial dose was 160 mg in the group of 20 patients, while the remaining patients received lower doses. Of 31 patients, one had partial response and ten had stable disease.107 The most common hematological AE was anemia (8.9%), and nonhematological AE was hand–foot skin reaction (26.7%).107
Efficacy and safety of regorafenib has been also confirmed for treatment of gastrointestinal stromal tumor after progression or toxicity during imatinib and sunitinib treatment.108,109
Safety of regorafenib as second-line therapy for intermediate or advanced HCC was assessed in the multicenter, Phase II study, by Bruix et al.110 Thirty-six patients were enrolled and received 160 mg dose once daily during 3 weeks on and 1 week off treatment. Treatment was interrupted when disease progression (n=10), unacceptable toxicity (n=20), or death (n=1) occurred. Treatment-related AEs were: hand–foot skin reaction, diarrhea, fatigue, hypothyroidism, anorexia, hypertension, nausea, and voice changes. Partial response was achieved in one patient and stable disease in 25. Median time to progression and overall survival were 4.3 and 13.8 months, respectively110 (Table 2).
A larger, international, multicentric Phase III trial (RESORCE) concluded that regorafenib is the only systemic treatment found to provide survival benefit in HCC patients progressing on sorafenib treatment.111 During the study period, 573 patients were randomized (379 to regorafenib and 194 to placebo). Regorafenib group experienced longer median survival compared to placebo group (10.6 vs 7.8 months).111 The most common grade 3 or 4 treatment-related AEs were hypertension, hand–foot skin reaction, fatigue, and diarrhea, similar to sorafenib AE profile. Mortality rate was 13% in the regorafenib group and 20% in the placebo group. Seven deaths (2%) were associated with study drug treatment and two (1%) in the placebo group were related with liver failure.111
The approval of regorafenib for this indication is expected during 2017.106
Immunotherapy for HCC
Immune checkpoint inhibitors – antibody-based therapy
The main goal of previous cancer immunotherapy has been to enhance immune cell activity to kill the cancer cells. However, this does not result in actual activation of the immune system because of the inhibition signal by checkpoint molecules.
Under physiologic conditions, the liver is an organ of relative immune tolerance, and checkpoint pathways play a key role in this process. This function is very significant as the liver is persistently exposed to food-borne and microbial antigens delivered from the gut. HCC immune tolerance is mediated through decreased co-stimulatory or increased inhibitory checkpoint signaling that leads into immunosuppression. Human cytotoxic T-lymphocyte antigen 4 (CTLA-4) and a programmed death 1 (PD-1) and its ligand (PD-L1) are among the several inhibitory checkpoints that have been extensively investigated, as well as related with defective immune process in HCC. CTLA-4 is expressed constitutively on regulatory T cells (Tregs) and transiently on a broad range of T cells at the early activation stage. In the anticancer immunity, CTLA-4 can inhibit the activation and proliferation of T cells that recognize cancer antigens.112
PD-1 is an immune coinhibitory receptor expressed on T cells, B cells, NK cells, and myeloid cells. On T cells, PD-1 suppresses antigen-specific T-cell activation through interactions with its ligands PD-L1 and PD-L2.113–115 PD-L1 is expressed on hepatocytes,116 Kupffer cells,117 and liver sinusoidal endothelial cells.118–120 Currently, there are several immune checkpoint monoclonal antibody blockers, such as ipilimumab, tremelimumab, nivolumab, MED14736, anti-LAG-3 (BMS-986016), and pembrolizumab.
Several studies show that higher levels of circulating PD-1+/CD8+ T cells correlate with disease progression;121 as well, increased PD-L1 levels in HCC patients were associated with poorer prognosis.122
Less data from preclinical and translational studies exist regarding the mechanistic role of CTLA-4 in HCC.123 It is known that high levels of CTLA-4 expression on hepatic dendritic cells have been linked with inhibition of T-cell proliferation and T-cell apoptosis in HCC,124 while high levels of CTLA-4 expression by Tregs correlate with decreased production of cytotoxic enzymes by effector T cells in HCC.113
Several clinical trials of immune checkpoint inhibitors in HCC are ongoing. Sangro et al in a Phase II trial (ClinicalTrials. gov: NCT01008358) evaluated the efficacy and safety of anti-CTLA-4 antibodies (tremelimumab) for the treatment of HCC.120 Patients with unresectable HCC and chronic hepatitis C received tremelimumab as a monotherapy. Treatment was generally well tolerated. The patients most frequently developed skin rash, fatigue, and anorexia. Severe immune-mediated AEs were not recorded. Among treated patients, there was a 76.4% disease control rate and 17.6% partial response. The median time to progression was 6.5 months.125
Regarding the PD-1 therapies, in a Phase I/II trial (MEDI4736), a fully human anti-PD-L1 monoclonal antibody is being evaluated in multiple advanced cancers, including HCC (ClinicalTrials.gov: NCT01938612).126 Until now, only a relatively favorable safety profile has been reported. Preliminary results of a Phase I/II trial of nivolumab, a fully human IgG4 anti-PD-1 monoclonal antibody, were reported (ClinicalTrials.gov: NCT01658878).127
This study included eleven patients with HCV and ten patients with HBV infection, while 50% of patients were unaffected. Approximately 19% of patients responded to nivolumab, two patients achieved complete response, while six patients had partial response. Overall survival was 72% at 6 months and 62% at 12 months. Of note, 68% of patients had received prior sorafenib therapy, and responses were seen in both patients with and without viral hepatitis. AEs that included transaminase and lipase elevations along with anemia and fatigue were observed in 19% of patients.127
Immune checkpoint inhibition can induce immune-related AEs such as hyperthyroidism, hypothyroidism, Type 1 diabetes mellitus, myasthenia gravis, rheumatoid arthritis, or Addison’s disease, along with dry mouth, hepatitis, dermopathy, pancreatitis, pneumonitis, adrenal insufficiency, and enteritis. Many of these AEs can be controlled by withdrawal of immune checkpoint inhibitors and initiation of steroid therapy. Immune-related AEs are least frequent in patients treated with anti-PD-L1 antibodies and most frequent in patients treated with anti-CTLA-4 antibodies.128
Having in mind multiple mechanisms by which HCC evades the immune system, in addition to anti-CTLA-4 and PD-1 monotherapy, combination therapy may be necessary to achieve better therapeutic outcomes than the corresponding monotherapies. In November 2015, a Phase I/II study (ClinicalTrials.gov: NCT01658878) was modified and included a nivolumab/ipilimumab combination arm.127 One other trial of tremelimumab with MEDI4736 (a PD-L1 antagonist) for patients with unresectable HCC (ClinicalTrials.gov:NCT02519348) is ongoing. Also, the combination of checkpoint inhibitor with sorafenib appears promising, as sorafenib exerts immunomodulatory effect.129
Peptide vaccines
Another approach to immunotherapy involves peptide-based vaccines. GPC3 belongs to the glypican family of heparan sulfate proteoglycans, which are linked to the outer surface of the cell membrane through a glycosylphosphatidylinositol anchor.130 Since it is specifically overexpressed in HCC (72%–81%), GPC3 is an ideal target for antigen-specific immunotherapy against HCC.131,132
A nonrandomized, open-label, Phase I clinical trial of a GPC3-derived peptide vaccine for advanced HCC patients was conducted in Japan.133 In this trial, 33 patients with advanced HCC received peptide vaccines. This trial demonstrated that the vaccine was well tolerated, and for the first time this study showed that the frequency of GPC3-derived, peptide-specific cytotoxic T lymphocytes (CTLs) was correlated with overall survival in patients with HCC receiving a peptide vaccine.134
The same group conducted a Phase II clinical trial of the GPC3-derived peptide vaccine in the adjuvant setting (UMIN-CTR: 000002614).134 Forty-one patients with initial HCC who had undergone surgery or radiofrequency ablation were enrolled in this Phase II, open-label, single-arm trial. Sawada et al investigated case–control subjects, where selected patients treated surgically during the same period were analyzed.134 Six patients received radiofrequency ablation therapy while 35 received surgery. The recurrence rate tended to be lower in the 35 patients treated with surgery plus vaccination compared to 33 patients who underwent surgery alone. Twenty-five patients treated with surgery and vaccination had GPC3-positive tumors; the recurrence rate in this group was significantly lower compared to that in 21 GPC3-positive patients who received surgery only.134 The GPC3 peptide vaccine improved the 1-year recurrence rate in patients with GPC3-positive tumors.
MicroRNA-based therapies
Nucleic acid-based drugs such as microRNAs (miRNAs) have promising therapeutic potential for HCC treatment. miRNAs are an abundant class of endogenous, short (20–25 nucleotides in length) non-coding RNAs. miRNAs control the stability and translation of targeted messenger RNAs (mRNAs) through complementary interaction with the 3′ untranslated region of target genes (or of complementary sequences).
miRNAs are involved in fundamental cellular processes, like embryonic development, differentiation, cell cycle, metabolism, and in carcinogenesis and tumor progression.135–139 Accumulating evidence has addressed that miRNAs are involved in oncogene and tumor-suppressor pathways.140–143
Several studies have shown that expression of miRNA is deregulated in HCC in comparison with normal liver tissue. Thus, for example, miRNAs are both upregulated (miR-21, miR-221, miR-222, miR-224, and miR-17-92) and downregulated (miR-29, miR-122, miR-200, miR-123, miR-199a, and miR-199b, let-7) in association with HCC.144,145
Cellular miRNA, like miR-196, plays a protective role in HCV-induced HCC by upregulating hemeoxygenase 1 (HMOX1) expression and inhibiting HCV transcription.146 On the other hand, miR-217 could promote ethanol-induced fat accumulation in hepatocytes,147 while miR-126 was found decreased in alcohol-related HCC.148 miR-21 and miR-155 are also involved in the pathogenesis of NASH. Results from some studies showed a correlation between modulation of cellular miRNAs abundance and the sensitivity of HCC cancer cells to chemotherapeutic drugs. Thus, for example, overexpression and restoration of miR-122 increases the sensitivity of HCC cells to chemotherapeutic agents, such as doxorubicin or sorafenib.149,150
The therapeutic application of miRNAs involves two strategies. The first aims to inhibit oncogenic miRNAs by using miRNA antagonists, such as antimiRs, or antagomiRs.151 miRNA antagonists are single-stranded RNA molecules, which act through complementary base-pairing with miRNAs. The second strategy, miRNA replacement, involves the reintroduction of a tumor-suppressor miRNA mimetic to restore a loss of function.152
However, two main problems for miRNA-based therapeutics are off-target effects, since one miRNA can target multiple genes and pathways, and their delivery. One possibility to overcome these is usage of the adeno-associated viral vectors with lower risk of vector-related toxicities and good gene transfer efficacy.
Kota et al administered miR-26a, which is frequently downregulated in HCC and acts as tumor suppressor, in a mouse model using adeno-associated virus. They found that ectopic expression of miR-26a results in inhibition of cancer cell proliferation and induction of tumor-specific apoptosis.153 Also, a Phase I trial which investigates the role of MRX34, a liposome-based miR-34 mimic, is currently ongoing.154
Conclusion
The rising incidence of HCC occurrence in patients with underlying chronic liver disease, initial presentation in advanced stage in the majority of cases, and high mortality rates demonstrate an urgent need to develop effective treatment and prevention strategies for this complex disease. Although multimodal management is clearly needed for the majority of HCC patients, appropriate randomized clinical trials or meta-analyses addressing this problem are lacking. The consensus for successful management of advanced HCC patients using a multimodal approach should be further established.
Today, sorafenib is the first-line and regorafenib is the second-line therapy for HCC patients with PS 1 or 2 and/or macrovascular invasion or extrahepatic spread, providing a median gain in overall survival of <3 months. The genetic instability makes HCC commonly resistant to chemotherapy and additional management strategies are needed.
The introduction of checkpoint regulators has opened a new horizon for cancer immunotherapy. As indicated by several trials, immunotherapeutics are very promising therapeutic tools for HCC treatment. In future, HCC immunotherapy should be rationally combined with other treatment modalities like surgery, thermal ablation, and cytotoxic agents to maximize its therapeutic efficacy.
Therapeutic interventions are challenging for this disease, although the majority of them are providing palliative treatment only. In the upcoming months, data of several ongoing first-line and second-line trials will become available and might further change the care of patients with advanced HCC.
Disclosure
The authors report no conflicts of interest in this work.
References
Fact Sheets by Cancer [webpage on the Internet]. All Cancers (Excluding Non-Melanoma Skin Cancer) Estimated Incidence, Mortality and Prevalence Worldwide in 2012. Available from: http://globocan.iarc.fr/Pages/fact_sheets_cancer.aspx. Accessed June 12, 2016. | ||
National Cancer Institute [webpage on the Internet]. A Snapshot of Liver Cancers. Available from: http://www.cancer.gov/research/progress/snapshots/liver. Accessed August 23, 2016. | ||
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118–1127. | ||
Mortality Database. WHO Statistical Information System. Geneva: World Health Organization; 2008. Available from: http://www.who.int/whosis. Accessed June 26, 2017. | ||
Ferlay J, Shin H, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008:GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. | ||
Altekruse SF, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009;27:1485–1491. | ||
El-Serag HB, Lau M, Eschbach K, Davila J, Goodwin J. Epidemiology of hepatocellular carcinoma in Hispanics in the United States. Arch Intern Med. 2007;167:1983–1989. | ||
Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47(Suppl):S2–S6. | ||
White DL, Thritt AP, Kanwal F et al. Incidence of Hepatocellular Carcinoma in All 50 United States, From 2000 Through 2012. Gastroenterology. 2016;152(4):812–820. | ||
Nordenstedt H, White DL, El-Serag HB. The changing pattern of epidemiology in hepatocellular carcinoma. Dig Liver Dis. 2010;42(Suppl 3):S206–S214. | ||
Cabibbo G, Enea M, Attanasio M, Bruix J, Craxì A, Cammà C. A meta-analysis of survival rates of untreated patients in randomized clinical trials of hepatocellular carcinoma. Hepatology. 2010;51:1274–1283. | ||
Kew MC. Hepatocellular carcinoma: epidemiology and risk factors. J Hepatocell Carcinoma. 2014;1:115–125. | ||
Kanwal F, Hoang T, Kramer JR, et al. Increasing prevalence of HCC and cirrhosis in patients with chronic hepatitis C virus infection. Gastroenterology. 2011;140:1182–1188. | ||
Sherman M. Hepatocellular carcinoma: epidemiology, surveillance, and diagnosis. Semin Liver Dis. 2010;30:3–16. | ||
Galun D, Basaric D, Zuvela M, et al. Hepatocellular carcinoma: from clinical practice to evidence-based treatment protocols. World J Hepatol. 2015;7(20):2274–2291. | ||
Marrero JA, Fontana RJ, Barrat A, et al. Prognosis of hepatocellular carcinoma: comparison of 7 staging systems in an American cohort. Hepatology. 2005;41:707–716 | ||
Kim BK, Kim SU, Park JY, et al. Applicability of BCLC stage for prognostic stratification in comparison with other staging systems: single centre experience from long-term clinical outcomes of 1717 treatment-naïve patients with hepatocellular carcinoma. Liver Int. 2012;32:1120–1127 | ||
Pascual S, Zapater P, Such J, et al. Comparison of staging systems to predict survival in hepatocellular carcinoma. Liver Int. 2006;26:673–679. | ||
Santambrogio R, Salceda J, Costa M, et al. External validation of a simplified BCLC staging system for early hepatocellular carcinoma. Eur J Surg Oncol. 2013;39(8):850–857. | ||
Hsu CY, Hsia CY, Huang YH, et al. Selecting an optimal staging system for hepatocellular carcinoma: comparison of 5 currently used prognostic models. Cancer. 2010;116:3006–3014. | ||
EASL–EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2012;56:908–943. | ||
Graf D, Vallböhmer D, Knoefel WT, et al. Multimodal treatment of hepatocellular carcinoma. Eur J Int Med. 2014;25:430–437. | ||
Han KH, Kudo M, Ye SL, et al. Asian consensus workshop report: expert consensus guideline for the management of intermediate and advanced hepatocellular carcinoma in Asia. Oncology. 2011;81(Suppl 1):158–164. | ||
Park HC, Seong J, Tanaka M, et al. Multidisciplinary management of nonresectable hepatocellular carcinoma. Oncology. 2011;81(Suppl 1):134–140. | ||
Huitzil-Melendez FD, Capanu M, O’Reilly EM, et al. Advanced hepatocellular carcinoma: which staging systems best predict prognosis? J Clin Oncol. 2010;28:2889–2895. | ||
Choi C, Choi GH, Kim TH, Tanaka M, Meng MB, Seong J. Multimodality management for Barcelona clinic liver cancer stage C hepatocellular carcinoma. Liver Cancer. 2014;3:405–416. | ||
Deng G-L, Zeng S, Shen H. Chemotherapy and target therapy for hepatocellular carcinoma: new advances and challenges. World J Hepatol. 2015;7(5):787–798. | ||
Llovet JM, Ricci S, Mazzaferro V, et al. SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. | ||
Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25–34. | ||
Kokudo N, Makuuchi M. Evidence-based clinical practice guidelines for hepatocellular carcinoma in Japan: the J-HCC guidelines. J Gastroenterol. 2009;44(Suppl 19):119–121. | ||
Korean Liver Cancer Study Group and National Cancer Center, Korea: Practice guidelines for management of hepatocellular carcinoma 2009. Korean J Hepatol. 2009;15:391–423. | ||
Lee JM, Han KH. Positioning and indication of sorafenib in the treatment algorithm and real practice setting: Western and eastern approach—Asian perspective. Oncology. 2010;78(Suppl 1):167–171. | ||
Kudo M, Ueshima K. Positioning of a molecular-targeted agent, sorafenib, in the treatment algorithm for hepatocellular carcinoma and implication of many complete remission cases in Japan. Oncology. 2010;78(Suppl 1):154–166. | ||
Omata M, Lesmana LA, Tateishi R, et al. Asian Pacific Association for the Study of the Liver consensus recommendations on hepatocellular carcinoma. Hepatol Int. 2010;4:439–474. | ||
Kim KM, Kim JH, Park IS, et al. Reappraisal of repeated transarterial chemoembolization in the treatment of hepatocellular carcinoma with portal vein invasion. J Gastroenterol Hepatol. 2009;24:806–814. | ||
Kudo M. Treatment of advanced hepatocellular carcinoma with emphasis on hepatic arterial infusion chemotherapy and molecular targeted therapy. Liver Cancer. 2012;1:62–70. | ||
Jihye C, Jinsil S. Application of radiotherapeutic strategies in the BCLC-defined stages of hepatocellular carcinoma. Liver Cancer. 2012;1:216–225. | ||
Hsu HC, Chen TY, Chiu KW, et al. Three-dimensional conformal radiotherapy for the treatment of arteriovenous shunting in patients with hepatocellular carcinoma. Br J Radiol. 2007;80:38–42. | ||
Han KH, Seong J, Kim JK, Ahn SH, Lee DY, Chon CY. Pilot clinical trial of localized concurrent chemoradiation therapy for locally advanced hepatocellular carcinoma with portal vein thrombosis. Cancer. 2008;113:995–1003. | ||
Meng MB, Cui YL, Lu Y, et al. Transcatheter arterial chemoembolization in combination with radiotherapy for unresectable hepatocellular carcinoma: a systematic review and meta-analysis. Radiother Oncol. 2009;92:184–194. | ||
Yu W, Gu K, Yu Z, et al. Sorafenib potentiates irradiation effect in hepatocellular carcinoma in vitro and in vivo. Cancer Lett. 2013;329:109–117. | ||
Lee S, Kim BK, Kim SU, et al. Efficacy of sorafenib monotherapy versus sorafenib-based loco-regional treatments in advanced hepatocellular carcinoma. PLoS ONE. 2013;8:e77240. | ||
Liapi E, Geschwind JF. Combination of local transcatheter arterial chemoembolization and systemic antiangiogenic therapy for unresectable hepatocellular carcinoma. Liver Cancer. 2012;1:201–215. | ||
Deng GL, Zeng S, Shen H. Chemotherapy and target therapy for hepatocellular carcinoma: new advances and challenges. World J Hepatol. 2015;7(5):787–798. | ||
Petraccia L, Onori P, Sferra R, et al. MDR (multidrug resistance) in hepatocarcinoma clinical-therapeutic implications. Clin Ter. 2003;154:325–335. | ||
Teicher BA. Acute and chronic in vivo therapeutic resistance. Biochem Pharmacol. 2009;77:1665–1673. | ||
Luo D, Wang Z, Wu J, Jiang C, Wu J. The role of hypoxia inducible factor-1 in hepatocellular carcinoma. Biomed Res Int. 2014;2014:409272. | ||
van Zijl F, Zulehner M, Petz M, et al. Epithelial–mesenchymal transition in hepatocellular carcinoma. Future Oncol. 2009;5:1169–1179. | ||
Zhang H, Mizumachi T, Carcel-Trullols J, et al. Targeting human 8-oxoguanine DNA glycosylase (hOGG1) to mitochondria enhances cisplatin cytotoxicity in hepatoma cells. Carcinogenesis. 2007;28:1629–1637. | ||
Berridge M, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. | ||
Hu J, Qin K, Zhang J, et al. Downregulation of transcription factor Oct4 induces an epithelial-to-mesenchymal transition via enhancement of Ca2+influx in breast cancer cells. Biochem Biophys Res Commun. 2011;411:786–791. | ||
Davis FM, Peters AA, Grice DM, et al. Non-stimulated, agonist-stimulated and store-operated Ca2+ influx in MDA-MB-468 breast cancer cells and the effect of EGF-induced EMT on calcium entry. PLoS One. 2012;7:e36923. | ||
Davis FM, Azimi I, Faville RA, et al. Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene. 2014;33:2307–2316. | ||
Wen L, Liang C, Chen E, et al. Regulation of multi-drug resistance in hepatocellular carcinoma cells isTRPC6/calcium dependent. Sci Rep. 2016;6:23269. | ||
Harmer NJ, Pellegrini L, Chirgadze D, Fernandez-Recio J, Blundell TL. The crystal structure of fibroblast growth factor (FGF) 19 reveals novel features of the FGF family and offers a structural basis for its unusual receptor affinity. Biochemistry. 2004;43:629–640. | ||
Zhao H, Lv F, Liang G, et al. FGF19 promotes epithelial–mesenchymal transition in hepatocellular carcinoma cells by modulating the GSK3beta/beta-catenin signaling cascade via FGFR4 activation. Oncotarget. 2016;7:13575–13586. | ||
Miura S, Mitsuhashi N, Shimizu H, et al. Fibroblast growth factor 19 expression correlates with tumor progression and poorer prognosis of hepatocellular carcinoma. BMC Cancer. 2012;12:56. | ||
Sawey ET, Chanrion M, Cai C, et al. Identification of a therapeutic strategy targeting amplified FGF19 in liver cancer by oncogenomic screening. Cancer Cell. 2011;19:347–358. | ||
Repana D, Ross P. Targeting FGF19/FGFR4 pathway: a novel therapeutic strategy for hepatocellular carcinoma. Diseases. 2015;3:294–305. | ||
Kan Z, Zheng H, Liu X, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422–1433. | ||
Gao L, Wang X, Tang Y, Huang S, Hu CA, Teng Y.. FGF19/FGFR4 signaling contributes to the resistance of hepatocellular carcinoma to sorafenib. J Exp Clin Cancer Res. 2017;36:8. | ||
Smalley KS, Xiao M, Villanueva J, et al. CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations. Oncogene. 2009;28(1):85–94. | ||
Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7(10):3129–3140. | ||
Keating GM, Santoro A. Sorafenib: a review of its use in advanced hepatocellular carcinoma. Drugs. 2009;69(2):223–240. | ||
Zhang Y. Screening of kinase inhibitors targeting BRAF for regulating autophagy based on kinase pathways. J Mol Med Rep. 2014;9(1):83–90. | ||
Kudo M. Treatment of advanced hepatocellular carcinoma with emphasis on hepatic arterial infusion chemotherapy and molecular targeted therapy. Liver Cancer. 2012;1(2):62–70. | ||
Choi GH, Shim JH, Kim MG, et al. Sorafenib alone versus sorafenib combined with transarterial chemoembolization for advanced-stage hepatocellular carcinoma: results of propensity score analyses. Radiology. 2013;269(2):603–611. | ||
Iavarone M, Cabibbo G, Piscaglia F, et al. Field-practice study of sorafenib therapy for hepatocellular carcinoma: a prospective multicenter study in Italy. Hepatology. 2011;54(6):2055–2063. | ||
Amar S, Wu KJ, Tan WW. Sorafenib-induced pancreatitis. Mayo Clin Proc. 2007;82:516–522. | ||
Autier J, Escudier B, Wechsler J, Spatz A, Robert C. Prospective study of the cutaneous adverse effects of sorafenib, a novel multikinase inhibitor. Arch Dermatol. 2008;144:886–892. | ||
Product Information. Nexavar (sorafenib). Bayer Pharmaceutical Inc, West Haven, CT. | ||
Alexandrescu DT, McClure R, Farzanmehr H, Dasanu CA. Secondary erythrocytosis produced by the tyrosine kinase inhibitors sunitinib and sorafenib. J Clin Oncol 2008;26:4047–4048. | ||
Lencioni R, Kudo M, Ye SL, Bronowicki JP, Chen XP, Dagher L, et al. First interim analysis of the GIDEON (Global Investigation of therapeutic DEcisions in hepatocellular carcinoma and Of its treatment with sorafeNib) non-interventional study. Int J Clin Pract. 2012;66:675–683. | ||
Bruix, J, Reig, M, and Sherman, M. Evidence-based diagnosis, staging, and treatment of patients with hepatocellular carcinoma. Gastroenterology. 2016;150:835–853. | ||
Zhu AX, Rosmorduc O, Evans TR, et al. SEARCH: a phase III, randomized, double-blind, placebo-controlled trial of sorafenib plus erlotinib in patients with advanced hepatocellular carcinoma. J Clin Oncol. 2015;33(6):559–566. | ||
Abou-Alfa GK, Johnson P, Knox JJ, e al. Doxorubicin plus sorafenibvs doxorubicin alone in patients with advanced hepatocellularcarcinoma: a randomized trial. JAMA. 2010;304:2154–2160. | ||
Jackson R, Psarelli EE, Berhane S, et al. Impact of viral status on survival in patients receiving sorafenib for advanced hepatocellular cancer: a meta-analysis of randomized phase III trials. J Clin Oncol. 2017;35(6):622–633. | ||
Bruix J, Raoul JL, Sherman M, et al. Efficacy and safety of sorafenib in patients with advanced hepatocellular carcinoma: subanalyses of a phase III trial. J Hepatol. 2012;57: 821–829. | ||
Galati G, Dell’Unto C, Vespasiani-Gentilucci U, et al. Hepatocellular carcinoma in alcoholic liver disease: current management and recent advances. Rev Recent Clin Trials. 2016;11(3):238–252. | ||
Weinmann A, Alt Y, Koch S, et al. Treatment and survival of non-alcoholic steatohepatitis associated hepatocellular carcinoma. BMC Cancer. 2015;15:210. | ||
Di Costanzo GG, Tortora R, Morisco, et al. Impact of diabetes on outcomes of sorafenib therapy for hepatocellular carcinoma. Target Oncol. 2017;12(1): 61–67. | ||
Gardini AC, Marisi G, Scarpi E, et al. Effects of metformin on clinical outcome in diabetic patients with advanced HCC receiving sorafenib. Expert Opin Pharmacother. 2015;16(18):2719–2725. | ||
Bruix J, Takayama T, Mazzaferoo V, et al. STORM: A phase III randomized, double-blind, placebo-controlled trial of adjuvant sorafenib after resection or ablation to prevent recurrence of hepatocellular carcinoma (HCC). J Clin Oncol. 2014;32:5s. | ||
Lencioni, R, Llovet, JM, Han, G et al. Sorafenib or placebo plus TACE with doxorubicin-eluting beads for intermediate stage HCC: the SPACE trial. J Hepatol. 2016;64:1090–1098. | ||
Munshi N, Jeay S, Li Y, et al. ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Mol Cancer Ther. 2010;9(6):1544–1553. | ||
Cooper CS, Park M, Blair DG, et al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984;311(5981):29–33. | ||
Boccaccio C, Comoglio PM. Invasive growth: a MET-driven genetic programme for cancer and stem cells. Nat Rev Cancer. 2006;6:637–645. | ||
Gherardi E, Birchmeier W, Birchmeier C, et al. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12(2):89–103. | ||
Basilico C, Pennacchietti S, Vigna E, Vande Woude G. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Am Assoc Cancer Res. 2013;19(9):2381–2392. | ||
Xiang Q, Zhen Z, Deng DY, et al. Tivantinib induces G2/M arrest and apoptosis by disrupting tubulin polymerization in hepatocellular carcinoma. J ExpClin Cancer Res. 2015;34:118. | ||
Pievsky D, Pyrsopoulos N. Profile of tivantinib and its potential in the treatment of hepatocellular carcinoma: the evidence to date. J hepatocell Carcinoma. 2016;3:69–76. | ||
Aoyama A, Katayama R, Oh-hara T, Sato S, Okuno Y, Fujita N. Tivantinib (ARQ 197) exhibits antitumor activity by directly interacting with tubulin and overcomes abc transporter–mediated drug resistance. Am Assoc Cancer Res. 2014;13(12):2978–2990. | ||
Katayama R, Aoyama A, Yamori T, et al. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res. 2013;73(10):3087–3096. | ||
Remsing Rix LL, Kuenzi BM, Luo Y, et al. GSK3 alpha and beta are new functionally relevant targets of tivantinib in lung cancer cells. ACS Chem Biol. 2014;9(2):353–358. | ||
Lu S, Rizzani A, Kolligs F, et al. Antitumor efficacy of the c-met inhibitor tivantinib (ARQ-197) in liver cancer cells: effects on apoptosis and cell cycle. Abstract to be presented at the 2012 GASL conference. | ||
Eathiraj S, Palma R, Volckova E, et al. Discovery of a novel mode of protein kinase inhibition characterized by the mechanism of inhibition of human mesenchymal–epithelial transition factor (c-Met) protein autophosphorylation by ARQ 197. J Biol Chem. 2011;286(23):20666–20676. | ||
Santoro A, Simonelli M, Rodriguez-Lope C, et al. A Phase-1b study of tivantinib (ARQ 197) in adult patients with hepatocellular carcinoma and cirrhosis. Br J Cancer. 2013;108(1):21–24. | ||
Simonelli M, Rodriguez-Lope C, Rimassa L, et al. Biomarker results from ARQ 197-114: a phase 1b trial evaluating the c-MET inhibitor ARQ 197 in cirrhotic patients (pts) with hepatocellular carcinoma (HCC). Ann Oncol. 2010;21(8s): abstract 196p. | ||
Chen C-R, Szwaya J, Rojnuckarin A, et al. Abstract #820: combination studies of tyrosine kinase inhibitors (TKIs): assessment of potential cytotoxic synergy of ARQ 197 with sorafenib or sunitinib. Cancer Res. 2009;69(9 suppl):820. | ||
Santoro A, Rimassa L, Borbath I, et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo controlled phase 2 study. Lancet Oncol. 2013;14(1):55–63. | ||
Puzanov I, Sosman J, Santoro A, et al. Phase 1 trial of tivantinib in combination with sorafenib in adult patients with advanced solid tumors. Invest New Drugs. 2015;33(1):159–168. | ||
Chai F, Abbadessa G, Savage R, et al. Phase 1 experience of tivantinib in patients with hepatocellular carcinoma (HCC) or biliary tract cancer (BTC). Ann Oncol. 2012;23(9 Suppl):ix245. | ||
Santoro A, Porta C, Rimassa L, et al. Metiv-HCC: a phase III clinical trial evaluating tivantinib (ARQ 197), a MET inhibitor, versus placebo as second-line in patients (pts) with MET-high inoperable hepatocellular carcinoma (HCC). ASCO Meet Abstr. 2013;31(15_suppl):TS4159. | ||
METIV-HCC_Interim_Analysis_Press_Release__cs.pdf. Available from: http://www.businesswire.com/news/home/20170327005077/en/. Accessed October 2, 2016. | ||
Strumberg D, Schultheis B. Regorafenib for cancer. Expert Opin Investig Drugs. 2012;21(6):879–889. | ||
Trojan J., Waidmann O. Role of regorafenib as second-line therapy and landscape of investigational treatment options in advanced hepatocellular carcinoma. J Hepatocellular Carcinoma. 2016;3:31–36. | ||
Lam KO, Lee KC, Chiu J, et al. The real-world use of regorafenib for metastatic colorectal cancer: multicentre analysis of treatment pattern and outcomes in Hong Kong. Postgraduate Med J. 2016. Epub 2016 Nov 11. | ||
Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295–302. | ||
Son MK, Ryu MH, Park JO, et al. Efficacy and safety of regorafenib in Korean patients with advanced gastrointestinal stromal tumor after failure of imatinib and sunitinib: a multicenter study based on the management access program. Cancer Res Treat. 2017;49(2):350–357. | ||
Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412–3419. | ||
Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56–66. | ||
Engelhardt JJ, Sullivan TJ, Allison JP. CTLA-4 overexpression inhibits T cell responses through a CD28-B7-dependent mechanism. J Immunol. 2006;177(2):1052–1061. | ||
Ishida Y, Agata Y, Shibahara, et al. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11(11):3887–3895. | ||
Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–1034. | ||
Keir ME, Liang SC, Guleria I, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203(4):883–895. | ||
Wahl C, Bochtler P, Chen L, Schirmbeck R, Reimann J. B7-H1 on hepatocytes facilitates priming of specific CD8 T cells but limits the specific recall of primed responses. Gastroenterology. 2008;135(3): 980–988. | ||
Kassel R, Cruise MW, Iezzoni JC, Taylor NA, Pruett TL, Hahn YS. Chronically inflamed livers up-regulate expression of inhibitory B7 family members. Hepatology. 2009;50(5):1625–1637. | ||
Carambia A, Frenzel C, Bruns OT, et al. Inhibition of inflammatory CD4 T cell activity by murine liver sinusoidal endothelial cells. J. Hepatol. 2013;58(1):112–118. | ||
Diehl L, Schurich A, Grochtmann R, Hegenbarth S, Chen L, Knolle PA. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ T cell tolerance. Hepatology. 2008;47(1): 296–305. | ||
Yu MC, Chen CH, Liang X, et al. Inhibition of T-cell responses by hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice. Hepatology. 2004;40(6):1312–1321. | ||
Shi F, Shi M, Zeng Z, et al. PD-1 and PD-L1 upregulation promotes CD8(+) T-cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int. J. Cancer. 2011;128(4): 887–896. | ||
Gao Q, Wang XY, Qiu SJ, et al. Overexpression of PD-L1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin Cancer Res. 2009;15(3):971–979. | ||
Hato T, Goyal L, Greten TF, Duda DG, Zhu AX. Immune checkpoint blockade in hepatocellular carcinoma: current progress and future directions. Hepatology. 2014;60(5):1776–1782. | ||
Han Y, Chen Z, Yang Y, et al. Human CD14+ CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine- 2,3-dioxygenase production in hepatocellular carcinoma. Hepatology. 2014;59(2):567–579. | ||
Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81–88. | ||
Segal NH, Hamid O, Hwu W, et al. A Phase I multi-arm dose-expansion study of the anti-programmed cell death-ligand-1 (PD-L1) antibody MEDI4736: preliminary data. Ann Oncol. 2014;25(Suppl. 4):iv365. | ||
El-Khoueiry AB, Melero I, Crocenzi TS, et al. Phase I/II safety and antitumor activity of nivolumab in patients with advanced hepatocellular carcinoma (HCC): CA209-040. Presented at: 51st Annual Meeting of American Society Of Clinical Oncology; May 29–June 2; 2015; Chicago, IL, USA. | ||
Michot JM, Bigenwald C, Champiat S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139–148. | ||
Zhao W, Gu YH, Song R, Qu BQ, Xu Q. Sorafenib inhibits activation of human peripheral blood T cells by targeting LCK phosphorylation. Leukemia. 2008;22(6):1226–1233. | ||
Filmus J. The contribution of in vivo manipulation of gene expression to the understanding of the function of glypicans. Glycoconj J. 2002;19:319–323. | ||
Nakatsura T, Yoshitake Y, Senju S, et al. Glypican-3, overexpressed specifically in human hepatocellular carcinoma, is a novel tumor marker. Biochem Biophys Res Commun. 2003;306:16–25. | ||
Capurro M, Wanless IR, Sherman M, et al. Glypican-3: a novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology. 2003;125:89–97. | ||
Sawada Y, Yoshikawa T, Nobuoka D, et al. Phase I trial of a glypican-3-derived peptide vaccine for advanced hepatocellular carcinoma: immunologic evidence and potential for improving overall survival. Clin Cancer Res. 2012;18:3686–3696. | ||
Sawada Y, Yoshikawa T, Ofuji K, et al. Phase II study of the GPC3-derived peptide vaccine as an adjuvant therapy for hepatocellular carcinoma patients. Oncoimmunology. 2016; 5(5):e1129483. | ||
Bushati N, Cohen SM. MicroRNA functions. Annu Rev Cell Dev Biol. 2007;23:175–205. | ||
Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116: 281–297. | ||
Carleton M, Cleary MA, Linsley PS. MicroRNAs and cell cycle regulation. Cell Cycle. 2007;6:2127–2132. | ||
Boehm M, Slack FJ. MicroRNA control of lifespan and metabolism. Cell Cycle. 2006;5:837–840. | ||
Harfe BD. MicroRNAs in vertebrate development. Curr Opin Genet Dev. 2005;15:410–415. | ||
Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:15524–15529. | ||
He L, Thomson JM, Hemann MT, et al. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. | ||
Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. | ||
Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21:1025–1030. | ||
Negrini M, Gramantieri L, Sabbioni S, Croce CM. microRNA involvement in hepatocellular carcinoma. Anticancer Agents Med Chem. 2011;11(6):500–552. | ||
Yang N, Ekanem NR, Sakyi CA, Ray SD. Hepatocellular carcinoma and microRNA: new perspectives on therapeutics and diagnostics. Advanced Drug Delivery Reviews. 2015;81:62–74. | ||
Hou W, Tian Q, Zheng J, Bonkovsky HL. MicroRNA-196 represses Bach1 protein and hepatitis C virus gene expression in human hepatoma cells expressing hepatitis C viral proteins. Hepatology. 2010;51:1494–1504. | ||
Yin H, Hu M, Zhang R, Shen Z, Flatow L, You M. MicroRNA-217 promotes ethanol-induced fat accumulation in hepatocytes by down-regulating SIRT1. J Biol Chem. 2012;287:9817–9826. | ||
Ladeiro Y, Couchy G, Balabaud C, et al. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology. 2008;47:1955–1963. | ||
Bai S, Nasser MW, Wang B, et al. MicroRNA-122 inhibits tumorigenic properties of hepatocellular carcinoma cells and sensitizes these cells to sorafenib. J Biol Chem. 2009;284(46):32015–32027. | ||
Fornari F, Gramantieri L, Giovannini C, et al. MiR-122/cyclin G1 interaction modulates p53 activity and affects doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res. 2009;69(14):5761–5767. | ||
Krützfeldt J, Kuwajima S, Braich R, et al. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007;35:2885–2892. | ||
Bader AG, Brown D, Winkler M. The promise of microRNA replacement therapy. Cancer Res. 2010;70:7027–7030. | ||
Kota, J, Chivukula RR, O’Donnell KA, et al. Therapeutic microRNA delivery suppresses tumourigenesis in a murine liver cancer model. Cell. 2009;137:1005–1017. | ||
Ling H, Fabbri M, Calin GA. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov. 2013;12:847–865. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.